

Abstract
BK channels are activated by physiological concentrations of intracellular Ca2+ and Mg2+ in a variety of cells. Previous studies have identified two sites important for high-affinity Ca2+ sensing between [Ca2+]i of 0.1–100 μM and a site important for Mg2+ sensing between [Mg2+]i of 0.1–10 mM. BK channels can be also activated by Ca2+ and Mg2+ at concentrations >10 mM so that the steady-state conductance and voltage (G-V) relation continuously shifts to more negative voltage ranges when [Mg2+]i increases from 0.1–100 mM. We demonstrate that a novel site is responsible for metal sensing at concentrations ≥10 mM, and all four sites affect channel activation independently. As a result, the contributions of these sites to channel activation are complex, depending on the combination of Ca2+ and Mg2+ concentrations. Here we examined the effects of each of these sites on Ca2+ and Mg2+-dependent activation and the data are consistent with the suggestion that these sites are responsible for metal binding. We provide an allosteric model for quantitative estimation of the contributions that each of these putative binding sites makes to channel activation at any [Ca2+]i and [Mg2+]i.
INTRODUCTION
BK type large conductance K+ channels contain multiple binding sites for divalent cations that allow intracellular Ca2+ and Mg2+ to activate the channel at physiological concentrations ([Ca2+]i, 0.1–100 μM; [Mg2+]i, 0.4–3 mM) (Marty, 1981; Pallotta et al., 1981; Flatman, 1984; Gupta et al., 1984; Corkey et al., 1986; Golowasch et al., 1986; Flatman, 1991). Ca2+-dependent activation of the BK channel is key to its functions in a variety of physiological processes including neural excitation (Adams et al., 1982; Lancaster and Nicoll, 1987; Storm, 1987; Roberts et al., 1990; Robitaille and Charlton, 1992; Robitaille et al., 1993; Raffaelli et al., 2004; Sun et al., 2004), muscle contraction (Nelson et al., 1995; Tanaka et al., 1997; Perez et al., 1999; Brenner et al., 2000b; Pluger et al., 2000; Wellman and Nelson, 2003), hearing (Hudspeth and Lewis, 1988a, 1988b; Wu et al., 1995; Fettiplace and Fuchs, 1999; Ricci et al., 2000; Samaranayake et al., 2004), and immunity (Ahluwalia et al., 2004). The physiological function of Mg2+ dependence in BK channel activation has not been explored extensively. However, pharmacological studies have shown that Mg2+ may be an effective therapeutic agent following neurotrauma to improve survival and motor outcome and to alleviate cognitive deficits (Vink and Cernak, 2000). Magnesium supplements are also important in the prevention and management of cardiovascular diseases that predispose to hypertension or congestive heart failure (Laurant and Touyz, 2000; Seelig, 2000; Delva, 2003a,b,c; Touyz, 2003). Given the importance of BK channels in neurotransmitter release and vessel tone (Petersen and Maruyama, 1984; Stretton et al., 1992; Robitaille et al., 1993; Perez and Toro, 1994; Wang et al., 1994; Kume et al., 1995; Yazejian et al., 1997), Mg2+ modulation of BK channels may play a significant role in these pathophysiological processes.
Two sites important for Ca2+ sensing and one site important for Mg2+ sensing have been identified in the intracellular domain of the α subunit of BK channels, which is encoded by slo1 genes (Atkinson et al., 1991; Adelman et al., 1992; Butler et al., 1993; Dworetzky et al., 1994; Pallanck and Ganetzky, 1994; Tseng-Crank et al., 1994; Moss et al., 1996a,b; Schreiber and Salkoff, 1997; Schreiber et al., 1999; Bian et al., 2001; Braun and Sy, 2001; Zhang et al., 2001; Bao et al., 2002, 2004; Shi et al., 2002; Xia et al., 2002, 2004; Zeng et al., 2005). One site important for Ca2+ sensing is located in “Ca2+ bowl,” a motif that contains many Asp residues (Moss et al., 1996a,b; Schreiber and Salkoff, 1997; Schreiber et al., 1999; Bian et al., 2001; Braun and Sy, 2001; Xia et al., 2002; Bao et al., 2004). The other is located in the RCK1 domain (Bao et al., 2002; Xia et al., 2002), which is similar to a structural domain found in prokaryotic K+ channels important for regulating K+ conductance (Jiang et al., 2001, 2002). Mutations in Ca2+ bowl, such as the change of five consecutive Asp residues (D898 –902) into Asn, known as 5D5N and mutations in RCK1 such as M513I or D367A each reduces part of Ca2+ sensitivity of the channel (Schreiber and Salkoff, 1997; Bao et al., 2002; Shi et al., 2002; Xia et al., 2002). The site important for Mg2+ sensing was identified to contain E374, Q397, and E399 in the RCK1 domain of mslo1 (Shi et al., 2002; Xia et al., 2002). These previous results, primarily derived from mutational studies, suggest that these sites may be the candidates for metal binding sites. Previous studies have revealed complex effects of Ca2+ or Mg2+ binding on channel activation. Ca2+ at concentrations beyond physiological conditions can bind to the Mg2+ site, with a similar affinity as Mg2+, to activate the channel (Shi and Cui, 2001; Zhang et al., 2001); while Mg2+ at physiological concentrations can bind to Ca2+ sites and compete with Ca2+ (Shi and Cui, 2001). These complex effects of Ca2+ and Mg2+ on BK channel activation determine physiological functions of the channel and have been a major subject of structure–function studies in recent years (Bian et al., 2001; Braun and Sy, 2001; Shi and Cui, 2001; Zhang et al., 2001; Bao et al., 2002, 2004; Piskorowski and Aldrich, 2002; Shi et al., 2002; Xia et al., 2002; Qian and Magleby, 2003; Zeng et al., 2005). However, uncertainties regarding metal-binding sites and the effects of Ca2+ and Mg2+ binding still exist. Among these uncertainties, it was reported that the channel with a triple mutation 5D5N + D362A:D367A + E399A, which should have destroyed the three candidate metal binding sites, can be activated by high [Mg2+]i (≥10 mM) (Xia et al., 2002).
In this paper we investigate BK channel activation at various [Mg2+]i, [Ca2+]i, and voltages in order to examine the effects of this novel Mg2+ dependence on channel activation at physiological [Mg2+]i and [Ca2+]i. We find that the effects of the two Mg2+-dependent activation at [Mg2+]i around 10 mM overlap, and thus this novel Mg2+ dependence interferes with the study of Mg2+-dependent activation at physiological concentrations. On the other hand, we found that an additional metal binding site may contribute to this activation and residue D362 may be involved in channel activation at high metal concentrations. The mechanism of this novel Mg2+-dependent activation is independent from channel activation due to metal binding to other sites.
An abstract of this work has been presented at the 49th Annual Meeting of Biophysical Society.
MATERIALS AND METHODS
Clones, Mutagenesis, and Channel Expression
All channel constructs were made from the mbr5 clone of mslo1 (Cui et al., 1997) by using PCR with Pfu polymerase (Stratagene). The PCR-amplified regions of all mutants were verified by sequencing. RNA was transcribed in vitro with T3 polymerase (Ambion). We injected 0.05–50 ng of RNA into each Xenopus laevis oocyte 2–6 d before recording.
Electrophysiology
Macroscopic currents were recorded from inside-out patches formed with borosilicate pipettes of 1∼2 MΩ resistance. Data were acquired using an Axopatch 200-B patch-clamp amplifier (Axon Instruments, Inc.) and Pulse acquisition software (HEKA Electronik). Records were digitized at 20-μs intervals and low-pass filtered at 10 kHz with the 4-pole Bessel filter built in the amplifier. The pipette solution contained the following (in mM): 140 potassium methanesulfonic acid, 20 HEPES, 2 KCl, and 2 MgCl2, pH 7.20. The basal internal solution (with buffer) contains the following (in mM): 140 potassium methanesulfonic acid, 20 HEPES, 2 KCl and 1 EGTA, pH 7.20. The basal internal solution (without buffer) contains the following (in mM): 140 potassium methanesulfonic acid, 2 KCl, pH 7.20. Methanesulfonic acid was purchased from Sigma-Aldrich. The “0 [Ca2+]i” solution was the same as the basal internal solution (with buffer) except that it contained 5 mM EGTA (Fabiato and Fabiato, 1979). MgCl2 was added to these internal solution (with buffer) to give the appropriate free [Mg2+]i. For solutions with [Ca2+]i < 100 μM, CaCl2 was added to the basal internal solutions (with buffer) with amounts calculated using a program similar to published (Cox et al., 1997b; Shi and Cui, 2001) to give rise various free [Ca2+]i. For solutions with [Ca2+]i ≥ 100 μM, CaCl2 was added to the basal internal solutions (without buffer) with calculated amounts. The free [Ca2+]i was measured with a calcium-sensitive electrode (Orion Research Inc.) with the same procedure as previously described (Shi et al., 2002; Xia et al., 2002). The calcium-sensitive electrode was always calibrated right before measurements, and then recalibrated immediately after measurements. The results of calibration and recalibration were the same, indicating that the electrode was stable during measurements. 18-crown-6-tetracarboxylic acid (18-C-6-T; Sigma-Aldrich) was added to internal solutions to prevent Ba2+ block. For solutions with [Ca2+]i ≤ 100 μM, 50 μM 18-C-6-T was added; for solutions with higher [Ca2+]i, 100–150 μM 18-C-6-T was added. Experiments were conducted at room temperature (22°C–24°C).
Analysis and Model Fitting
Relative conductance was determined by measuring tail current amplitudes at −50 mV for WT and E374A:E399N mutant channels and at −120 mV for 5D5N + D367A + E399N and 5D5N + D362A:D367A + E399N mutant channels. The conductance–voltage (G-V) relations of the WT and mutant mslo1 channels were fitted with Boltzmann equation:
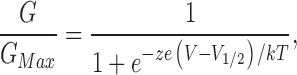 |
(1) |
where z is the number of equivalent charges, V1/2 is the voltage for channel in half activation, e is the elementary charge, k is Boltzmann's constant, and T is the absolute temperature.
Eqs. 2, 3, and 4 are derived from the MWC models based on hypotheses 1, 2, and 3 respectively.
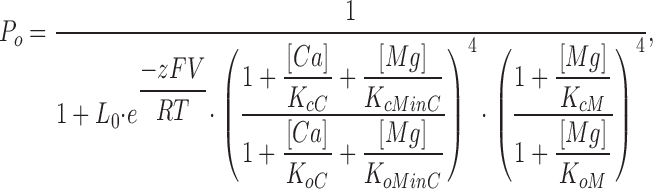 |
(2) |
where Po is the probability of the channel being open; L0 is the equilibrium constant between the closed and open states at the voltage of 0 mV when no Ca2+ or Mg2+ is bound (L0 = [C0]/[O0]); z is the number of equivalent gating charges; KcC and KoC are the dissociation constants of Ca2+ binding to the Ca2+ site on each subunit when the channel is at closed and open states, respectively; KcMinC and KoMinC are the dissociation constants of Mg2+ binding to the Ca2+ site on each subunit when the channel is at closed and open states, respectively; KcM and KoM are the dissociation constants of Mg2+ binding to the Mg2+ site on each subunit when the channel is at closed and open states, respectively; and V, T, F, and R have their usual meanings.
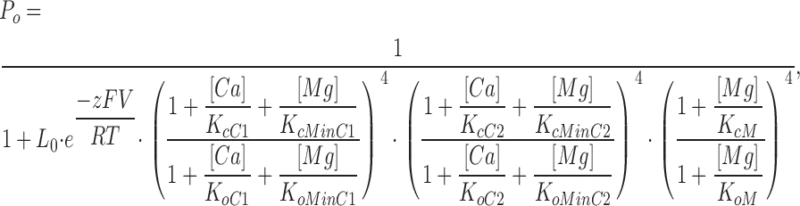 |
(3) |
where KcC1, KcC2, KoC1, and KoC2 are the dissociation constants of Ca2+ binding to the first and second Ca2+ sites on each subunit when the channel is at closed and open states, respectively; KcMinC1, KcMinC2, KoMinC1, and KoMinC2 are the dissociation constants of Mg2+ binding to the first and second Ca2+ sites on each subunit when the channel is at closed and open states, respectively, and KcMinC1 = KoMinC1; and other parameters have the same meaning as in Eq. 2.
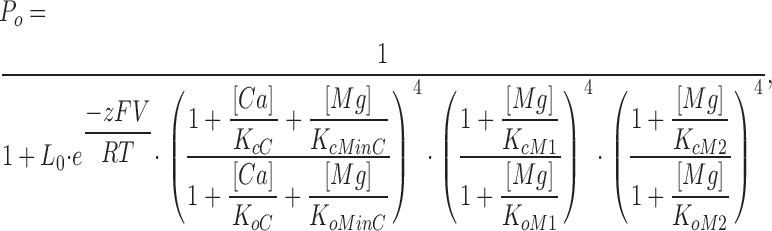 |
(4) |
where KcM1, KcM2, KoM1, and KoM2 are the dissociation constants of Mg2+ binding to the first and second Mg2+ sites on each subunit when the channel is at closed and open states, respectively, and other parameters have the same meaning as in Eq. 2 (KcMinC = KoMinC).
In fitting these models, the G-V relations of the WT and E374A:E399N mutant channels at various [Ca2+]i and [Mg2+]i were fitted with Eqs. 2–4 respectively using LSQCURVEFIT program in Matlab 6.1 (the MathWorks, Inc.). The parameters defining the models were acquired step by step through the model fittings as described in RESULTS. Rational lower and upper bounds of the parameters were set during model fitting. The equivalent charge, z, was assumed to be the same for both the WT and E374A:E399N mutant channels under all ionic conditions. The means of the data were obtained by averaging from 4 to 16 patches and error bars represent SEMs.
RESULTS
Activation of BK Channels at High (≥10 mM) and Low (0–10 mM) [Mg2+]i
Previous studies have suggested that the residues E374 and E399 in the RCK1 domain of the mslo1 BK channels are important for Mg2+ sensing in the RCK1 domain of the mslo1 BK channels (Shi et al., 2002; Xia et al., 2002). Consistent with these studies, the double mutation E374A:E399N dramatically reduced the Mg2+ sensitivity when [Mg2+]i increased from 0 to 10 mM (Fig. 1), indicating that the mutation has destroyed the ability of Mg2+ sensing mediated by this site. Fig. 1 A shows the WT and E374A:E399N mutant channel macroscopic currents recorded in inside-out patches with and without 10 mM [Mg2+]i. The outward currents at positive voltages decreased in 10 mM [Mg2+]i due to a fast, voltage dependent block of the channel (Ferguson, 1991; Laver, 1992; Shi and Cui, 2001). Upon the return of membrane potential to −50 mV, the block is relieved rapidly and the tail current of the WT mslo1 channel has a slower time course and a larger maximum amplitude in the presence of 10 mM [Mg2+]i than at 0 [Mg2+]i (5.5 vs. 3.7 nA), revealing that the channel is activated by Mg2+ (Shi and Cui, 2001). To the contrary, for the mutant channel, 10 mM [Mg2+]i did not have obvious effects on the tail current. The effect of the mutation on Mg2+-dependent activation is more obviously shown by the conductance–voltage relation (G-V relation). Fig. 1 (B and C) shows that 10 mM [Mg2+]i shifts the G-V relation for the WT channel by about −67 mV, indicating that the channels can be activated in more negative voltages with Mg2+ binding. However, for the mutant channel the shift of the G-V relation is reduced to about −14 mV.
Figure 1.

Mutation E374A:E399N reduces Mg2+ sensitivity. (A) Current traces of WT mslo1 channels (left) and E374A:E399N mutant channels (right) recorded at indicated [Mg2+]i and [Ca2+]i. Testing potentials were −20 to 240 mV with 20-mV increments. The holding and repolarizing potentials were −80 and −50 mV, respectively. (B) Mean G-V relations of WT mslo1 (gray symbols) and the mutant channels (black symbols) at 0 or 10 mM [Mg2+]i and 0 [Ca2+]i. G-V relations of these channels are fitted with the Boltzmann relation (solid lines, see MATERIALS AND METHODS). (C) Shift of G-V relations from 0 to 10 mM [Mg2+]i at 0 [Ca2+]i for WT mslo1 and the mutant channels. V1/2 is the voltage where the G-V relation is at half maximum.
Although the destruction of the site at E374 and E399 reduces Mg2+ sensitivity of channel activation dramatically, a leftward shift of G-V relation is consistently observed in response to 10 mM [Mg2+]i as shown in Fig. 1 C, indicating that the mutant channel remains Mg2+ sensitive. The remaining Mg2+ sensitivity of channel activation becomes more prominent when [Mg2+]i increases to 100 mM (Fig. 2). Fig. 2 B shows G-V relations of the WT and E374A:E399N mutant channels at 0 and 100 mM [Mg2+]i. Consistent with the previous observation (Shi and Cui, 2001), 100 mM [Mg2+]i shifts the G-V relation of the WT channel by about −124 mV, −57 mV more than the leftward shift caused by 10 mM [Mg2+]i (Fig. 2 C). For the mutant channel, 100 mM [Mg2+]i shifts the G-V relation by about −57 mV, −43 mV more than the leftward shift by 10 mM [Mg2+]i. The large G-V shift caused by 100 mM Mg2+ on the E374A:E399N mutant channel suggests that Mg2+ is still able to affect channel activation even though the mutation abolishes the ability of Mg2+ sensing mediated by this site. This result is consistent with a previous report that when E399 was mutated the channel was still sensitive to high concentrations of Mg2+ (Xia et al., 2002).
Figure 2.

Mg2+ sensitivity of WT mslo1 and mutant channels at high [Mg2+]i. (A) Current traces of WT mslo1 channels (left) and E374A:E399N mutant channels (right) recorded at indicated [Mg2+]i and [Ca2+]i. Testing potentials were −70 to 190 mV with 20-mV increments for WT mslo1 channels at 100 mM [Mg2+]i and 0 [Ca2+]i; and were −20 to 240 mV with 20-mV increments for others. The holding and repolarizing potentials were −80 and −50 mV, respectively. (B) Mean G-V relations of WT mslo1 (gray symbols) and E374A:E399N mutant channels (black symbols) at 0 or 100 mM [Mg2+]i and 0 [Ca2+]i, and fits with the Boltzmann relation (solid lines). (C) G-V shifts at 0 [Ca2+]i from 0 to 10 mM [Mg2+]i (gray bars) and from 0 to 100 mM [Mg2+]i (black bars) of WT mslo1 and mutant channels E374A:E399N, E374Q and E399Q.
We considered the possibility that the mutation might not have fully removed the ability of this site in mediating Mg2+ sensing so that Mg2+ in high concentration could still activate the channel. Fig. 2 C compares Mg2+ sensitivity of the double mutant E374A:E399N with two single mutants E374Q and E399Q. If the double mutant could not completely destroy Mg2+ sensing and left some residual Mg2+ sensitivity then each single mutant may destroy the site to a less extent and leave a higher Mg2+ sensitivity. However, contrary to this prediction, ΔV1/2 (V1/2 is the voltage where the G-V relation is half maximum) caused by 10 and 100 mM [Mg2+]i are similar for both single mutations and the double mutation. In addition, although the activation of all mutant channels by Mg2+ at low concentration of 0–10 mM is largely reduced (Fig. 1), ΔV1/2 between 10 and 100 mM [Mg2+]i is similar among the WT and mutant channels (Fig. 2 C, subtracting gray bars from the black bars), indicating that the mutations affect Mg2+ sensitivity primarily in the concentration range between 0 and 10 mM but not between 10 and 100 mM. These results suggest that the ability of mediating Mg2+ sensing by the site at E374 and E399 may have been fully removed by the double and single mutations and the remaining Mg2+ sensitivity must be the consequence of some other mechanism.
The differences between Mg2+-dependent activation at low (0–10 mM) and high [Mg2+]i (10–100 mM) are also reflected on the kinetics of the channel gating. The left panels in Fig. 3 (A–E) show the current traces of the WT channel elicited by depolarization testing pulses in different [Mg2+]is. Although the steady-state currents were reduced by Mg2+ block of the channel pore, the blocking was so fast as compared with the time course of channel activation (Ferguson, 1991; Laver, 1992; Shi and Cui, 2001) that the current trace could still be fitted with a single exponential function (Fig. 3, A–E). The corresponding activation constants as the function of testing voltages (τ-V relations) are shown on the right panels. The averaged τ-V curves at various [Mg2+]i are plotted in Fig. 3 F. Unlike the shift of G-V relations caused by [Mg2+]i changes from 0 to 10 mM, the τ-V relation does not shift as much (Fig. 3 F) (Horrigan, 2005; Zeng et al., 2005). However, τ-V relation shifts to the left as [Mg2+]i increases to 30 and 100 mM (Fig. 3 F), similar to the shift of G-V relations in response to the same [Mg2+]i changes (Fig. 2).
Figure 3.

Voltage and Mg2+ dependence of activation time courses of mslo1 channels. (A–E) Left panels, current traces recorded from a single membrane patch in response to depolarizing membrane voltages of 20 mM increments, and fitted with a single exponential function (thin curves). [Mg2+]i was as indicated. Right panels, activation time constants as a function of test potential, and fits with the function τ = Ae −qFV/RT + b (solid curves). (F) Semi-log plots of mean activation time constants as a function of both voltage and [Mg2+]i. Solid lines represent fits to the same function as in A–E.
The effects of high [Mg2+]i on channel activation appear differently at different [Ca2+]i. Fig. 4 shows that in the presence of 100 μM [Ca2+]i, the shift of G-V relations by 100 mM [Mg2+]i is reduced for both WT and E374A:E399N mslo1 channels as compared with the shift at 0 [Ca2+]i (Fig. 2). For WT channels, 100 mM [Mg2+]i shifts the G-V relation to a more negative voltage range by about −70 mV. However, for the mutant channel, the G-V does not shift to more negative voltages at all, or even shifts slightly to more positive voltages (Fig. 4, B and C). The lack of G-V shift in response to 100 mM [Mg2+]i at 100 μM [Ca2+]i is the same for both single mutants E374Q and E399Q and the double mutant E374A:E399N (Fig. 4 C).
Figure 4.

Effects of 100 μM [Ca2+]i on channel activation at high [Mg2+]i. (A) Current traces of WT mslo1 channels (left) and E374A:E399N mutant channels (right) recorded at indicated [Mg2+]i and [Ca2+]i. Dashed lines indicate zero current. Testing potentials were −200 to 140 mV with 20-mV increments. The holding and repolarizing potentials were −80 and −100 mV, respectively. (B) Mean G-V relations of WT mslo1 (gray symbols) and E374A:E399N mutant channels (black symbols) at 0 or 100 mM [Mg2+]i and 100 μM [Ca2+]i, and fits with the Boltzmann relation (solid lines). (C) G-V shifts from 0 to 100 mM [Mg2+]i at 100 μM [Ca2+]i of WT mslo1 and mutant channels E374A:E399N, E374Q, and E399Q.
Activation of BK Channels at High [Mg2+]i and Physiological [Ca2+]i
In this section, we examine if a high affinity Ca2+ binding site may contribute to channel activation at high [Mg2+]i. Although two sites important for high affinity Ca2+ sensing have been suggested as candidates for Ca2+ binding sites by mutational studies, the ultimate identification of the binding sites may rely on additional structural data that are not yet available. Further, it is not clear whether any unidentified Ca2+ binding sites besides the two candidates also contribute to BK channel activation at physiological [Ca2+]i (Piskorowski and Aldrich, 2002; Qian and Magleby, 2003). Therefore, here we address this question by studying BK channels at various [Ca2+]i and [Mg2+]i without presuming the identity of Ca2+ binding sites.
It has been previously demonstrated that Mg2+ can bind to high affinity Ca2+ binding sites and compete with Ca2+ (Shi and Cui, 2001). An increase of [Mg2+]i from 0 to 10 mM shifts the G-V relation of the WT mslo1 by about −60 mV at 0 or 100 μM [Ca2+]i (Fig. 1) but much less at 3.7 μM [Ca2+]i (Fig. 5) because the activation due to Mg2+ binding to its site is compensated by the loss of Ca2+-dependent activation due to such competition (Shi and Cui, 2001). We examined such competition further by measuring the G-V shift of E374A:E399N mutant channels under the same ionic conditions (Fig. 5). Since this mutation destroys Mg2+ sensitivity at low concentrations, an increase of [Mg2+]i from 0 to 10 mM should only result in a small activation of the channel (see Fig. 1) but the loss of Ca2+-dependent activation due to competition should remain the same as in the WT mslo1. Therefore, instead of activating the channel, a [Mg2+]i increase from 0 to 10 mM should make the channel less active. This prediction is confirmed in Fig. 5 (B and C), which shows that the G-V relation of the mutant channel shifted to a more positive voltage range in response to [Mg2+]i increase, demonstrating that Mg2+ can bind to high affinity Ca2+ binding sites. These experiments, however, do not show if the binding of Mg2+ to Ca2+ binding sites can also activate the channel.
Figure 5.

Mg2+ competes with Ca2+ by binding to Ca2+ binding sites. (A) Current traces of WT mslo1 channels (left) and E374A:E399N mutant channels (right) recorded at indicated [Mg2+]i and [Ca2+]i. Testing potentials were −140 to 180 mV with 20-mV increments. The holding and repolarizing potential were −80 and −50 mV, respectively. (B) Mean G-V relations of WT mslo1 (gray symbols) and the mutant channels (black symbols) at 0 or 10 mM [Mg2+]i and 3.7 μM [Ca2+]i, and fits with the Boltzmann relation (solid lines). (C) G-V shifts from 0 to 10 mM [Mg2+]i at 3.7 μM [Ca2+]i of WT mslo1 and the mutant channels.
To answer this question we measured the activation of the WT and mutant E374A:E399N mslo1 channels at various [Ca2+]i and [Mg2+]i (Figs. 6–8). We then fit these data with MWC models of channel activation, which have been successfully used to describe Ca2+ and Mg2+-dependent activation of BK channels in previous studies (McManus and Magleby, 1991; Cox et al., 1997a; Shi and Cui, 2001; Zhang et al., 2001; Xia et al., 2002). The fitting was based on the general hypothesis that each subunit of the channel contains two high affinity Ca2+ binding sites (Schreiber and Salkoff, 1997; Bao et al., 2002, 2004; Xia et al., 2002; Zeng et al., 2005), which have similar Ca2+ affinities (Bao et al., 2002). We then assume in the following hypotheses two, one, or none of the high affinity Ca2+ binding sites are responsible for channel activation at high [Mg2+]i to examine if any hypothesis results in a good fit of all the data.
Figure 6.

Voltage, Ca2+, and Mg2+-dependent activation of WT and E374A:E399N mutant channels and fittings with the hypothesis 1 model. (A–C) G-V relations of WT mslo1 and E374A:E399N mutant channels at indicated [Mg2+]i and [Ca2+]i. The data were fitted (solid lines) with Eq. 2. The parameters obtained from fittings in A and B are listed in Table I. In the fits in C, KcC = 8 μM and KoC = 1 μM.
Figure 8.

Voltage, Ca2+, and Mg2+-dependent activation of WT and E374A:E399N mutant channels and fittings with the hypothesis 3 model. (A–F) G-V relations of WT mslo1 and E374A:E399N mutant channels at indicated [Mg2+]i and [Ca2+]i. The data were fitted (solid lines) with Eq. 4. The parameters obtained from fittings in A-C are listed in Table III.
Hypothesis 1
Mg2+ at high concentration activates the channel by binding to both Ca2+ binding sites. Since the two Ca2+ binding sites are similar, for simplicity, we assume that each BK channel subunit contains one Ca2+ binding site and one Mg2+ binding site. Ca2+ at our experimental concentrations (0, 3.7, and 100 μM) can cause little activation by binding to the Mg2+ binding site (Shi and Cui, 2001) so that it is assumed to only bind to the Ca2+ binding site to activate the channel. Mg2+ can bind to both Ca2+ and Mg2+ binding sites to activate the channel.
Hypothesis 2
Mg2+ at high concentration activates the channel by binding to one of the Ca2+ binding sites, but cannot activate the channel by binding to the other Ca2+ binding site. In this model, Ca2+ binds to the two Ca2+ binding sites to activate the channel. Mg2+ can bind to all three binding sites but only one of the Ca2+ binding sites can contribute to channel activation by Mg2+ binding. Mg2+ binds to the other Ca2+ site with the same affinity at open and closed states and thus cannot activate the channel.
Hypothesis 3
Mg2+ at high concentration activates the channel by binding to a novel low affinity Mg2+ binding site. In this model, we assume that each BK channel subunit contains one Ca2+ binding site since the two Ca2+ binding sites are similar, and two Mg2+ binding sites including the novel low affinity site. Ca2+ binds to the Ca2+ site to activate the channel. Mg2+ can bind to all three sites, but it binds to the Ca2+ site with the same affinity at open and closed states and thus cannot activate the channel. Eqs. 2, 3, and 4 based on these hypotheses are given in MATERIALS AND METHODS. The model fitting proceeded in four steps and is described as follows.
Step 1
The models are fitted to the data from E374A:E399N mutant channels at 0 [Ca2+]i, with [Mg2+]i of 0, 0.1, 0.5, 1, 5, 10, 30, and 100 mM. Under these conditions there is no activation due to Ca2+ binding to the Ca2+ binding site(s) or Mg2+ sensing at low [Mg2+]i. Therefore, the fitting generates parameters for channel activation only at high [Mg2+]i. The experimental data show that at 0 [Mg2+]i and all [Ca2+]i tested (0, 3.7, and 100 μM) the G-V relation of E374A:E399N mutant channels is similar to that of the WT mslo1 (Fig. 7 or 8), indicating that the mutation only destroys Mg2+ sensing at low [Mg2+]i without affecting Ca2+-dependent activation significantly. The mutation does not significantly affect Mg2+ sensing at high [Mg2+]i either (Fig. 2). Therefore, the parameters obtained in this step for channel gating at high [Mg2+]i are also used for the WT mslo1 channel.
Figure 7.

Voltage, Ca2+, and Mg2+-dependent activation of WT and E374A:E399N mutant channels and fittings with the hypothesis 2 model. (A–F) G-V relations of WT mslo1 and E374A:E399N mutant channels at indicated [Mg2+]i and [Ca2+]i. The data were fitted (solid lines) with Eq. 3. The parameters obtained from fittings in A–C are listed in Table II.
Step 2
The models are fitted to the data from the WT channel at 0 [Ca2+]i, with [Mg2+]i of 0, 0.1, 0.5, 1, 5, 10, 30, and 100 mM. Under these conditions the channel is activated by Mg2+ at both low and high concentrations, but without Ca2+ binding to the Ca2+ site(s). The parameters obtained in step 1 for channel gating at high [Mg2+]i will be used in the fitting, which allows us to obtain the parameters for channel gating at low [Mg2+]i.
Step 3
The models are fitted to the data from E374A:E399N mutant channels at 3.7 μM [Ca2+]i, with [Mg2+]i of 0, 10, 30, and 100 mM. Under these conditions the channel is activated by Mg2+ at high concentrations, and by Ca2+ binding to the Ca2+ site(s). The parameters obtained in step 1 will be used in the fitting, which allows us to obtain the parameters for Mg2+ and Ca2+ binding to the Ca2+ site(s). The fittings in steps 1–3 have allowed us to obtain all the parameters needed to define the models.
Step 4
With all the parameters acquired in steps 1–3, we examine if the models can fit the rest of data to verify the validity of each hypothesis. The data fitted in this step include (a) WT channels at 3.7 μM [Ca2+]i, with [Mg2+]i of 0, 10, 30, and 100 mM; (b) E374A:E399N mutant channels at 100 μM [Ca2+]i, with [Mg2+]i of 0, 10, 30, and 100 mM; and (c) WT channels at 100 μM [Ca2+]i, with [Mg2+]i of 0, 0.1, 0.5, 1, 5, 10, 30, and 100 mM.
Fig. 6 shows the model fitting of data based on hypothesis 1, i.e., each subunit contains one Ca2+ and one Mg2+ binding site for Ca2+ and Mg2+-dependent activation at all concentrations. During the step 1 and step 2 fittings, the data of the WT and mutant channels at 0 [Ca2+]i and various [Mg2+]i can be well fitted by the model (Fig. 6, A and B). The parameters generated from these fittings are listed in Table I. However, in the step 3 fitting, the model cannot fit the data of the mutant channel at 3.7 μM [Ca2+]i (Fig. 6 C). This result can be understood by the analysis as follows. The experimental results demonstrate that Mg2+ at 10 mM can bind to the Ca2+ binding site(s) and effectively compete with Ca2+ to reduce the activation of the channel, shifting the G-V relation to the right by ∼35 mV (Fig. 5 and Fig. 6 C). This result indicates that the affinity of the Ca2+ binding site for Mg2+ should be high enough to allow a significant binding of Mg2+ at 10 mM. This requirement is contradictory to the model fitting in Fig. 6 A, in which the affinity of the Ca2+ binding site for Mg2+ is low, being 136 mM at closed states and 40 mM at open states (Table I), in order to allow Mg2+ binding at high concentrations to open the channel. Because a single Ca2+ binding site or two Ca2+ binding sites with similar properties cannot satisfy the requirements of both activation and competition results (Fig. 6, A and C) hypothesis 1 is not valid.
TABLE I.
Parameters for Model Fitting of Hypothesis 1
| WT mslo1 | E374A:E399N | |
|---|---|---|
| L0 | 9334 | 4360 |
| z | 1.25 | 1.25 |
| KcMinC | 136.1 | 136.1 |
| KoMinC | 40.1 | 40.1 |
| KcM | 5.46 | – |
| KoM | 2.25 | – |
Unlike hypothesis 1, hypothesis 2 assumes that the two Ca2+ binding sites are not equal in their affinity for Mg2+. Fig. 7 shows the model fitting of data based on hypothesis 2, i.e., each subunit contains two Ca2+ and one Mg2+ binding site for Ca2+ and Mg2+-dependent activation at all concentrations. During step 1–3 fittings, we limited the affinity of the first Ca2+ site for Mg2+ binding (KcMinC1 and KoMinC1) between 1 and 30 mM and the affinity of the second Ca2+ site for Mg2+ binding (KcMinC2 and KoMinC2) between 30 and 200 mM. The data of the WT and mutant channels at 0 and 3.7 μM [Ca2+]i and various [Mg2+]i can be well fitted by the model (Fig. 7, A–D). The parameters generated from these fittings are listed in Table II. However, the model fails to fit the data of both WT and mutant channels at 100 μM [Ca2+]i (Fig. 7, E and F). It happens because in the model Mg2+ competes with Ca2+ at both Ca2+ binding sites and causes too much loss of Ca2+-dependent activation. On one hand the binding of Mg2+ to the second Ca2+ binding site activates the channel, but on the other hand the channels lose activation due to less Ca2+ binding to both Ca2+ sites. The loss of Ca2+-dependent activation is larger than the activation by Mg2+ binding, thus at high [Mg2+]i (≥10 mM) the model predicts a larger right shift of G-V than the experimental data (Fig. 7, E and F). The adverse effects of Mg2+-dependent activation and loss of Ca2+-dependent activation due to Mg2+ binding to Ca2+ sites also happen at 3.7 μM [Ca2+]i, which can be accounted for by the model during the step 3 fitting (Fig. 7 C). However, the parameters obtained from the fitting at 3.7 μM [Ca2+]i cannot be used to fit the data at 100 μM [Ca2+]i. Thus, hypothesis 2 is not valid.
TABLE II.
Parameters for Model Fitting of Hypothesis 2
| WT mslo1 | E374A:E399N | |
|---|---|---|
| L0 | 9334 | 4360 |
| z | 1.25 | 1.25 |
| KcM | 5.24 | – |
| KoM | 2.12 | – |
| KcMinC1 | 3.18 | 3.18 |
| KoMinC1 | 3.18 | 3.18 |
| KcMinC2 | 142.43 | 142.43 |
| KoMinC2 | 42.02 | 42.02 |
| KcC1 | 9.04 | 9.04 |
| KoC1 | 1.95 | 1.95 |
| KcC2 | 1.90 | 1.90 |
| KoC2 | 0.84 | 0.84 |
The above results indicate that Mg2+ at high concentrations cannot activate the channel by binding to Ca2+ binding sites. Fig. 8 shows the model fitting of data based on hypothesis 3, i.e., a novel low affinity Mg2+ binding site is responsible for such activation. The parameters generated from the step 1–3 fittings (Fig. 8, A–C) are listed in Table III. These parameters are then used in the step 4 fittings (Fig. 8, D–F). Under all ionic conditions the model fits well to the experimental data of both the WT and mutant channels (Fig. 8), supporting the hypothesis that a novel low affinity Mg2+ binding site is responsible for channel activation at high [Mg2+]i. These results also suggest that Mg2+ can bind to Ca2+ binding sites and compete with Ca2+ but the binding does not activate the channel. Due to Mg2+ competition both WT and E374A:E399N mutant channels lose Ca2+-dependent activation at 3.7 or 100 μM [Ca2+]i and high [Mg2+]i (Fig. 8, C–F). Such competition may explain the results in Fig. 4 such that at 100 μM [Ca2+]i and 100 mM [Mg2+]i the loss of Ca2+-dependent activation due to Mg2+ competitive binding to the Ca2+ sites is compensated by Mg2+-dependent activation through the novel low affinity Mg2+ site. Therefore, for the E374A:E399N mutant channels, in which Mg2+ sensing at low [Mg2+]i is destroyed, 100 mM [Mg2+]i does not change the G-V relation, while the WT mslo1 100 mM [Mg2+]i shifts the G-V relation to more negative voltage ranges due to Mg2+ sensing mediated by the site at E374/E399.
TABLE III.
Parameters for Model Fitting of Hypothesis 3
| WT mslo1 | E374A:E399N | |
|---|---|---|
| L0 | 9334 | 4360 |
| z | 1.25 | 1.25 |
| KcM1 | 5.46 | – |
| KoM1 | 2.25 | – |
| KcM2 | 136.1 | 136.1 |
| KoM2 | 40.1 | 40.1 |
| KcMinC | 5.36 | 5.36 |
| KoMinC | 5.36 | 5.36 |
| KcC | 8.38 | 8.38 |
| KoC | 0.81 | 0.81 |
BK Channel Activation at High [Mg2+]i Is Independent from Activation at Low [Ca2+]i and [Mg2+]i
In above fittings we use an independent MWC model to describe channel activation due to each metal binding site (Eqs. 2–4). The fitting results in Fig. 8 are reasonably good, suggesting that channel activation mediated by the novel low affinity Mg2+ binding site is independent from that mediated by metal binding at high affinity Ca2+ binding sites and Mg2+ sensing mediated by the site at E374/E399. To verify this conclusion we studied two triple mutations 5D5N + D367A + E399N and 5D5N + D367A:D362A + E399N, which destroy metal sensing at low concentrations of Ca2+ and Mg2+ (Xia et al., 2002). If BK channel activation at high [Mg2+]i is independent from activation at low [Ca2+]i and [Mg2+]i the remaining activation of these mutant channels by high Mg2+ should be mediated only by the novel low affinity Mg2+ binding site, which should be intact from the mutations, and consequently, the above model should still be able to fit the Mg2+ dependence of channel activation.
This prediction is confirmed as shown in Fig. 9. For both mutant channels, the G-V relations do not change much between 0 and 1 mM [Mg2+]i. However, at high [Mg2+]i (10–100 mM), the G-V relation of both mutants shifts to more negative voltage ranges (Fig. 9 B), a total of about −50 mV. Such shift is comparable to the G-V shift of the WT mslo1 in response to the same [Mg2+]i increase (Fig. 2). The G-V relations of both mutants can be fitted (Fig. 9 B, solid curves) by the MWC model with only the low affinity Mg2+ binding site being intact and the same parameters as listed in Table III except for L0.
Figure 9.

Voltage and Mg2+-dependent activation of WT, 5D5N + D367A + E399N, and 5D5N + D362A:D367A + E399N mutant channels. (A) Current traces of WT mslo1 channels (left), 5D5N + D367A + E399N mutant channels (middle), and 5D5N + D362A:D367A + E399N mutant channels (right) recorded at indicated [Mg2+]i. Testing potentials were −200 to 140 mV with 20-mV increments. The holding and repolarizing potentials were −80 and −120 mV, respectively. (B) Mean G-V relations of 5D5N + D367A + E399N (left) and 5D5N + D362A:D367A + E399N mutant channels (right) in the presence of 0–100 mM [Mg2+]i and fittings with the hypothesis 3 model. The symbols represent [Mg2+]i (in mM) at 0 (open circle), 0.1 (closed circle), 1 (open triangle), 10 (closed triangle), 30 (open diamond), and 100 (closed diamond). The data were fitted (solid lines) with Eq. 4. The parameters were fixed with the numbers in Table III, except L0, which were set free. In the fits, L0 for 5D5N + D367A + E399N and 5D5N + D362A:D367A + E399N is 3516 and 4908, respectively.
DISCUSSION
Previous studies have identified three sites in mslo1 BK channels important for metal sensing, which are located in Ca2+ bowl (Schreiber and Salkoff, 1997; Bian et al., 2001; Braun and Sy, 2001; Bao et al., 2004), RCK1 domain (Bao et al., 2002; Xia et al., 2002), and at E374/E399/Q397 (Shi et al., 2002; Xia et al., 2002). These sites mediate metal-dependent activation at physiological Ca2+ and Mg2+ concentrations. In this paper we have studied steady-state and kinetic properties of WT and mutant mslo1 channels in various voltage ranges, Ca2+ and Mg2+ concentrations, and demonstrated that a novel low affinity metal binding site is responsible for channel activation at metal concentrations beyond normal physiological conditions (Figs. 1–3 and 6–8). Metal sensing mediated by any of these sites independently activates the channel (Fig. 8 and 9). Nevertheless, the contributions of these sites to channel activation are complex, depending on the combination of [Ca2+]i and [Mg2+]i (Figs. 1, 2, 4, 5, and 8). The reason, as our results demonstrated, is that both Ca2+ and Mg2+ can bind to all metal binding sites to modulate channel activation (Figs. 5, 8, 9, and 10). We examined the effects of Ca2+ and Mg2+ binding to each of metal binding sites on channel activation and provide an allosteric model for quantitative estimation of the contributions that each binding site makes to channel activation at any [Ca2+]i and [Mg2+]i (Eq. 4).
Figure 10.

Voltage and Ca2+-dependent activation of WT, 5D5N + D367A + E399N, and 5D5N + D362A:D367A + E399N mutant channels. (A) Current traces of WT mslo1 channels (left), 5D5N + D367A + E399N mutant channels (middle), and 5D5N + D362A:D367A + E399N mutant channels (right) recorded at indicated [Ca2+]i. Dashed lines indicate zero current. Testing potentials for WT at 0.1 mM [Ca2+]i were −150 to 190 mV with 20-mV increments. Testing potentials for WT at 1, 10, 30, and 100 mM [Ca2+]i were −200 to 140 mV with 20-mV increments. Testing potentials for WT at 0 [Ca2+]i and 5D5N + D367A + E399N and 5D5N + D362A:D367A + E399N mutant channels were −20 to 240 mV with 20-mV increments. The holding and repolarizing potentials were −80 and −120 mV, respectively. (B) Mean G-V relations of 5D5N + D367A + E399N mutant channels (left) and 5D5N + D362A:D367A + E399N mutant channels (right) in the presence of 0–100 mM [Ca2+]i The symbols represent [Ca2+]i (in mM) at 0 (open circle), 0.1 (inverted open triangle), 1 (inverted closed triangle), 10 (open diamond), 30 (closed diamond) and 100 (open square). G-V relations of these channels are fitted with the Boltzmann relation (solid lines, see MATERIALS AND METHODS). (C) V1/2 versus [Mg2+]i (closed symbols) or [Ca2+]i (open symbols) for WT (circle), E374A:E399N (diamond), 5D5N + D367A + E399N (triangle), and 5D5N + D362A:D367A + E399N (inverted triangle) mutant channels.
Based on this model, we calculated the contribution of the novel low affinity metal binding site to the mslo1 channel activation at physiological [Mg2+]i between 0.3 and 3 mM (Flatman, 1984, 1991; Gupta et al., 1984; Corkey et al., 1986). At the physiological [Ca2+]i between 0.1 and 100 μM (Roberts et al., 1990; Robitaille et al., 1993; Roberts, 1994; Yazejian et al., 1997; Marrion and Tavalin, 1998; Berridge et al., 2000; Yazejian et al., 2000), 3 mM [Mg2+]i only shifts the V1/2 for about −4.1 mV due to Mg2+ binding to this site, indicating that the contribution of this site is quite small. However, the physiological relevance of this binding site is not fully understood because the effects of β subunits on the function of this site are not explored yet. Since slo1 proteins often associate with various β subunits to form BK channels in vivo (Tseng-Crank et al., 1996; Cox et al., 1997a; Jiang et al., 1999; Riazi et al., 1999; Wallner et al., 1999; Behrens et al., 2000; Brenner et al., 2000b; Meera et al., 2000; Weiger et al., 2000) and it is known that β1 and β2 subunits affect apparent Ca2+ sensitivity of the channel (McManus et al., 1995; Meera et al., 1996, 2000; Tseng-Crank et al., 1996; Jiang et al., 1999; Nimigean and Magleby, 1999; Wallner et al., 1999; Brenner et al., 2000a,b; Cox and Aldrich, 2000; Weiger et al., 2000; Orio and Latorre, 2005), it will be interesting to examine if Mg2+-dependent activation is also affected by β subunits. The studies here provide a useful basis for such future studies.
The identity of this novel low affinity Mg2+ binding site is still unknown. A previous study reported that a triple mutation 5D5N + D362A:D367A + E399A not only destroyed Ca2+ and Mg2+-dependent activation at low concentrations (0–10 mM) but also significantly altered Ca2+-dependent activation at high concentrations (10–100 mM) (Xia et al., 2002). We noticed that among the mutated residues in the triple mutation D362 might not be important in either Ca2+ or Mg2+-dependent activation at low concentrations, because Ca2+ and Mg2+ sensitivity of mslo1 channels changed little when it was mutated individually (Xia et al., 2002). Therefore, this result seems to suggest that D362 is important for channel activation at high [Ca2+]i.
To verify this suggestion we compared Ca2+ dependence of the triple mutation 5D5N + D367A:D362A + E399N with 5D5N + D367A + E399N that left D362 intact (Fig. 10). At [Ca2+]i ≥ 1 mM, the currents at depolarized voltages are smaller than the currents at lower [Ca2+]i, probably due to a rapid block of the channel pore by Ca2+ (Oberhauser et al., 1988; Cox et al., 1997a), and the current reduction is more prominent in mutant channels than in the WT mslo1 (Fig. 10 A). For the WT mslo1 channels, the block by Ca2+ and Mg2+ is voltage dependent, and the rate of unblock is very rapid at voltages more negative than the K+ equilibrium potential such that the inward macroscopic tail current upon repolarization is not affected by the block (Cox et al., 1997b; Shi and Cui, 2001). However, at 100 mM [Ca2+]i, most mutant channels are blocked not only at depolarized voltages but also at the repolarization of −120 mV (also see Xia et al., 2002), possibly due to a more rapid deactivation time course (Fig. 10 A). Fig. 10 B shows the G-V relations of the two mutant channels at various [Ca2+]i from 0 to 100 mM. The mutations destroy channel activation at low [Ca2+]i so that the G-V relations do not differ much between 0 and 1 mM [Ca2+]i for both mutant channels. However, at high [Ca2+]i (10–100 mM) the G-V relation of 5D5N + D367A + E399N shifts to more negative voltage ranges, and at 100 mM [Ca2+]i the shift is about −36 mV from the G-V at 10 mM [Ca2+]i, comparable to the G-V shift of the WT mslo1 caused by the same [Ca2+]i increase (Fig. 10 C). On the other hand, the G-V relation of 5D5N + D362A:D367A + E399N is not shifted as much, only −17 mV when [Ca2+]i increases from 10 to 100 mM. These results indicate that D362 may be involved in channel activation at high [Ca2+]i.
However, Fig. 9 shows that neither of the triple mutations 5D5N + D367A + E399N or 5D5N + D367A:D362A + E399N affected Mg2+ dependence at high concentration, indicating that D362A is not part of the Mg2+ binding site. The different effects of D362A on Ca2+ and Mg2+-dependent activation at high concentrations is striking that the mutation 5D5N + D362A:D367A + E399N reduces the leftward shift of G-V relations at high [Ca2+]i but does not affect channel activation at high [Mg2+]i (Figs. 9 and 10). This result may suggest that Mg2+ at high concentrations binds to a different site than Ca2+ at high concentrations in activating the channel and D362 may only affect Ca2+ binding. However, this possibility seems unlikely because for 5D5N + D367A + E399N mutant channels, Ca2+ and Mg2+ at high concentrations have similar effects on channel activation (Fig. 10 C), suggesting that they may bind to the same site. Since other higher affinity metal sites bind both Mg2+ and Ca2+ in mslo1 (Figs. 4 and 8), it is hard to imagine that the low affinity binding site can have a high selectivity to distinguish between Mg2+ and Ca2+. Therefore, an alternative possibility is that D362 may not be part of the metal binding site but be part of the structure that links metal binding to channel opening. The mutation D362A may affect Mg2+ and Ca2+-dependent activation differently because the two ions have different sizes and hence cause different conformational changes at the binding site.
As shown in Figs. 9 and 10, 5D5N + D362A:D367A + E399N mutant channels are mostly blocked at high [Ca2+]i and [Mg2+]i. Such block was observed previously in a similar triple mutant channel (Xia et al., 2002). The nature of the block is not studied in detail here. It is possible that the block may interfere with our measurements of channel activation so that the observed mutational effects on channel activation in Figs. 9 and 10 may derive from this block but not the intrinsic property of channel activation. However, 5D5N + D367A + E399N and 5D5N + D362A:D367A + E399N mutant channels are similarly blocked by high [Ca2+]i but Ca2+-dependent activation of these mutant channels is different (Fig. 10). This result gave us some confidence to believe that D362A may indeed affect channel activation at high [Ca2+]i. We also notice that although the triple mutation 5D5N + D362A:D367A + E399N reduces the leftward shift of G-V relations at high [Ca2+]i it is unlike another very similar triple mutation 5D5N + D362A:D367A + E399A that reversed Ca2+ dependence of channel activation by causing rightward G-V shifts at high [Ca2+]i (Xia et al., 2002). The cause of this discrepancy is not known.
It has been shown that extracellular Mg2+ can shift the G-V relation of BK channels by screening electric charges on the channel protein (MacKinnon et al., 1989). While the mechanism of D362A in reducing G-V shift at high [Ca2+]i (Fig. 10) is not clear, the different effects of D362A on Ca2+ and Mg2+-dependent activation at high concentrations suggest that D362 may not be responsible for any screening effect of intracellular cations on BK channels, because Ca2+ and Mg2+ should have caused a similar screening effect had the mutation D362A reduced the surface charge. However, D362A does not completely abolish the G-V shift induced by the [Ca2+]i increase from 10 to 100 mM, leaving a residual ∼−17 mV shift (Fig. 10) as compared with a ∼−40 mV shift in the WT channels induced by a similar [Mg2+]i increase (Fig. 2). This residual G-V shift can be caused by Ca2+ or Mg2+ screening some other charges on the surface of the channel protein or the inner membrane lipids. To estimate possible contributions of the screening effect to G-V shifts induced by increases of [Ca2+]i or [Mg2+]i, we calculated surface potentials under our experimental conditions using the Gouy-Chapman model (Grahame, 1947; Hille et al., 1975; McLaughlin, 1977; MacKinnon et al., 1989) (Fig. 11). The surface potential depends on charge densities on the surface of the channel protein or the inner membrane lipids (Fig. 11). The charge density on the inner surface of the plasma membrane is ∼0.14 charges/nm2 (Chandler and Meves, 1965; Hille et al., 1975). The charge density on the intracellular surface of the BK channel protein that affects its voltage-dependent gating is not known. It would be ∼0.02 charges/nm2 if we assume a similar charge density at both extracellular and intracellular surface (MacKinnon et al., 1989). Hence, the screening effect may contribute ∼−9 mV to the shift in G-V relations due to increases of [Ca2+]i or [Mg2+]i from 10 to 100 mM (Fig. 11 B). Therefore, the −40 mV shift in G-V relations in response to increases of [Ca2+]i or [Mg2+]i from 10 to 100 mM largely resulted from these ions' binding to the novel metal binding site. Even if the charge density at the intracellular surface of BK channels is 20 times higher than at the extracellular surface half of the G-V shift is contributed by ions' binding to the novel metal binding site.
Figure 11.

Effects of Mg2+ on the membrane surface potential due to surface charge screening. (A) Surface potentials at 1, 10, and 100 mM [Mg2+]i as a function of surface charge density. The calculation is based on the Grahame equation (Grahame, 1947). The solution contains 140 mM K+. (B) Change of surface potential as [Mg2+]i increases from 1 to 10 mM and from 10 to 100 mM.
We use MWC models to describe channel activation due to metal binding. The MWC model is chosen because it is the simplest kinetic scheme that represents the allosteric mechanism of metal-dependent BK channel activation and it described experimental data very well in various studies (McManus and Magleby, 1991; Cox et al., 1997a; Shi and Cui, 2001; Zhang et al., 2001; Xia et al., 2002). A more complex model has been proposed to describe the dual-allosteric mechanism of both voltage and Ca2+-dependent activation of BK channels (Horrigan and Aldrich, 2002). This model includes an allosteric interaction between voltage and Ca2+-dependent activation mechanisms (described by the E factor) that accounts for the change of G-V slopes and shifts at various [Ca2+]i that slightly differ from the prediction of the MWC model (Horrigan and Aldrich, 2002). However, the primary difference between the two models lies at the description of voltage-dependent mechanism while their descriptions on Ca2+ dependence of channel activation are similar. Both models predict a dissociation constant of Ca2+ binding at the closed conformation around 10 μM and an allosteric factor C around 10 (Cox et al., 1997a; Horrigan and Aldrich, 2002). In the dual-allosteric model the coupling between voltage and Ca2+-dependent mechanism is weak (E is small) (Horrigan and Aldrich, 2002). Previous experimental results also demonstrated that the mechanisms of voltage and Ca2+-dependent activation do not affect each other significantly (Cui and Aldrich, 2000; Shi et al., 2002; Qian and Magleby, 2003). Therefore, among existing models, the MWC model is not only the simplest but also adequate in describing metal-dependent activation of BK channels. In this study the MWC model is able to fit experimental data under a broad range of ionic and voltage conditions although only a subset of the data is required to define the parameters of the model (Fig. 8). The conclusion based on model fittings (Fig. 8) is consistent with mutational studies (Fig. 9). These results indicate that the model provides a quite precise description for mslo1 channel activation within our tested experimental conditions.
Acknowledgments
The mslo1 clone was provided to us by Larry Salkoff (Washington University, St. Louis, MO).
This work was supported by National Institutes of Health grant R01-HL70393, the American Heart Association, and the Whitaker Foundation (J. Cui). J. Cui is Associate Professor of Biomedical Engineering on the Spencer T. Olin Endowment.
Olaf S. Andersen served as editor.
References
- Adams, P.R., A. Constanti, D.A. Brown, and R.B. Clark. 1982. Intracellular Ca2+ activates a fast voltage-sensitive K+ current in vertebrate sympathetic neurones. Nature. 296:746–749. [DOI] [PubMed] [Google Scholar]
- Adelman, J.P., K.Z. Shen, M.P. Kavanaugh, R.A. Warren, Y.N. Wu, A. Lagrutta, C.T. Bond, and R.A. North. 1992. Calcium-activated potassium channels expressed from cloned complementary DNAs. Neuron. 9:209–216. [DOI] [PubMed] [Google Scholar]
- Ahluwalia, J., A. Tinker, L.H. Clapp, M.R. Duchen, A.Y. Abramov, S. Pope, M. Nobles, and A.W. Segal. 2004. The large-conductance Ca2+-activated K+ channel is essential for innate immunity. Nature. 427:853–858. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Atkinson, N.S., G.A. Robertson, and B. Ganetzky. 1991. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science. 253:551–555. [DOI] [PubMed] [Google Scholar]
- Bao, L., C. Kaldany, E.C. Holmstrand, and D.H. Cox. 2004. Mapping the BKCa channel's “Ca2+ bowl”: side-chains essential for Ca2+ sensing. J. Gen. Physiol. 123:475–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, L., A.M. Rapin, E.C. Holmstrand, and D.H. Cox. 2002. Elimination of the BKCa channel's high-affinity Ca2+ sensitivity. J. Gen. Physiol. 120:173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens, R., A. Nolting, F. Reimann, M. Schwarz, R. Waldschutz, and O. Pongs. 2000. hKCNMB3 and hKCNMB4, cloning and characterization of two members of the large-conductance calcium-activated potassium channel β subunit family. FEBS Lett. 474:99–106. [DOI] [PubMed] [Google Scholar]
- Berridge, M.J., P. Lipp, and M.D. Bootman. 2000. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1:11–21. [DOI] [PubMed] [Google Scholar]
- Bian, S., I. Favre, and E. Moczydlowski. 2001. Ca2+-binding activity of a COOH-terminal fragment of the Drosophila BK channel involved in Ca2+-dependent activation. Proc. Natl. Acad. Sci. USA. 98:4776–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, A., and L. Sy. 2001. Contribution of potential EF hand motifs to the calcium-dependent gating of a mouse brain large conductance, calcium-sensitive K+ channel. J. Physiol. 533:681–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner, R., T.J. Jegla, A. Wickenden, Y. Liu, and R.W. Aldrich. 2000. a. Cloning and functional characterization of novel large conductance calcium-activated potassium channel β subunits, hKCNMB3 and hKCNMB4. J. Biol. Chem. 275:6453–6461. [DOI] [PubMed] [Google Scholar]
- Brenner, R., G.J. Perez, A.D. Bonev, D.M. Eckman, J.C. Kosek, S.W. Wiler, A.J. Patterson, M.T. Nelson, and R.W. Aldrich. 2000. b. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature. 407:870–876. [DOI] [PubMed] [Google Scholar]
- Butler, A., S. Tsunoda, D.P. McCobb, A. Wei, and L. Salkoff. 1993. mSlo, a complex mouse gene encoding “maxi” calcium-activated potassium channels. Science. 261:221–224. [DOI] [PubMed] [Google Scholar]
- Chandler, W.K., and H. Meves. 1965. Voltage clamp experiments on internally perfused giant axons. J. Physiol. 180:788–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corkey, B.E., J. Duszynski, T.L. Rich, B. Matschinsky, and J.R. Williamson. 1986. Regulation of free and bound magnesium in rat hepatocytes and isolated mitochondria. J. Biol. Chem. 261:2567–2574. [PubMed] [Google Scholar]
- Cox, D.H., and R.W. Aldrich. 2000. Role of the β1 subunit in large-conductance Ca2+-activated K+ channel gating energetics. Mechanisms of enhanced Ca2+ sensitivity. J. Gen. Physiol. 116:411–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, D.H., J. Cui, and R.W. Aldrich. 1997. a. Allosteric gating of a large conductance Ca2+-activated K+ channel. J. Gen. Physiol. 110:257–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, D.H., J. Cui, and R.W. Aldrich. 1997. b. Separation of gating properties from permeation and block in mslo large conductance Ca2+-activated K+ channels. J. Gen. Physiol. 109:633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, J., and R.W. Aldrich. 2000. Allosteric linkage between voltage and Ca2+-dependent activation of BK-type mslo1 K+ channels. Biochemistry. 39:15612–15619. [DOI] [PubMed] [Google Scholar]
- Cui, J., D.H. Cox, and R.W. Aldrich. 1997. Intrinsic voltage dependence and Ca2+ regulation of mslo large conductance Ca2+-activated K+ channels. J. Gen. Physiol. 109:647–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delva, P. 2003. a. Magnesium and cardiac arrhythmias. Mol. Aspects Med. 24:53–62. [DOI] [PubMed] [Google Scholar]
- Delva, P. 2003. b. Magnesium and coronary heart disease. Mol. Aspects Med. 24:63–78. [DOI] [PubMed] [Google Scholar]
- Delva, P. 2003. c. Magnesium and heart failure. Mol. Aspects Med. 24:79–105. [DOI] [PubMed] [Google Scholar]
- Dworetzky, S.I., J.T. Trojnacki, and V.K. Gribkoff. 1994. Cloning and expression of a human large-conductance calcium-activated potassium channel. Brain Res. Mol. Brain Res. 27:189–193. [DOI] [PubMed] [Google Scholar]
- Fabiato, A., and F. Fabiato. 1979. Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J. Physiol. (Paris). 75:463–505. [PubMed] [Google Scholar]
- Ferguson, W.B. 1991. Competitive Mg2+ block of a large-conductance, Ca2+-activated K+ channel in rat skeletal muscle. Ca2+, Sr2+, and Ni2+ also block. J. Gen. Physiol. 98:163–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fettiplace, R., and P.A. Fuchs. 1999. Mechanisms of hair cell tuning. Annu. Rev. Physiol. 61:809–834. [DOI] [PubMed] [Google Scholar]
- Flatman, P.W. 1984. Magnesium transport across cell membranes. J. Membr. Biol. 80:1–14. [DOI] [PubMed] [Google Scholar]
- Flatman, P.W. 1991. Mechanisms of magnesium transport. Annu. Rev. Physiol. 53:259–271. [DOI] [PubMed] [Google Scholar]
- Golowasch, J., A. Kirkwood, and C. Miller. 1986. Allosteric effects of Mg2+ on the gating of Ca2+-activated K+ channels from mammalian skeletal muscle. J. Exp. Biol. 124:5–13. [DOI] [PubMed] [Google Scholar]
- Grahame, D.C. 1947. The electrical double layer and the theory of electrocapillarity. Chem. Rev. 41:441–501. [DOI] [PubMed] [Google Scholar]
- Gupta, R.K., P. Gupta, and R.D. Moore. 1984. NMR studies of intracellular metal ions in intact cells and tissues. Annu. Rev. Biophys. Bioeng. 13:221–246. [DOI] [PubMed] [Google Scholar]
- Hille, B., A.M. Woodhull, and B.I. Shapiro. 1975. Negative surface charge near sodium channels of nerve: divalent ions, monovalent ions, and pH. Philos. Trans. R. Soc. Lond. B Biol. Sci. 270:301–318. [DOI] [PubMed] [Google Scholar]
- Horrigan, F.T. 2005. Mg2+ increases the coupling of voltage-sensor activation to channel opening to BK potassium channels. Biophys. J. 88:100A. [Google Scholar]
- Horrigan, F.T., and R.W. Aldrich. 2002. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J. Gen. Physiol. 120:267–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudspeth, A.J., and R.S. Lewis. 1988. a. Kinetic analysis of voltage- and ion-dependent conductances in saccular hair cells of the bull-frog, Rana catesbeiana. J. Physiol. 400:237–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudspeth, A.J., and R.S. Lewis. 1988. b. A model for electrical resonance and frequency tuning in saccular hair cells of the bull-frog, Rana catesbeiana. J. Physiol. 400:275–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Pico, M. Cadene, B.T. Chait, and R. MacKinnon. 2001. Structure of the RCK domain from the E. coli K+ channel and demonstration of its presence in the human BK channel. Neuron. 29:593–601. [DOI] [PubMed] [Google Scholar]
- Jiang, Z., M. Wallner, P. Meera, and L. Toro. 1999. Human and rodent MaxiK channel β-subunit genes: cloning and characterization. Genomics. 55:57–67. [DOI] [PubMed] [Google Scholar]
- Kume, H., K. Mikawa, K. Takagi, and M.I. Kotlikoff. 1995. Role of G proteins and KCa channels in the muscarinic and β-adrenergic regulation of airway smooth muscle. Am. J. Physiol. 268:L221–L229. [DOI] [PubMed] [Google Scholar]
- Lancaster, B., and R.A. Nicoll. 1987. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J. Physiol. 389:187–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurant, P., and R.M. Touyz. 2000. Physiological and pathophysiological role of magnesium in the cardiovascular system: implications in hypertension. J. Hypertens. 18:1177–1191. [DOI] [PubMed] [Google Scholar]
- Laver, D.R. 1992. Divalent cation block and competition between divalent and monovalent cations in the large-conductance K+ channel from Chara australis. J. Gen. Physiol. 100:269–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon, R., R. Latorre, and C. Miller. 1989. Role of surface electrostatics in the operation of a high-conductance Ca2+-activated K+ channel. Biochemistry. 28:8092–8099. [DOI] [PubMed] [Google Scholar]
- Marrion, N.V., and S.J. Tavalin. 1998. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 395:900–905. [DOI] [PubMed] [Google Scholar]
- Marty, A. 1981. Ca-dependent K channels with large unitary conductance in chromaffin cell membranes. Nature. 291:497–500. [DOI] [PubMed] [Google Scholar]
- McLaughlin, S. 1977. Electrostatic potentials at membrane-solution interfaces. Curr. Top. Membr. Transp. 9:71–144. [Google Scholar]
- McManus, O.B., L.M. Helms, L. Pallanck, B. Ganetzky, R. Swanson, and R.J. Leonard. 1995. Functional role of the β subunit of high conductance calcium-activated potassium channels. Neuron. 14:645–650. [DOI] [PubMed] [Google Scholar]
- McManus, O.B., and K.L. Magleby. 1991. Accounting for the Ca2+-dependent kinetics of single large-conductance Ca2+-activated K+ channels in rat skeletal muscle. J. Physiol. 443:739–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meera, P., M. Wallner, Z. Jiang, and L. Toro. 1996. A calcium switch for the functional coupling between α (hslo) and β subunits (KV,Ca β) of maxi K channels. FEBS Lett. 382:84–88. [DOI] [PubMed] [Google Scholar]
- Meera, P., M. Wallner, and L. Toro. 2000. A neuronal β subunit (KCNMB4) makes the large conductance, voltage- and Ca2+-activated K+ channel resistant to charybdotoxin and iberiotoxin. Proc. Natl. Acad. Sci. USA. 97:5562–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss, G.W., J. Marshall, and E. Moczydlowski. 1996. a. Hypothesis for a serine proteinase-like domain at the COOH terminus of Slowpoke calcium-activated potassium channels. J. Gen. Physiol. 108:473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss, G.W., J. Marshall, M. Morabito, J.R. Howe, and E. Moczydlowski. 1996. b. An evolutionarily conserved binding site for serine proteinase inhibitors in large conductance calcium-activated potassium channels. Biochemistry. 35:16024–16035. [DOI] [PubMed] [Google Scholar]
- Nelson, M.T., H. Cheng, M. Rubart, L.F. Santana, A.D. Bonev, H.J. Knot, and W.J. Lederer. 1995. Relaxation of arterial smooth muscle by calcium sparks. Science. 270:633–637 (see comments). [DOI] [PubMed] [Google Scholar]
- Nimigean, C.M., and K.L. Magleby. 1999. The β subunit increases the Ca2+ sensitivity of large conductance Ca2+-activated potassium channels by retaining the gating in the bursting states. J. Gen. Physiol. 113:425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberhauser, A., O. Alvarez, and R. Latorre. 1988. Activation by divalent cations of a Ca2+-activated K+ channel from skeletal muscle membrane. J. Gen. Physiol. 92:67–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orio, P., and R. Latorre. 2005. Differential Effects of β1 and β2 subunits on BK channel activity. J. Gen. Physiol. 125:395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallanck, L., and B. Ganetzky. 1994. Cloning and characterization of human and mouse homologs of the Drosophila calcium-activated potassium channel gene, slowpoke. Hum. Mol. Genet. 3:1239–1243. [DOI] [PubMed] [Google Scholar]
- Pallotta, B.S., K.L. Magleby, and J.N. Barrett. 1981. Single channel recordings of Ca2+-activated K+ currents in rat muscle cell culture. Nature. 293:471–474. [DOI] [PubMed] [Google Scholar]
- Perez, G., and L. Toro. 1994. Differential modulation of large-conductance KCa channels by PKA in pregnant and nonpregnant myometrium. Am. J. Physiol. 266:C1459–C1463. [DOI] [PubMed] [Google Scholar]
- Perez, G.J., A.D. Bonev, J.B. Patlak, and M.T. Nelson. 1999. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J. Gen. Physiol. 113:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, O.H., and Y. Maruyama. 1984. Calcium-activated potassium channels and their role in secretion. Nature. 307:693–696. [DOI] [PubMed] [Google Scholar]
- Piskorowski, R., and R.W. Aldrich. 2002. Calcium activation of BKCa potassium channels lacking the calcium bowl and RCK domains. Nature. 420:499–502. [DOI] [PubMed] [Google Scholar]
- Pluger, S., J. Faulhaber, M. Furstenau, M. Lohn, R. Waldschutz, M. Gollasch, H. Haller, F.C. Luft, H. Ehmke, and O. Pongs. 2000. Mice with disrupted BK channel β1 subunit gene feature abnormal Ca2+ spark/STOC coupling and elevated blood pressure. Circ. Res. 87:E53–E60. [DOI] [PubMed] [Google Scholar]
- Qian, X., and K.L. Magleby. 2003. β1 subunits facilitate gating of BK channels by acting through the Ca2+, but not the Mg2+, activating mechanisms. Proc. Natl. Acad. Sci. USA. 100:10061–10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaelli, G., C. Saviane, M.H. Mohajerani, P. Pedarzani, and E. Cherubini. 2004. BK potassium channels control transmitter release at CA3-CA3 synapses in the rat hippocampus. J. Physiol. 557:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazi, M.A., P. Brinkman-Mills, A. Johnson, S.L. Naylor, S. Minoshima, N. Shimizu, A. Baldini, and H.E. McDermid. 1999. Identification of a putative regulatory subunit of a calcium-activated potassium channel in the dup(3q) syndrome region and a related sequence on 22q11.2. Genomics. 62:90–94. [DOI] [PubMed] [Google Scholar]
- Ricci, A.J., M. Gray-Keller, and R. Fettiplace. 2000. Tonotopic variations of calcium signalling in turtle auditory hair cells. J. Physiol. 524(Pt 2):423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, W.M. 1994. Localization of calcium signals by a mobile calcium buffer in frog saccular hair cells. J. Neurosci. 14:3246–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, W.M., R.A. Jacobs, and A.J. Hudspeth. 1990. Colocalization of ion channels involved in frequency selectivity and synaptic transmission at presynaptic active zones of hair cells. J. Neurosci. 10:3664–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille, R., and M.P. Charlton. 1992. Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. J. Neurosci. 12:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille, R., M.L. Garcia, G.J. Kaczorowski, and M.P. Charlton. 1993. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 11:645–655. [DOI] [PubMed] [Google Scholar]
- Samaranayake, H., J.C. Saunders, M.I. Greene, and D.S. Navaratnam. 2004. Ca2+ and K+ (BK) channels in chick hair cells are clustered and colocalized with apical-basal and tonotopic gradients. J. Physiol. 560:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber, M., and L. Salkoff. 1997. A novel calcium-sensing domain in the BK channel. Biophys. J. 73:1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber, M., A. Yuan, and L. Salkoff. 1999. Transplantable sites confer calcium sensitivity to BK channels. Nat. Neurosci. 2:416–421. [DOI] [PubMed] [Google Scholar]
- Seelig, M.S. 2000. Interrelationship of magnesium and congestive heart failure. Wien. Med. Wochenschr. 150:335–341. [PubMed] [Google Scholar]
- Shi, J., and J. Cui. 2001. Intracellular Mg2+ enhances the function of BK-type Ca2+-activated K+ channels. J. Gen. Physiol. 118:589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, J., G. Krishnamoorthy, Y. Yang, L. Hu, N. Chaturvedi, D. Harilal, J. Qin, and J. Cui. 2002. Mechanism of magnesium activation of calcium-activated potassium channels. Nature. 418:876–880. [DOI] [PubMed] [Google Scholar]
- Storm, J.F. 1987. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J. Physiol. 385:733–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stretton, D., M. Miura, M.G. Belvisi, and P.J. Barnes. 1992. Calcium-activated potassium channels mediate prejunctional inhibition of peripheral sensory nerves. Proc. Natl. Acad. Sci. USA. 89:1325–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, X.P., B. Yazejian, and A.D. Grinnell. 2004. Electrophysiological properties of BK channels in Xenopus motor nerve terminals. J. Physiol. 557:207–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, Y., P. Meera, M. Song, H.G. Knaus, and L. Toro. 1997. Molecular constituents of maxi KCa channels in human coronary smooth muscle: predominant α + β subunit complexes. J. Physiol. 502:545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz, R.M. 2003. Role of magnesium in the pathogenesis of hypertension. Mol. Aspects Med. 24:107–136. [DOI] [PubMed] [Google Scholar]
- Tseng-Crank, J., C.D. Foster, J.D. Krause, R. Mertz, N. Godinot, T.J. DiChiara, and P.H. Reinhart. 1994. Cloning, expression, and distribution of functionally distinct Ca2+- activated K+ channel isoforms from human brain. Neuron. 13:1315–1330. [DOI] [PubMed] [Google Scholar]
- Tseng-Crank, J., N. Godinot, T.E. Johansen, P.K. Ahring, D. Strobaek, R. Mertz, C.D. Foster, S.P. Olesen, and P.H. Reinhart. 1996. Cloning, expression, and distribution of a Ca2+-activated K+ channel β-subunit from human brain. Proc. Natl. Acad. Sci. USA. 93:9200–9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink, R., and I. Cernak. 2000. Regulation of intracellular free magnesium in central nervous system injury. Front. Biosci. 5:D656–D665. [DOI] [PubMed] [Google Scholar]
- Wallner, M., P. Meera, and L. Toro. 1999. Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: a transmembrane β-subunit homolog. Proc. Natl. Acad. Sci. USA. 96:4137–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X., T. Inukai, M.A. Greer, and S.E. Greer. 1994. Evidence that Ca2+-activated K+ channels participate in the regulation of pituitary prolactin secretion. Brain Res. 662:83–87. [DOI] [PubMed] [Google Scholar]
- Weiger, T.M., M.H. Holmqvist, I.B. Levitan, F.T. Clark, S. Sprague, W.J. Huang, P. Ge, C. Wang, D. Lawson, M.E. Jurman, et al. 2000. A novel nervous system β subunit that downregulates human large conductance calcium-dependent potassium channels. J. Neurosci. 20:3563–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman, G.C., and M.T. Nelson. 2003. Signaling between SR and plasmalemma in smooth muscle: sparks and the activation of Ca2+-sensitive ion channels. Cell Calcium. 34:211–229. [DOI] [PubMed] [Google Scholar]
- Wu, Y.C., J.J. Art, M.B. Goodman, and R. Fettiplace. 1995. A kinetic description of the calcium-activated potassium channel and its application to electrical tuning of hair cells. Prog. Biophys. Mol. Biol. 63:131–158. [DOI] [PubMed] [Google Scholar]
- Xia, X.M., X. Zeng, and C.J. Lingle. 2002. Multiple regulatory sites in large-conductance calcium-activated potassium channels. Nature. 418:880–884. [DOI] [PubMed] [Google Scholar]
- Xia, X.M., X. Zhang, and C.J. Lingle. 2004. Ligand-dependent activation of Slo family channels is defined by interchangeable cytosolic domains. J. Neurosci. 24:5585–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazejian, B., D.A. DiGregorio, J.L. Vergara, R.E. Poage, S.D. Meriney, and A.D. Grinnell. 1997. Direct measurements of presynaptic calcium and calcium-activated potassium currents regulating neurotransmitter release at cultured Xenopus nerve-muscle synapses. J. Neurosci. 17:2990–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazejian, B., X.P. Sun, and A.D. Grinnell. 2000. Tracking presynaptic Ca2+ dynamics during neurotransmitter release with Ca2+-activated K+ channels. Nat. Neurosci. 3:566–571. [DOI] [PubMed] [Google Scholar]
- Zeng, X.H., X.M. Xia, and C.J. Lingle. 2005. Divalent cation sensitivity of BK channel activation supports the existence of three distinct binding sites. J. Gen. Physiol. 125:273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X., C.R. Solaro, and C.J. Lingle. 2001. Allosteric regulation of BK channel gating by Ca2+ and Mg2+ through a nonselective, low affinity divalent cation site. J. Gen. Physiol. 118:607–636. [DOI] [PMC free article] [PubMed] [Google Scholar]