

Abstract
Cefditoren is the active form of cefditoren pivoxil, an oral cephalosporin antibiotic used for the treatment of respiratory tract infections and otitis media caused by bacteria such as Streptococcus pneumoniae, Haemophilus influenzae, Streptococcus pyogenes, Klebsiella pneumoniae, and methicillin-susceptible strains of Staphylococcus aureus. β-Lactam antibiotics, including cefditoren, target penicillin-binding proteins (PBPs), which are membrane-associated enzymes that play essential roles in the peptidoglycan biosynthetic process. To envision the binding of cefditoren to PBPs, we determined the crystal structure of a trypsin-digested form of PBP 2X from S. pneumoniae strain R6 complexed with cefditoren. There are two PBP 2X molecules (designated molecules 1 and 2) per asymmetric unit. The structure reveals that the orientation of Trp374 in each molecule changes in a different way upon the formation of the complex, but each forms a hydrophobic pocket. The methylthiazole group of the C-3 side chain of cefditoren fits into this binding pocket, which consists of residues His394, Trp374, and Thr526 in molecule 1 and residues His394, Asp375, and Thr526 in molecule 2. The formation of the complex is also accompanied by an induced-fit conformational change of the enzyme in the pocket to which the C-7 side chain of cefditoren binds. These features likely play a role in the high level of activity of cefditoren against S. pneumoniae.
Cefditoren is the active form of cefditoren pivoxil (Fig. 1), an oral cephalosporin antibiotic used to treat respiratory tract infections and otitis media (1, 11, 24, 30, 32, 33). Cefditoren has a broad spectrum of activity against gram-positive and gram-negative bacteria, including common respiratory pathogens such as Streptococcus pneumoniae, Haemophilus influenzae, Streptococcus pyogenes, Klebsiella pneumoniae, and methicillin-susceptible strains of Staphylococcus aureus (4, 8, 14, 27-29).
FIG. 1.

Chemical structures of cefditoren and cefditoren pivoxil with the atomic numbering scheme for the cephem skeleton.
Penicillin-binding proteins (PBPs) are enzymes that catalyze the polymerization and cross-linking of peptidoglycan precursors in bacterial cell wall biosynthesis (9, 17). The PBPs have been divided into three classes. High-molecular-weight (hmw) class A PBPs are bifunctional enzymes with transglycosylase and transpeptidase activities. Hmw class B PBPs act only as transpeptidases. The low-molecular-weight (lmw) PBPs generally act as dd-carboxypeptidases. S. pneumoniae contains six PBPs: the hmw class A PBPs 1A, 1B, and 2A; the hmw class B PBPs 2B and 2X; and lmw PBP 3. In the cross-linking reaction, the transpeptidase or dd-carboxypeptidase must bind to the first of two peptidoglycan substrates called the donor strand. The active-site serine residue then attacks the carbonyl carbon atom of the C-terminal d-Ala-d-Ala peptide bond, leading to an acyl-enzyme complex, with the subsequent release of the C-terminal d-Ala. The transient acyl-enzyme then has two possible fates: hydrolysis, which releases the shortened peptidoglycan strand (dd-carboxypeptidation), or cross-link formation with an acceptor strand from a neighboring peptidoglycan polymer (transpeptidation) (20). The β-lactam antibiotics inhibit transpeptidase and dd-carboxypeptidase activities by acylating the active-site serine of PBPs. The active site of PBPs is bordered by three conserved motifs: Ser-X-X-Lys (SXXK), which includes the catalytic serine; Ser-X-Asn (SXN); and Lys-Thr/Ser-Gly (KT/SG) (12). Alterations of the PBPs reduce their affinities for β-lactam antibiotics, resulting in drug resistance. In S. pneumoniae, it has been documented that PBP 2X is frequently associated with cephalosporin resistance (2, 3, 6, 12, 25).
The relative antimicrobial activities of various cephalosporins with a 2-alkyloxyimino-2-(2-aminothiazol-4-yl)-acetamido moiety as the C-7 side chain of the cephem skeleton depend mainly on the different structures of the C-3 side chains. However, the crystal structure of PBP 2X in complex with a cephalosporin that retains its original C-3 side chain has not yet been determined (10). Therefore, to elucidate the role of the C-3 side chain in the complex with cefditoren, we determined the crystal structure of it complexed with S. pneumoniae R6 PBP 2X.
S. pneumoniae R6 PBP 2X is composed of a short cytoplasmic region, a transmembrane region, and a periplasmic unit containing three domains: the N-terminal, transpeptidase, and C-terminal domains. A soluble form of PBP 2X without the cytoplasmic and transmembrane regions has been crystallized in two crystal forms. The hexagonal and orthorhombic crystals diffracted to 3.5-Å and 2.4-Å resolutions, respectively (10, 23). However, it took several months to grow the latter crystals. To improve the quality and reproducibility of these crystals, the soluble PBP 2X protein was subjected to limited trypsin digestion to reduce the conformational heterogeneity, as described in earlier structure determinations of PBP 1A and PBP 1B (5, 18).
MATERIALS AND METHODS
Protein expression and purification.
The gene for the soluble PBP 2X protein (amino acids 49 to 750) from S. pneumoniae R6 was amplified by PCR and cloned into the pET-15b vector (Novagen). The resulting plasmid was transformed into Escherichia coli BL21(DE3)pLysS (Novagen). Protein expression was induced by using 1 mM isopropyl-β-d-thiogalactopyranoside at 310 K for 3 h. The cells were harvested, resuspended in lysis buffer (20 mM Tris-HCl, pH 7.9, 500 mM NaCl, 5 mM imidazole, 1 mM phenylmethylsulfonyl fluoride, 1 μM leupeptin, 1 μM pepstatin A, 0.4 mg/ml lysozyme [Sigma], 5 μg/ml RNase A, and 5 mU/ml DNase I [Takara Bio]), and then sonicated. The crude lysate was centrifuged, and the resulting supernatant was applied to a HiTrap chelating column (GE Healthcare Biosciences) with Ni2+ ions that was preequilibrated in a binding buffer (20 mM Tris-HCl, pH 7.9, 500 mM NaCl, 5 mM imidazole) and then eluted with a linear gradient of 20 to 500 mM imidazole. Pooled fractions were dialyzed against 20 mM Tris-HCl, pH 7.9, and 1 mM EDTA. The dialyzed PBP 2X was then digested with 0.53 mg/ml trypsin (Sigma) for 1 h at 298 K. The protein was subsequently purified on a POROS HQ column (PerSeptive Biosystems) in the same buffer used for dialysis, but with a linear gradient of from 0 to 300 mM NaCl. Pooled peak fractions were then dialyzed against 5 mM potassium phosphate buffer, pH 6.8, and then applied to a Macro-Prep ceramic hydroxyapatite type I column (Bio-Rad) and eluted with a linear gradient of from 5 to 500 mM potassium phosphate. Pooled peak fractions were then dialyzed against 50 mM sodium acetate buffer, pH 5.0, applied to a MonoS column (GE Healthcare Biosciences), and eluted with a linear gradient of from 0 to 1 M NaCl. The eluted peak fractions were then concentrated and loaded onto a Superdex 75 column (GE Healthcare Biosciences) equilibrated with 20 mM Tris, pH 7.5-150 mM NaCl. The resulting peak fractions were pooled and concentrated to a final protein concentration of 10 mg/ml. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis showed that the trypsin-digested PBP 2X consisted primarily of three fragments with molecular weights of about 42,000 (Leu241-Lys625), 16,000 (Thr71-Lys238), and 13,000 (Ala626-Asp750). The identities of these fragments were verified by N-terminal sequencing and electrospray ionization mass spectrometry.
Crystallization and data collection.
Uncomplexed PBP 2X crystals were grown by the hanging-drop vapor-diffusion method at 293 K. Two microliters of protein solution was mixed with an equal volume of reservoir solution containing 10 to 25% (wt/vol) polyethylene glycol 4000, 200 mM ammonium sulfate, and 100 mM sodium acetate and equilibrated against 0.5 ml reservoir solution with 250 μl Al's oil (Hampton Research) barrier. Crystals of the cefditoren complex were prepared by soaking uncomplexed PBP 2X crystals for 105 min in a solution containing 5 mg/ml cefditoren sodium. The resulting crystals were cryoprotected by using 25% (wt/vol) glycerol. Data were collected at 100 K at a wavelength of 1.0 Å on the BL32B2 beamline at the SPring-8 synchrotron facility. All diffraction data were integrated and scaled by using the CrystalClear software package (Rigaku Corporation).
Structure determination and refinement.
A molecular replacement solution was found by using the program Molrep (31) and by using the structure of the soluble PBP 2X as a model (Protein Data Bank code 1QME). Two trypsin-digested PBP 2X molecules (designated molecules 1 and 2) were present in the asymmetric unit. Molecules 1 and 2 consisted of chains A to C and D to F, respectively, as described in the coordinate file. The value of the Matthews coefficient (VM) was 2.8 Å3 Da−1, which corresponds to a solvent content of 55% (19). Crystallographic refinement was performed by using the programs Refmac5 (21) and CNX (Accelrys, Inc.). Model building was performed by using the programs Coot (7), Turbo-Frodo (AFMB-CNRS), and DS Modeling (Accelrys, Inc.). All structures were validated by using the PROCHECK program (13). A summary of statistics from the data collection and refinement is given in Table 1. The final model of the uncomplexed PBP 2X protein includes 1,277 residues, one sulfate ion, and 50 water molecules. The final model of the PBP 2X-cefditoren complex structure includes 1,234 residues, one sulfate ion, and 93 water molecules.
TABLE 1.
Data collection and refinement statistics
| Data set | Uncomplexed PBP 2X | PBP 2X-cefditoren complex |
|---|---|---|
| Space group | P21212 | P21212 |
| Unit cell parameters | ||
| a (Å) | 106.88 | 107.74 |
| b (Å) | 171.71 | 171.20 |
| c (Å) | 89.36 | 89.15 |
| Resolution (Å) | 2.85 (2.95-2.85)a | 2.60 (2.69-2.60) |
| Total no. of reflections | 220,996 | 196,510 |
| No. of unique reflections | 39,032 | 46,229 |
| Completeness (%) | 99.8 (99.8) | 89.9 (90.5) |
| Redundancy | 5.7 (5.5) | 4.3 (4.0) |
| Rmergeb (%) | 6.1 (23.2) | 5.4 (21.5) |
| <I/σ(I)>c | 17.1 (5.7) | 16.7 (5.5) |
| Refinement | ||
| Total no. of atoms | 9,852 | 9,617 |
| Rcrystd (%) | 22.4 | 22.1 |
| Rfreee (%) | 28.6 | 26.7 |
| RMSD from ideal | ||
| Bond lengths (Å) | 0.007 | 0.007 |
| Bond angles (degrees) | 1.1 | 1.0 |
| Avg B valuesf (Å2) | 66.6 | 46.4 |
| Ramachandran plot | ||
| Most favored (%) | 87.7 | 89.6 |
| Additional allowed (%) | 11.9 | 10.2 |
| Generously allowed (%) | 0.4 | 0.0 |
| Disallowed (%) | 0.1 | 0.2 |
Values in parentheses are for the highest-resolution shell.
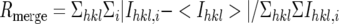 , where I is the observed intensity and <I> is the average intensity for multiple observations.
, where I is the observed intensity and <I> is the average intensity for multiple observations.
Average of the diffraction intensities divided by their standard deviations.
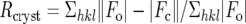 , where |Fo| and |Fc| are the observed and calculated structure factor amplitudes for reflection hkl, respectively.
, where |Fo| and |Fc| are the observed and calculated structure factor amplitudes for reflection hkl, respectively.
Rfree is the same as Rcryst for a random 5% of reflections excluded from refinement.
Average of the isotropic temperature factors.
Protein structure accession numbers.
Atomic coordinates of the PBP 2X and PBP 2X-cefditoren complex structures have been deposited in the Protein Data Bank with accession codes 2Z2L and 2Z2M, respectively.
RESULTS AND DISCUSSION
Uncomplexed structure of the trypsin-digested PBP 2X.
There were two trypsin-digested PBP 2X molecules (designated molecules 1 and 2) per asymmetric unit. The overall architecture of the trypsin-digested PBP 2X molecule was similar to that of the soluble PBP 2X (10, 23), consisting of three domains: an N-terminal domain (residues 49 to 265), a transpeptidase domain (residues 266 to 616), and a C-terminal domain (residues 635 to 750). Trypsin cleavage sites (following Arg70, Lys238, and Lys625) corresponded to flexible regions that were far from the active site of the soluble PBP 2X molecule (Fig. 2). Therefore, it is thought that the conformational changes caused by trypsin digestion do not influence the structure of the active site. The models of molecules 1 and 2 in the trypsin-digested PBP 2X were similar to each other except in the loop region (residues 376 to 386), with a root mean standard deviation (RMSD) of 0.39 Å for 340 Cα positions (residues 266 to 375 and 387 to 616). The different conformation of this loop region is presumably due to crystal packing interactions with the neighboring molecule. An additional difference between the two molecules in the trypsin-digested PBP 2X was observed in the N-terminal domain (Fig. 2). The N-terminal domains of molecules 1 and 2 had completely different orientations that were stabilized by different crystal packing interactions.
FIG. 2.
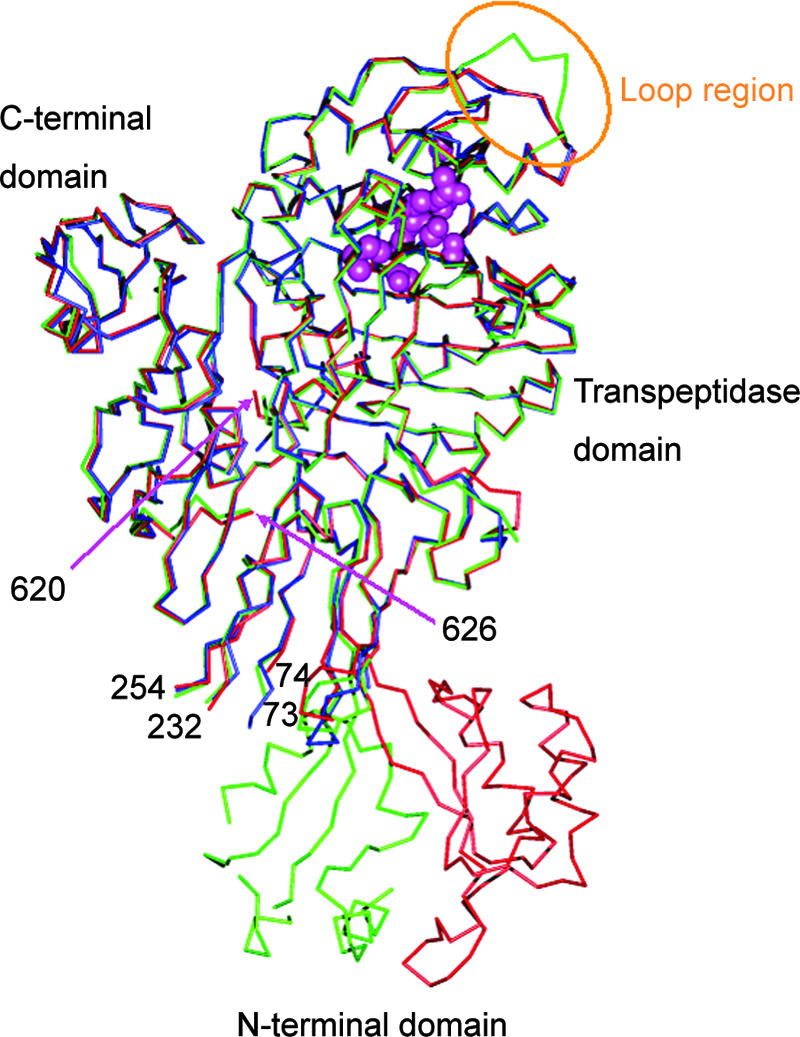
Structural comparison between the trypsin-digested PBP 2X and the soluble PBP 2X from S. pneumoniae. The Cα models of molecule 1 (residues 74 to 232, 254 to 620, and 626 to 750) and molecule 2 (residues 73 to 101, 113 to 120, 133 to 169, 173 to 232, 254 to 620, and 626 to 750) of the trypsin-digested PBP 2X are colored red and green, respectively. The Cα model of the soluble PBP 2X (residues 71 to 92, 183 to 232, 254 to 620, and 632 to 750) is colored blue, with spheres (in magenta) representing side chains in the conserved motifs (337STMK, 395SSN, and 547KSG). The residue numbers of amino acids near the trypsin digestion sites are also shown. The loop region (residues 376 to 386), which assumes a different conformation in molecule 2 of the trypsin-digested PBP 2X, is indicated by an ellipse (yellow).
PBP 2X-cefditoren complex structure.
The electron density maps within the active sites of each of the trypsin-digested PBP 2X molecules (designated molecules 1 and 2) in the asymmetric unit revealed cefditoren molecules covalently bound to Ser337 (Fig. 3A and B). The transpeptidase domains of molecules 1 and 2 in the PBP 2X-cefditoren complex showed a gross structural similarity close to those of the uncomplexed structure, as indicated by the Cα RMSDs of 0.57 Å for molecule 1 (residues 266 to 555 and 560 to 616) and 0.64 Å for molecule 2 (residues 266 to 376 and 382 to 616). However, striking differences were noticeable in two regions, specifically, where the C-3 and C-7 side chains of cefditoren interact (Fig. 3C and D).
FIG. 3.

Cefditoren-PBP 2X complex structure and conformational changes. (A and B) Omit Fo-Fc difference density maps (contoured at 3 σ) covering the cefditoren moiety covalently attached to Ser337 of the trypsin-digested PBP 2X in molecules 1 and 2, respectively. Cefditoren-acylated Ser residues are shown as stick models with carbon atoms colored green, oxygens red, nitrogens blue, and sulfurs yellow. (C and D) Superposition of the active-site region from the uncomplexed (light blue) and cefditoren complex (orange) structures in molecules 1 and 2, respectively. Cefditoren and side chains of selected residues are shown as thick stick renderings. Atoms are colored as described for panel A, except that the carbon atoms of selected residues in the uncomplexed and cefditoren complex structures are colored light blue and orange, respectively. Covalent binding of cefditoren to the trypsin-digested PBP 2X requires conformational changes in the active site.
At the interaction sites for the C-3 side chain in cefditoren, the Cα atoms of Trp374 in molecules 1 and 2 were displaced 1.1 and 1.7 Å, respectively, toward the opening of a distinct cleft in the molecular surface compared to their positions in the uncomplexed structures. Moreover, additional hydrophobic interaction sites were formed due to conformational changes of the Trp374 side chains in both molecules to avoid a steric clash with the C-3 side chain of cefditoren, but these changes differed. In forming a complex with cefditoren, the 4-methylthiazole group in the C-3 side chain bound to the hydrophobic pocket formed by Trp374, His394, and Thr526 in molecule 1 and by His394, Asp375, and Thr526 in molecule 2 when Trp374 shifted its position (Fig. 4A and B). Schematic representations of these interactions are shown in Fig. 4C and D. Although the different interactions in molecules 1 and 2 were assumed to be caused by the different crystal packing around Trp374 and the loop region (residues 379 to 386 in molecule 1 and residues 376 to 386 in molecule 2), it is difficult to assert which binding mode, that seen in molecule 1 or that seen in molecule 2, represents the physiologically relevant interactions between cefditoren and PBP 2X in bacteria.
FIG. 4.

Interaction of the trypsin-digested PBP 2X with cefditoren. (A and B) Stereo view of the active sites of the cefditoren-PBP 2X complex structures in molecules 1 and 2, respectively. Atoms are colored as described in the legend to Fig. 3. Hydrogen bonds are represented by thin lines (magenta). (C and D) Schematic diagrams of cefditoren binding mode in molecules 1 and 2, respectively. Hydrogen bonds are represented by broken lines (magenta), and hydrophobic contacts are shown as arcs (green). The atomic numbering scheme of the cephem skeleton is also shown.
The active pockets in molecules 1 and 2 of the complexed structure were also more open around the sites of interaction with the C-7 side chains of cefditoren than in the uncomplexed structure, as seen in the complex formed by soluble PBP 2X with cefuroxime (Fig. 3C and D) (10). The Cα atoms (residues 550 to 555 and 560 to 569 in molecule 1 and residues 550 to 569 in molecule 2) became displaced 1.0 to 2.1 Å and 1.0 to 2.7 Å from their uncomplexed positions, respectively. Interactions between the C-7 side chains of cefditoren and the trypsin-digested PBP 2X were quite similar in molecules 1 and 2 (Fig. 4A to D). Amido moieties in the C-7 side chains formed hydrogen bonds to carbonyl oxygens of Thr550 and to the side chains of Asn397. The aminothiazole moieties in the C-7 side chains were stabilized by hydrogen bonds with Gln552 and Glu334 and by hydrophobic interactions with Ala551, Gln452, and Tyr561. A similar induced-fit mechanism involving the formation of hydrogen bonds between the amido moiety of the C-7 side chain in cefditoren and PBP 2X has been seen in the structures of S. pneumoniae PBP 1A and PBP 1B in complex with cefotaxime (5, 18).
Comparison with PBPs in complex with peptidoglycan mimetic compounds.
To further investigate the role of the C-7 side chain of cefditoren, the PBP 2X-cefditoren complex structure was superposed on the structure of Bacillus subtilis PBP 4A in complex with d-α-aminopimelyl-ɛ-d-alanyl-d-alanine, which mimics the C-terminal end of the peptidoglycan stem peptide (the donor strand) (26). The binding sites for the C-7 side chain of cefditoren and for the adduct, d-α-aminopimelyl-ɛ-d-alanine, in PBP 4A overlapped; and the hydrogen bonds between the amido moiety of the C-7 side chain of cefditoren, Thr550 and Asn397 in PBP 2X corresponded to those between the amido moiety of the mimetic peptide, Ser414 and Asn301 in PBP 4A. This observation supports the established theory that β-lactam antibiotics mimic the conformation of the acyl-d-Ala-d-Ala portion of the donor strand (15, 20, 22). The induced-fit conformational change from the uncomplexed PBP 2X is also expected to be required for substrate binding, in which a hydrogen bond forms between the amido moiety of the substrate, Thr550 and Asn397. Previous structural analyses of S. pneumoniae PBP 1B have revealed not only the “closed” active-site conformation but also the “open” active-site conformation in the absence of β-lactam antibiotics (16). In conclusion, the C-7 side chains of cephalosporins apparently contribute high-affinity interactions with PBPs by mimicking the donor strand, and they also take advantage of flexibility in the active site to accommodate the substrate.
On the other hand, superposition of the PBP 2X-cefditoren complex structure with the Streptomyces sp. strain R61 dd-peptidase/transpeptidase in complex with a cephalosporin that mimics the approaching nucleophile, diaminopimelate, from the second strand of the peptidoglycan (the acceptor strand) (15) revealed that the C-3 side chain of cefditoren overlapped a part of the diaminopimelate surrogate. However, the shape of the binding pocket in PBP 2X differed from that in the Streptomyces sp. strain R61 PBP. Moreover, the conformation of the C-3 side chain of cefditoren is considered to be so rigid as to induce the conformational change of Trp374 upon binding, although the substrate is expected to be flexible enough to adopt various conformations. Actually, the conformation of the C-3 side chain of cefditoren in the PBP 2X-cefditoren complex is similar to that seen in a crystal structure of cefditoren pivoxil, whose β-lactam ring is intact (34). Although the flexibility of Trp374 might contribute to recognition of the substrate, further investigation is required to determine whether the binding of the acceptor strand to PBP 2X causes a conformational change of Trp374, as seen in cefditoren binding.
Conclusion.
Cefditoren has a unique structure at the C-3 side chain of the cephem skeleton. The PBP 2X-cefditoren complex structure revealed that the C-3 side chain of cefditoren interacts with an additional hydrophobic pocket formed by a conformational change of Trp374. In addition, the C-7 side chain of cefditoren is stabilized by hydrogen bonds and hydrophobic interactions with the active site in which an induced-fit conformational change occurs upon binding. These features are likely to play a role in the high level of activity of cefditoren against S. pneumoniae.
Acknowledgments
We thank Yoshio Katsuya and Izumi Wada for help with data collection at the SPring-8 BL32B2 beamline. We thank Kazue Nagano for technical assistance with protein expression, purification, and crystallization. We also thank Kunio Atsumi and Takashi Ida for discussions and comments on the manuscript.
Footnotes
Published ahead of print on 27 August 2007.
REFERENCES
- 1.Alvarez-Sala, J. L., P. Kardos, J. Martinez-Beltran, P. Coronel, and L. Aguilar. 2006. Clinical and bacteriological efficacy in treatment of acute exacerbations of chronic bronchitis with cefditoren-pivoxil versus cefuroxime-axetil. Antimicrob. Agents Chemother. 50:1762-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asahi, Y., Y. Takeuchi, and K. Ubukata. 1999. Diversity of substitutions within or adjacent to conserved amino acid motifs of penicillin-binding protein 2X in cephalosporin-resistant Streptococcus pneumoniae isolates. Antimicrob. Agents Chemother. 43:1252-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barcus, V. A., K. Ghanekar, M. Yeo, T. J. Coffey, and C. G. Dowson. 1995. Genetics of high level penicillin resistance in clinical isolates of Streptococcus pneumoniae. FEMS Microbiol. Lett. 126:299-303. [DOI] [PubMed] [Google Scholar]
- 4.Clark, C. L., K. Nagai, B. E. Dewasse, G. A. Pankuch, L. M. Ednie, M. R. Jacobs, and P. C. Appelbaum. 2002. Activity of cefditoren against respiratory pathogens. J. Antimicrob. Chemother. 50:33-41. [DOI] [PubMed] [Google Scholar]
- 5.Contreras-Martel, C., V. Job, A. M. Di Guilmi, T. Vernet, O. Dideberg, and A. Dessen. 2006. Crystal structure of penicillin-binding protein 1a (PBP1a) reveals a mutational hotspot implicated in beta-lactam resistance in Streptococcus pneumoniae. J. Mol. Biol. 355:684-696. [DOI] [PubMed] [Google Scholar]
- 6.Dowson, C. G., T. J. Coffey, and B. G. Spratt. 1994. Origin and molecular epidemiology of penicillin-binding-protein-mediated resistance to beta-lactam antibiotics. Trends Microbiol. 2:361-366. [DOI] [PubMed] [Google Scholar]
- 7.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126-2132. [DOI] [PubMed] [Google Scholar]
- 8.Fenoll, A., M. J. Gimenez, O. Robledo, P. Coronel, M. Gimeno, J. Casal, and L. Aguilar. 2007. Activity of cefditoren against clinical isolates of Streptococcus pneumoniae showing non-susceptibility to penicillins, cephalosporins, macrolides, ketolides or quinolones. Int. J. Antimicrob. Agents 29:224-226. [DOI] [PubMed] [Google Scholar]
- 9.Fisher, J. F., S. O. Meroueh, and S. Mobashery. 2005. Bacterial resistance to beta-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 105:395-424. [DOI] [PubMed] [Google Scholar]
- 10.Gordon, E., N. Mouz, E. Duee, and O. Dideberg. 2000. The crystal structure of the penicillin-binding protein 2x from Streptococcus pneumoniae and its acyl-enzyme form: implication in drug resistance. J. Mol. Biol. 299:477-485. [DOI] [PubMed] [Google Scholar]
- 11.Granizo, J. J., M. J. Gimenez, J. Barberan, P. Coronel, M. Gimeno, and L. Aguilar. 2006. The efficacy of cefditoren pivoxil in the treatment of lower respiratory tract infections, with a focus on the per-pathogen bacteriologic response in infections caused by Streptococcus pneumoniae and Haemophilus influenzae: a pooled analysis of seven clinical trials. Clin. Ther. 28:2061-2069. [DOI] [PubMed] [Google Scholar]
- 12.Hakenbeck, R. 1998. Mosaic genes and their role in penicillin-resistant Streptococcus pneumoniae. Electrophoresis 19:597-601. [DOI] [PubMed] [Google Scholar]
- 13.Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283-291. [Google Scholar]
- 14.Lee, M. Y., K. S. Ko, W. S. Oh, S. Park, J. Y. Lee, J. Y. Baek, J. Y. Suh, K. R. Peck, N. Y. Lee, and J. H. Song. 2006. In vitro activity of cefditoren: antimicrobial efficacy against major respiratory pathogens from Asian countries. Int. J. Antimicrob. Agents 28:14-18. [DOI] [PubMed] [Google Scholar]
- 15.Lee, W., M. A. McDonough, L. Kotra, Z. H. Li, N. R. Silvaggi, Y. Takeda, J. A. Kelly, and S. Mobashery. 2001. A 1.2- Å snapshot of the final step of bacterial cell wall biosynthesis. Proc. Natl. Acad. Sci. USA 98:1427-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lovering, A. L., L. De Castro, D. Lim, and N. C. Strynadka. 2006. Structural analysis of an “open” form of PBP1B from Streptococcus pneumoniae. Protein Sci. 15:1701-1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Macheboeuf, P., C. Contreras-Martel, V. Job, O. Dideberg, and A. Dessen. 2006. Penicillin binding proteins: key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol. Rev. 30:673-691. [DOI] [PubMed] [Google Scholar]
- 18.Macheboeuf, P., A. M. Di Guilmi, V. Job, T. Vernet, O. Dideberg, and A. Dessen. 2005. Active site restructuring regulates ligand recognition in class A penicillin-binding proteins. Proc. Natl. Acad. Sci. USA 102:577-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matthews, B. W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33:491-497. [DOI] [PubMed] [Google Scholar]
- 20.McDonough, M. A., J. W. Anderson, N. R. Silvaggi, R. F. Pratt, J. R. Knox, and J. A. Kelly. 2002. Structures of two kinetic intermediates reveal species specificity of penicillin-binding proteins. J. Mol. Biol. 322:111-122. [DOI] [PubMed] [Google Scholar]
- 21.Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240-255. [DOI] [PubMed] [Google Scholar]
- 22.Nicola, G., S. Peddi, M. Stefanova, R. A. Nicholas, W. G. Gutheil, and C. Davies. 2005. Crystal structure of Escherichia coli penicillin-binding protein 5 bound to a tripeptide boronic acid inhibitor: a role for Ser-110 in deacylation. Biochemistry 44:8207-8217. [DOI] [PubMed] [Google Scholar]
- 23.Pares, S., N. Mouz, Y. Petillot, R. Hakenbeck, and O. Dideberg. 1996. X-ray structure of Streptococcus pneumoniae PBP2x, a primary penicillin target enzyme. Nat. Struct. Biol. 3:284-289. [DOI] [PubMed] [Google Scholar]
- 24.Sakagami, K., K. Atsumi, A. Tamura, T. Yoshida, K. Nishihata, and S. Fukatsu. 1990. Synthesis and oral activity of ME1207, a new orally active cephalosporin. J. Antibiot. (Tokyo) 43:1047-1050. [DOI] [PubMed] [Google Scholar]
- 25.Sanbongi, Y., T. Ida, M. Ishikawa, Y. Osaki, H. Kataoka, T. Suzuki, K. Kondo, F. Ohsawa, and M. Yonezawa. 2004. Complete sequences of six penicillin-binding protein genes from 40 Streptococcus pneumoniae clinical isolates collected in Japan. Antimicrob. Agents Chemother. 48:2244-2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sauvage, E., C. Duez, R. Herman, F. Kerff, S. Petrella, J. W. Anderson, S. A. Adediran, R. F. Pratt, J. M. Frere, and P. Charlier. 2007. Crystal structure of the Bacillus subtilis penicillin-binding protein 4a, and its complex with a peptidoglycan mimetic peptide. J. Mol. Biol. 371:528-539. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu, A., K. Maebashi, M. Niida, T. Mikuniya, M. Hikida, and K. Ubukata. 2007. In vitro activities of oral cephem and telithromycin against clinical isolates of major respiratory pathogens in Japan. J. Korean Med. Sci. 22:20-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soriano, F., J. J. Granizo, P. Coronel, M. Gimeno, E. Rodenas, M. Gracia, C. Garcia, R. Fernandez-Roblas, J. Esteban, and I. Gadea. 2004. Antimicrobial susceptibility of Haemophilus influenzae, Haemophilus parainfluenzae and Moraxella catarrhalis isolated from adult patients with respiratory tract infections in four southern European countries. The ARISE project. Int. J. Antimicrob. Agents 23:296-299. [DOI] [PubMed] [Google Scholar]
- 29.Soriano, F., J. J. Granizo, A. Fenoll, M. Gracia, R. Fernandez-Roblas, J. Esteban, I. Gadea, P. Coronel, M. Gimeno, E. Rodenas, and F. Santos. 2003. Antimicrobial resistance among clinical isolates of Streptococcus pneumoniae isolated in four southern European countries (ARISE project) from adult patients: results from the cefditoren surveillance program. J. Chemother. 15:107-112. [DOI] [PubMed] [Google Scholar]
- 30.Tamura, A., R. Okamoto, T. Yoshida, H. Yamamoto, S. Kondo, M. Inoue, and S. Mitsuhashi. 1988. In vitro and in vivo antibacterial activities of ME1207, a new oral cephalosporin. Antimicrob. Agents Chemother. 32:1421-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vagin, A., and A. Teplyakov. 1997. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30:1022-1025. [Google Scholar]
- 32.Wellington, K., and M. P. Curran. 2004. Cefditoren pivoxil: a review of its use in the treatment of bacterial infections. Drugs 64:2597-2618. [DOI] [PubMed] [Google Scholar]
- 33.Wellington, K., and M. P. Curran. 2005. Spotlight on cefditoren pivoxil in bacterial infections. Treat. Respir. Med. 4:149-152. [DOI] [PubMed] [Google Scholar]
- 34.Yasui, K., M. Onodera, M. Sukegawa, T. Watanabe, Y. Yamamoto, Y. Murai, and K. Iinuma. Feb. 2003. A crystalline substance of cefditoren pivoxyl and the production of the same. Japan patent 3403206.