

Abstract
TGFβ induces hepatocyte apoptosis via reactive oxygen species (ROS) generation, the mitochondrial permeability transition (MPT), and caspase activation. The role of the Smad pathway in these events is unknown. In this study primary hepatocytes were isolated from Smad3 wild-type (+/+) and knockout (−/−) mice, and were treated with TGFβ (5 ng/ml) and/or trolox (2 mM). ROS generation, MPT, TGFβ-dependent transcription, and apoptosis were assessed in the presence or absence of Smad3 wild-type (WT) and dominant-negative (DN) plasmids. With TGFβ treatment, Smad3 (−/−) hepatocytes did not generate ROS activity, exhibit MPT, activate caspases, or undergo apoptosis when compared to Smad 3 (+/+) hepatocytes. Similarly, transfection of Smad3 (+/+) hepatocytes with DN-Smad3 inhibited TGFβ-mediated transcription, ROS generation, MPT, and apoptosis. However, Smad3 (−/−) cells transfected with WT-Smad3 and treated with TGFβ demonstrated increased transcriptional activity, the MPT, and TGFβ-induced apoptosis. TGFβ-mediated ROS generation occurred through an NADPH–like oxidase pathway since diphenyleneiodonium chloride inhibited ROS induction. In conclusion, TGFβ-induced hepatocyte apoptosis occurs through Smad3 dependent activation of ROS with subsequent activation of the MPT and caspases.
Keywords: transforming growth factor beta, hepatocyte, apoptosis, Smad3, reactive oxygen species
1. Introduction
Transforming growth factor beta 1 (TGFβ) is a multifunctional cytokine, which mediates hepatocellular differentiation, growth, and apoptosis [1]. After partial hepatectomy, TGFβ has an integral role in terminating liver regeneration secondary to its hepatocyte anti-proliferative effects. In vitro and in vivo studies have also shown that TGFβ controls hepatocyte growth directly by inducing apoptosis [2-4]. Furthermore, TGFβ-induced apoptosis occurs through ROS generation, the mitochondrial permeability transition (MPT), and caspase activation [5]. Moreover, Smad3-dependent cleavage of the mitochondrial, pro-apoptotic protein, BAD, results in TGFβ-induced apoptosis in FaO hepatoma cells and suggests a distinct role for Smad3 [2]. Thus, these findings suggest that the TGFβ apoptotic pathway includes ROS generation, Smad3 activation, mitochondria involvement, and caspase cleavage.
Several mechanisms by which Smad3 initiates cell death signals have been examined in a number of studies. GADD45β has been identified as a Smad-dependent, early response mediator of TGFβ-induced apoptosis [6]. In this study, AML12 murine hepatocytes underwent apoptosis in response to GADD45β activation of p38. Furthermore, GADD45β activation was dependent on Smad2, Smad3, and Smad4 activation of the proximal portion of the GADD45β promoter [6]. Additional studies have also identified a TGFβ-responsive enhancer in the third intron of GADD45β [7]. The enhancer is responsive to Smad3 and Smad4, but not Smad2. Collectively, these studies show that GADD45β expression is mediated at multiple genetic loci, and is an important Smad-dependent mediator of TGFβ-induced apoptosis that is affected through p38 expression. In addition, Smad3 regulates hepatocyte responsiveness to apoptosis by the down-regulation of the anti-apoptotic protein, Bcl-2 [8]. In a mouse model of carcinogen-induced hepatocellular cancer, forced expression of Smad3 decreased expression of Bcl-2 with resultant apoptosis and decreased cancer cell formation. Interestingly, this study also showed that expression of p38 was required for apoptosis. Furthermore, Akt has been identified as an integral regulator of Smad3 phosphorylation and, subsequently, TGFβ-mediated apoptosis [9]. Akt sequestered unphosphorylated Smad3 in the cytoplasm and, thus inhibited nuclear translocation and transcription, ultimately decreasing TGFβ-induced apoptosis. The ratio of Smad3 to Akt is an important mechanism that controls TGFβ-induced apoptosis, but not growth inhibition.
In addition to altering effector signaling pathways, TGFβ also induces ROS. Both early [5] and late activation of ROS in response to TGFB have been identified [10; 11]. In lung fibroblasts, TGFβ induced NADH oxidase production of hydrogen peroxide begins 8 hours after treatment with peak activity at 16 hours [10]. Additionally, Sanchez et al. demonstrated that TGFβ induced ROS occurred 4 hours after TGFβ stimulation in fetal rat hepatocytes [11]. Both studies demonstrated that ROS generation was dependent on new protein synthesis. Other studies have demonstrated that early (90 minutes) activation of ROS is important for TGFβ-induced apoptosis [5]. However, no study has demonstrated the importance of Smad activation in ROS generation following TGFβ stimulation. Moreover, a recent investigation of hepatocyte specific deletion of Smad2 suggests that this TGFβ-induced protein is not required for apoptosis [12].
Although Smad proteins have been associated with TGFβ-induced hepatocyte growth control, the relationship between ROS generation and activation of Smad proteins has not been examined. Importantly, how ROS and Smad3 affect TGFβ-induced apoptosis has not been determined. The purpose of this study was to determine if Smad3 and ROS are required for TGFβ-induced apoptosis, and, if so, how these signaling factors influence TGFβ-induced cell death. Herein, the role of Smad3 in the TGFβ apoptotic pathway was determined in Smad3 wild-type (+/+) and knockout (−/−) primary murine hepatocytes by evaluating the mediators of the apoptotic pathway: ROS, MPT, and caspase activation.
2. Materials and Methods
2.1 Hepatocyte Harvest and Culture
Smad3 (+/+) and Smad3 (−/−) mice were kindly obtained from David Brenner, M.D. [13]. These Smad3 deficient mice have a deletion in exon 1 of the Smad3 gene [14]. Hepatocytes from Smad3 (+/+) and Smad3 (−/−) mice were isolated by retrograde collagenase perfusion [15]. Briefly, the liver was perfused with 100 ml of Krebs-Ringer-Hepes (KRH) buffer containing 0.25 M HEPES, 115 mM NaCl, 50 mM KCl, 10 mM KH2PO4, and 0.5 mM EGTA at pH 7.4. A collagenase buffer (200 ml) containing 1 mM CaCl2 and 0.4 mg/ml type 1 collagenase was perfused into the liver. The liver was excised and gently combed to manually disperse hepatocytes. Hepatocytes were then selected by differential centrifugation. Cell viability was assessed by trypan blue exclusion (>90% for experiments). Hepatocytes were plated in Waymouth's media supplemented with 10% fetal calf serum, 5 μg/ml insulin, and 100 nmol/L dexamethasone for four hours. The medium was changed overnight to a serum-free, hormonally-defined medium (HDM) consisting of RPMI 1640 containing insulin (5 μg/ml), transferrin (5 μg/ml), selenium (3 μM), and free fatty acid (1.5 μM). Subsequent hepatocyte experiments were performed under these conditions.
In select experiments, hepatocytes were pretreated with trolox (2 mM), actinomycin D (1 μg/ml), cycloheximide, (10 mg/ml) or diphenyleneiodonium chloride (DPI; 1 mM) before TGFβ (5 ng/ml) or tumor necrosis factor alpha (TNFα; 30 ng/ml) treatment was initiated.
2.2 JAR Cell Culture
Smad3 deficient JAR cell line was purchased from ATCC (HTB-144) and cultured in RPMI 1640 medium containing 2 mM L-glutamine, 17.8 mM NaHCO3, 30 mM glucose, 10 mM HEPES, 1 mM sodium pyruvate, and 10% fetal bovine serum in humidified 5% CO2 / 95% air at 37°C [16].
2.3 Morphologic Assessment of Apoptosis
Hepatocytes were fixed and permeabilized in a 3:1 methanol/acetic acid for 10 minutes at 4°C, and stained with 2 mg/ml propidium iodide (PI) following 0, 24, 36, and 48 hours of incubation with TGFβ. Hepatocytes were visualized under green UV light using an IX-70 Olympus microscope (Olympus, Tokyo, Japan), and the number of condensed nuclei indicative of apoptotic hepatocytes was quantified in 5 high powered fields (hpf) at 400× magnification [17].
2.4 Caspase-3 and Caspase-8 Activity
As described previously, caspase-3 and caspase-8 activities were determined with the substrates DETD-AFC and LEVD-AFC, respectively, by fluorometric analysis on a Perkin Elmer Luminescence Spectrometer LS50B (Perkin Elmer; Norwalk, CT) [3]. Hepatocytes were plated at a density of 1.5 × 106 in a 60 mm dish and lysed in a 1× lysis buffer (10 mM HEPES, 2 mM EDTA, 0.1% CHAPS, 5 mM DTT, 1 mM PMSF, 10 μg/ml pepstatin A, 10 μg/ml aprotinin, 20 μg/ml leupeptin) by five rapid freeze/thaw cycles. Lysates were incubated in the dark at 37°C for four hours with 25 μM of the caspase substrate. The rate of caspase cleavage was determined by a standard curve, and then normalized for protein concentration.
2.5 Reactive Oxygen Species Activity
Hepatocytes and JAR cells were plated at 1.8 × 105 per well in a 12-well plate, rinsed with PBS, and incubated with 10 μM 2',7'-dichlorodihydrofluorescein diacetate (Molecular Probes, Eugene, OR) for 20 minutes at 37°C in the presence of TGFβ (5 ng/ml), trolox (2 mM), actinomycin D (1 μg/ml), and/or cycloheximide (10 mg/ml). Fluorescence activity was determined on a fluormetric BMG Labtechnologies plate reader (Durham, NC) with a gain of 30 nm and 529 nm emission and 485 nm excitation wavelengths, respectively.
2.6 Mitochondrial Permeability Transition (MPT)
The MPT was assessed by confocal microscopy with the dyes tetramethylrhodamine methyl ester (250 nM; TMRM, Molecular Probes) and calcein (1 μM Molecular Probes) as described previously [5]. Hepatocytes (1 × 106 in a 60 mm dish) plated on collagen-coated coverslips were loaded with the dyes for 20 minutes at 37°C, and then rinsed with PBS. Calcein and TMRM fluorescence was captured with an argon laser at 488 nm and 568 nm, respectively, using a 60× objective.
2.7 Plasmid Transient Transfections
Hepatocytes were plated at 1.8 × 105 per well on 6-well plates, and transfection with 1 μg DNA accomplished with 2.5 μl of Targafect F-1 (Targeting Systems, San Diego, CA) in 1 ml Opti-Mem1 (or DMEM for apoptosis experiments) for 2 hours. We assessed transfection efficiency using Targafect F-1 and a GFP expressing plasmid with 30% transfection efficiency.
Full-length human FLAG-tagged Smad3 and dominant negative Smad3 cloned into a pRK5 vector were generous gifts from Dr. Xing Zhang (UCSF and Genentech, South San Francisco, CA). DN-Smad3 is truncated at the carboxy terminus. The polylinker region of pcDNA3.1(-) (Invitrogen, Carlsbad, CA) was modified to introduce a unique ClaI site by inserting a novel oligonucleotide pair with BamHI and HindIII overhangs into the BamHI and HindIII sites. The sequences of the oligonucleotides were as follows: 5' GAT CCG CAA TCG ATG CAG CAA 3'. The oligos were annealed at 65°C for 10 minutes and ligated into the BamHI and HindIII digested vector. FLAG-tagged Smad3 in pRK5 was digested with ClaI and HindIII, gel purified and cloned into the ClaI/HindIII digested vector. FLAG-tagged DN-Smad3 in pRK5 was digested with EcoRI and HindIII, gel purified and cloned into the EcoRI/HindIII digested pcDNA3.1.
2.8 Transcriptional Activity
TGFβ-induced transcriptional activity was assessed with the luciferase assay system (Promega, Madison, WI) by transiently transfecting the p3TP-luciferase reporter plasmid containing multiple Smad binding elements in the promoter region. Hepatocytes were treated with TGFβ for 24 hours and luciferase activity was assessed via a luminometer.
2.9 Statistical Analysis
Each experiment was performed with hepatocytes from a minimum of three mice. Assays were performed in triplicate. Outcomes are reported as the mean plus or minus the standard error of the mean. Statistical analysis was determined using the Student's t-test and significance was set at p<0.05.
3. Results
3.1 Smad3 (−/−) Hepatocytes Resist TGFβ-Induced Apoptosis
To determine the necessity of Smad3 in TGFβ-induced apoptosis, Smad3 (+/+) and Smad3 (−/−) hepatocytes were treated with or without TGFβ for 48 hours, and apoptosis was assessed by PI stain. After 48 hours of treatment, 43% ± 0.06% of Smad3 (+/+) hepatocytes had morphologic evidence of apoptosis (Fig. 1A & 1B). However, Smad3 (−/−) hepatocytes treated with TGFβ showed only 7.6% ± 0.01% apoptosis (p<0.05), which was similar to untreated Smad3 hepatocytes (Fig. 1A & 1B).
Figure 1.
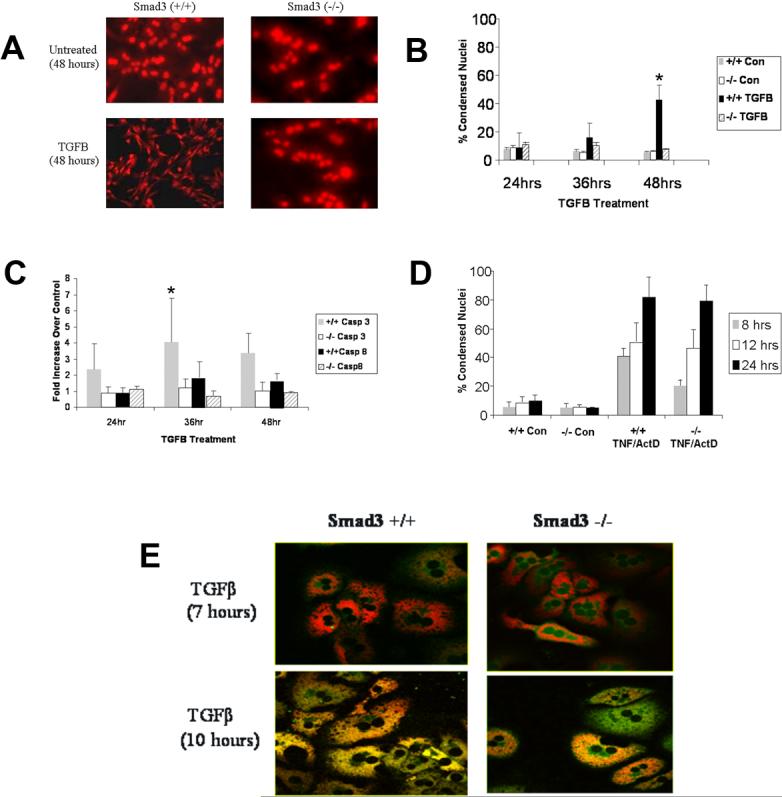
Smad3 (+/+) or (−/−) hepatocytes were treated with or without TGFβ (5 ng/ml), TNFα (30 ng/ml), or actinomycin D (1 μg/ml). Apoptosis was determined morphologically by propidium iodide staining and biochemically by caspase activity. (A) Smad3 (+/+) and (−/−) hepatocytes were untreated or treated with TGFβ and morphology was assessed at 48 hours by PI staining. Smad3 (+/+) hepatocytes displayed condensed nuclei and cell shrinkage consistent with apoptosis whereas Smad3 (−/−) hepatocytes appeared similar to controls. (B) Hepatocyte apoptosis was quantified by counting the number of condensed nuclei per 5 high power fields and expressed as percent condensed nuclei over time. Smad3 (+/+) hepatocytes had significantly increased (*p<0.05) apoptosis at 48 hours of TGFβ treatment compared to Smad3 (−/−) hepatocytes. (C) Apoptosis was assessed by caspase activity assay for caspase-3 and caspase-8 activation expressed as fold increase over control at 24, 36, and 48 hours. Caspase-3 activity was increased significantly (*p<0.05) in Smad3 (+/+) hepatocytes compared to Smad (−/−) hepatocytes at 36 hours. Caspase-8 activity demonstrated a similar pattern, but was less robust. (D) To determine if inhibition of apoptosis in Smad3 (−/−) mice was specific for TGFβ, Smad3 (+/+) and (−/−) hepatocytes were treated with or without TNFα and actinomycin D (ActD) and apoptosis was determined. Both Smad3 (+/+) hepatocytes and Smad3 (−/−) hepatocytes readily underwent TNFα-induced apoptosis (no significant difference). (E) To determine if Smad3 (+/+) and (−/−) hepatocytes underwent MPT in response to TGFβ treatment; these hepatocytes were loaded with the fluorophores TMRM (red) and calcein (green). After 10 hours of treatment, Smad3 (+/+) hepatocytes demonstrated mitochondrial loss of TMRM and uptake of calcein consistent with the MPT. Smad3 (−/−) hepatocytes, however, maintained mitochondrial integrity indicating resistance to TGFβ-induced MPT.
Caspase-3 and caspase-8 activities were measured to confirm TGFβ-induced apoptosis in Smad3 (+/+) hepatocytes, and to assess caspase activation in Smad3 (−/−) hepatocytes. Smad3 (+/+) hepatocytes treated with TGFβ had a 3.4-fold ± 1.2 increase (p<0.05) in caspase-3 activity at 36 hours as compared to untreated Smad3 (+/+) hepatocytes (Fig. 1C). Smad3 (−/−) hepatocytes did not generate caspase-3 activity over control values following treatment with TGFβ, thus confirming the lack of caspase-induced apoptosis (Fig. 1C). Similarly, caspase-8 activity was increased in Smad3 (+/+) hepatocytes with a 1.7-fold ± 0.41 increase at 36 hours as compared to the control treated Smad3 (+/+) hepatocytes. Smad3 (−/−) hepatocytes had no caspase-8 activity induction with TGFβ treatment as compared to untreated controls (Fig. 1C). These findings suggest that Smad3 (−/−) hepatocytes do not undergo apoptosis in response to TGFβ treatment because of lack of activation of the caspase pathway.
To determine if Smad3 (−/−) hepatocyte resistance to apoptosis was specific to TGFβ, we treated Smad3 (+/+) and Smad3 (−/−) hepatocytes with TNFα following inhibition of NFκB with actinomycin D. Both Smad3 (+/+) and Smad3 (−/−) hepatocytes were susceptible to TNFα-induced apoptosis. At 12 hrs, TNFα/ActD-treated Smad3 (+/+) hepatocytes showed 50.9% ± 13.3% apoptosis versus 8.4% ± 4.2% for untreated controls, and Smad3 (−/−) hepatocytes exhibited 46.3% ± 13% apoptosis versus 5.7% ± 1.7% for untreated controls (Fig. 1D). These findings suggest that the requirement for Smad3 is specific for TGFβ-induced hepatocyte apoptosis.
Typically, TGFβ causes hepatocyte MPT prior to apoptosis [5]. In these experiments, MPT was assessed by confocal microscopy using the dyes TMRM and calcein. Seven to 10 hours after TGFβ treatment, Smad3 (+/+) hepatocytes underwent MPT, visualized by TMRM exiting the mitochondria and calcein entering the mitochondria with the opening of the conductance pore. Untreated Smad3 (+/+) hepatocytes (data not shown) did not undergo the MPT nor did Smad3 (−/−) hepatocytes treated with TGFβ (Fig. 1E).
3.2 Role of ROS in TGFβ-Mediated Apoptosis
Following TGFβ treatment, ROS generation was examined in Smad3 (+/+) hepatocytes by fluorometric analysis for cleavage of the probe H2DCFDA. Compared to untreated Smad3 (+/+) cells, those treated with TGFβ demonstrated a 39% to 81% increase (p<0.05) in ROS with maximal activity between 30 min and one hour of treatment. Smad3 (−/−) hepatocytes treated with TGFβ had no increase in ROS, but demonstrated up to a 51% decrease in ROS activity at 60 minutes, suggesting that ROS increase after TGFβ is dependent on Smad3 (Fig. 2A). Furthermore, ROS activity remained increased in Smad3 (+/+) hepatocytes for 4 hours (Fig. 2A).
Figure 2.

The effect of ROS generation in Smad3 (+/+) and (−/−) was determined from 30−240 minutes following TGFβ treatment. (A) Smad3 (+/+) hepatocytes demonstrated consistently increased ROS activity compared to Smad3 (−/−) hepatocytes (*p<0.05 for the curves). A consistent burst of ROS activity was noted at 30−60 minutes after treatment, but the magnitude of the activity was variable. (B) ROS generation in Smad3 deficient JAR cells was fluorometrically determined. In contrast to Smad (+/+) hepatocytes, JAR cells did not increase ROS after TGFβ treatment (p<0.05). (C) Smad3 (+/+) hepatocytes were treated with the antioxidant, trolox, alone or in combination with TGFβ to determine if TGFβ-induced ROS activity could be inhibited. At 60 minutes, TGFβ treatment increased ROS generation significantly, and pretreatment with trolox attenuated markedly (*p<0.05) this burst of activity compared to TGFβ alone. (D) To determine the effect of inhibition of TGFβ-induced ROS on apoptosis, Smad3 (+/+) hepatocytes were treated with trolox, TGFβ, or trolox and TGFβ and apoptosis was assessed morphologically at 48 hours. Inhibition of ROS generation by trolox decreased significantly (*p<0.05) TGFβ-induced apoptosis. (E) The dependence of Smad3-induced ROS generation on transcription and translation was determined by pretreating Smad3 (+/+) hepatocytes with either actinomycin D (10 μg/ml) or cycloheximide (10 mg/ml) and measuring H2DCFDA cleavage at 60 minutes. Neither actinomycin D nor cycloheximide decreased early ROS generation suggesting that gene transcription or protein synthesis is not required for ROS activity following TGFβ treatment. (F) The involvement of NADPH oxidase-like system in ROS increase was investigated in the presence of diphenyleneiodonium chloride (DPI). Hepatocytes were treated with 1 μM of DPI for 30 min prior to TGFβ administration and ROS generation was determined by H2DCFDA fluorometry. DPI prevented TGFβ-induced increase in ROS. (G) Smad3 (+/+) and (−/−) hepatocytes were transiently transfected with a TGFβ-responsive luciferase reporter plasmid, p3TPLuc, and subsequently treated with 5 ng/ml TGFβ. In additional experiments, following transfection of the reporter plasmid, Smad3 (+/+) hepatocytes were transfected with a DN-Smad3 plasmid and Smad3 (−/−) hepatocytes transfected with a WT-Smad3 plasmid and luciferase measured after stimulation with TGFβ. Treatment of Smad3 (+/+) hepatocytes with TGFβ alone resulted in significantly increased luciferase activity compared to Smad3 (−/−) hepatocytes. However, transfection of Smad3 (+/+) hepatocytes with the DN-Smad3 followed by TGFβ treatment (light gray bar) decreased markedly transcriptional activity. Conversely, transfection of Smad3 (−/−) hepatocytes with WT-Smad3 followed by TGFβ treatment significantly increased luciferase activity (light gray bar).
The importance of Smad3 in TGFβ-mediated ROS increase was also investigated in Smad3 deficient JAR cells [7]. JAR cells did not increase ROS in the presence of TGFβ (Fig. 2B), further suggesting the need of Smad3 for TGFβ-induced ROS generation.
In addition, Smad3 (+/+) hepatocytes were pretreated with the anti-oxidant, trolox (2 mM), and ROS activity and apoptosis were assessed. Smad3 (+/+) hepatocytes pretreated with trolox with subsequent TGFβ administration showed no increase in ROS compared to TGFβ-only treated hepatocytes, which had a 1.38 ± 0.15-fold increase in ROS activity at 60 minutes (Fig. 2C). Apoptosis was assessed in Smad3 (+/+) hepatocytes pretreated with trolox, and only 6.4% ± 1.5% of the cells were apoptotic in response to TGFβ as compared to 55% ± 11% apoptosis in Smad3 (+/+) hepatocytes that were not pretreated with trolox (Fig. 2D). Trolox also decreased TGFβ-induced caspase-3 activity by 55% at 48 hours (data not shown).
Because it appeared that Smad3 is required for TGFβ-induced acute generation of ROS, we wished to determine if this finding was dependent on protein synthesis. Smad3 (+/+) hepatocytes were pretreated with actinomycin D (1−10 μg/ml) or cycloheximide (10 μg/ml) one hour before TGFβ treatment and ROS generation was determined. A dose-response study revealed that lower concentrations of actinomycin D (1−10 μg/ml) and cycloheximide (1−10 μg/ml) did not compromise hepatocyte viability, whereas higher concentrations were cytotoxic (data not shown). At the 10 μg/ml doses used in this study, hepatocytes were viable and multiple previous studies show that transcription and translation are inhibited at these concentrations in hepatocytes [18; 19]. Smad3 (+/+) hepatocytes pretreated with actinomycin D prior to TGFβ treatment did not have decreased ROS following treatment (Fig 2E). This observation demonstrated that Smad3 transcriptional activity is not necessary for TGFβ-induced ROS (Fig. 2E). Similar findings were obtained with cycloheximide pretreatment.
In order to investigate whether a NADPH-like system is involved in Smad3-dependent ROS increase after TGFβ treatment, ROS formation was fluorometrically evaluated in the presence and absence of 1 μM of diphenyleneiodonium chloride (DPI), a flavoprotein inhibitor of NADPH oxidase [20]. Pretreatment of hepatocytes with DPI prevented TGFβ-induced ROS increase, suggesting that this system is required for TGFβ-induced ROS generation (Fig. 2F).
To demonstrate Smad3 involvement in TGFβ-induced gene transcription and assess the function of wild-type and dominant-negative Smad3 plasmids, Smad3 hepatocytes were transiently transfected with p3TP-Luc, a TGFβ responsive reporter plasmid that contains multiple Smad binding elements. TGFβ treatment induced transcriptional activity of p3TP-Luc was 6.21 ± 2.08-fold in TGFβ-treated Smad3 (+/+) hepatocytes as compared with untreated cells (Fig. 2G). Smad3 (−/−) hepatocytes showed a decreased response as compared to Smad3 (+/+) hepatocytes to transfection with the reporter plasmid.
Transfection of Smad3 (+/+) hepatocytes with a dominant-negative Smad3 (DN) plasmid inhibited transcriptional activity in response to TGFβ treatment (Fig. 2G). In Smad3 (−/−) hepatocytes, transfection with Smad3 WT-plasmid transfection increased TGFβ responsive reporter activity compared to untreated and TGFβ treated Smad3 (−/−) hepatocytes (Fig. 2G).
3.3 DN-Smad3 Plasmid Inhibits TGFβ-Induced Apoptosis in Smad3 (+/+) Hepatocytes
To confirm that the TGFβ apoptotic signaling pathway is Smad3 dependent, Smad3 (+/+) hepatocytes were transiently transfected with a DN-Smad3 expressing plasmid and apoptosis was subsequently determined. After 48 hours of TGFβ treatment, Smad3 (+/+) hepatocytes transiently transfected with the DN-Smad3 plasmid demonstrated 22.7% ± 6.1% apoptosis versus 56.6% ± 15.8% apoptosis in those cells transiently transfected with the control vector (Fig. 3A).
Figure 3.
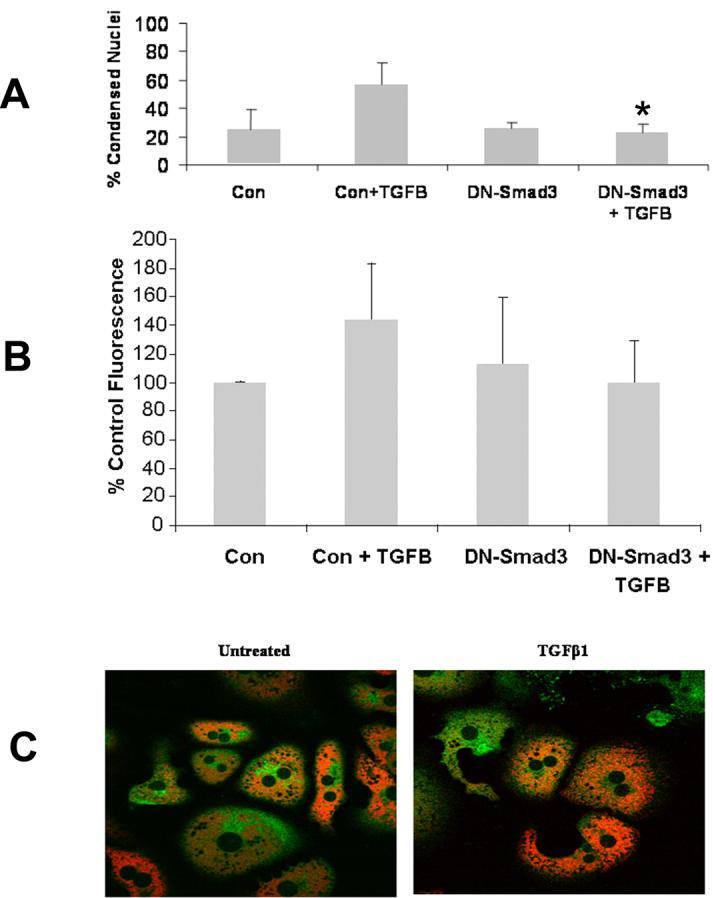
Smad3 (+/+) hepatocytes were transiently transfected with control vector or the DN-Smad3 vector and subsequently treated with or without TGFβ prior to assessment of apoptosis, ROS generation, and MPT. (A) Smad3 (+/+) hepatocytes treated with 5 ng/ml TGFβ following transfection with DN-Smad3 demonstrated significantly decreased apoptosis (*p<0.05) as compared to identical cells transfected with the control vector and treated with TGFβ. (B) TGFβ-induced ROS in Smad3 (+/+) hepatocytes transfected with DN-Smad3 exhibited ROS generation similar to controls. (C) The MPT was assessed by confocal microscopy in Smad3 (+/+) hepatocytes transfected with the DN-Smad3 vector and subsequently treated with TGFβ. Transfection with DN-Smad3 inhibited the MPT following TGFβ treatment in Smad3 (+/+) hepatocytes.
Furthermore, Smad3 (+/+) hepatocytes transiently transfected with the DN-Smad3 plasmid blocked important mediators of the TGFβ apoptotic pathway including ROS generation and occurrence of MPT. Smad3 (+/+) hepatocytes transfected with the control vector and treated with TGFβ had a 56% increase in ROS activity at 60 minutes, whereas Smad3 (+/+) hepatocytes transfected with DN-Smad3 did not differ from controls (Fig. 3B). Moreover, the MPT, which normally occurs between 7 and 10 hours of TGFβ treatment in Smad3 (+/+) hepatocytes was not present through 12 hours of TGFβ treatment in hepatocytes transfected with DN-Smad3 (Fig. 3C).
3.4 WT-Smad3 Plasmid Mediates TGFβ-Induced Apoptosis in Smad3 (−/−) Hepatocytes
To determine if Smad3 replacement was sufficient enough to permit TGFβ-induced apoptosis in Smad3 (−/−) hepatocytes, these cells were transiently transfected with WT-Smad3 plasmid and apoptosis was then determined. Smad3 (−/−) hepatocytes transiently transfected with WT-Smad3 plasmid alone had 22.7% ± 7.3% apoptosis after 48 hours, while the addition of TGFβ increased the percent of apoptosis to 40.6% ± 4.6% (Fig. 4A).
Figure 4.
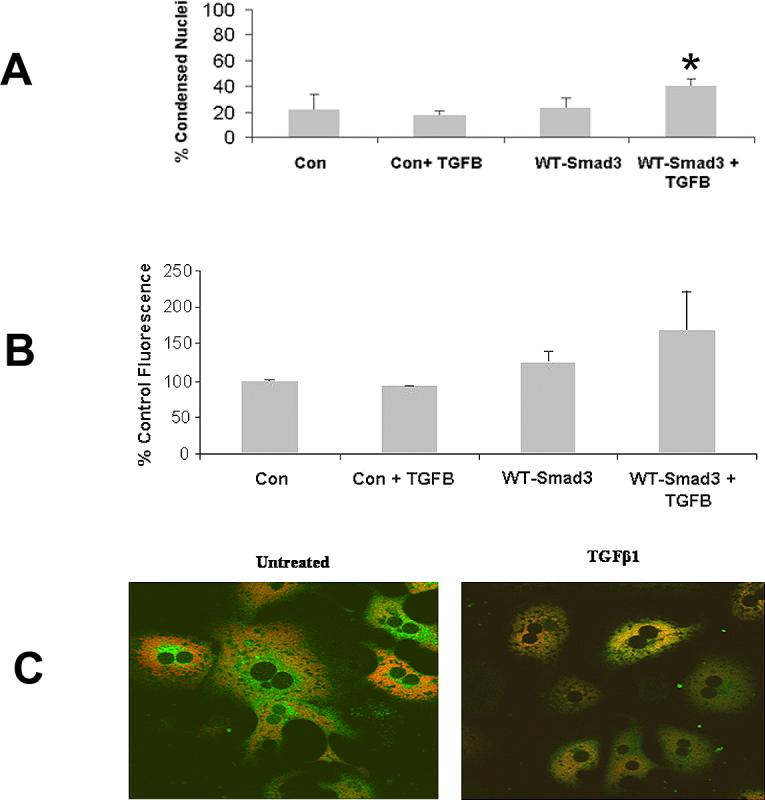
Smad3 (−/−) hepatocytes were transiently transfected with either control vector or vector expressing WT-Smad3. Subsequently, apoptosis, ROS generation, and MPT were determined. (A) Smad3 (−/−) hepatocytes transiently transfected with WT-Smad3 and subsequently treated with TGFβ demonstrated significantly increased apoptosis (*p<0.05) compared to identical cells transfected with control vector and treated with TGFβ. (B) ROS was determined in Smad3 (−/−) hepatocytes transiently transfected with WT-Smad3 and subsequently treated with TGFβ. Transfection with the WT-Smad3 partially restored ROS generation in these hepatocytes. (C) Smad3 (−/−) hepatocytes transiently transfected with WT-Smad3 and treated with TGFβ showed MPT 12 hours following treatment. Control transfected Smad3 (−/−) hepatocytes treated with TGFβ failed to undergo to MPT.
To determine if ROS and MPT were altered by the WT-Smad3 plasmid, Smad3 (−/−) hepatocytes were transiently transfected with WT-Smad3 and ROS, and MPT was subsequently assessed. As expected, Smad3 (−/−) hepatocytes did not generate increased ROS activity with TGFβ treatment, but the introduction of WT-Smad3 into hepatocytes treated with TGFβ increased ROS activity by 55% as compared to cells treated with the control vector (Fig. 4B).
Similar to non-transfected Smad3 (−/−) hepatocytes, those cells transiently transfected with the control vector did not undergo MPT with or without TGFβ treatment (data not shown). However, TGFβ treatment for 12 hours induced MPT in Smad3 (−/−) hepatocytes transfected with the WT-Smad3 plasmid, but cells treated with TGFβ only did not exhibit the MPT (Fig. 4C).
4. Discussion
Hepatocyte apoptosis is one of the mechanisms by which TGFβ regulates liver growth. Alterations in the TGFβ signal transduction pathway, including Smad defects, may result in carcinogenic mutations. Examining the role of the Smad protein family in TGFβ-induced apoptosis may provide further insight into the growth capabilities of abnormal cells and cancers. In the present study, a direct relationship between Smad3 and mediators of the TGFβ apoptotic pathway were examined in primary murine hepatocytes from Smad3 (+/+) and Smad3 (−/−) mice. These data support a critical role for Smad3 in TGFβ-induced apoptosis because Smad3 (−/−) hepatocytes failed to undergo programmed cell death. Furthermore, Smad3 appears to be critical in the initiation of apoptosis since caspase-8 activity was absent in the Smad3 (−/−) hepatocytes. Smad3 (−/−) hepatocytes also failed to generate ROS, a necessary intermediary for TGFβ-induced hepatocyte apoptosis. Importantly, these early events were transcription independent and could be reversed by introducing Smad3 into Smad3 (−/−) hepatocytes with a wild-type Smad3 expressing plasmid. Therefore, Smad3 appears to play a crucial role in the TGFβ-induced hepatocyte apoptotic pathway.
Previous work has demonstrated that TGFβ-induced hepatocyte apoptosis occurs in a ROS, MPT, and caspase-dependent apoptotic pathway that has no clear association with the Smad signal transduction pathway [5]. These experiments provide evidence that the TGFβ apoptotic pathway is dependent on Smad3. Smad3 (+/+) hepatocytes treated with TGFβ displayed ROS generation, MPT, caspase activation, and apoptosis while Smad3 (−/−) hepatocytes did not. In addition, Smad3 (+/+) hepatocytes transiently transfected with DN-Smad3 showed decreased apoptosis compared to controls whereas Smad3 (−/−) hepatocytes transiently transfected with WT-Smad3 exhibited an increase in apoptosis, thus indicating that Smad3 is critical in the TGFβ-induced apoptotic pathway. These findings are also consistent with previous studies demonstrating a role for Smad3 in apoptosis in non-primary cell lines [2; 21].
Although the necessity of Smad3 in TGFβ-induced apoptosis has been established, the mechanism of action or point of interaction in the apoptotic pathway has not been previously elucidated. Smad3 appears to be critical to the generation of ROS because Smad3 (−/−) hepatocytes did not demonstrate acute generation of oxygen intermediates. Furthermore, in JAR cells that lack Smad3, there was no generation of ROS in response to TGFβ suggesting that Smad3 is required for ROS generation in response to this cytokine. ROS generation is an early upstream event in TGFβ-induced apoptosis, and inhibition of ROS with trolox prevents apoptosis, MPT, and caspase activity [5]. Oxidative stress is a known mediator of apoptosis, including TGFβ-induced apoptosis, and to more closely exam a potential source of ROS we treated hepatocytes with DPI, an inhibitor of the NADPH-like oxidase system. DPI inhibited the generation of ROS in response to TGFβ suggesting that Smad3 interacts with the NADPH-like oxidase system to produce ROS after treatment with TGFβ. The mechanism(s) through which Smad3 interacts with NADPH-like oxidase remain unknown, but previous work has demonstrated that Smad2/3, NADPH-oxidase, and ERK1/2 may interact to alter ROS [22]. These findings are also supported by previous work that demonstrates the rule of the NADPH-like oxidase system in hepatocyte ROS generation [20]. Furthermore, in TGFβ-mediated apoptosis, ROS generation in L1210 leukemic cells occurs at 60 minutes, similar to our findings in primary murine hepatocytes [23]. Likewise, Herrera et al. found an increase in ROS production by confocal microscopy at 8 hours of TGFβ treatment in fetal hepatocytes [24].
Acute ROS generation is an early event (30 minutes) in this model of TGFβ-induced apoptosis. Importantly, this work also demonstrates that ROS generation 30−60 minutes following TGFβ treatment is independent of new protein synthesis. Our experiments with actinomycin D and cycloheximide demonstrate that the early ROS peak after TGFβ treatment persists with inhibition of new protein synthesis. This finding differs from the previous work of other authors [10; 11], which demonstrates that ROS production required protein synthesis. However, these two studies examined ROS generation from 4−16 hours after treatment, and this time lag may explain, in part, why the current study differs from the previously published work. Additional work in our laboratory demonstrates that new protein synthesis is not required for early TGFβ mediated transcriptional activation. However, protein synthesis is required for sustained TGFβ induced Smad3 expression. Nonetheless, the time course of apoptosis is long and likely dependent on additional factors. In addition, because MPT occurs at ∼10 hours and caspase activation is not evident until 36−48 hours after treatment; other factors are likely to be necessary for the convergence of ROS generation, MPT, and caspase activation. Therefore, the time lapse between ROS generation, MPT, and cytochrome c release may be due to mitochondrial-dependent events.
5. Conclusion
In summary, Smad3 is a primary mediator of TGFβ-induced hepatocyte apoptosis through ROS generation with subsequent mitochondrial depolarization and caspase activation. Smad3-dependent activities do not require protein synthesis, and ROS generation appears to be through a NADPH-like oxidase pathway. These findings suggest interactions between the traditional apoptotic pathway including receptor activation, mitochondrial depolarization, and caspase activation. The TGFβ-induced Smad signal transduction pathways are complex and may involve direct interactions rather than transcriptionally-regulated events.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Massague J. TGF-beta signal transduction. Annu.Rev.Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 2.Kim BC, Mamura M, Choi KS, Calabretta B, Kim SJ. Transforming growth factor beta 1 induces apoptosis through cleavage of BAD in a Smad3-dependent mechanism in FaO hepatoma cells. Mol.Cell Biol. 2002;22:1369–1378. doi: 10.1128/mcb.22.5.1369-1378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schrum LW, Bird MA, Salcher O, Burchardt ER, Grisham JW, Brenner DA, Rippe RA, Behrns KE. Autocrine expression of activated transforming growth factor-beta(1) induces apoptosis in normal rat liver. Am.J.Physiol Gastrointest.Liver Physiol. 2001;280:G139–G148. doi: 10.1152/ajpgi.2001.280.1.G139. [DOI] [PubMed] [Google Scholar]
- 4.Shima Y, Nakao K, Nakashima T, Kawakami A, Nakata K, Hamasaki K, Kato Y, Eguchi K, Ishii N. Activation of caspase-8 in transforming growth factor-beta-induced apoptosis of human hepatoma cells. Hepatology. 1999;30:1215–1222. doi: 10.1002/hep.510300503. [DOI] [PubMed] [Google Scholar]
- 5.Black D, Bird MA, Samson CM, Lyman S, Lange PA, Schrum LW, Qian T, Lemasters JJ, Brenner DA, Rippe RA, Behrns KE. Primary cirrhotic hepatocytes resist TGFbeta-induced apoptosis through a ROS-dependent mechanism. J.Hepatol. 2004;40:942–951. doi: 10.1016/j.jhep.2004.02.031. [DOI] [PubMed] [Google Scholar]
- 6.Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ, Jr., Liebermann DA, Bottinger EP, Roberts AB. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J.Biol.Chem. 2003;278:43001–43007. doi: 10.1074/jbc.M307869200. [DOI] [PubMed] [Google Scholar]
- 7.Major MB, Jones DA. Identification of a gadd45beta 3' enhancer that mediates SMAD3- and SMAD4-dependent transcriptional induction by transforming growth factor beta. J.Biol.Chem. 2004;279:5278–5287. doi: 10.1074/jbc.M311517200. [DOI] [PubMed] [Google Scholar]
- 8.Yang YA, Zhang GM, Feigenbaum L, Zhang YE. Smad3 reduces susceptibility to hepatocarcinoma by sensitizing hepatocytes to apoptosis through downregulation of Bcl-2. Cancer Cell. 2006;9:445–457. doi: 10.1016/j.ccr.2006.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conery AR, Cao Y, Thompson EA, Townsend CM, Jr., Ko TC, Luo K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat.Cell Biol. 2004;6:366–372. doi: 10.1038/ncb1117. [DOI] [PubMed] [Google Scholar]
- 10.Thannickal VJ, Fanburg BL. Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor beta 1. J.Biol.Chem. 1995;270:30334–30338. doi: 10.1074/jbc.270.51.30334. [DOI] [PubMed] [Google Scholar]
- 11.Sanchez A, Alvarez AM, Benito M, Fabregat I. Cycloheximide prevents apoptosis, reactive oxygen species production, and glutathione depletion induced by transforming growth factor beta in fetal rat hepatocytes in primary culture. Hepatology. 1997;26:935–943. doi: 10.1002/hep.510260420. [DOI] [PubMed] [Google Scholar]
- 12.Ju W, Ogawa A, Heyer J, Nierhof D, Yu L, Kucherlapati R, Shafritz DA, Bottinger EP. Deletion of Smad2 in mouse liver reveals novel functions in hepatocyte growth and differentiation. Mol.Cell Biol. 2006;26:654–667. doi: 10.1128/MCB.26.2.654-667.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schnabl B, Kweon YO, Frederick JP, Wang XF, Rippe RA, Brenner DA. The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology. 2001;34:89–100. doi: 10.1053/jhep.2001.25349. [DOI] [PubMed] [Google Scholar]
- 14.Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF. Targeted disruption of Smad3 reveals an essential role in transforming growth factor beta-mediated signal transduction. Mol.Cell Biol. 1999;19:2495–2504. doi: 10.1128/mcb.19.4.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arias I, Boyer J, Fausto N. The liver: biology and pathobiology. Raven Press; New York: 1994. [Google Scholar]
- 16.Pattillo RA, Ruchert A, Hussa R, Bernstein R, Des E. The Jar cell line-continuous human multihormone production and control. In Vitro. 1971;6:398–399. [Google Scholar]
- 17.Sit KH, Yin L, Paramanantham R. Apoptotic condensations in M-phase cells. Anat.Rec. 1997;248:149–158. doi: 10.1002/(SICI)1097-0185(199706)248:2<149::AID-AR1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 18.Hatano E, Bradham CA, Stark A, Iimuro Y, Lemasters JJ, Brenner DA. The mitochondrial permeability transition augments Fas-induced apoptosis in mouse hepatocytes. J.Biol.Chem. 2000;275:11814–11823. doi: 10.1074/jbc.275.16.11814. [DOI] [PubMed] [Google Scholar]
- 19.Leist M, Gantner F, Bohlinger I, Germann PG, Tiegs G, Wendel A. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-alpha requires transcriptional arrest. J.Immunol. 1994;153:1778–1788. [PubMed] [Google Scholar]
- 20.Herrera B, Murillo MM, varez-Barrientos A, Beltran J, Fernandez M, Fabregat I. Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-beta in fetal rat hepatocytes. Free Radic.Biol.Med. 2004;36:16–26. doi: 10.1016/j.freeradbiomed.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 21.Jang CW, Chen CH, Chen CC, Chen JY, Su YH, Chen RH. TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase. Nat.Cell Biol. 2002;4:51–58. doi: 10.1038/ncb731. [DOI] [PubMed] [Google Scholar]
- 22.Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, Karwande SV, Stringham JC, Bull DA, Gleich M, Kennedy TP, Hoidal JR. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am.J.Physiol Lung Cell Mol.Physiol. 2006;290:L661–L673. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- 23.Schuster N, Krieglstein K. Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res. 2002;307:1–14. doi: 10.1007/s00441-001-0479-6. [DOI] [PubMed] [Google Scholar]
- 24.Herrera B, Alvarez AM, Sanchez A, Fernandez M, Roncero C, Benito M, Fabregat I. Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor (beta) in fetal hepatocytes. FASEB J. 2001;15:741–751. doi: 10.1096/fj.00-0267com. [DOI] [PubMed] [Google Scholar]