

Abstract
Developmental programming is defined as the process by which gene–environment interaction in the developing organism leads to permanent changes in phenotype and function. Numerous reports of maternal nutrient restriction during pregnancy demonstrate altered renal development. Typically this alteration manifests as a reduction in the total number of glomeruli in the mature kidney of the offspring, and suggests that predisposition to develop chronic renal disease may include an in utero origin. In a previous study, we defined the transcriptome in the kidney from fetuses of control (CON, fed ad libitum) and nutrient-restricted (NR, fed 70% of CON starting at 0.16 gestation (G)) pregnancies at half-way through gestation (0.5G), and established transcriptome and morphological changes in NR kidneys compared to CON. One goal of the present study was to use transcriptome data from fetal kidneys of CON and NR mothers at 0.5G with histological data to identify the molecular mechanisms that may regulate renal development. A second goal was to identify mechanisms by which NR elicits its affect on fetal baboon kidney. We have used an end-of-pathway gene expression analysis to prioritize and identify key pathways regulating the 0.5G kidney phenotype in response NR. From these data we have determined that the mammalian target of rapamycin (mTOR) signalling pathway is central to this phenotype.
Mammalian fetal development proceeds according to a genetic programme encoded in the transcriptome. The programme is determined at the moment of fertilization and is immediately susceptible to gene–environment interaction (epigenetic modification) and alteration of subsequent phenotype. For example, phenotype at birth is altered by the epigenetic influence of variation in culture media prior to implantation (Drake & Walker, 2004). Several mechanisms are likely to play a role in mediating change in phenotype, including DNA methylation (for review see Miozzo & Simoni, 2002), DNA acetylation (Fu et al. 2004), degree of transcription factor abundance and transcription factor post-translational modification. Significant effort has been invested in determining the expression of targeted genes during normal fetal development. Increasing effort has focused on situations in which epigenetic events may modify the expression of key genes and thereby influence eventual phenotype (Lillycrop et al. 2005). The recent explosion of interest in developmental programming, defined as the process by which gene–environment interaction in the developing organism leads to permanent changes in phenotype and function, highlights the need for the study of changes in the transcriptome in response to challenges during development (Armitage et al. 2004).
Application of methods such as Northern analysis, ribonuclease protection assay and quantitative real-time polymerase chain reaction (QRT-PCR) has provided significant information on the level of expression of individual genes. In order to gain a deeper understanding of overall phenotype, a more comprehensive picture of gene expression is required. Powerful whole-genome expression profiling techniques are now available to characterize expression of multiple components of the transcriptome. Using Affymetrix genechips (Affymetrix, Santa Clara, CA, USA), or similar techniques such as Illumina beadchips, it is possible to quantify expression of large numbers of genes in small amounts of tissue. In addition to measuring the degree of expression of a lengthy list of genes relative to the genome, information obtained utilizing these approaches allows for the analysis of transcriptome function. One powerful method of obtaining a more complete picture from multiple gene expression profiling of the transcriptome is to place profile data into biological context using information technology methods, such as the Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway analysis (Kanehisa et al. 2004), that relate the expression profile to biochemical pathways such as cell signalling, protein synthesis and apoptosis.
In a previous study (Cox et al. 2006), we defined the transcriptome in the kidney from fetuses of control (CON, fed ad libitum) and nutrient-restricted (NR, fed 70% of CON starting at 0.16 gestation (G)) mothers at half-way through gestation (0.5G), using Affymetrix genechips. By examining the gene expression profiles for each treatment group in the context of functional classification, we identified group-specific gene ontologies that were active above baseline. As we also demonstrated histological changes in kidney phenotype of fetuses exposed to decreased nutrient availability, we identified genes within the NR animals that were significantly differentially expressed, either up-regulated or down-regulated, when compared to CON animals (Cox et al. 2006).
In our previous study, we described a decrease in proximal tubule density and decreased vascular endothelial growth factor (VEGF) gene expression, detected by both the Affymetrix gene chip and QRT-PCR, at 0.5G following NR (Cox et al. 2006). These changes, together with changes in transcriptome expression such as decreased transcription and decreased translation, are used as an index of NR-induced change in renal phenotype in the present study. We hypothesize that an alteration in overall activity of pathways that have a significant impact on organ phenotype should be accompanied by altered gene expression at the end of those pathways (downstream) relevant to the phenotype. If so, the mechanism(s) by which epigenetic events affect a biological process can be examined by first investigating downstream steps in key pathways and then, step-by-step, moving upstream. We further hypothesize that if no change can be demonstrated at the end of a pathway, it is unlikely that the pathway plays a central role in the molecular mechanisms responsible for eliciting the effects of NR on kidney development because events downstream of the pathway are unlikely to be activated. Using this end-of-pathway gene expression approach to biological pathway analysis of our differentially expressed gene data, we have prioritized pathways that are relevant to the effects of NR on the fetal primate kidney, and identified critical molecular mechanisms that require further investigation. While most approaches used during development to date have targeted specific genes such as those controlling receptors (Freeman et al. 2004) or early post-receptor binding steps (Ozanne et al. 1997), our analytic approach has revealed the importance of newly annotated entire pathways such as mammalian target of rapamycin (mTOR) and hedgehog signalling.
In summary, we have analysed and compared gene expression in kidneys of fetuses of CON and NR mothers using pathway analysis of whole-genome expression profiles. As a result of these analyses and those from our previous study (Cox et al. 2006), we have analysed all pathways that showed significant alterations in activity in the context of decreased nutrient availability. The data obtained in this way have led us beyond ontological gene grouping and have facilitated the identification of key biochemical pathways influenced by decreased nutrient availability. Because mTOR signalling is upstream of overall translation into protein, and plays a role in nutrient sensing (Marshall, 2006), mTOR signalling is a promising candidate for examining the mechanisms by which maternal NR affects renal development.
Methods
Animal care and maintenance
All procedures were approved by the South west Foundation for Biomedical Research (SFBR) Institutional Animal Care and Use Committee and conducted in Association with Assessment and Accreditation of Laboratory Animal Care approved facilities. A total of 12 female baboons, six baboons from each of two independently housed groups each consisting of 16 females housed with a single male, were studied. Each group was housed in a cage with a floor area of 37 m2 and height of 3.5 m. Details of housing structure and environmental enrichment provided have been published in detail (Schlabritz-Loutsevitch et al. 2004). Maternal morphometric measurements were made prior to pregnancy to ensure homogeneity of weight and general measurements in the females in the two groups.
System for controlling and recording individual feeding
Once a day prior to feeding all baboons were run into individual feeding cages. Baboons passed along the chute, over a weighing scale and into one of the individual feeding cages. Once in the individual cages they were fed either between 07.00 h and 09.00 h or 11.00 h and 13.00 h as previously described (Schlabritz-Loutsevitch et al. 2004). Food was provided as Purina Monkey Diet 5038 standard biscuits. Water was continuously available in the feeding cage through individual lixits and at several locations in the group housing.
Formation of stable grouping for the NR study
Each group of 16 females was initially housed with a vasectomized male to establish a stable social group (Schlabritz-Loutsevitch et al. 2004). All female baboons were observed twice a day for well-being and three times a week for turgescence (sex skin swelling) and signs of vaginal bleeding to enable timing of pregnancy (Hendrickx, 2001). At the end of a 30 day period of adaptation to the feeding system, a fertile male was introduced into each breeding cage. Pregnancy was dated initially by following the changes in the swelling of the sex skin and confirmed at 30 days of gestation by ultrasonography.
Diet and food consumption
The Purina Monkey Diet 5038 is described by the vendor as ‘a complete life-cycle diet for all Old World Primates.’ The biscuit contains stabilized vitamin C as well as all other required vitamins. Its basic composition is crude protein not less than 15%, crude fat not less than 5%, crude fibre not more than 6%, ash not more than 5% and added minerals not more than 3% (Schlabritz-Loutsevitch et al. 2004). At the start of the feeding period, each baboon was given 60 biscuits in the feeding tray. At the end of the 2 h feeding period after the baboons had returned to the group cage, the biscuits remaining in the tray, on the floor of the cage and in the pan were counted. For all animals, food consumption, weight and health status were recorded daily. The weight of each baboon was obtained as she crossed the electronic scale system (GSE 665; GSE Scale Systems, MI, USA). A commercial software application designed to capture weight data was modified to permit the recording of 50 individual measurements over 3 s. If the standard deviation of the weight measurement was greater than 0.01 of the mean weight, the weight was automatically discarded and the weighing procedure begun again. All baboons were fed ad libitum until 30 days of gestation (0.16G) when six CON baboons continued to feed ad libitum and six NR baboons were fed 70% of feed consumed by CON on a weight-adjusted basis.
Caesarean sections and tissue collection
Caesarean sections were performed at 90 days of gestation (0.5G) under isoflurane anaesthesia (2%, 2 l min−1) to obtain the fetus and placenta. Each experimental group was composed of three male and three female fetuses. Techniques used and postoperative maintenance have been previously described in detail (Schlabritz-Loutsevitch et al. 2004). Analgesia was provided with buprenorphine hydrochloride 0.015 mg kg−1 day−1 during three postoperative days (Buprenex Injectable, Reckitt Benckiser Healthcare (UK) Ltd, Hull, UK). Fetal morphometric measurements were obtained at the time of Caesarean section (Cox et al. 2006). In addition, kidneys were collected, divided into aliquots, immediately snap-frozen in liquid nitrogen and stored at −80°C until used for RNA extractions.
RNA isolation from tissue
RNA was isolated from tissue using Trizol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. For kidney RNA samples, approximately 100 mg section of frozen kidney was cut from one pole of a longitudinally sliced kidney half. The tissue was homogenized in 1 ml Trizol Reagent using a Power Gen Homogenizer (Omni International, Wilmington, DE, USA). Genomic DNA in the sample was sheared by passing the homogenate three times through a 22 gauge needle attached to a 1 ml syringe. The homogenized samples were incubated for 5 min at 25°C. Chloroform (200 μl) was added to each sample, the samples were shaken vigorously by hand for 15 s and incubated at 25°C for 3 min. Samples were then centrifuged at 12 000 g for 15 min at 4°C. The aqueous phase containing RNA was transferred to a fresh tube and the RNA precipitated by addition of 0.5 ml isopropyl alcohol. Samples were incubated for 10 min at 25°C and then centrifuged at 12 000 g for 10 min at 4°C. The RNA precipitate was washed with 1 ml 75% ethanol and centrifuged at 7500 g for 5 min at 4°C. The RNA was resuspended in 100 μl diethylpyrocarbonate-treated water and stored at −80°C.
Preparation of cRNA probe and genechip interrogation
RNA was extracted from six CON and six NR 0.5G baboon kidney samples and analysed by genechip (Affymetrix U133A 2.0). Total RNA samples were shipped on dry ice to Genome Explorations Inc. (Memphis, TN, USA) for RNA quality check, cRNA synthesis, and determination of gene expression profiles for each RNA sample using the Affymetrix Human Genome U133 Plus 2.0 Array. RNA quality was checked using an Agilent Bioanalyser 2100 ‘Laboratory on a Chip’ system. RNA concentrations were confirmed by quantification using a dual-beam spectrophotometer on approximately 200 ng of each RNA sample. Complementary RNA was synthesized and biotin-labelled at Genome Explorations Inc. using the MessageAmp aRNA Kit (Ambion, Austin, TX, USA) according to the manufacturer's instructions. Total RNA was used for first and second strand cDNA synthesis followed by an in vitro transcription step to synthesize biotin-labelled cRNA. The cRNA was quality checked and then hybridized to the Human Genome U133A 2.0 Array. Gene expression was detected using GCOS software (Affymetrix).
Statistical analysis
Data from male and female fetuses were combined within dietary treatment groups for all analyses. Genechip data were analysed with GeneSifter (GeneSifter.Net, VizX Laboratories, Seattle, WA, USA) using a ‘custom baboon array’ as described previously (Cox et al. 2006). Array data were all-median normalized and log2 transformed. Statistical analyses of array data for individual RNA samples were performed by unpaired t test (GeneSifter.Net, VizX Laboratories).
Pathway analysis
Pathway analyses were performed using a ‘custom baboon array’ as described previously (Cox et al. 2006). Z-score calculations defining significant gene categories and pathways were based on the total number of genes on the array that gave a detectable quality signal using baboon RNA-generated probes (16 186 of the 22 227 genes on the Affymetrix U133A 2.0 genechip comprise the ‘custom baboon array’). Thus, z-scores were calculated based on these 16 186 genes.
Array data for significantly differently expressed genes were overlaid onto KEGG pathways (http://www.genome.jp/kegg/) (Kanehisa et al. 2004) using GeneSifter (GeneSifter.Net). Pathways were considered significantly different among normal tissue gene expression profiles or significantly different between normal (CON) and NR gene expression profiles if the z-score for that pathway was less than −2.0 or greater than 2.0. Z-scores were calculated in GeneSifter using the following formula: z-score =[r−n(R/N)]/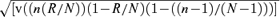 : where R = total number of genes meeting selection criteria, N = total number of genes measured, r = number of genes meeting selection criteria with the specified gene ontology (GO) term, and n = total number of genes measured with the specific GO term (Doniger et al. 2003). Pathway z-scores differ from those previously published (Cox et al. 2006) because of continuous updating of KEGG pathway content.
: where R = total number of genes meeting selection criteria, N = total number of genes measured, r = number of genes meeting selection criteria with the specified gene ontology (GO) term, and n = total number of genes measured with the specific GO term (Doniger et al. 2003). Pathway z-scores differ from those previously published (Cox et al. 2006) because of continuous updating of KEGG pathway content.
Immunohistochemistry
Sections (5 μm) were deparaffinized with three changes of xylene for 10 min each. Sections were hydrated in two changes of 100% ethanol for 2 min each, 95% ethanol for 1 min each then rinsed in water. Antigen retrieval was performed for 5 min using sodium citrate buffer (10 mm sodium citrate, pH 6.0) in a pressure cooker, allowed to cool for 20 min then rinsed three times in automation buffer (100 mm Tris-HCl, 100 mm Tris, 3% polyethylene(23) lauryl ether (BRIJ 35); pH 7.5). Non-specific blocking was performed for 20 min using 10% ovalbumin in autobuffer (50 mm Tris, 0.3% BRIJ 35, pH 7.5). To minimize artifact from endogenous biotin, streptavidin–biotin blocking was performed using a kit (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer's directions. Sections were incubated in primary antibody (VEGF monoclonal anti-mouse; Abcam, catalogue no. ab16883) at appropriate dilutions (1% bovine serum albumin in Tris-buffered saline (TBS)) overnight at 4°C. Sections were rinsed in three changes of automation buffer and allowed to stand for 5 min in the third change. Biotinylated secondary antibodies were placed on the slides for 30 min (swine anti-rabbit DAKO catalogue no. E0353; DakoCytomation, Glostrup, Denmark) and rinsed again in automation buffer. Proteins of interests were visualized by incubating a tertiary strepavidin horseradish peroxidase (DAKO catalogue no. PO397; DakoCytomation)–antibody for 30 min followed by rinsing in three changes of automation buffer (and allowed to for stand 5 min during the third change). Diaminobenzidine was applied to the sections for 10 min, followed by three quick changes of phosphate buffered saline (PBS), incubation in 0.2% osmium tetroxide for 1 min, rinsing in PBS and counterstaining with methyl green (0.5% in distilled water) for 5 min at room temperature (25–28°C). Samples were then dehydrated in a series of ethanol washes and xylene washes and a coverslip applied. Negative control slides were developed for each antibody used and revealed no staining.
End-of-pathway gene expression analysis
For each significantly different KEGG pathway, the gene(s) at the end of the KEGG pathway were analysed for the presence of a signal on the baboon array and for differential expression between CON and NR kidney RNA samples. Gene expression profiles for end-of-pathway genes were assessed in the context of the 30% NR 0.5G kidney IHC of VEGF and the morphometric data (Cox et al. 2006). Data are provided as means ±s.e.m.
Access to gene array data
All array data from CON and NR RNA samples are available on South west National Primate Research Center Primate Genomics Database website (http://www.sfbr.org/pages/snprc_projects.php?P=63).
Results
KEGG pathway analysis of 0.5G CON and NR kidney gene expression
Differentially expressed genes were overlaid onto KEGG pathways to identify up-regulated and down-regulated biological pathways in response to 30% maternal NR. Table 1 lists the nine pathways that were up-regulated and the 15 pathways that were down-regulated. Included in Table 1 are the number of differentially expressed genes, the number of up-regulated genes, the number of down-regulated genes, the total number of genes in that pathway that give a signal with baboon RNA on the genechip, and the z-score for the listed pathway.
Table 1.
KEGG pathway analysis of gene expression in nutrient-restricted (NR) versus control (CON) kidney half-way through gestation
| Diff genes | Up | Down | Array | Z-score | |
|---|---|---|---|---|---|
| Up pathways | |||||
| C21-Steroid hormone metabolism | 2 | 2 | 0 | 8 | 2.6 |
| Complement and coagulation cascades | 7 | 5 | 2 | 59 | 2.6 |
| Dorso-ventral axis formation | 6 | 3 | 3 | 28 | 3.7 |
| Gap junction | 15 | 6 | 9 | 87 | 2.3 |
| Haematopoietic cell lineage | 5 | 4 | 1 | 52 | 2.1 |
| Maturity onset diabetes of the young | 3 | 2 | 1 | 17 | 2.2 |
| mTOR signalling pathway | 5 | 3 | 2 | 37 | 2.1 |
| O-Glycan biosynthesis | 4 | 3 | 1 | 15 | 3.3 |
| SNARE interactions in vesicular transport | 6 | 4 | 2 | 26 | 2.2 |
| Down pathways | |||||
| Adherens junction | 15 | 3 | 12 | 65 | 2.0 |
| Apoptosis | 3 | 0 | 3 | 83 | −1.7 |
| Arginine and proline metabolism | 7 | 0 | 7 | 43 | 2.3 |
| ATP synthesis | 7 | 1 | 6 | 34 | 2.3 |
| Axon guidance | 19 | 3 | 16 | 101 | 3.3 |
| Cytokine–cytokine receptor interaction | 11 | 6 | 5 | 154 | −2.0 |
| Focal adhesion | 34 | 9 | 25 | 182 | 2.7 |
| Glycosylphosphatidylinositol (GPI)-anchor biosynthesis | 3 | 0 | 3 | 14 | 2.0 |
| Hedgehog signalling pathway | 7 | 2 | 5 | 30 | 2.0 |
| Huntington's disease | 6 | 1 | 5 | 24 | 2.5 |
| Long-term potentiation | 15 | 3 | 12 | 63 | 3.6 |
| Neuroactive ligand–receptor interaction | 7 | 4 | 3 | 152 | −2.6 |
| Oxidative phosphorylation | 16 | 1 | 15 | 99 | 3.0 |
| Regulation of actin cytoskeleton | 28 | 8 | 20 | 163 | 2.5 |
| Wnt signalling pathway | 21 | 3 | 18 | 106 | 2.3 |
‘Up pathways’ denote KEGG pathways that are signficantly up-regulated in NR compared with CON samples; ‘Down pathways’ denote KEGG pathways that are signficantly down-regulated in NR compared with CON samples; ‘Diff genes’ denotes the total number of differentially expressed genes between CON and NR kidney RNA on the array. ‘Up’ denotes the number of genes up-regulated in NR compared with CON; ‘Down’ denotes the number of genes down-regulated in NR compared with control; ‘Array’ denotes the total number of genes in this pathway that are included on the array and give a signal with baboon RNA; and ‘Z-score’ denotes the z-score for the list pathway.
Immunohistochemistry (IHC) for VEGF expression
Kidney sections from 0.5G CON and NR animals were immunostained for VEGF. The antibody used for VEGF recognizes VEGF121, VEGF165, VEGF189 and VEGF206. VEGF staining was localized within the extracellular space in association with the proximal and distal tubules (Fig. 1). Light staining within glomeruli was also noted. Staining was more diffuse in the fetuses of the NR mothers.
Figure 1.

Staining for vascular endothelial growth factor (VEGF) in kidneys from a fetus of control fed (CON; A) and a nutrient-restricted (NR; B) pregnant baboon half-way through gestation VEGF staining (100 × magnification) is seen closely associated with the proximal and distal tubules, as indicated by the white circles, with more diffuse staining in the NR animals. The white arrows indicate glomeruli with light staining.
Analysis of differential KEGG pathways based on 30% NR kidney phenotype
End-of-pathway genes were identified for each differential KEGG pathway and are shown in Table 2. For each end-of-pathway gene, expression for NR compared with CON animals and the downstream event are shown.
Table 2.
End-of-pathway gene expression analysis of nutrient-restricted (NR) versus control (CON) kidney half-way through gestation
| Pathway and end-of-pathway gene(s) | Gene ID | NR versus Con | Downstream event |
|---|---|---|---|
| Adherens junction (down, 2.0) | |||
| Cadherin 1, type 1, E-cadherin | CDH1 | NC | Actin polymerization |
| Wiskott-Aldrich syndrome-like | WASL (NWASP) | NC | Actin polymerization |
| Wiskott-Aldrich syndrome | WAS (WASP) | NC | Actin polymerization |
| IQ motif containing GTPase activating protein 1 | IQGAP1 | D | Actin polymerization |
| WAS protein family, member 1 | WASF1 | D | Actin polymerization |
| BAI1-associated protein 2 | BAIAP2 (IRSp53) | NC | Actin polymerization |
| Apoptosis (down, −1.7) | |||
| Caspase 3 | CASP3 | ND | Cleavage of caspase substrate |
| BCL2-associated X protein | BAX | NC | Cleavage of caspase substrate |
| Caspase 6 | CASP6 | ND | Cleavage of caspase substrate |
| DNA fragmentation factor, 45 kDa | DFFA | NC | DNA fragmentation |
| DNA fragmentation factor, 40 kDa | DFFB | NC | DNA fragmentation |
| Tumour protein p53 | TP53 | NC | Cell cycle |
| BCL2-antagonist of cell death | BAD | NC | Survival |
| B-cell CLL/lymphoma 2 | BCL2 | NC | Survival |
| BCL2-like 1 | BCL2L1 | NC | Survival |
| BaculoviralI AP repeat-containing 2 | BIRC2 (IAP) | NC | Survival |
| Arginine and proline metabolism (down, 2.3) | |||
| Arginyl-tRNA synthetase | RARS | D | Protein synthesis |
| Glutamic-oxaloacetictransaminase 1 | GOT1 | NC | Glyoxylate to pyruvate |
| Glycine amidinotransferase | GATM | NC | Glutamate metabolism |
| Aldehyde dehydrogenase 9 family, member A1 | ALDH9A1 | NC | Glutamate metabolism |
| Aldehyde dehydrogenase 2 family (mitochondrial) | ALDH2 | NC | Glutamate metabolism |
| Nitric oxide synthase 1 | NOS1 | NC | Nitric oxide |
| ATP synthesis (down, 2.3) | |||
| F type ATPase components | ATP5C1, ATP5F1, ATP5J2 | 3 D | ATP synthesis |
| F type ATPase components | ATP5G2 | 1 U | ATP synthesis |
| F type ATPase components | ATP5A1, ATP5B, ATP5D, ATP5E, ATP5G3, ATP5II, ATP5I, ATP5J, ATP5L | 9 NC | ATP synthesis |
| F type ATPase components | ATP5G1, ATP5S | 2 ND | ATP synthesis |
| V type ATPase components | ATP6V0A2, ATP6V0E, ATP6V1H | 3 D | ATP synthesis |
| V type ATPase components | ATP6V0A1, ATP6V0B, ATP6V0C, ATP6V0D1, ATP6V1B1, ATP6V1B2, ATP6V1D, ATP6V1F, ATP6V1G1 | 9 NC | ATP synthesis |
| Axon guidance (down, 3.3) | |||
| Actin binding LIM protein 1 | ABLIM1 | D | Regulation of actin cytoskeleton |
| Actin binding LIM proteinfamily, member 3 | ABLIM3 | D | Regulation of actin cytoskeleton |
| Nuclear factor of activated T cells | NFAT | NC | Transcription |
| Rho-associated, coiled-coil containing protein kinase | ROCK | NC | Transcription |
| Cell division cycle 42 | CDC42 | D | Regulation of actin cytoskeleton |
| Cofilin 1 (non-muscle) | CFL1 | D | Regulation of actin cytoskeleton |
| P21 (CDKN1A)-activated kinase 4 | PAK4 | D | Regulation of actin cytoskeleton |
| Mitogen-activated protein kinase1 | MAPK | NC | Regulation of actin cytoskeleton |
| Dihydropyrimidinase-like 2 | DPYSL2 (CRMP) | D | Microtubule reorganization |
| Dihydropyrimidinase-like 5 | DPYSL5 (CRAM) | ND | Microtubule reorganization |
| C21-steroid hormone metabolism (up 2.6) | |||
| 3-α-Hydroxysteroid dehydrogenase | AKR1C4 | NC | Trihydroxy-5B-pregnane-11-one |
| Hydroxysteroid(11-β)dehydrogenase 1 | HSD11B1 | U | Inducer of apoptosis |
| Hydroxysteroid(11-β)dehydrogenase 1 | HSD11B1 | U | Cortisone |
| Complement and coagulation cascades (up, 2.6) | |||
| Bradykinin receptor B | BDKRB1 | ND | Inflammation |
| Complement component 3a receptor 1 | C3AR1 | NC | Inflammation |
| Complement component 5a receptor 1 | C5AR1 | NC | Inflammation |
| Complement component receptor 2 | CR2 | ND | B cell receptor signalling |
| Complement component receptor 1 | CR1 | U | B cell receptor signalling |
| Coagulation factor II (thrombin) receptor | F2R | NC | Endothelial cell and smooth cell activation |
| Cytokine–cytokine receptor interaction (down, −2.0) | |||
| Chemokines: CXC family | 3 receptors | NC | Cytokine–cytokine receptor interaction |
| Chemokines: CXC family | 2 receptors | ND | Cytokine–cytokine receptor interaction |
| Chemokines: C subfamily | 1 receptor | ND | Cytokine–cytokine receptor interaction |
| Chemokines: CX3C family | 1 receptor | NC | Cytokine–cytokine receptor interaction |
| Chemokines: CC family | 6 receptors | NC | Cytokine–cytokine receptor interaction |
| Chemokines: CC family | 3 receptors | ND | Cytokine–cytokine receptor interaction |
| Haematopoietins | 20 receptors/subunits | NC | Cytokine–cytokine receptor interaction |
| Haematopoietins | 2 receptors/subunits | U | Cytokine–cytokine receptor interaction |
| Haematopoietins | 3 receptors/subunits | ND | Cytokine–cytokine receptor interaction |
| PDGF family | 19 receptors/subunits | NC | Cytokine–cytokine receptor interaction |
| PDGF family | 3 receptors/subunits | U | Cytokine–cytokine receptor interaction |
| PDGF family | 2 receptors/subunits | U | Cytokine–cytokine receptor interaction |
| Interferon family | 2 receptor subunits | NC | Cytokine–cytokine receptor interaction |
| IL-10 family | 2 receptor subunits | NC | Cytokine–cytokine receptor interaction |
| IL-10 family | 2 receptor subunits | ND | Cytokine–cytokine receptor interaction |
| TNF family | 17 receptors | NC | Cytokine–cytokine receptor interaction |
| TNF family | 12 receptors | ND | Cytokine–cytokine receptor interaction |
| TGFB family | 6 receptor subunits | NC | Cytokine–cytokine receptor interaction |
| TGFB family | 2 receptor subunits | U | Cytokine–cytokine receptor interaction |
| TGFB family | 2 receptor subunits | ND | Cytokine–cytokine receptor interaction |
| Dorso-ventral axis formation (up, 3.7) | |||
| v-ets erythroblastosis virus E26 oncogene homologue 1 | ETS1 (Pnt) | NC | Dorso-ventral axis formation |
| Ets variant gene 6 | ETV6 (Yan) | NC | Dorso-ventral axis formation |
| Focal adhesion (down, 2.7) | |||
| Actin, β | ACTB | NC | Actin polymerization |
| Actin, α | ACTC | NC | Actin polymerization |
| Actin, γ | ACTG | NC | Actin polymerization |
| Cyclin D1 | CCND1 | D | Cell cycle |
| Cyclin D2 | CCND2 | D | Cell cycle |
| B-cell CLL/lymphoma2 | BCL2 | NC | Cell survival |
| Gap junction (up, 2.3) | |||
| Gap junction protein, α1 | GJA1 | NC | Gap junction channels |
| Connexin-36 | CX36 | NC | Gap junction channels |
| Glycosylphosphatidylinositol anchor biosynthesis (down, 2.0) | |||
| GPI deacylase | PGAP1 | NC | GPI anchor biosynthesis |
| Hedgehog signalling pathway (down, 2.0) | |||
| Wingless-type MMTV integration site family, member 5A | WNT5A | D | wnt signalling |
| Bone morphogenetic protein | BMP | NC | TGF signalling |
| Haematopoietic cell lineage (up, 2.1) | |||
| No genes encoding mature haematopoietic cells were differentially expressed | — | — | Mature haematopoietic cells |
| Huntington's disease (down, 2.5) | |||
| Transglutaminase 2 | TGM2 (tTGase) | U | Apoptosis |
| Caspase 3 | CASP3 | ND | Apoptosis |
| Long-term potentiation (down, 3.6) | |||
| Activating transcription factor 4 | ATF (CREB) | NC | Long-term potentiation |
| Maturity-onset diabetes of the young (up, 2.2) | |||
| Pyruvate kinase | PKLR | NC | Insulin signalling and diabetes mellitus |
| Solute carrier family 2 | SLC2A2 | NC | Insulin signalling and diabetes mellitus |
| Insulin | INS | ND | Insulin signalling and diabetes mellitus |
| Glucokinase | GCK | ND | Insulin signalling and diabetes mellitus |
| Islet amyloid polypeptide | IAPP | NC | Insulin signalling and diabetes mellitus |
| mTOR signalling pathway (up, 2.1) | |||
| Vascular endothelial growth factor | VEGF | D | VEGF signalling |
| Eukaryotic translation initiation factor 4B | EIF4B | NC | Translation |
| Ribosomal protein S6 | RPS6 | NC | Translation |
| Eukaryotic translation initiation factor 4E | EIF4E | D | Translation |
| unc-51-like kinase 1 | ULK1, ATG1 | U | Autophagy |
| V-raf murine sarcoma viral oncogene homolog B1 | BRAF | U | Differentiation |
| O-Glycan biosynthesis (up, 3.3) | |||
| α-2,6-Sialyl transferase | ST6GALNAC1 | ND | Protein glycosylation |
| Glucosaminyl (N-acetyl) transferase | GCNT1 | ND | Protein glycosylation |
| Oxidative phosphorylation (down, 3.0) | |||
| Cytochrome c oxidase subunit Vb | COX5B | D | Complex I: NADH to NAD+ H+ |
| NADH dehydrogenase 1 α subcomplex,5,13 kDa | NDUFA5 | D | Complex I: NADH to NAD+ H+ |
| NADH dehydrogenase 1 α subcomplex,7,14.5 kDa | NDUFA7 | D | Complex I: NADH to NAD+ H+ |
| NADH dehydrogenase 1 β subcomplex,7,18 kDa | NDUFB7 | D | Complex I: NADH to NAD+ H+ |
| NADH dehydrogenase Fe-S protein 6,13 kDa | NDUFS6 | D | Complex I: NADH to NAD+ H+ |
| NADH dehydrogenase Fe-S protein 8,23 kDa | NDUFS8 | D | Complex I: NADH to NAD+ H+ |
| Succinate dehydrogenase complex, subunit A, flavoprotein | SDHA | D | Complex II: succinate to fumarate |
| Ubiquinol-cytochrome c reductase core protein I | UQCR | D | Complex III: cytochrome bc1 complex |
| Cytochrome c oxidase subunit IV isoform1 | COX4I1 | D | Complex IV: ½O2 to H2O |
| ATP synthase, H+ transporting, mitochondrial F0 complex, subunitB1 | ATP5F1 | D | Complex V |
| ATP synthase, H+ transporting, mitochondrial F0 complex, subunitC2 | ATP5G2 | U | Complex V |
| ATP synthase, H+ transporting, mitochondrial F0 complex, subunitF2 | ATP5J2 | D | Complex V |
| ATP synthase, H+ transporting, mitochondrial F1 complex, γ 1 | ATP5C1 | D | Complex V |
| ATPase, H+/K + exchanging α polypeptide | ATP4A | NC | Complex V |
| ATPase, H+–K+ exchanging β polypeptide | ATP4B | NC | Complex V |
| Regulation of actin cytoskeleton (down, 2.5) | |||
| IQ motif containing GTPase activating protein 1 | IQGAP2 | D | Adherens junction |
| Vinculin | VCL | D | Focal adhesion |
| Actinin, α1 | ACTN1 | D | Focal adhesion |
| Gelsolin (amyloidosis, Finnish type) | GSN | D | Focal adhesion |
| Paxillin | PXN | NC | Focal adhesion |
| Moesin | MSN (ERM) | NC | Focal adhesion |
| β Actin | ACTB | NC | Actin polymerization |
| Myosin, heavy polypeptide | MYH | NC | Actomysin assembly |
| Diaphanous homolog 1 | DIAPH1 | D | Actin polymerization |
| Diaphanous homolog 2 | DIAPH2 | U | Actin polymerization |
| SNARE interactions in vesicular transport (up, 2.2) | |||
| Syntaxin4 | stx4 | U | Transport to apical membrane |
| Synaptosomal-associated protein | SNAP23 | NC | Transport to apical membrane |
| Syntaxin7 | stx7 | NC | Transport to late endosome |
| Syntaxin8 | stx8 | NC | Transport to late endosome |
| Vesicle transport through interaction with t-SNAREs homolog 1 | vti1 | ND | Transport to late endosome |
| Wnt signalling pathway (down, 2.3) | |||
| v-myc myelocytomatosis viral oncogene homolog | MyC | NC | Cell cycle |
| v-jun sarcomavirus 17 oncogene homolog | JUN | NC | Cell cycle |
| FOS-like antigen1 | FOSL1 (fra-1) | ND | Cell cycle |
| Cyclin D1 | CCND1 | D | Cell cycle |
| Cyclin D2 | CCND2 | D | Cell cycle |
| Peroxisome proliferative activated receptor, δ | PPARD | NC | Cell cycle |
| Matrix metallopeptidase 7 | MMP7 (uterine) | NC | Cell cycle |
The ‘Pathway and end-of-pathway gene(s)’ column lists each significantly different pathway, the direction of change for that pathway, and the z-score for that pathway. Under the pathway heading, all genes at the end of the pathway are listed. ‘Gene ID’ denotes the gene identification for the gene and, if the KEGG pathway is annotated with a different name, the KEGG gene name is shown in parentheses. The third column indicates if the end-of-pathway gene is not detected on the array (ND), if gene expression is detected but does not change in NR compared with CON (NC), if gene expression is up-regulated in NR compared with CON, (U) or if gene expression is down-regulated in NR compared with CON (D). ‘Downstream event’ denotes annotated activity downstream of the listed end-of-pathway gene. For pathways with numerous subunits or receptors, the end-of-pathway genes are grouped according to the number of ND, NC, U and D regulated genes.
Sequential analysis of the mTOR signalling pathway
The mTOR pathway was analysed step by step beginning with the end-of-pathway genes. The annotated mTOR pathway with the order of analysis 1–6 is shown in Fig. 2.
Figure 2.

Mammalian target of rapamycin (mTOR) signalling pathway for gene expression in the kidney of fetuses of nutrient-restricted (NR) versus control (CON) mothers half-way through gestation Green boxes denote genes down-regulated in NR samples; red boxes denote genes up-regulated in NR samples; grey/lack boxes denote genes that were similarly expressed between CON and NR samples; black/white boxes denote genes not detected on the human genechip with baboon RNA generated probe. Pathway steps requiring protein phosphorylation are denoted by ‘+p’ in a yellow circle. Pathway activation steps are denoted by arrows and pathway inhibition steps are indicated by a vertical line at the end of the pathway line. The order of analysis of pathway components is denoted by circled numbers 1, 2, 3, 4, 5, 6a, 6b, 6c and 6d (the mTOR pathway was modified from http://www.KEGG.org).
(1) The end-of-pathway genes for mTOR signalling include VEGF, eukaryotic translation initiation factor 4B (eIF4B), ribosomal protein S6 (S6) and eukaryotic translation initiation factor 4E (eIF4E). Consistent with VEGF protein (IHC) data, VEGF gene expression was decreased in NR kidney RNA compared with CON; VEGF expression is upstream of VEGF signalling cascades. In addition, eIF4E gene expression was decreased in NR kidney RNA compared with CON; eIF4E expression is upstream of translational signalling cascades that influence cell growth. Neither eIF4B nor ribosomal subunit S6 were different between CON and NR kidney RNA samples. (2) Hypoxia inducible factor 1 (HIF1a) gene expression was not different between CON and NR kidney RNA samples; genes encoding the heterodimer S6 kinase 1/S6 kinase 2 were decreased in NR kidney RNA compared with CON; eukaryotic translation initiation factor 4E binding protein 1 (4EBP1) was not detected on the array; unc-51-like kinase 1 (ATG1), which is upstream of programmed cell death by autophagy, was up-regulated in NR compared with CON samples. (3) Subunits G protein beta subunit-like (GBL) and raptor were not detected on the gene array; mTOR was not differentially expressed between CON and NR samples. (4) Subunits GBL and rictor were not detected on the gene array; mTOR and ras homolog enriched in brain (RheB) were not differentially expressed between CON and NR samples. (5) Protein kinase, AMP-activated (AMPK) and tuberous sclerosis 2 (TSC2) were not detected on the array. AKT and tuberous sclerosis 1 (TSC1) were not differentially expressed between CON and NR samples. (6a) Phosphoinositide-3-kinase (PI3K), downstream of the insulin signalling pathway, was up-regulated in NR compared with CON samples; 3-phosphoinositide-dependent protein kinase-1 (PDK1) was not differentially expressed between CON and NR samples. (6b) Genes encoding the mitogen-activated protein kinase 1 (MAPK1, ERK1)–mitogen-activated protein kinase 2 (MAPK2, ERK2) complex of the MAPK signalling pathway were not differentially expressed between CON and NR samples; ribosomal protein S6 kinase, 90 kDa, polypeptide 6 (RSK) was up-regulated in NR compared with CON samples. (6c) Leptin receptor (LEPR) and janus kinase (JAK) were up-regulated in NR compared with CON samples. Signal transducer and activator of transcription 3 (STAT3) was not differentially expressed and leptin was not detected. (6d) In the serine/threonine protein kinase-11–serine/threonine-protein kinase pak-β–calcium binding protein 39 (LKB1–STRAD–MO25) complex, LKB1 was not detected and STRAD and MO25 were not differentially expressed.
Discussion
The goals of this study were to use transcriptome data from fetal kidneys of CON and NR mothers at 0.5G to create a NR kidney expression profile, to use previously documented proximal tubule density and VEGF gene expression measurements as an index of altered kidney structural phenotype, and to use these data together to identify the molecular mechanisms that may down-regulate renal development of the NR baboon fetal kidney.
To prioritize pathways and factors in pathways that will define molecular mechanisms affected by NR in 0.5G kidneys, we assessed the structural phenotype and the transcriptome expression phenotype of 0.5G CON and NR baboon kidneys. To further define the structural phenotype, we performed IHC for VEGF in kidneys of both dietary groups. In addition, we combined these data with previous morphometric data for CON and NR 0.5G kidneys (Cox et al. 2006) showing that CON and NR kidney size were similar. A preliminary assessment of glomerular number at 0.5G also showed no difference with treatment. Tubule density was decreased suggesting that the 0.5G NR kidney tubules are shorter than controls (Cox et al. 2006). From studies in mice it is clear that VEGF is critical for glomerular development as intraperitoneal injection of VEGF antibodies into newborns results in decreased nephron number and the appearance of avascular glomeruli (Kitamoto et al. 1997). Of the nine known isoforms of VEGF, VEGF165 predominates. VEGF121 is entirely soluble, VEGF165 exhibits a low heparin-binding affinity and is 50–70% extracellular matrix (ECM) bound (Houck et al. 1992). VEGF189 and VEGF206 exhibit strong heparin-binding affinity and are consequently fully sequestered in the ECM (Ferrara & Davis-Smyth, 1997). The extracellular localization of immunoreactivity shown in Fig. 1 is therefore likely to represent the ECM-bound isoforms. Decreased VEGF expression is also associated with decreased peritubular capillary density (Yuan et al. 2003). The more diffuse immunoreactivity in the NR compared to the CON kidney sections may therefore represent a decrease in VEGF protein abundance in association with impaired peritubular capillary development. The present study was intended to provide clues as to where to begin more in-depth analysis. VEGF protein expression and signalling require focused future analysis.
We used gene expression data and phenotype data to prioritize critical molecular mechanisms that require further investigation. We hypothesized that a pathway may only be relevant to the tissue phenotype if gene expression profiles at the end of the pathway were consistent with that phenotype. If factors at the end of a pathway did not change, we questioned the relevance of the pathway to downstream processes that change fetal renal phenotype. We therefore contend that evaluation of factors at the end of a pathway is important for defining molecular mechanisms that regulate the response in expression of the pathway to epigenetic challenges. Once significant changes at the end of the pathway have been defined, the next ‘upstream’ step in the pathway can be examined to define mechanisms regulating altered pathway activity. By working upstream from the end of a pathway to each event in the pathway, it is possible to define the critical steps that are likely to be rate limiting in the pathway response to the challenge. It is therefore also possible to define the signalling events that act as input to the pathway, thereby identifying mechanism of action.
For the nine up-regulated and 15 down-regulated KEGG pathways, we identified genes at the end of each pathway and assessed whether the expression profile was consistent with the tissue phenotype of decreased VEGF expression and proximal tubule density. To do so we categorized pathways as follows: (1) the end-of-pathway gene expression differed from the tissue phenotype or the end-of-pathway gene expression was similar between the CON and NR groups; (2) the expression phenotype was consistent with the tissue phenotype; however, the pathway was probably downstream of the NR responsive mechanism; and (3) the expression phenotype was consistent with the tissue phenotype and the pathway was probably responsive directly to NR.
An example of a significantly different pathway in the first category (i.e. pathways that do not show an expression phenotype consistent with the tissue phenotype or for which insufficient data are available to determine whether the expression phenotype is consistent with the tissue phenotype) is the apoptosis signalling pathway. Apoptosis was down-regulated in NR kidney RNA samples compared with CON samples; however, the detected end-of-pathway genes were not differentially expressed between CON and NR samples. It should also be noted that both pro- and anti-apoptotic genes were down-regulated. These data suggest that transcriptional regulation of the apoptosis signalling pathway is not central to the NR response. Similar conclusions could be drawn about several other pathways such as wingless type MMTV integration site (Wnt) signalling, axon guidance and focal adhesion (Table 2).
An example of a significantly different pathway in the second category (i.e. pathways that show an expression phenotype consistent with the tissue phenotype but are not likely to provide information on the molecular nutrient sensing mechanism) is the O-glycan biosynthesis pathway. Up-regulation of O-glycan biosynthesis genes α-2,6-sialyltransferase and glucosaminyl (N-acetyl) transferase is consistent with increased availability of monosaccharides from salvage pathways in the NR kidneys (Wopereis et al. 2006). However, this pathway is downstream of programmed cell death and nutrient sensing pathways and is consequently probably downstream from events precipitating the NR phenotype. Similar conclusions could be drawn about several other pathways such as adherens junctions and oxidative phosphorylation (Table 2).
The only pathway that fits the third category in which the expression phenotype is consistent with the tissue phenotype is mTOR signalling. It has been suggested that mTOR signalling plays a central role in sensing and responding to intracellular nutrient availability (Marshall, 2006). As such, this pathway is likely to be involved in the cellular responses to maternal NR. Following the rationale that important information is contained in downstream portions of important cellular pathways, we identified the mTOR signalling pathway as the significantly different pathway that shows an expression phenotype consistent with the tissue phenotype and is most likely to provide information on the molecular response to NR. The downstream events for end-of-pathway mTOR genes include VEGF signalling, translation and programmed cell death by autophagy. For VEGF signalling, VEGF array data, IHC data and QRT-PCR data (Cox et al. 2006) all indicate a down-regulation in VEGF signalling. For translation, eIF4E is down-regulated in NR samples. The eIF4B and S6 genes are not differentially expressed; however, their gene products are activated by the heterodimer S6K1/S6K2 via phosphorylation. S6K1/2 was down regulated in NR. The decreased expression of eIF4E, decreased expression of ribosomal subunits (Cox et al. 2006) and decreased tubule density in the 0.5G NR kidney (Cox et al. 2006) suggest that translation is down-regulated in the NR kidney. Furthermore, ATG1 expression which is upstream of caspase-independent programmed cell death (autophagy) is up-regulated in NR kidney which is also consistent with the tissue phenotype.
Investigation of the mTOR signalling pathway as currently determined by gene array pathway analysis indicates that at least four signalling events upstream of mTOR can influence mTOR signalling: abundance of hormones, AMP, amino acids and growth factors. The mTOR pathway regulatory mechanisms are complex and include protein phosphorylation, protein localization and control of mTOR activity via binding to auxiliary cytosolic proteins raptor, rictor and GBL. These regulatory mechanisms are under the control of a number of sensors that detect changes in nutrient availability (e.g. lipids, amino acids and carbohydrates), intracellular energy levels and the external environment (Marshall, 2006). To determine whether gene expression profiles for events upstream in the mTOR pathway were consistent with the tissue phenotype and to assess primary signalling events relevant to the NR kidney phenotype upstream of the mTOR pathway, we analysed each step from the end back to the beginning of the pathway. By investigating each step in the mTOR pathway, it was then possible to define the transcriptional regulatory mechanisms in mTOR signalling that are affected by NR during renal development and to define events upstream of mTOR signalling that specifically drive it (e.g. insulin signalling via insulin and insulin-like growth factors and adipocytokine signalling via leptin and leptin receptors). For this analysis, each segment of the pathway was assigned a ‘stage’.
As discussed above, the end-of-pathway genes comprising stage 1 are consistent with the NR tissue phenotype. Stage 2 is composed of HIF1a, the S6 kinase1/2 heterodimer and 4EBP1. The S6 kinase1/2 heterodimer, which was down-regulated in the NR kidney samples, activates eIF4B and S6 via phosphorylation which leads to translation. HIF1a, which was similarly expressed between CON and NR, is activated by phosphorylation via the GBL–mTOR–raptor complex. ATG1 expression, which leads to autophagy is inhibited by the GBL–mTOR–raptor complex. Taken together, the down-regulation of the S6 kinase1/2 heterodimer and the up-regulation of ATG1 support the decreased activity of the GBL–mTOR–raptor complex in NR kidneys.
Stage 3 includes GBL–mTOR–raptor complex. The GBL–mTOR–raptor complex is activated by phosphorylation via AKT, which is activated by RheB, which in turn is activated by amino acids, and which is inhibited by AMP-activated protein kinase α 1 catalytic subunit (AMPK) via phosphorylation. Although two of the three genes encoding this complex are not detected and one gene is not differentially expressed, the phosphorylation-dependent activity of this complex indicates that the gene expression data are compatible with the tissue phenotype. Stage 4 genes include RheB and genes encoding the GBL–mTOR–rictor complex. As with stage 3 genes, two genes are not detected on the array and two are not differentially expressed; however, expression of these factors is phosphorylation dependent.
In stage 5, AMPK and the TSC2/TSC1 heterodimer form two major hubs in the regulation of mTOR signalling. The TSC1/TSC2 complex integrates information from insulin signalling, AMPK signalling, MAPK signalling pathways and extracellular amino acids. TSC2/TSC1 is a negative regulator of cell growth. Analysis of gene expression upstream of the TSC1/TSC2 complex shows up-regulation of PI3K from the insulin signalling pathway (6a), up-regulation of RSK (6b) and up-regulation of LEPR and JAK (6c). Up-regulation of AKT and up-regulation of RSK should inhibit TSC1/TSC2 complex-dependent inhibition of cell growth. Therefore, this upstream pathway is unlikely to be the NR-responsive mechanism. Up-regulation of LEPR and JAK should result in inhibition of AMPK which subsequently activates the TSC1/TSC2 complex. Thus, the gene expression data suggest that the leptin signalling pathway may be a NR-responsive mechanism in the 0.5G NR kidney. In addition to being downstream of leptin signalling, AMPK is the centre for the energy stress response and AMP response. Gene expression data do not provide insights into the activity of these factors upstream of AMPK (6d).
Summary
Use of gene array data is often considered a fishing expedition in search of a new ‘favourite’ gene. Our data indicate that appropriately used whole-genome expression data can help to define cellular pathway changes that direct the investigation without any preconceived ideas of what may be important. When placed in the context of biological pathways, the data provide a snap-shot of the transcriptome and more clearly direct the course of future studies. Once biological pathways have been used to describe the biological system (i.e. an expression phenotype), the data must be prioritized to lead the investigation, defining mechanisms regulating the expression phenotype and the relationship with the tissue or organism phenotype. As many biological pathways are connected to other biological pathways, prioritization of pathways can rapidly become complex. Therefore, we hypothesize that an essential step for defining relevant pathways for the tissue phenotype is evaluation of factors at the end of a pathway. If factors at the end of a pathway do not change, then it is difficult to justify the relevance of this pathway to downstream processes. The ‘end-of-pathway phenotype’ can be further refined by including investigation of expression for the gene, the protein and the post-translationally modified protein at the end of the pathway. If this factor is modified at the level of the mature protein (abundance, modifications) and the modification impacts downstream pathways, then the pathway phenotype is established. Subsequent analysis includes evaluation of molecular mechanisms regulating the genes at the end of the pathway including transcriptional, post-transcriptional, translational and post-translational events. Once mechanisms regulating the factor at the end of the pathway have been defined, then the next ‘upstream’ step in the pathway can be examined to define mechanisms regulating that factor. This process of working upstream from the end of a pathway to each event in the pathway will allow the definition of critical steps affected by the environmental challenge. From this approach we have determined that the mTOR signalling pathway is central to the phenotype expressed in the 0.5G NR kidney of the fetal baboon.
Acknowledgments
We would like to acknowledge contributions by Greg Langone for VEGF immunohistochemistry and grant support from NIH grants: 1 C06 RR13556 and HD21350.
References
- Armitage JA, Khan IY, Taylor PD, Nathanielsz PW, Poston L. Developmental programming of the metabolic syndrome by maternal nutritional imbalance: how strong is the evidence from experimental models in mammals? J Physiol. 2004;561:355–377. doi: 10.1113/jphysiol.2004.072009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LA, Nijland MJ, Gilbert JS, Schlabritz-Loutsevitch NE, Hubbard GB, McDonald TJ, et al. Effect of 30 per cent maternal nutrient restriction from 0.16 to 0.5 gestation on fetal baboon kidney gene expression. J Physiol. 2006;572:67–85. doi: 10.1113/jphysiol.2006.106872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlor SC, Conklin BR. MAPP Finder: using gene ontology and genMAPP to create global gene-expression profile from microarray data. Gen Biol. 2003;4:R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake AJ, Walker BR. The intergenerational effects of fetal programming: non-genomic mechanisms for the inheritance of low birth weight and cardiovascular risk. J Endocrinol. 2004;180:1–16. doi: 10.1677/joe.0.1800001. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- Freeman AI, Munn HL, Lyons V, Dammermann A, Seckl JR, Chapman KE. Glucocorticoid down-regulation of rat glucocorticoid receptor does not involve differential promoter regulation. J Endocrinol. 2004;183:365–374. doi: 10.1677/joe.1.05773. [DOI] [PubMed] [Google Scholar]
- Fu Q, McKnight RA, Wang X, Yu L, Callaway CW, Lane RH. Uteroplacental insufficiency induces site-specific changes in histone H3 covalent modifications and affects DNA-histone H3 positioning in day 0 IUGR rat liver. Physiol Genomics. 2004;20:108–116. doi: 10.1152/physiolgenomics.00175.2004. [DOI] [PubMed] [Google Scholar]
- Hendrickx AG. The menstrual cycle of the baboon as determined by vaginal smear, vaginal biopsy, and perineal swelling. In: Vagtborg H, editor. The Baboon in Medical Research (Proc 2nd Int Symp on the Baboon and its use as an experimental animal) Austin: University of Texas Press; 1967. pp. 437–459. [Google Scholar]
- Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem. 1992;267:26031–26037. [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto Y, Tokunaga H, Tomita K. Vascular endothelial growth factor is an essential molecule for mouse kidney development: glomerulogenesis and nephrogenesis. J Clin Invest. 1997;99:2351–2357. doi: 10.1172/JCI119416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutrition. 2005;135:1382–1386. doi: 10.1093/jn/135.6.1382. [DOI] [PubMed] [Google Scholar]
- Marshall S. Role of insulin, adipocyte hormones, and nutrient-sensing pathways in regulating fuel metabolism and energy homeostasis: a nutritional perspective of diabetes, obesity, and cancer. Sci STKE. 2006;2006:re7. doi: 10.1126/stke.3462006re7. [DOI] [PubMed] [Google Scholar]
- Miozzo M, Simoni G. The role of imprinted genes in fetal growth. Biol Neonate. 2002;81:217–228. doi: 10.1159/000056752. [DOI] [PubMed] [Google Scholar]
- Ozanne SE, Nave BT, Wang CL, Shepherd PR, Prins J, Smith GD. Poor fetal nutrition causes long-term changes in expression of insulin signaling components in adipocytes. Am J Physiol Endocrinol Metab. 1997;273:E46–E51. doi: 10.1152/ajpendo.1997.273.1.E46. [DOI] [PubMed] [Google Scholar]
- Schlabritz-Loutsevitch NE, Howell K, Rice K, Glover EJ, Nevill CH, Jenkins SL, et al. Development of a system for individual feeding of baboons maintained in an outdoor group social environment. J Med Primatol. 2004;33:117–126. doi: 10.1111/j.1600-0684.2004.00067.x. [DOI] [PubMed] [Google Scholar]
- Wopereis S, Lefeber DJ, Morava E, Wevers RA. Mechanisms in protein O-glycan biosynthesis and clinical and molecular aspects of protein O-glycan biosynthesis defects: a review. Clin Chem. 2006;52:574–600. doi: 10.1373/clinchem.2005.063040. [DOI] [PubMed] [Google Scholar]
- Yuan HT, Li XZ, Pitera JE, Long DA, Woolf AS. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1 α. Am J Pathol. 2003;163:2289–2301. doi: 10.1016/s0002-9440(10)63586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]