

Abstract
The Na+/glucose cotransporter (SGLT1) is an archetype for the SLC5 family, which is comprised of Na+-coupled transporters for sugars, myo-inositol, choline, and organic anions. Application of the reducing agent dithriothreitol (DTT, 10 mM) to oocytes expressing human SGLT1 affects the protein's presteady-state currents. Integration of these currents at different membrane potentials (Vm) produces a Q-V curve, whose form was shifted by +25 mV due to DTT. The role of the 15 endogenous cysteine residues was investigated by expressing SGLT1 constructs, each bearing a single mutation for an individual cysteine, in Xenopus oocytes, using two-microelectrode voltage-clamp electrophysiology and fluorescent labeling. 12 of the 15 mutants were functional and could be separated into three distinct groups based on the effect of the mutation on the Q-V curve: four mutants did not perturb the transferred charge, six mutants shifted the Q-V curve towards negative potentials, and two mutants (C255A and C511A) produced a shift in the positive direction that was identical to the shift produced by DTT on the wild-type (wt) SGLT1. The double mutant C255,511A confirms that the effects of each single mutant on the Q-V curve were not additive. With respect to wt SGLT1, the apparent affinities for α-methylglucose (αMG) were increased in a similar manner for the single mutants C255A and C511A, the double mutant C255,511A as well as for wt SGLT1 treated with DTT. When exposed to a maleimide-based fluorescent probe, wt SGLT1 was not significantly labeled but mutants C255A and C511A could be clearly labeled, indicating an accessible cysteine residue. These residues are presumed to be C511 and C255, respectively, as the double mutant C255,511A could not be labeled. These results strongly support the hypothesis that C255 and C511 form a disulfide bridge in human SGLT1 and that this disulfide bridge is involved in the conformational change of the free carrier.
INTRODUCTION
The Na+/glucose cotransporter SGLT1, responsible for intestinal absorption and renal reabsorption of glucose, was first postulated by Crane four decades ago (Crane, 1965). In 1987, the cDNA of a first Na+/glucose cotransporter was cloned from rabbit small intestine (Hediger et al., 1987), which has enabled detailed characterization using expression in Xenopus oocytes with two-electrode voltage-clamp and cut-open oocyte electrophysiology. Exploration of steady-state and presteady-state kinetics (i.e., phlorizin-sensitive presteady-state currents) led to a simple kinetic model explaining ligand binding and conformational changes but the identification of the amino acids forming the actual binding sites for Na+, glucose, and phlorizin (Pz) is still a matter of some debate (Parent et al., 1992a,b; Panayotova-Heiermann et al., 1994; Chen et al., 1995; Panayotova-Heiermann et al., 1995, 1996; X.Z. Chen et al., 1996, 1997; Wright et al., 1998; Lo and Silverman, 1998a; Wright and Turk, 2004).
Cysteine residues involved in disulfide bridges generally play an important role in stabilizing a protein's three dimensional structure. Studies identifying essential cysteine and potential disulfide bridges in transporters have been performed in the past and have led to the identification of some initial three dimensional constraints, which are generally difficult to obtain for membrane proteins (van Iwaarden et al., 1991; Sahin-Toth et al., 1994; J.G. Chen et al., 1997; Sur et al., 1997; Lambert et al., 2000a,b, 2001; Kohler et al., 2003; Murer et al., 2004). In some studies, cysteine replacements were shown to affect the correct trafficking or stability of the protein in the plasma membrane but there was no evidence that this effect implied breaking of a disulfide bridge (J.G. Chen et al., 1997; Sur et al., 1997; Pajor et al., 1999; Tanaka et al., 2004).
The effect of the reducing agent dithiothreitol (DTT) on Na+/glucose cotransport (probably via SGLT2) was explored in 1983 on rabbit renal outer cortical brush border membranes (Turner and George, 1983, 1984) and the authors proposed that at least two disulfide bridges were important for Pz binding. Unfortunately, no extensive mutagenesis study on the endogenous cysteine residues of SGLT1 (or SGLT2) has been published. However, a few observations suggest important roles for some cysteine residues in SGLT1 (Martin et al., 1996; Wright, 1998; Xia et al., 2003, 2004). The mutations C292Y or C355S in human SGLT1 produce the hereditary disease glucose-galactose malabsorption syndrome (Martin et al., 1996; Wright, 1998). Cysteines in positions 560 and 608 (in rabbit SGLT1) have been proposed to form a disulfide bridge in the purified isolated loop between the last two transmembrane segments (Xia et al., 2003, 2004). Recently, the mutations C351A and C361A have been shown to impair proper trafficking to the plasma membrane that was, however, present in the double mutant (C351A/C361A) (Xia et al., 2005). Finally, endogenous cysteine residues in SGLT1 have been shown to be insensitive to alkylation by NEM or MTS reagents, which suggests that they are either physically inaccessible or involved in disulfide bridges (Loo et al., 1998; Lo and Silverman, 1998a; Gagnon et al., 2005).
The aim of the present study is to identify the cysteine residues involved in the functional effects caused by treating SGLT1 with DTT, using mutagenesis, electrophysiology, and fluorescent labeling after expressing SGLT1 mutants in Xenopus oocytes. We mutated each endogenous cysteine residue individually into an alanine residue. Our results indicate that cysteine residues C255 and C511 form a disulfide bridge in SGLT1, which explains the effect of DTT on SGLT1. To our knowledge, the identification of this disulfide bridge represents the first three-dimensional constraint to the simple membrane topology proposed for SGLT1.
MATERIALS AND METHODS
Molecular Biology
Mutant forms of SGLT1 were constructed using the human Na+/glucose cotransporter with an NH2-terminal myc epitope in expression vector pBS (pBS-myc-hSGLT1) (Bissonnette et al., 1999). Site-directed mutations were created following the method of Fisher and Pei (1997) and were confirmed by dideoxy DNA sequencing (Sheldon Biotechnology). QIAGEN mini-prep kits were used to extract DNA, and after EcoR1 digestion, the cDNA insert was transcribed in vitro using the T3 mMessage mMachine kit (Ambion).
Oocyte Preparation and Injection
Oocytes were surgically removed from Xenopus laevis frogs, dissected, and defolliculated as described previously (Bissonnette et al., 1999; Gagnon et al., 2004). 1 d after defolliculation, oocytes were injected with 46 nl of mRNA solution (0.1 and 0.25 μg/μl for wt SGLT1 and mutants, respectively) in order to obtain maximal protein expression. Oocytes were maintained in Barth's solution (in mM: 90 NaCl, 3 KCl, 0.82 MgSO4, 0.41 CaCl2, 0.33 Ca(NO3)2, 5 HEPES, pH 7.6) supplemented with 5% horse serum, 2.5 mM Na+-pyruvate, 100 U/ml penicillin, and 0.1 mg/ml streptomycin for 4–7 d before being used in an experiment.
Electrophysiology
The saline solution normally used in our electrophysiological experiments is composed of (in mM) 90 NaCl, 3 KCl, 0.82 MgCl2, 0.74 CaCl2, 10 HEPES, and the pH was adjusted to 7.6 with NaOH. Two electrode voltage-clamp experiments were performed using an Oocyte Clamp OC-725 (Warner Instrument Corp.) and a data acquisition system (Digidata 1322A and Clampex 8.2; Axon Instruments Inc.). Current and voltage microelectrodes were filled with 1 M KCl and had a resistance of 2–5 MΩ. The bath current electrode was a Ag-AgCl pellet, and the reference electrode was a 1 M KCl agar bridge. The oocytes were clamped to a membrane potential of −50 mV, and three repetitions of membrane potential steps between +70 and −170 mV (by increments of 20 mV, 300 ms duration, no series resistance compensation used) were applied with an interval of 1.7 s between each step. Data was obtained with a sampling frequency of 10 kHz and the three repetitions were averaged for each experiment. DTT (Sigma-Aldrich) treatments were performed by exposing oocytes for 20–30 min at their resting potential to 2.5 mM DTT for steady-state measurements (α-methyl-glucose [αMG] affinity measurements) or to 10 mM for presteady-state and Na+ leak measurements. As the effects of DTT were rapidly reversible, DTT was also present in all experimental solutions after the initial treatment.
Data Analysis
Current versus membrane potential curves (I-V curves) were obtained by averaging the current data points at the end of each membrane potential step (between 150 and 300 ms, except for mutants in Group B [see below], which were measured between 250 and 300 ms). Determination of apparent affinity for αMG (Km α MG) was done as described before (Bissonnette et al., 1999). Using the same pulse protocol, presteady-state current analysis (Q-V curves) was performed by subtracting the integrated baseline-corrected currents in phlorizin solution (200 μM) from similar currents in saline solution. In general, the correction for steady-state currents was done by removing the mean current measured between 50 and 80 ms after the initiation of a voltage pulse. This was done in order to minimize the contribution of slower time-dependent increases in measured current that were sometimes observed at extreme membrane potentials. For some mutated proteins that presented significantly slower presteady-state currents, as compared with wt SGLT1 (Group B), the mean currents were taken at the end of the pulse (from 270 to 300 ms). A simple Boltzmann equation was fitted to the Q-V curve to estimate V1/2 (the voltage at which half of the charge is transferred), Qmax (the amplitude of the total charge transferred), and z (the valence of the transferred charge) (Bissonnette et al., 1999). The time constants were evaluated by fitting a double exponential on the Itransit (Isaline − IPz) with the Clampfit 8.2 program (Axon Instruments Inc.).
Modeling of Presteady-state Currents
The presteady-state current time constants and transferred charges were calculated using a four-state kinetic model that we have proposed in a previous publication (Chen et al., 1996) (see Appendices A and B and discussion for details). The numerical simulations were performed using Matlab 6.5.0 software (The Math Works Inc.). The transferred charges were analyzed by fitting a Boltzmann curve to the calculated data in order to extract the V1/2 and z parameters. The parameters of the model were adjusted by trial and error to obtain a satisfactory fit to the measured V1/2 and the slow time constants of the presteady-state currents.
Plasma Membrane Preparation of Oocytes and Western Blot Detection
Preparation of enriched plasma membrane fractions from oocytes and Western blot detection were performed as previously described (Bissonnette et al., 1999). In brief, 20 oocytes were rinsed and then homogenized in 1.5 ml homogenization solution (50 mM mannitol, 2 mM Tris, 5 mM EGTA supplemented with a protease inhibitor cocktail [Sigma-Aldrich], pH 7.0). Three consecutive centrifugations were performed (2 × 10 min at 1,000 g and 1 × 20 min at 10,000 g, at 4°C), and the final pellet was resuspended in 10 μl of homogenization solution. 10 μl of Laemmli solution was added and the samples were loaded onto SDS-polyacrylamide gels for electrophoresis. The quality of transfer onto nitrocellulose was verified using Ponceau red staining. For Western blots, the membrane was blocked against nonspecific binding (5% fat milk in TBS, 1 h at room temperature) and then incubated with the primary antibody (anti-myc, 1:500 in blocking solution, overnight at 4°C). The membrane was washed, blocked again, and then incubated with the secondary antibody (HRP-conjugated anti–mouse IgG, 1:1,000 in blocking solution) at room temperature for 1 h. After final washing steps, blots were revealed by enhanced chemiluminescence detection (Phototope-HRP; New England Biolabs).
Fluorescence
Fluorescence measurements were done as previously described (Gagnon et al., 2005). In brief, a 5–10 min fluorescent labeling was performed on ice in a saline solution containing 20 μM tetramethylrhodamine-5-maleimide fluorescent probe (TMR5M, Molecular Probes) at the oocyte's resting potential. Oocytes were preincubated 5–10 min on ice before the fluorescent probe was added. Fluorescence intensity was read, at the animal pole, using excitation and emission filters (10 nm bandwidth) centered at 500 nm and 600 nm, respectively.
Statistics
Experiments were performed on at least three oocytes obtained from a minimum of two different donors. Data are reported as mean ± SEM (except when otherwise noted) and are compared using unpaired Student's t test; statistical significance was set at P < 0.05.
RESULTS
DTT Effect on Wild-type SGLT1 Activity
We tested the effects of breaking disulfide bridges with DTT on steady-state parameters (cotransport current due to the addition of 5 mM αMG [Icotr], Na+ leak current [Ileak], apparent affinities for the sugar substrate [Km α MG]) and on presteady-state currents (Q-V curves) of the wt human SGLT1. Prior to DTT treatment, our measurements were fully consistent with those previously published for human SGLT (Parent et al., 1992a,b; Loo et al., 1993; Chen et al., 1996; Hirayama et al., 1996; Bissonnette et al., 1999). Fig. 1 illustrates the effects of DTT on steady-state parameters of wt SGLT1. The reducing treatment was found to decrease Ileak (Fig. 1 B) as well as the apparent affinities for αMG (panel C) without having any significant effect on Icotr (panel A).
Figure 1.

Effects of DTT exposure on steady-state parameters of wt Na+/glucose cotransporter (wt SGLT1). (A) Effect of DTT on αMG cotransport current (Icotr) of wt SGLT1. Estimation of the currents was done under control conditions (filled symbols) and in the presence of DTT (2.5 mM) (open symbols) on each oocyte (n = 3, paired experiments). (B) Effect of DTT on Na+ leak current (Ileak) of wt SGLT1. A 30-min exposure with 10 mM DTT was done before the experimental test of Ileak (done in the presence of DTT) (n = 6 for control conditions, filled symbols, n = 5 for DTT, open symbols, unpaired experiments). (C) Effect of DTT on apparent affinity for αMG (Km α MG) as a function of membrane potential (Vm). Estimation of the apparent affinity for αMG was done under control conditions (filled symbols) and in the presence of DTT (2.5 mM) (open symbols) on each oocyte (n = 4, paired experiments).
The effect of DTT treatment on the transferred charge curve, shown in Fig. 2, demonstrates a clear cut shift of +25 mV (see Fig. 2 B) from a V1/2 of −46 ± 3 mV under control conditions (n = 9) to −21 ± 1 mV in the presence of DTT (n = 6). As shown in Fig. 2 C, the effects of DTT on the presteady-state currents of wt SGLT1 are not only to shift the Q-V curve toward more positive potentials but also to accelerate the presteady-state current. Prior to the DTT treatment, the slow time constant of the presteady-state current is observed to plateau at 10 ms for all hyperpolarizing potentials, whereas in DTT the plateau is found around 4.5 ms. This effect requires ∼20–25 min to be complete, and is completely reversible when DTT is removed from bathing solutions (unpublished data). It is clear that the total charge transferred and the z parameter (related to the slope of the Q-V curve) are not affected by the reducing agent, which affects only the V1/2 parameter and the time constant.
Figure 2.

Effect of DTT exposure on presteady-state parameters of wt Na+/glucose cotransporter (wt SGLT1). (A) Presteady-state currents (Pz-sensitive currents in the absence of glucose) of a typical oocyte expressing wt SGLT1 under control conditions and after exposure to DTT (30 min preincubation with 10 mM DTT, with DTT present in subsequent experimental solutions). (B) Effect of DTT on transferred charges (Q-V curve) of wt SGLT1. Q-V curves were fitted with a Boltzmann equation (see MATERIALS AND METHODS), adjusted to 0 at hyperpolarizing voltages and normalized with respect to the extrapolated Qmax (n = 9 for control conditions, filled symbols, n = 6 for DTT, open symbols, unpaired experiments). (C) Time constant of the 10-ms component of presteady-state currents of wt SGLT1 compared with wt + DTT. The curves represent the time constants evaluated with a four-state model of presteady-state currents with parameters described in Table I.
As the presteady-state currents are thought to originate from the movement of a charged portion of SGLT1 in the membrane electric field and/or from the voltage-dependent Na+ binding step (Parent et al., 1992a,b), we hypothesized that one or more disulfide bridges were playing important roles in one or both of these steps. The presence of a disulfide bridge can be assessed by mutating each endogenous cysteine residue, which would potentially remove a disulfide bridge, and further tested by studying double mutants. If a cysteine residue plays an important functional role through a disulfide bridge, mutating either of the two cysteines involved should produce equivalent effects. In addition, mutating a cysteine residue involved in a disulfide bridge should not produce any effect if its partner cysteine has already been replaced (i.e., comparing a single with a double cysteine mutant).
Molecular Biology and Expression of Mutants SGLT1s
Fig. 3 illustrates the position of the 15 endogenous cysteine residues present in the human Na+/glucose cotransporter using a modified topologic model (Gagnon et al., 2005) based on the model originally proposed by Turk et al. (1996). Mutants were tested using two microelectrode electrophysiology. We first tested the three basic electrophysiological characteristics: Icotr (current due to the addition of 5 mM αMG), Ileak (currents in the saline solution minus the currents measured in the presence of 200 μM Pz), and the charge transferred (Q-V).
Figure 3.

A modified topological model of SGLT1. Transmembrane segments are identified by Roman numerals, amino acid positions of endogenous cysteine residues, depicted in white circles, are indicated and the extracellular and intracellular side are indicated by the words “out” and “in,” respectively.
Of the 15 single mutants generated, only three were nonfunctional: C351A, C355A, and C361A. These mutants displayed no presteady-state currents, no Ileak and no Icotr. The possibility that the sugar affinity could have been vastly decreased was investigated by applying 100 mM αMG to these mutants but still no cotransport currents could be detected (n = 3). Through Western blot detection against plasma membrane enriched subcellular fractions of oocytes expressing these mutant proteins, SGLT1 expression could be detected for C351A and C361A but not for mutant C355A (Fig. 4). In a few experiments, a very weak band could be observed for C355A, indicating that the protein was poorly translated or generated an unstable product. As preparations enriched in plasma membranes may also contain intracellular membranes, the three inactive mutants may have been trapped in some intracellular compartments or may be present at the plasma membrane but inactive.
Figure 4.

Western blot analysis of some selected mutants, compared with wt SGLT1, detected with an anti-myc antibody. Extracts enriched in plasma membrane were purified from noninjected oocytes (A), wt SGLT1–injected oocytes (B), and mutants C345A (C), C351A (D), C355A (E), and C361A (F) and double mutant C522,560A (G) and were loaded onto a polyacrylamide gel (equivalent to membranes from 20 oocytes), and protein equivalencies were confirmed by Ponceau Red staining. Molecular standard masses are indicated on the left (in kD) (New England Biolabs).
Based on analysis of their presteady-state currents, the 12 functional mutants could be separated into three categories: Group A, which exhibited no significant shift of the Q-V curve; Group B, which had a shift toward negative potentials; and Group C, which had a shift toward positive potential. These shifts are compared with V1/2 of the wt SGLT1, which averaged −46 ± 3 mV. Figs. 5–7 illustrate, for each of the three groups of mutants, the amplitude of the αMG cotransport current (Icotr), the Na+ leak current (Ileak) at −110 mV (panel A), the Pz-sensitive presteady-state currents (Isaline − IPz) for a selection of membrane potentials ranging from +70 to −170 mV (panel B), and the values of the measured V1/2 (panel C) for presteady-state currents, i.e., the voltage for which the mobile charge is equally balanced between inward-facing and outward-facing configurations.
Figure 5.

Electrophysiological characteristics of mutants classified in Group A, as compared with wt SGLT1. (A) Comparison of mutant SGLTs activities to that of wt SGLT1. Maximal αMG cotransport (Icotr at 5 mM αMG) and Na+ leak (Ileak) currents were measured at −110 mV. The dotted lines indicate the mean expression level of wt SGLT1. n ≥ 5 for each mutant. (B) Off response of presteady-state currents (Pz-sensitive currents in the absence of glucose) of typical oocytes expressing each of the mutant SGLT1s compared with an oocyte expressing wt SGLT1. (C) V1/2 of mutant SGLT1s as compared with that of wt SGLT1. A dotted box represents the interval for which V1/2 of a mutant would not be significantly different from that of wt SGLT1. Errors are SEM. Asterisks indicate the statistical significance with respect to wt SGLT1 (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001).
Figure 7.

Electrophysiological characteristics of mutants classified in Group C, as compared with wt SGLT1, with and without DTT. (A) Comparison of mutant SGLT1s activities to that of wt SGLT1 and to wt SGLT1 + DTT. Maximal αMG cotransport (Icotr at 5 mM αMG) and Na+ leak (Ileak) currents were measured at −110 mV. The dotted lines indicate the mean expression level of wt SGLT1. n ≥ 5 for each mutant. (B) Off response of presteady-state currents (Pz-sensitive currents in the absence of glucose) of typical oocytes expressing wt SGLT1 exposed to DTT and mutants C255A and C511A. (C) V1/2 of mutant SGLT1s as compared with that of wt SGLT1 (filled bars) and to wt SGLT1 and mutants exposed to DTT (open bars). The dotted box represents the interval for which V1/2 of a mutant would not be significantly different from that of wt SGLT1. (D) Time constants of the slow component of presteady-state currents for mutants of Group C. The curves represent the time constants evaluated with a four-state model of presteady-state currents with parameters described in Table I. Errors are SEM.
Figure 6.

Electrophysiological characteristics of mutants classified in Group B, as compared with wt SGLT1. (A) Comparison of mutant SGLT1 activities to that of wt SGLT1. Maximal αMG cotransport (Icotr at 5 mM αMG) and Na+ leak (Ileak) currents were measured at −110 mV. The dotted lines indicate the mean expression level of wt SGLT1. n ≥ 5 for each mutant. (B) I-V curve for αMG cotransport and Na+ leak currents of C292A. (C) Off response of presteady-state currents (Pz-sensitive currents in the absence of glucose) of typical oocytes expressing each of the mutant SGLT1s. (D) V1/2 of mutant SGLT1s as compared with that of wt SGLT1. A dotted box represents the interval for which V1/2 of a mutant would not be significantly different from that of wt SGLT1. Errors are SEM. All V1/2 values for this series of mutants were significantly different (P < 0.001 in each case) from the V1/2 of wt SGLT1.
Mutants C317A, C345A, C517A, and C659A form Group A (Fig. 5). Aside from mutant C345A, which has a very low expression level (Icotr of −107 ± 39 nA, Ileak of −13 ± 5 nA, and Qmax of 2.66 ± 0.09 nC, n = 4, see Fig. 4 for the detection of C345A in Western blots), the expression levels of mutants C317A, C517A, and C659A were comparable to that normally reached by wt SGLT1. The V1/2 of these four mutants ranged between −40 and −56 mV and, aside from the V1/2 for mutant C659A, were not significantly different from the V1/2 for wt SGLT1. Mutant C659A had a V1/2 of −56 ± 2 mV (n = 5) and was barely more negative than that of wt SGLT1 (P = 0.011), but because of the small difference and the considerable variability normally observed within a series of measurements of V1/2, mutant C659A was classified into Group A.
Group B (Fig. 6) is composed of single mutants that have a V1/2 shifted toward negative membrane potentials as compared with that of wt SGLT1. The V1/2 values for mutants C292A, C301A, C314A, C522A, C560A, and C610A were measured at −84 ± 4 (n = 7), −81 ± 2 (n = 6), −66 ± 2 (n = 7), −67 ± 4 (n = 7), −75 ± 3 (n = 6) and −121 ± 6 (n = 6) mV, respectively. As observed for three of the Group A mutants, mutants C301A, C314A, and C610A display Icotr and Ileak values quite comparable to those for wt SGLT1. Mutants C522A and C560A were both characterized by a reduced Icotr (−174 ± 29 nA and −307 ± 43 nA, respectively), which might be caused by a low expression level as their Qmax (2.7 ± 0.2 and 9.2 ± 0.5 nC, respectively) values were also reduced with respect to that for wt SGLT1 (Qmax wt = 25 ± 5 nC). Cotransport through the last mutant in Group B, C292A, appears to be severely perturbed since its Icotr is only 20% of the wt Icotr but its Ileak is twice as large. As shown in Fig. 6 B, the leak current of C292A becomes larger than its cotransport current at potentials more negative than −90 mV. As the reduced Icotr seen for C292A, C522A, and C560A could be due to a large decrease in sugar affinity, the Km α MG of these mutants were measured. They were found to be even smaller than that for wt SGLT1 (in mM: 0.27 ± 0.01 for C292A, 0.71 ± 0.06 for C522A, 0.38 ± 0.1 for C560A, and 0.97 ± 0.1 mM for wt SGLT1). We tested the effect of DTT on mutants C560A and C610A, and their V1/2 was also shifted by approximately +25 mV for mutant C560A and about +45 mV for C610A, indicating that these two cysteines were not responsible for the observed effect of DTT on V1/2 of wt SGLT1.
Finally, the two remaining mutants, C255A and C511A, comprise Group C. As shown in Fig. 7 A, these mutants share the characteristics of wt SGLT1 in the presence of DTT, such as a reduced Ileak. The V1/2 of mutant C255A was −24 ± 2 mV (n = 8) and −27 ± 2 mV (n = 4) for C511A, which is not significantly different from the V1/2 of wt SGLT1 in the presence of DTT (−21 ± 1 mV, n = 6). The two mutants were insensitive to DTT treatment with respect to V1/2 (no further shift) and Ileak (remained identical to the Ileak observed before DTT treatment). The time constants, shown in Fig. 7 D, are characterized by their plateau value at ∼4.5 ms. These values are remarkably similar to the time constants characterizing wt SGLT1 exposed to DTT (see Fig. 2 C).
αMG Affinities of Certain Mutants
Other functional characteristics can be used to help identify cysteine residues involved in disulfide bridges. Echoing Fig. 1 C, Fig. 8 shows that the DTT increased the Km α MG of wt SGLT1 from 0.97 ± 0.1 mM to 1.6 ± 0.1 mM (as measured at −150 mV). Interestingly, the two mutants of Group C (C255A and C511A) have Km α MG values very similar to that of DTT-treated wt SGLT1 (in mM: 1.5 ± 0.1 for C255A and 1.6 ± 0.2 for C511A).
Figure 8.

Apparent affinity for αMG (Km α MG) for oocytes expressing mutants C255A, C511A, and C255,511A compared with wt SGLT1, with and without DTT. The application of DTT to wt SGLT1 was performed as described in MATERIALS AND METHODS (paired experiments). The affinity was assessed with data measured at −150 mV. The dotted box represents the interval for which the Km α MG of a mutant would not be significantly different from that of wt SGLT1. The number of individual oocytes used per value is indicated in parentheses. Errors are SEM. Stars indicate the statistical significance (see Fig 5 legend).
Identification of Pairs of Cysteine Residues Possibly Forming a Disulfide Bridge
The data shown in Figs. 6 and 7 already suggest specific pairs of cysteine residues that could be involved in disulfide bridges. The most obvious pair is from Group C where replacing C255 and C511 generate functional effects that closely mimic the effect of DTT. This is true for the positive shift in V1/2, the acceleration of the presteady-state currents and the reduced proportion between Ileak and Icotr. Within Group B, the identification of interesting cysteine residues pairs is less straightforward. Mutants C314A, C522A, and C560A had very similar V1/2 values. Of these three mutants, C522A and C560A also exhibited similar reduction in levels of expression. Consequently, double mutants were produced involving the pairs of amino acids residues 314,560 and 522,560. The pair 560,610 was also studied because these two cysteines have previously been postulated to form a disulfide bridge in an isolated loop in rabbit SGLT1 (Xia et al., 2003, 2004) and this hypothesis had not yet been tested using the complete and functional protein. We also studied other pairs of mutants under the assumption that C610 could be forming a disulfide bridge with a different partner than the one proposed by Xia et al. (2003, 2004). The most logical partners (292 and 301) were the cysteines that had the most important shift in V1/2 as compared with wt SGLT1.
Double Mutants
In creating double mutants to check for the presence of a disulfide bridge, we sought instances where the effect of a single cysteine replacement on a given parameter would vanish if its partner cysteine residue had already been replaced. Six double mutants were generated. Unfortunately, the double mutant C522,560A was not functional even though it was present as observed by Western blot (Fig. 4, lane G). The other double mutants (C255,511A, C314,560A, C560,610A, C292,610A, and C301,610A) were functional and the amplitude of their αMG cotransport currents (Icotr) and Na+ leak currents (Ileak) at −110 mV (Fig. 9 A), as well as their V1/2 values (Fig. 9 C) were compared with those of wt SGLT1. (The name of double mutants was shortened for clarity in figures. For example, the symbol C255,511A means that the cysteines in positions 255 and 511 were both mutated into an alanine residue.)
Figure 9.

Electrophysiological characteristics of the double mutants as compared with wt SGLT1. (A) Comparison of mutant SGLT1 activities to that of wt SGLT1. Maximal αMG cotransport (Icotr at 5 mM αMG) and Na+ leak (Ileak) currents were measured at −110 mV. The dotted lines indicate the mean expression level of wt SGLT1. n ≥ 5 for each mutant. (B) I-V curve for αMG cotransport and Na+ leak currents of the double mutant C292,610A. (C) V1/2 of double mutant SGLTs as compared with that of wt SGLT1 and to single mutants. Each double mutant bar (black) is preceded and followed by the two bars representing the V1/2 values for the corresponding single mutants (light gray). The dotted box represents the interval for which V1/2 of a mutant would not be significantly different from that of wt SGLT1. (D) Time constants of the slow component of presteady-state currents for the double mutant C255,511A (see Fig 7). (n = 5). The curve represents the time constants evaluated with a four-state model of presteady-state currents with parameters described in Table I. Errors are SEM.
In two cases, the effects of each mutations on V1/2 were roughly additive. This is the case of C292,610A and C301,610A. Analysis of Fig. 9 C indicates that mutating C610 to an alanine residue produces a powerful negative shift in V1/2 by ∼75 mV independent of the presence of C292 or C301. The double mutant C292,610A retains the large Ileak of the individual, single mutants while the Icotr is completely abolished for 5 mM αMG (Fig. 9 B) and no current could be induced by the addition of either 25 or 100 mM αMG (not depicted). In contrast, when C560 is mutated to an alanine, replacing C610 only produces a shift of V1/2 by 50 mV. To put it another way, the single C560A mutation produced a shift of V1/2 by −30 mV when compared with wt SGLT1 but the effect of the double mutant is only −6 mV when compared with the single mutant C610A. A qualitatively similar comparison can be done with mutations C314A and C560A. The effect of introducing either mutation is significantly reduced when they are performed after the other cysteine residue has been replaced. In these two cases (C560,610A and C314,560A), other parameters have to be considered before suggesting the presence of disulfide bridges. Fig. 6 A shows that replacing C560 produces a reduction of Icotr by 63% while the Icotr reached with C314A and C610A are equal or larger than the wt level. It appears that the effect of C560 replacement on the activity level is conserved when it is performed alongside alanine substitution of C314 or of C610 (80% and 75% reduction with respect to C314A and C610A, respectively, see Fig. 6 A and Fig. 9 A). This is not what would be expected if the effect of C560 replacement was due to the disruption of disulfide bridges with C314 or C610.
Finally, the double mutant C255,511A had an activity level (Fig. 9 A), a time constant for presteady-state currents (Fig. 9 D), and a V1/2 (Fig. 9 C) similar to the simple mutants C255A and C511A. With respect to the effect on V1/2, it is clear that the effect of introducing either of the single mutants (a shift by +20 mV) is lost when the other cysteine residue has already been replaced (compare the effect of C255,511A to the effect of each of the single mutants in Fig. 9 C). Moreover, C255A, C511A, and C255,511A are insensitive to DTT as their V1/2 values remain unchanged by treatment with 10 mM DTT (Fig. 7 C for simple mutant, not shown for C255,511A). The presence of a disulfide bridge between C255 and C511 is also supported by the observation that, in the double mutant, Km α MG was the same as that for each of the simple mutants C255A and C511A, which is also very similar to that of wt SGLT1 in the presence of DTT (see Fig. 8).
Fluorescent Labeling of C255 and C511
To complement studies examining the function of SGLT1 mutants and to further test the hypothesis that C255 and C511 could form a disulfide bridge, we used fluorescent labeling as an independent test of available cysteine residues in the mutant proteins. We incubated the C255A- and C511A-expressing oocytes with saline solution containing the fluorescent probe tetramethylrhodamine-5-maleimide (TMR5M, see MATERIALS AND METHODS). Fig. 10 shows the fluorescent signal of labeled oocytes expressing mutant C511A (TMR5M putatively attached to C255) and of mutant C255A (TMR5M putatively attached to C511) as compared with wt SGLT1 and to the double mutant C255,511A in which no cysteine appears to be available for labeling. This indicates that the two simple mutations (C255A and C511A) allow the fluorescent probe to access a cysteine that is not accessible in the double mutant (C255,511A) or in wt SGLT1 and strongly supports the hypothesis of a disulfide bridge between these two cysteine residues in SGLT1.
Figure 10.

Fluorescent labeling of oocytes expressing mutants C255A, C511A, and C255,511A as compared with wt SGLT1. See MATERIALS AND METHODS for the procedure of fluorescent labeling with the TMR5M fluorescent probe. The fluorescence signal is indicated in arbitrary units (a.u.). NI indicates noninjected oocytes. The number of oocytes used per sample is indicated in parentheses. A cartoon shows the availability of cysteine residues in each situation. Stars indicate the statistical significance for unpaired Student's t test against wt SGLT1 data (see Fig 5 legend). Errors are SEM.
DISCUSSION
In SGLT1 only 5 of the 15 endogenous cysteine residues are located in putative transmembrane segments (TMS VII, VIII, and XIV), with the majority located in extracellular loops (loop 6–7, loop 8–9, loop 12–13) and in the special, putatively reentrant loop 13–14 (Lin et al., 1999; Gagnon et al., 2005). Eight of these cysteine residues (C255, C292, C345, C351, C355, C361, C511, and C610) are conserved amongst SGLT1, SGLT2, SGLT3, SMIT1, and SMIT2. Amongst those, C351 is conserved across nearly the entire SLC5 family. Some other residues are conserved among the SGLT1, 2, and 3 isoforms (C301, C314, C317, C517, C522, and C560). In contrast, C659 is present only within the SGLT1 isoforms. This high degree of cysteine conservation suggests that they may play a role either in a specific function or in the general structure (or both) of the Na+-coupled cotransporters of SLC5 family. It has been shown before that wt SGLT1 is not sensitive to alkylation (NEM nor MTS reagents) (Loo et al., 1998; Lo and Silverman, 1998a; Gagnon et al., 2005). This means that these reagents can bind to some cysteine residues without producing any functional effect or, alternatively, that no cysteine residues are available to these reagents because the residues are buried in the membrane, involved in disulfide bridges or inaccessible to the solvent. The fact that the degree of fluorescent labeling of wt SGLT1 with TMR5M is identical to the basal fluorescent labeling obtained with noninjected oocytes supports the latter possibility. Although the roles of the endogenous cysteine residues in SGLT1 have never been studied in detail, a few reports have suggested important roles for, at least, some of them in protein trafficking (Martin et al., 1996; Wright, 1998; Xia et al., 2003, 2004).
Presteady-state currents involving SGLT1 can be observed in the absence of glucose. They were first observed shortly after the cloning of SGLT1 which, through expression in Xenopus oocytes, enabled its precise characterization using the two microelectrode voltage-clamp technique (Umbach et al., 1990; Parent et al., 1992a,b). The model proposed by Parent et al. (1992a,b) was revisited using the cut-open oocyte technique, providing the observation of a faster component of presteady-state currents (Chen et al., 1996). A slower time constant was also postulated recently (Krofchick and Silverman, 2003; Loo et al., 2005). As illustrated in Fig. 11, presteady-state currents arise from the voltage-dependent empty carrier translocation (C1↔C2↔C3) and from voltage-dependent Na+ binding–debinding (C3↔C4(Na+)). Integration of these currents corresponds to the quantity of transferred charge that can be approximated (as a function of membrane potential) by a Boltzmann equation. Two parameters of presteady-state currents were examined to compare normal and mutant SGLT1: V1/2, the voltage at which half of the charge is transferred and the ∼10 ms time constant (described by Loo et al., 1993; Chen et al., 1996) arising from the modulated transition between C1 to C4(Na+). The faster time constant could not be accurately resolved by two-microelectrode voltage-clamp.
Figure 11.

Kinetic model of the Na+/glucose cotransporter states for the estimation of presteady-state currents. The model was previously described in Chen et al. (1996). See Appendices A and B for details.
DTT Effect on wt SGLT1 Activity
We report in the present study the sensitivity of SGLT1 steady-state and presteady-state currents to a reducing agent, DTT, which is assumed to break all disulfide bridges even if they are buried within the membrane (Cleland, 1964). The steady-state parameters (Ileak and Icotr) were not similarly affected by DTT exposure: Ileak was reduced whereas Icotr was not affected at all. The Km α MG was increased by 60% without any change in its voltage dependence (Fig. 1 C). According to the current model (see below), these effects are consistent with a shift in the conformational equilibrium toward an outward facing configuration of the transporter. As the affinity for sugar depends on a large number of parameters, it is difficult at this point to propose any specific change produced by DTT in the transporter mechanism to explain the rise of Km α MG. In contrast, presteady-state currents were greatly affected by DTT as the Q-V curve was shifted by +25 mV (Fig. 2 B) and the time constants were accelerated (Fig. 2 C). Compared with a parameter such as KmN α+, the V1/2 of the Q-V curve depends on a smaller set of rate constants and was therefore chosen as our principal criterion for classifying the mutated forms of SGLT1 for identifying the disulfide bridge(s) responsible for the effects of DTT.
Nonfunctional Mutants
Mutants C351A, C355A, and C361A, located in the extracellular loop between TMS VIII and IX, and the double mutant C522,560A were nonfunctional: they exhibited no presteady-state currents, no Ileak nor Icotr (5 mM αMG or 100 mM αMG, unpublished data). However, we could detect the presence of SGLT1 on Western blots using preparations enriched in plasma membranes. As mutants C351A and C361A were shown not to be trafficked to the plasma membrane (Xia et al., 2005), we suggest that these mutants are also retained within intracellular compartments and confirm that C351 and C361 are important in the correct trafficking to the plasma membrane.
Expression of Single Mutants
The characteristics of the single mutants presented in this study suggest that endogenous cysteine residues in SGLT1 are involved in important aspects of protein function: proper trafficking to the plasma membrane (as suggested by the nonfunctional mutants and by some weakly expressed mutants) and maintaining the voltage sensor in either the inward or outward configurations (as suggested by mutants in Group B and C). Only three cysteines could be removed without any prominent effect on the function of the transporter (C317A in TMS 8, C517A in loop 12–13, and C659A in the last TMS). These results are similar to those obtained with other cotransporters, which also suggested a role for endogenous cysteines in the proper trafficking of proteins to the plasma membrane (J.G. Chen et al., 1997; Sur et al., 1997; Pajor et al., 1999; Tanaka et al., 2004). However, none of the endogenous cysteines previously studied in other cotransporters has been reported to play a role in presteady-state currents. The first round of mutation identified C255 and C511 as being potentially involved in a disulfide bridge.
Expression of Double Mutants
To identify a disulfide bridge, the mutants of Group A were discarded because the V1/2 of their Q-V curve was not significantly different from that of wt SGLT1 under control conditions. Then mutants whose V1/2 was affected were classified into two categories: those with Q-V curves shifted toward negative potentials (Group B) and those which were shifted toward positive potentials (Group C).
As a first observation, all mutants in Group B display distinctly different characteristics, except for C522A and C560A, which were indistinguishable in terms of V1/2, Icotr, and Ileak. However, the double mutant C522,560A did not generate any cotransport activity. As both C522A and C560A presented reduced activity in terms of Icotr and Ileak (20–30% of wt SGLT1), the absence of functional activity for the double mutant could be interpreted as though the effects of the two mutations were additive. This is not what would be expected if C522 and C560 were linked through a disulfide bridge.
The additive effect can also be seen with other mutants; for instance, the mutant C314,560A displays half the Icotr seen with the single mutant C314A while the Ileak and the V1/2 remain normal, similar to the effects of the single mutant C560A. This reduced Icotr also occurred in the double mutant C560,610A. This was the first double mutant produced in this study because these two cysteines have been postulated to be involved in a disulfide bridge in an isolated loop in rabbit SGLT1 (Xia et al., 2003, 2004). Our results do not support this hypothesis as the V1/2 associated with the double mutant seems to be controlled by the C610A mutation while the activity level seems to be controlled by the C560A mutation. Moreover, the Q-V curves of these two mutants are still shifted toward positive potential by DTT, similarly to wt SGLT1.
The double mutant C292,610A is very interesting since it has no αMG cotransport whereas its Ileak is as large as those of the singly mutated forms of SGLT1. The absence of Icotr was not due to a simple decrease of αMG affinity for its binding site since the Km α MG values for the single mutants were similarly low (0.27 ± 0.01 mM for C292A and 0.28 ± 0.02 mM for C610A). Interestingly, Pz still inhibits the Ileak and presteady-state currents. We cannot state that these cysteines form a disulfide bridge because of the effects observed with the double mutant. However, it suggests that these two cysteines, together, somehow play a role in coupling Na+ and glucose transport.
Disulfide Bridge between C255 and C511
To date, intramolecular disulfide bridges have been identified for only a few transporters. The endogenous cysteine residues of Na+/Pi cotransporter type IIa have been studied in detail using mutagenesis, Western blot, and by examining the effects of the reagents TCEP and MTS on transport (Lambert et al., 2000; Kohler et al., 2003); an essential disulfide bridge between two cysteines, which are located in the same extracellular loop, has been identified and another was postulated. These are responsible for the TCEP (another reducing agent) sensitivity of the transporter. The serotonin transporter (SERT) contains 16 endogenous cysteines of which three are conserved throughout the NaCl-coupled neurotransmitter transporter family (Chen et al., 1997; Sur et al., 1997). Characterization of the transport properties of SERT mutants have identified functionally important cysteines as well as a disulfide bridge between two conserved cysteines, also located on the same extracellular loop.
The similarity of the V1/2 values of the mutants in Group C (C255A and C511A, located in extracellular loops 6–7 and 12–13, respectively) with that of reduced wt SGLT1 is remarkable (Fig. 2 D, Fig. 7 D, and Fig. 9 D). It suggests that these two cysteines are, in fact, covalently bound by a disulfide bridge and that its breakage affects the V1/2. The V1/2 of both of these mutants are also DTT insensitive. The double mutant C255,511A supports this hypothesis as its V1/2 is the same as those of the simple mutants, and it is also insensitive to DTT. As another functional test, we measured the apparent affinity for αMG for these mutants. The three mutants C255A, C511A, and C255,511A also exhibit a Km α MG value similar to that of wt SGLT1 under reducing conditions. We showed that SGLT1 (when C511 had been mutated into alanine or when C255 had been mutated into alanine) could be labeled by a fluorescent probe and that, in the absence of both cysteines (in mutant C255,511A), no labeling could be achieved (Fig. 10). Taken together, these results strongly support the disulfide bridge hypothesis. We also demonstrated that the disruption of the bridge affects the function of the protein, changing the equilibrium voltage for the proposed negatively charged binding site (V1/2) and thus affecting the Na+ current in the absence of glucose (Ileak) and the apparent affinity for glucose (Km α MG).
Kinetic Model of Presteady-state Currents
The simple four-state kinetic model proposed by Chen et al. (1996) and illustrated in Fig. 11 (see also Table I for kij values and for deduced V1/2), was used to model the time constants and transferred charges. The principal equations describing this model are given in Appendices A and B. The model explains the time constants of presteady-state currents (curves in Fig. 2 C for wt SGLT1, Fig. 7 D for C255A and C511A, and Fig. 9 D for C255,511A) and transferred charges, allowing the fit of a theoretical Q-V curve with a simple Boltzmann curve to extract V1/2. Parameter values proposed by Chen et al. (1996) were slightly modified to fit wt SGLT1 time constant and Q-V curves obtained in double microelectrode electrophysiology. The values of zi used for the equivalent charge moving across the entire membrane electrical field in the step between state “i” and “i+1” were −0.4, −0.35, −0.6, and 0, while the values for αi describing the asymmetry of the energy barrier were 0.3, 0, 0.4, and 0 for i going from 1 to 4, respectively. If it is assumed that one negative charge is attached to the binding site moving in the membrane electrical field, these values of zi would indicate that the charge is crossing 40% of the electrical field in the reaction linking C1 to C2 and 35% between C2 and C3. In the Na+ binding reaction, the equivalent of one positive charge would cross 60% of the membrane electrical field. This could be due to the movement of Na+, the movement of the negative charge attached to the binding site, or a combination of both. The set of proposed rate constants for wt SGLT1 in Table I would lead to occupancy probability (Ci) of 0.07, 0.31, 0.31, and 0.31 (from i = 1 to 4) at −50 mV.
TABLE I.
Rate Constant of the Four-state Kinetic Model Used for the Calculations of Time Constants and Transferred Charges of wt SGLT1 and Mutants
| k120 a | k210 | k230 | k320 | k340 | k430 | k410 b | V1/2 c | |
|---|---|---|---|---|---|---|---|---|
| s −1 | s −1 | s −1 | s −1 | M −1 s −1 | s −1 | s −1 | mV | |
| wt SGLT1 | 790 | 370 | 100 | 180 | 8000 | 2300 | 10 | −39 |
|
wt ± DTT,
C255A, C511A, and C255,511A |
790 | 370 | 230 | 180 | 8000 | 2300 | 10 | −20 |
The constant k120 (as well as other kij0) represents the value of k12 (and kij) at 0 mV.
The constant k140 (in s−1M−1) was calculated in order to respect the micro reversibility as described in Appendix A.
The V1/2 was obtained from a fit of a Boltzmann relation to the Q-V curve calculated with the model as described in Appendix B. The experimental values are cited in the text.
We thus achieve a V1/2 of −39 mV for Q-V curve with satisfying time constants as a function of Vm for wt SGLT1. To obtain the one of wt SGLT1 in the presence of DTT, we had to increase k230 by a factor 2.3 to account for the faster time constants observed for the presteady-state current at hyperpolarizing potentials. This sole change affected both the plateau of the time constants at hyperpolarizing potential and the V1/2, reaching −20 mV, without any change in the apparent valence of the Boltzmann curve (unpublished data), which is in agreement with the observations. These changes also correctly fit to the mutants characteristics.
Even though this disulfide bridge is not essential for the function of SGLT1, since removing it did not prevent the transporter from being functional at the plasma membrane, it greatly influenced the equilibrium position of transferred charges. The disulfide bridge described in other transporters has had no such important role.
Conclusion
With the presence of 15 endogenous cysteine residues in SGLT1, 105 different combinations exist for creating a disulfide bridge. In the present study, we have presented an approach that allows one to positively identify a specific disulfide bridge without having to test all of the possibilities. Our results suggest important roles for cysteine in SGLT1: proper trafficking to the plasma membrane and maintenance of the voltage sensing part of the protein in the inward or outward facing configuration. We also identified a functionally important disulfide bridge, between C255 and C511 of human SGLT1, which is responsible for the effects of DTT on the transferred charges of wt SGLT1, and which affects the voltage-independent conformation change from state C2 to C3. The identification of a disulfide bridge in SGLT1 (and possibly in SGLT2 and 3 and in SMIT1 and 2 where these residues are conserved) provides a direct, three-dimensional constraint to the structure of the transporter and to the organization of some transmembrane segments. If amino acids 255 and 511 are closer than 2.05 Å from each other (Loerger, 2005), it indicates that TMS VI and VII are close to TMS XI, XII, and XIII. To our knowledge, this is the first direct experimental evidence of a physical constraint between two distant amino acid positions in SGLT1.
Acknowledgments
We thank Michael Coady for valuable discussion and for his comments on the manuscript.
This work was supported by the Canadian Institutes of Health Research (grant MOP-10580). D.G. Gagnon is a NSERC and FRSQ postgraduate scholar.
Lawrence G. Palmer served as editor.
APPENDIX
A. Expressions for Rate Constant and Time Constants
The four-state kinetic model from Chen et al. (1996) was used to model the effects of DTT on wt SGLT1 and to examine the characteristics of mutants C255A, C511A, and the double mutant C255,511A (see Figs. 2, 7, and 9 for experimental and simulated curves, Fig. 11 for the scheme of the model, and Table I for the values of kij0 and of the deduced V1/2). For all of the simulations, [Na+ out] and [Na+ in] were fixed at 90 mM and 7 mM, respectively. See DISCUSSION for other values.
The following equations were used to calculate time constants occurring for different membrane potentials:
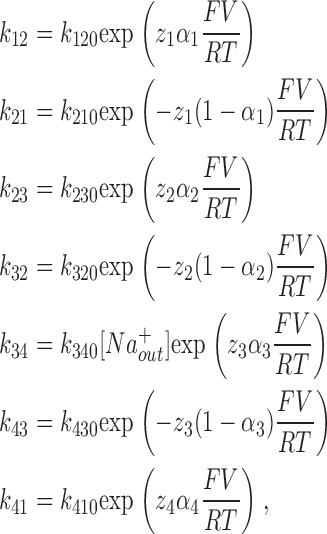 |
(1) |
where zi represents the charge valence (equivalent charge moving across the whole membrane), αi (ranging between 0 and 1) describes the asymmetry of the energy barriers, V is the membrane potential, and F, T, and R have their usual meanings. To assure microscopic reversibility, the last constant k140 was calculated to have no net transport at 0 mV with no Na+ gradient:
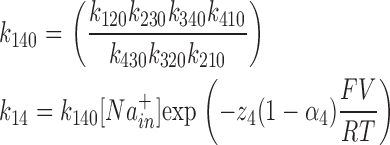 |
(2) |
Certain approximations, satisfied in our case and described in Chen et al. (1996), allowed us to establish the slow time constant and to determine the following expression for τslow:
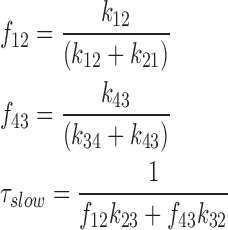 |
(3) |
B. Expressions for Transferred Charges
Variations in time of the probability of finding transporters in each state are calculated as usual, for the desired membrane potential, with a given first value:
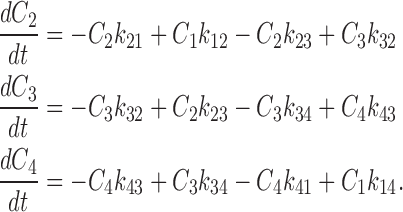 |
They were multiplied by the time increment and this variation was added to the previous value:
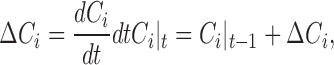 |
(4) |
where Ci represents the probability of occupation of state i in the model (see Fig. 11), but to conserve the total probability of transporters at unity, C1 is calculated as followed:
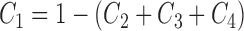 |
(5) |
We set the charge to be zero at extreme hyperpolarizing potentials (as C4 = 1 and C3 = C2 = C1 = 0) by use of the following equation:
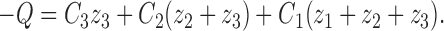 |
(6) |
Abbreviations used in this paper: αMG, α-methyl-glucose; DTT, dithriothreitol; Pz, phlorizin; SGLT1, Na+/glucose cotransporter; TMR5M, tetramethylrhodamine-5-maleimide; TMS, transmembrane segment; wt, wild-type.
References
- Bissonnette, P., J. Noel, M.J. Coady, and J.Y. Lapointe. 1999. Functional expression of tagged human Na+-glucose cotransporter in Xenopus laevis oocytes. J. Physiol. 520(Pt 2):359–371. [DOI] [PMC free article] [PubMed]
- Chen, J.G., S. Liu-Chen, and G. Rudnick. 1997. External cysteine residues in the serotonin transporter. Biochemistry. 36:1479–1486. [DOI] [PubMed] [Google Scholar]
- Chen, X.Z., M.J. Coady, F. Jackson, A. Berteloot, and J.Y. Lapointe. 1995. Thermodynamic determination of the Na+: glucose coupling ratio for the human SGLT1 cotransporter. Biophys. J. 69:2405–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X.Z., M.J. Coady, and J.Y. Lapointe. 1996. Fast voltage clamp discloses a new component of presteady-state currents from the Na+-glucose cotransporter. Biophys. J. 71:2544–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X.Z., M.J. Coady, F. Jalal, B. Wallendorff, and J.Y. Lapointe. 1997. Sodium leak pathway and substrate binding order in the Na+-glucose cotransporter. Biophys. J. 73:2503–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland, W.W. 1964. Dithiothreitol, a new protective reagent for Sh groups. Biochemistry. 3:480–482. [DOI] [PubMed] [Google Scholar]
- Crane, R.K. 1965. Na+-dependent transport in the intestine and other animal tissues. Fed. Proc. 24:1000–1006. [PubMed] [Google Scholar]
- Fisher, C.L., and G.K. Pei. 1997. Modification of a PCR-based site-directed mutagenesis method. Biotechniques. 23:570–571, 574. [DOI] [PubMed]
- Gagnon, D.G., A. Holt, F. Bourgeois, B. Wallendorff, M.J. Coady, and J.Y. Lapointe. 2005. Membrane topology of loop 13-14 of the Na+/glucose cotransporter (SGLT1): A SCAM and fluorescent labelling study. Biochim. Biophys. Acta. 1712:173–184. [DOI] [PubMed] [Google Scholar]
- Gagnon, M.P., P. Bissonnette, L.M. Deslandes, B. Wallendorff, and J.Y. Lapointe. 2004. Glucose accumulation can account for the initial water flux triggered by Na+/glucose cotransport. Biophys. J. 86:125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hediger, M.A., M.J. Coady, T.S. Ikeda, and E.M. Wright. 1987. Expression cloning and cDNA sequencing of the Na+/glucose co-transporter. Nature. 330:379–381. [DOI] [PubMed] [Google Scholar]
- Hirayama, B.A., M.P. Lostao, M. Panayotova-Heiermann, D.D. Loo, E. Turk, and E.M. Wright. 1996. Kinetic and specificity differences between rat, human, and rabbit Na+-glucose cotransporters (SGLT-1). Am. J. Physiol. 270:G919–G926. [DOI] [PubMed] [Google Scholar]
- Kohler, K., I.C. Forster, G. Stange, J. Biber, and H. Murer. 2003. Essential cysteine residues of the type IIa Na+/Pi cotransporter. Pflugers Arch. 446:203–210. [DOI] [PubMed] [Google Scholar]
- Krofchick, D., and M. Silverman. 2003. Investigating the conformational states of the rabbit Na+/glucose cotransporter. Biophys. J. 84:3690–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert, G., I.C. Forster, J. Biber, and H. Murer. 2000. a. Cysteine residues and the structure of the rat renal proximal tubular type II sodium phosphate cotransporter (rat NaPi IIa). J. Membr. Biol. 176:133–141. [DOI] [PubMed] [Google Scholar]
- Lambert, G., M. Traebert, J. Biber, and H. Murer. 2000. b. Cleavage of disulfide bonds leads to inactivation and degradation of the type IIa, but not type IIb sodium phosphate cotransporter expressed in Xenopus laevis oocytes. J. Membr. Biol. 176:143–149. [DOI] [PubMed] [Google Scholar]
- Lambert, G., I.C. Forster, G. Stange, K. Kohler, J. Biber, and H. Murer. 2001. Cysteine mutagenesis reveals novel structure-function features within the predicted third extracellular loop of the type IIa Na+/P(i) cotransporter. J. Gen. Physiol. 117:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, J., J. Kormanec, D. Homerova, and R.K. Kinne. 1999. Probing transmembrane topology of the high-affinity sodium/glucose cotransporter (SGLT1) with histidine-tagged mutants. J. Membr. Biol. 170:243–252. [DOI] [PubMed] [Google Scholar]
- Lo, B., and M. Silverman. 1998. a. Cysteine scanning mutagenesis of the segment between putative transmembrane helices IV and V of the high affinity Na+/glucose cotransporter SGLT1. Evidence that this region participates in the Na+ and voltage dependence of the transporter. J. Biol. Chem. 273:29341–29351. [DOI] [PubMed] [Google Scholar]
- Loerger, T.R. 2005. Automated detection of disulfide bridges in electron density maps using linear discriminant analysis. J. Appl. Cryst. 38:121–125. [Google Scholar]
- Loo, D.D., A. Hazama, S. Supplisson, E. Turk, and E.M. Wright. 1993. Relaxation kinetics of the Na+/glucose cotransporter. Proc. Natl. Acad. Sci. USA. 90:5767–5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo, D.D., B.A. Hirayama, E.M. Gallardo, J.T. Lam, E. Turk, and E.M. Wright. 1998. Conformational changes couple Na+ and glucose transport. Proc. Natl. Acad. Sci. USA. 95:7789–7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo, D.D., B.A. Hirayama, A. Cha, F. Bezanilla, and E.M. Wright. 2005. Perturbation analysis of the voltage-sensitive conformational changes of the Na+/glucose cotransporter. J. Gen. Physiol. 125:13–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M.G., E. Turk, M.P. Lostao, C. Kerner, and E.M. Wright. 1996. Defects in Na+/glucose cotransporter (SGLT1) trafficking and function cause glucose-galactose malabsorption. Nat. Genet. 12:216–220. [DOI] [PubMed] [Google Scholar]
- Murer, H., I. Forster, and J. Biber. 2004. The sodium phosphate cotransporter family SLC34. Pflugers Arch. 447:763–767. [DOI] [PubMed] [Google Scholar]
- Pajor, A.M., S.J. Krajewski, N. Sun, and R. Gangula. 1999. Cysteine residues in the Na+/dicarboxylate co-transporter, NaDC-1. Biochem. J. 344(Pt 1):205–209. [PMC free article] [PubMed]
- Panayotova-Heiermann, M., D.D. Loo, M.P. Lostao, and E.M. Wright. 1994. Sodium/d-glucose cotransporter charge movements involve polar residues. J. Biol. Chem. 269:21016–21020. [PubMed] [Google Scholar]
- Panayotova-Heiermann, M., D.D. Loo, and E.M. Wright. 1995. Kinetics of steady-state currents and charge movements associated with the rat Na+/glucose cotransporter. J. Biol. Chem. 270:27099–27105. [DOI] [PubMed] [Google Scholar]
- Panayotova-Heiermann, M., D.D. Loo, C.T. Kong, J.E. Lever, and E.M. Wright. 1996. Sugar binding to Na+/glucose cotransporters is determined by the carboxyl-terminal half of the protein. J. Biol. Chem. 271:10029–10034. [DOI] [PubMed] [Google Scholar]
- Parent, L., S. Supplisson, D.D. Loo, and E.M. Wright. 1992. a. Electrogenic properties of the cloned Na+/glucose cotransporter: I. Voltage-clamp studies. J. Membr. Biol. 125:49–62. [DOI] [PubMed] [Google Scholar]
- Parent, L., S. Supplisson, D.D. Loo, and E.M. Wright. 1992. b. Electrogenic properties of the cloned Na+/glucose cotransporter: II. A transport model under nonrapid equilibrium conditions. J. Membr. Biol. 125:63–79. [DOI] [PubMed] [Google Scholar]
- Sahin-Toth, M., B. Persson, J. Schwieger, P. Cohan, and H.R. Kaback. 1994. Cysteine scanning mutagenesis of the N-terminal 32 amino acid residues in the lactose permease of Escherichia coli. Protein Sci. 3:240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur, C., P. Schloss, and H. Betz. 1997. The rat serotonin transporter: identification of cysteine residues important for substrate transport. Biochem. Biophys. Res. Commun. 241:68–72. [DOI] [PubMed] [Google Scholar]
- Tanaka, K., F. Zhou, K. Kuze, and G. You. 2004. Cysteine residues in the organic anion transporter mOAT1. Biochem. J. 380:283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk, E., C.J. Kerner, M.P. Lostao, and E.M. Wright. 1996. Membrane topology of the human Na+/glucose cotransporter SGLT1. J. Biol. Chem. 271:1925–1934. [DOI] [PubMed] [Google Scholar]
- Turner, R.J., and J.N. George. 1983. Evidence for two disulfide bonds important to the functioning of the renal outer cortical brush-border membrane d-glucose transporter. J. Biol. Chem. 258:3565–3570. [PubMed] [Google Scholar]
- Turner, R.J., and J.N. George. 1984. Characterization of an essential disulfide bond associated with the active site of the renal brush-border membrane d-glucose transporter. Biochim. Biophys. Acta. 769:23–32. [DOI] [PubMed] [Google Scholar]
- Umbach, J.A., M.J. Coady, and E.M. Wright. 1990. Intestinal Na+/glucose cotransporter expressed in Xenopus oocytes is electrogenic. Biophys. J. 57:1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Iwaarden, P.R., J.C. Pastore, W.N. Konings, and H.R. Kaback. 1991. Construction of a functional lactose permease devoid of cysteine residues. Biochemistry. 30:9595–9600. [DOI] [PubMed] [Google Scholar]
- Wright, E.M. 1998. I. Glucose galactose malabsorption. Am. J. Physiol. 275:G879–G882. [DOI] [PubMed] [Google Scholar]
- Wright, E.M., and E. Turk. 2004. The sodium/glucose cotransport family SLC5. Pflugers Arch. 447:510–518. [DOI] [PubMed] [Google Scholar]
- Wright, E.M., D.D. Loo, M. Panayotova-Heiermann, B.A. Hirayama, E. Turk, S. Eskandari, and J.T. Lam. 1998. Structure and function of the Na+/glucose cotransporter. Acta Physiol. Scand. Suppl. 643:257–264. [PubMed] [Google Scholar]
- Xia, X., J.T. Lin, and R.K. Kinne. 2003. Binding of phlorizin to the isolated C-terminal extramembranous loop of the Na+/glucose cotransporter assessed by intrinsic tryptophan fluorescence. Biochemistry. 42:6115–6120. [DOI] [PubMed] [Google Scholar]
- Xia, X., C.T. Lin, G. Wang, and H. Fang. 2004. Binding of phlorizin to the C-terminal loop 13 of the Na+/glucose cotransporter does not depend on the [560-608] disulfide bond. Arch. Biochem. Biophys. 425:58–64. [DOI] [PubMed] [Google Scholar]
- Xia, X., G. Wang, Y. Peng, and J. Jen. 2005. Cys351 and Cys361 of the Na+/glucose cotransporter are important for both function and cell-surface expression. Arch. Biochem. Biophys. 438:63–69. [DOI] [PubMed] [Google Scholar]