

Abstract
The complexity of mammalian physiology requires a diverse array of ion channel proteins. This diversity extends even to a single family of channels. For example, the family of Ca2+-activated K channels contains three structural subfamilies characterized by small, intermediate, and large single channel conductances. Many cells and tissues, including neurons, vascular smooth muscle, endothelial cells, macrophages, and salivary glands express more than a single class of these channels, raising questions about their specific physiological roles. We demonstrate here a novel interaction between two types of Ca2+-activated K channels: maxi-K channels, encoded by the KCa1.1 gene, and IK1 channels (KCa3.1). In both native parotid acinar cells and in a heterologous expression system, activation of IK1 channels inhibits maxi-K activity. This interaction was independent of the mode of activation of the IK1 channels: direct application of Ca2+, muscarinic receptor stimulation, or by direct chemical activation of the IK1 channels. The IK1-induced inhibition of maxi-K activity occurred in small, cell-free membrane patches and was due to a reduction in the maxi-K channel open probability and not to a change in the single channel current level. These data suggest that IK1 channels inhibit maxi-K channel activity via a direct, membrane-delimited interaction between the channel proteins. A quantitative analysis indicates that each maxi-K channel may be surrounded by four IK1 channels and will be inhibited if any one of these IK1 channels opens. This novel, regulated inhibition of maxi-K channels by activation of IK1 adds to the complexity of the properties of these Ca2+-activated K channels and likely contributes to the diversity of their functional roles.
INTRODUCTION
Ca2+-activated K channels represent a subset of the super family of K+-selective ion channel proteins. First described in red blood cells (Gardos, 1958) where they are important for cell volume regulation, Ca2+-activated K channels are now known to have a wide spectrum of additional physiological roles, including the control of vascular tone and the regulation of neuronal firing rates. These channels display a large spectrum of functional properties, including their single channel conductance, Ca2+ sensitivity, and pharmacological profile (Latorre et al., 1989; Shieh et al., 2000; Stocker, 2004). Part of this functional variety arises from genetic diversity: there are three structural subfamilies of Ca2+-activated K channels and these are characterized by small, intermediate, and large single channel conductances. The prototypical member of the large conductance family (KCa1.1) is activated by voltage as well as by Ca2+ ions. This maxi-K channel is subject to considerable alternative splicing and can be associated with several β subunits, all of which adds to its functional diversity. The three small-conductance (SK) channels (KCa2.1, KCa2.2, and KCa2.3) share considerable amino acid identity and biophysical properties. The intermediate conductance channel (KCa3.1 or IK1) is only distantly related to the SK channels with ∼50% amino acid homology (Ishii et al., 1997; Logsdon et al., 1997), and has a different pharmacological profile. None of the small and intermediate conductance channels are voltage sensitive. Except for the channel pore region, there is no significant homology of the maxi-K channel with the smaller conductance SK and IK channels (Gutman et al., 2003).
In addition to the roles noted above, Ca2+-activated K channels are considered to be critical for sustained fluid secretion in secretory epithelia (Petersen and Maruyama, 1984; Melvin et al., 2005). Parotid acinar cells express both IK1 and maxi-K channels, raising questions about their specific roles (Nehrke et al., 2003; Takahata et al., 2003). To address these questions, a strain of mice was developed in which the expression of the IK1 channel was ablated (Begenisich et al., 2004). In the course of these investigations (Begenisich et al., 2004), we observed an entirely unexpected and novel behavior: activation of IK1 channels appeared to cause a concurrent loss of maxi-K channel activity. We report here the results of a study to investigate the properties of this functional interaction between these two Ca2+-activated K channels.
We found that the activation of IK1, either directly by perfusing the cell with elevated Ca2+ or via a muscarinic-agonist, caused a concomitant decrease in maxi-K channel activity. This was true even when the cell integrity and large cytoplasmic components were left intact with the perforated patch technique. The mechanism underlying this inhibition does not require elevated intracellular Ca2+ levels since activation of IK1 by the organic agonist DCEBIO (5,6-dichloro-1-ethyl-1,3-dihydro-2H-benzimidazol-2-one) inhibited maxi-K channels even at a fixed, low Ca2+ concentration. Furthermore, the mechanism appears to be a spatially localized one as the IK1-induced inhibition of maxi-K channels occurred in excised inside/out patches devoid of freely diffusible cytoplasmic components. We found that the maxi-K inhibition was due to a reduction in the channel open probability and not to a change in the single channel current. Finally, the interaction between the IK1 and maxi-K channels appears to be a property of the channels themselves and not due to some special property of parotid acinar cells, as we were able to recapitulate the inhibition in a heterologous expression system. A quantitative analysis of the interaction between these two types of channels indicates that each maxi-K channel may be surrounded by four IK1 channels and will be inhibited if any one of these IK1 channels opens.
These novel findings reveal a close interaction between the IK1 and maxi-K channel proteins. The activation of IK1 channels either directly or through a membrane-delimited intermediary inhibits maxi-K channels. This regulated inhibition of maxi-K channels by activation of IK1 adds to the complexity of the properties of these Ca2+-activated K channels and likely contributes to their functional diversity.
MATERIALS AND METHODS
Acinar Cell Preparation
Single parotid acinar cells were isolated from wild-type (Black Swiss 129) and KCa3.1-null mice, IK1(−/−). The details for the construction of the IK1(−/−) mice have been previously described (Begenisich et al., 2004). Since the background for the KCa3.1-null mice was C57, we tested our findings in this strain as well and found no differences from the results with the Black Swiss animals.
Mice were anesthetized with CO2 gas and killed by exsanguination via cardiac puncture. Parotid glands were minced in a solution consisting of Ca2+-free Eagle's minimum essential medium (SMEM) (Biofluids), 1% BSA, and 2 mM l-glutamine. The tissue was digested in this solution with 0.01% trypsin and 0.5 mM EDTA at 37°C for 8 min and then rinsed with SMEM + 1% BSA plus 0.2% trypsin inhibitor (TypeI-S, Sigma-Aldrich). Finally, the tissue was incubated in SMEM with 16 μg/ml Liberase RI enzyme (Roche Applied Sciences) plus 1% BSA and 2 mM l-glutamine at 37°C. All solutions were gassed continuously with 95% O2 + 5% CO2 throughout the procedure. After 40 min of digestion with Liberase, the cells were dispersed by pipette trituration and then resuspended in fresh digestion media and incubated for a further 20 min at 37°C. The cells were gently triturated and then washed with basal medium Eagle's (BME) containing 1% BSA before final resuspension in BSA-free BME containing 2 mM l-glutamine with 100 U penicillin G and 100 μg/ml streptomycin sulfate. The cells were attached to 5 mm diameter glass coverslips and maintained in the final resuspension media in a 5% CO2 incubator at 37°C until used (usually within 3 h after isolation). The procedures for animal handling, maintenance, and surgery were approved by the University of Rochester Committee on Animal Resources.
Electrophysiology
Whole-cell patch clamp recordings were done at room temperature (20–22°C) with an Axopatch 200B amplifier (Axon Instruments). Data acquisition was performed using a 12 bit analogue/digital converter controlled by a personal computer. Patch pipettes were constructed from either GC-150 glass (Warner Instruments Inc.) or quartz (Garner Glass Co.) and coated with sticky wax. For whole cell recordings the pipette resistance was ∼4–6 MΩ and the current records filtered at 5 kHz. For excised patches, the electrode tips were ∼1–2 μm diameter, and the current records were filtered at 2 kHz.
The external solution for whole-cell patch recordings consisted of (in mM) 135 Na-glutamate, 5 K-glutamate, 2 CaCl2, 2 MgCl2, 10 HEPES (pH 7.2). Single channel experiments used an external solution that consisted of (in mM) 135-K glutamate, 2 CaCl2, 2 MgCl2, and 10 HEPES (pH 7.2). We used internal solutions that consisted of 135 mM K-glutamate, 10 mM HEPES (pH 7.2), 5 mM EGTA, and with CaCl2 added to establish various Ca2+ concentrations (Bers et al., 1994) (see also http://www.stanford.edu/∼cpatton/maxc.html). In some single channel experiments 1 mM MgATP was included in the internal solution to minimize IK1 run down. In these experiments we examined the action of the IK1 channel activator DCEBIO on the maxi-K channel open probability, and this action was independent of the presence or absence of ATP.
Perforated patch experiments were performed by first front-filling the electrodes with (in mM) 135 K-glutamate, 2 CaCl2, 2 MgCl2, 10 HEPES (pH 7.2) and then back filling with the same solution containing 125 μg/ml nystatin (Sigma-Aldrich) and 0.1% of the fluorescent dye Lucifer yellow (Sigma-Aldrich). The Lucifer yellow was used as an indicator of the integrity of the cell membrane under the patch (Takeda et al., 2004). Cell fluorescence was monitored with epifluorescent microscopy with an EGFP filter cube. Only data from those cells in which the yellow dye was excluded from the cell interior were used. In addition, the high calcium in the pipette solution gave us an instant indication of a breach in the cell membrane integrity.
Heterologous Expression
The mouse parotid KCa1.1 variant (Parslo) and the KCa3.1 coding sequences were cloned from the mouse parotid gland and subcloned into mammalian expression vectors as described previously (Nehrke et al., 2003). Parslo is almost identical to the human KCa1.1 gene (Nehrke et al., 2003). The KCa3.1 was subcloned into the bicistronic pIRES2EGFP vector (CLONTECH Laboratories Inc.). The DNA was cotransfected (0.5 μg KCa3.1 and 1.5 μg Parslo) into CHO-K1 cells using a Nucleofector machine (Amaxa Biosystems) according to the manufacturer's instructions. Cells were used between 24 and 48 h after transfection. Transfected cells were identified using the EGFP fluorescence from the bicistronic pIRES2EGFP vector. The CHO-K1 cells were obtained from ATCC and grown in Ham's F12K media with 10% FBS and maintained in a CO2 incubator with 5% CO2.
Electrophysiological Analysis
For some of the analysis, K channel conductance was computed from the measured whole cell currents according to:
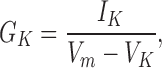 |
where GK is the conductance and IK is the K channel current. Vm and VK are the membrane potential at which the current was measured and the measured K channel reversal potential (−75 to −80 mV, quite close to the expected K+ Nernst potential of −82 mV). The computed conductance was normalized to measured cell capacitance in order to allow better comparison among cells of different sizes.
In some experiments we measured the activity of IK1 and maxi-K channels at the single channel level with inside/out patches. The mean current, <I>, from such a patch depends on the number of channels in the patch (N), the probability that the channel is open (Po), and on the single channel conductance (i): <I> = i × N × Po. We determined the NPo product by averaging large numbers of single channel records and dividing this average by the measured single channel current level.
RESULTS
Maxi-K and IK1 Currents in Parotid Acinar Cells
As noted in INTRODUCTION, parotid acinar cells express two types of Ca2+-activated K channels: maxi-K and IK1 channels encoded by the KCa1.1 and KCa3.1 genes, respectively (Nehrke et al., 2003; Begenisich et al., 2004). Maxi-K channels, both in native tissue and heterologously expressed, are strongly voltage gated and exhibit a marked time dependence (Latorre et al., 1989; Nehrke et al., 2003), whereas IK1 channels exhibit little or no time or voltage dependence in spite of the potential for activation by voltage-driven Ca influx (Ishii et al., 1997; Logsdon et al., 1997; Jensen et al., 1998; Nehrke et al., 2003; Takahata et al., 2003; Begenisich et al., 2004). Since both channels are activated by Ca2+, one would expect to see an increase in the activity of both channels shortly after achieving the whole-cell patch clamp configuration with a pipette solution containing free Ca2+ above the low (<100 nM; Foskett and Melvin, 1989; Foskett et al., 1989; Tojyo et al., 1998) level in the resting cell. That is, as the Ca2+ in the pipette dialyses into the cell, the current through both channels should increase. Such an experiment with a parotid acinar cell from a wild-type strain of mice is illustrated in Fig. 1 A.
Figure 1.

Maxi-K and IK1 currents in parotid acinar cells. (A) Parotid cell currents from a wild-type animal. (Inset) Raw currents just after (Early) and several minutes after (Later) achieving whole-cell mode. Currents in response to 40-ms test potentials to −110, −30, +10, and +50 mV from a −70 mV holding voltage. Calib: 1 nA and 10 ms. (Main) Currents measured at the end of the 40-ms test pulse to the indicated potentials just after (◯) and several minutes after (•) achieving whole-cell mode with 250 nM free Ca2+ in the pipette. Dashed lines represent linear extensions of the currents at the most negative potentials. (B) Parotid cell currents from an IK1-null animal recorded with methods identical to those used in the experiment of A. Calib: 0.5 nA/10 ms. “Later” records recorded with analogue capacitance compensation. (C) Block of maxi-K current component by paxilline. (Inset) Raw currents as above recorded in the absence (Control) and presence (Paxilline) of 100 nM paxilline. Calib: 1 nA and 10 ms. (Main) currents recorded at the end of the 40 ms pulses in the absence (•) and the presence (◯) of 100 nM paxilline. (D) Block of IK1 current component by clotrimazole. (Inset) Raw currents as above recorded in the presence of paxilline before and during application of 300 nM clotrimazole. Calib: 1 nA and 10 ms. (Main) Currents recorded at the end of the 40-ms pulses in the presence of paxilline before (•) and during (◯) application of 300 nM clotrimazole.
Shown in the top inset in Fig. 1 A are the K channel currents at several potentials obtained immediately after achieving the whole cell configuration with a pipette solution containing 250 nM of free Ca2+. Almost all the current had the pronounced time and voltage dependence characteristic of maxi-K channels with little or no indication of activity of the time- and voltage-independent IK1 channels. The voltage sensitivity of maxi-K channels allows activity even in low Ca2+ (Cui et al., 1997). When examined a short time (2–3 min) later, after sufficient time to allow the pipette Ca2+ to equilibrate with the cell cytoplasm, large, time- and voltage-independent IK1 currents were apparent (Fig. 1 A, bottom inset). However, quite surprisingly, while IK1 currents increased, the time- and voltage-dependent maxi-K currents decreased substantially. The time-dependent maxi-K component in response to the depolarization to +50 mV is indicated in the insets.
The large reduction in maxi-K current and the large increase in IK1 current that occurred when the pipette Ca2+ entered the cell are also apparent in the current–voltage relations in the main part of Fig. 1 A. We measured the current at the end of the 40 ms test pulse to the indicated potentials just after (◯) and 2–3 min after (•) achieving the whole cell condition. Initially, almost all the current was due to the voltage-sensitive maxi-K channels revealed by the strongly nonlinear current–voltage relation. After the pipette Ca2+ filled the cell, this current–voltage relation was dominated by the large, linear IK1 component with a very much reduced nonlinear, maxi-K component.
The large reduction in maxi-K channel activity seen after achieving whole-cell mode was due to the presence of IK1 channels since it did not occur in parotid acinar cells from mice without the IK1 gene. As illustrated in Fig. 1 B, maxi-K channels in cells from IK1-null mice actually increased a short time after the whole cell configuration was achieved, in contrast to the reduction seen in wild-type animals. This increase can be seen to be due to a shift of the current–voltage relation to lower voltages, a classic maxi-K channel property (Cui et al., 1997). In this example with 250 nM Ca2+ the maxi-K current at +50 mV increased by 48%; in a total of seven such experiments with cells from IK1-null mice, the mean increase was 28 ± 13% (SEM). In nine experiments with cells from wild-type animals expressing IK1 channels, the maxi-K current at +50 mV decreased by 78 ± 5%.
One possible interpretation of results like those in Fig. 1 A is that maxi-K channel activity did indeed increase but the channels lost their voltage and time dependence. Even if this possibility is remote, it is easy to show that only the time- and voltage-dependent currents are through maxi-K channels by treating the cell with the specific, maxi-K channel inhibitor paxilline (Knaus et al., 1994). The results illustrated in Fig. 1 C show that 100 nM paxilline blocked only the time- and voltage-dependent currents, clearly identifying these as the maxi-K components. In the presence of paxilline, essentially only the IK1 channel currents remained: the currents showed little or no time dependence (Fig. 1 C, inset), and the current–voltage relation was entirely linear. In addition, experiments like that illustrated in Fig. 1 D demonstrated that the linear, time-independent current left in the presence of paxilline was essentially entirely due to IK1 channels. Shown are the currents before and after treatment with 300 nM of the IK1 blocker clotrimazole (Alvarez et al., 1992; Ishii et al., 1997; Joiner et al., 1997; Logsdon et al., 1997). This concentration blocked ∼90% of the currents remaining in the presence of paxilline, an amount consistent with the known sensitivity of IK1 channels to this compound (Wulff et al., 2000; Nehrke et al., 2003). Similar results were obtained with TRAM-34, a clotrimazole analogue that, unlike the parent compound, does not block P450 enzymes (Wulff et al., 2000); in two experiments, 30 nM TRAM-34 blocked >80% of the time-independent current that remained after treatment with paxilline.
It has been reported that clotrimazole blocks maxi-K channels in smooth muscle cells (Rittenhouse et al., 1997). To address this question of specificity, we tested 300 nM clotrimazole on parotid maxi-K channels in IK1(−/−) animals. This concentration blocked only 4.1 ± 2.1% (N = 7) of the maxi-K current measured at +50 mV, confirming the IK1 specificity of this compound in our cells.
If, as suggested above, an increase in IK1 activity leads to a reduced maxi-K activity, then there should be an inverse relation between these two. Thus we measured IK1 activity by the conductance at −110 mV (normalized by cell capacitance) and the maxi-K channel activity as the time-dependent conductance at +50 mV in many cells patched with various Ca2+ concentrations. The pooled results of these experiments are illustrated in Fig. 2. It seems clear from these data that cells patched with higher intracellular Ca2+ generally resulted in larger IK1 conductance levels and, as predicted, smaller maxi-K levels.
Figure 2.

Reciprocal activity of maxi-K and IK1 channels. Maxi-K conductance (at +50 mV) as a function of IK1 conductance (at −110 mV) obtained from cells patched with 80 (▴), 160 (•), 250 (▾), and 400 nM Ca2+ (▪) and 250 Ca2+ with 2–10 μM DCEBIO (◯). Each data point represents a separate cell. Line: Eq. 1 with G0, a, and n values of 1.4 nS/pF, 0.18, and 4, respectively.
Muscarinic Stimulation of Maxi-K and IK1 Activity
The physiologically relevant Ca2+ increase in parotid acinar cells is produced by activation of muscarinic receptors. It is possible that the apparently paradoxical result of lower maxi-K channel activity in cells patched with higher Ca2+ levels (Fig. 2) was somehow a result of the introduction of the tightly buffered intracellular Ca2+ solution or due to the washing out of some critical cytoplasmic component. To address these possibilities, we used the perforated patch method, which does not disrupt the integrity of the cell membrane and, in these experiments, increased cytoplasmic Ca2+ with the muscarinic agonist carbachol (CCh). The ionophore in the pipette in this method (see MATERIALS AND METHODS) allows exchange only of small monovalent, cytoplasmic ions, leaving all other cytoplasmic elements entirely intact (Marty and Neher, 1995). The application of CCh to intact parotid acinar cells often induces an oscillatory increase in intracellular Ca2+ levels (Gray, 1988; Bruce et al., 2002; Harmer et al., 2004). These oscillatory increases in free Ca2+ should, simultaneously and synchronously, increase the activity of both IK1 and maxi-K channels.
To examine the muscarinic-induced IK1 and maxi-K channel activity, we measured IK1 current during a voltage pulse to −100 mV (at which no maxi-K channels activate) and estimated maxi-K activity as the time-dependent current during a 40-ms pulse to +30 mV. These currents were obtained every 0.5 s and converted to conductance (see MATERIALS AND METHODS). As shown in Fig. 3 A, there is little or no IK1 conductance in the absence of CCh, but, owing to the voltage dependence of maxi-K channels, there is a substantial constant maxi-K conductance at +30 mV even in the absence of CCh. Application of CCh (20 μM) caused oscillatory activity in both maxi-K and IK1 channel activity, as expected from an oscillatory increase in cytoplasmic Ca2+ levels. However, it is apparent from an expanded temporal view (Fig. 3 B) that there was a reciprocal relationship between the IK1 (▪) and Maxi-K (◯) conductances.
Figure 3.

Carbachol-induced maxi-K and IK1 channel activity in perforated patch experiments. (A) IK1 (top) and maxi-K (bottom) channel conductances at −100 and +30 mV, respectively, before, during, and after application of 20 μM CCh as indicated. (B) A 20-s expanded temporal view of the IK1 (▪) and maxi-K (◯) conductances from the data in A. Data points connected by spline functions.
Starting at the 60-s time point of the expanded view in Fig. 3 B, it can be seen that the maxi-K conductance at first increased, presumably as the cytoplasmic Ca2+ level began to increase above its resting level. Shortly after the maxi-K conductance began to increase, the IK1 conductance increased and the more the IK1 conductance increased, the more the maxi-K conductance decreased. When IK1 activity decreased, maxi-K conductance increased again. The net result was oscillatory, out of phase, activity of both conductances. Similar, robust, reciprocal oscillations of IK1 and maxi-K channel activity were observed in a total of six such experiments (including one with 5 μM CCh). In these experiments we measured a mean conductance oscillation frequency of 0.31 ± 0.039 Hz, quite similar to intracellular Ca2+ oscillation frequencies in these (Gray, 1988) and similar salivary gland cells (Harmer et al., 2004). Similar CCh applications elicited weak or no oscillations in three other cells, likely due to the known variability of the muscarinic-induced Ca2+ signal in these enzyme-dispersed native cells (Gray, 1988).
Chemical Activation of IK1
If, as suggested by the tight, reciprocal link between the conductances of the two channels, maxi-K channels are inhibited by increased activation of IK1 channels, then maxi-K activity should decrease regardless of the method by which IK1 channels are activated. Thus, we performed experiments in which IK1 channels were activated with the benzimidazolone DCEBIO (Singh et al., 2001), an example of which is illustrated in Fig. 4. In this experiment wild-type mouse parotid acinar cells were patched with a tightly buffered 80 nM Ca2+ solution. At this low intracellular Ca2+, only maxi-K channels were activated and so only time-dependent currents are apparent in the records shown in the inset of Fig. 4 A (Control). Under this condition, the current–voltage relation is strongly rectifying (Fig. 4 A, •). Addition of 10 μM DCEBIO produced a robust activation of IK1 channel current and, concomitantly, reduced maxi-K channel activity, as can be seen by the mostly time-independent current records (Fig. 4 A, inset) and in the almost entirely linear current–voltage relation (◯).
Figure 4.

Inhibition of maxi-K channel activity by DCEBIO activation of IK1. (A) Parotid acinar cell raw currents recorded in the absence (Control) and presence (DCEBIO) of 10 μM DCEBIO and currents recorded at the end of 40-ms pulses to the indicated potentials in the absence (•) and presence (◯) of 10 μM DCEBIO. (B) Maxi-K (◯) and IK1 (▪) conductances at +40 and −90 mV, respectively, during application of 10 μM DCEBIO. (C) Maxi-K conductance from data in B as a function of IK1 conductance. Line: Eq. 1 with G0, a, and n values of 0.8 nS/pF, 0.16, and 4, respectively. (D) Currents from a CHO cell transfected with KCa3.1 and Parslo DNA recorded at the end of 40-ms pulses to the indicated potentials in the absence (•) and presence (◯) of 10 μM DCEBIO. (E) ParSlo (maxi-K) (◯) and KCa3.1 (IK1) (▪) conductances at +50 and −100 mV, respectively, during application of 10 μM DCEBIO. (F) ParSlo (maxi-K) conductance from data in E as a function of KCa3.1 (IK1) conductance. Line: Eq. 1 with G0, a, and n values of 0.24 nS/pF, 0.5, and 4, respectively.
We determined the maxi-K and IK1 conductances during the DCEBIO application and these are shown in Fig. 4 B. The tight link between increased IK1 activity and decreased maxi-K activity is again apparent, in this case with no increase in intracellular Ca2+ levels. The tight, reciprocal link is further illustrated in Fig. 4 C, showing that the decline in maxi-K conductance depended only on the level of IK1 activity and did not require a change in intracellular Ca2+. In a total of eight similar experiments, the maxi-K conductance was reduced to 7.9 ± 1.5% of its value before DCEBIO application. DCEBIO had a negligible effect on maxi-K activity in the absence of IK1 channels: 10 μM of this compound reduced maxi-K current (at +50 mV) in cells from IK1(−/−) animals by an average of 6.9 ± 1.3% (N = 6).
IK1/Maxi-K Interaction in a Heterologous Expression System
The maxi-K channels in parotid acinar cells are composed of approximately equal amounts of two types: homotetramers of the mouse parotid KCa1.1 splice variant (ParSlo, see MATERIALS AND METHODS) and heteromers of this α subunit with a β subunit, most likely β4 (Nehrke et al., 2003). The presence of β subunits affects the pharmacology, Ca2+ sensitivity, and regulation of maxi-K channels (Dworetzky et al., 1996; Hanner et al., 1997; Behrens et al., 2000; Brenner et al., 2000; Meera et al., 2000; Santarelli et al., 2004), which raises the issue of their role in the observed interaction with IK1 channels. To address this question, we tested the interaction between these two channels in a heterologous expression system in which we could control the maxi-K channel composition.
We coexpressed ParSlo (maxi-K) and KCa3.1 (IK1) channels in CHO cells and patched these with a fixed, 80 nM Ca2+ pipette solution and activated IK1 channels with DCEBIO, exactly as was done in the native parotid cell experiments illustrated in Fig. 4(A–C). As with the native cells, in the absence of DCEBIO, only maxi-K channels were activated, as indicated by the nonlinear current–voltage relation of Fig. 4 D (•). Activation of IK1 channels with DCEBIO inhibited maxi-K channels, resulting in the purely linear current–voltage relation (Fig. 4 D, ◯). As in the experiments with the channels in native cells, there was a tight link between the decline in maxi-K conductance and the increased IK1 conductance (Fig. 4, E and F). In this example, the DCEBIO-induced activation of IK channels reduced the maxi-K conductance to 24% of its value before DCEBIO application. In a total of three such experiments, DCEBIO activated an average IK1 conductance between 0.95 and 3.1 nS/pF and the maxi-K conductance was reduced to 22 ± 11% of its control value. Thus, in this heterologous expression system, as in the native cells, IK1 activation produced a concomitant reduction in maxi-K channel activity. These results also illustrate that maxi-K β subunits are not required for the IK1-induced reduction in maxi-K activity.
IK1/Maxi-K Interaction at the Single Channel Level
As an additional step toward understanding the mechanism by which increased IK1 activity inhibited maxi-K channels, we examined this process at the single channel level in parotid acinar cells. An example of such experiments is illustrated in Fig. 5. In this experiment we measured single maxi-K and IK1 channel activity from an inside-out patch with the cytoplasmic side of the membrane exposed to a solution buffered to 80 nM free Ca2+. Both the cytoplasmic and external solutions contained 135 mM K, which allowed us to measure outward maxi-K channel activity at +50 mV (Fig. 5 A) and inward IK1 channel activity at −110 mV (Fig. 5 B). With this low cytoplasmic Ca2+ concentration, maxi-K channel activity could be observed at +50 mV (Fig. 5 A, inset) but no IK1 channel openings were observed (Fig. 5 B, inset; note scale differences). The inset of Fig. 5 A shows several single maxi-K channel records, including several instances when two channels were open at the same time. The main part of the left panel shows a histogram of the maxi-K current levels and reveals that there were at least three channels active in this patch, as indicated by the three peaks in current level (in addition to the peak of current at 0 pA, representing records with no channel openings). The single maxi-K channel level can be seen to be ∼10 pA, representing a 200 pS conductance at this potential, which is a typical value for these channels (Maruyama et al., 1983; Latorre et al., 1989; Nehrke et al.,2003). From these data, we computed the NPo value(see MATERIALS AND METHODS) for the maxi-K channels to be 1.5.
Figure 5.

Inhibition of maxi-K activity by IK1 activation at the single channel level. (A, inset) Single maxi-K channel records at +50 mV in an excised, inside-out patch from a parotid acinar cell exposed to 80 nM Ca2+. Dotted lines indicate current levels for zero, one, and two channel openings. Calib: 10 pA/5 ms. (Main) Channel current histogram indicating at least three maxi-K channels in the patch. (B) Raw data (with no channel openings) from the same patch recorded at −110 mV. Calib: 2 pA/5 ms. (C) Single maxi-K channel activity at +50 mV from the same patch exposed to 2 μM DCEBIO. Calib: 10 pA/5 ms. (D) Single IK1 channel activity at −110 mV from the same patch exposed to 2 μM DCEBIO. Calib: 2 pA/5 ms.
Treatment of this patch with DCEBIO induced the activity of several IK1 channels with a single channel current level near −2 pA, representing a conductance near 20 pS, a typical value for these channels (Nehrke et al., 2003) (Fig. 5 D). The data in Fig. 5 C reveal that activating IK1 channels with DCEBIO resulted in substantial loss of maxi-K channel activity with no alteration in the maxi-K single channel current level. The computed maxi-K NPo value was reduced 10-fold, to 0.15. In a total of five such experiments, DCEBIO activated IK1 channels and reduced the mean maxi-K NPo value to 18 ± 7.4% of the value before IK1 activation. In two additional patches, DCEBIO did not activate IK1 channels and maxi-K activity was unaffected. Thus, the chemical activation of IK1 channels in cell-free membrane patches significantly inhibited maxi-K channel activity.
DISCUSSION
The results of the studies presented above demonstrate that activation of IK1 channels produces a highly correlated reduction in maxi-K channel activity. This occurred whether the IK1 channels were activated by direct application of Ca2+, by muscarinic receptor stimulation, or by the exogenous chemical activator DCEBIO. These data and the associated analysis strongly suggest an intimate interaction between the IK1 and maxi-K channel proteins. In particular, the interaction occurred in cell-free patches, a result that eliminates a role for any freely diffusible cytosolic second messenger. The interaction also occurred with heterologously expressed channel proteins, showing that it does not depend on factors specific to parotid acinar cells, including maxi-K β subunits. Thus, these results suggest that the activation of IK1 channels may directly inhibit the activity of maxi-K channels, and the single channel experiments point to an inhibition of the maxi-K single channel open probability.
IK1/Maxi-K Interaction: a Model
As the data presented above reveal (especially Fig. 4, C and F), the reciprocal relationship between IK1 and maxi-K channel activity is not a precisely linear one, suggesting something other than a 1:1 stoichiometry between these two channel proteins. Thus, consider that each maxi-K channel is surrounded by n IK1 channels. If more than one IK1 channel had to activate before the maxi-K channel was inhibited, there would be a lag during which IK1 conductance would increase without a concomitant decrease in maxi-K conductance. No such lag was observed (see Fig. 2 and Fig. 4, C and F) so, apparently, only one of the n IK1 channels around each maxi-K channel needs to open in order to inhibit the maxi-K channel. In this case, the probability of the maxi-K channel being open will be given by: Pmaxi-K = Po × (probability of no IK1 channels open), where Po is the maxi-K channel open probability in the absence of IK1 channels. The probability that no IK1 channels are open can be computed from the binomial theorem leading to the following relationship between the maxi-K and IK1 channels open probabilities: Pmaxi-K = Po(1 − PIK1)n, where PIK1 is the IK1 single channel open probability.
To accurately measure the open channel probability in a patch with more than a single channel requires accurately determining the number of channels, a classically difficult problem (Horn, 1991). However, since the macroscopic conductances of these channels are proportional to their respective open probabilities, the maxi-K and IK1 conductances will be related as:
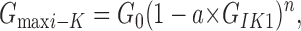 |
(1) |
where G0 is the maxi-K conductance in the absence of IK1 activation, and the proportionality between conductances and probabilities is contained in the parameter a. In our analysis we assume that this proportionality factor is a simple constant. This will be true as long as the single channel currents and the number of expressed channels remain fixed. The former was shown to be true (Fig. 5) and the latter seems reasonable for this simple analysis.
Note that if there were only a single IK1 channel that could influence each maxi-K channel, Eq. 1 predicts an inverse, linear relationship between the conductances which, as noted above, is not seen in the data. Thus, n, the number of IK1 channels able to influence each maxi-K channel, is larger than 1 but, unfortunately, the form of Eq. 1 does not allow an unambiguous determination of the precise number. Nevertheless, in order to see if this model is consistent with the data we fit this equation to the data of Fig. 2 and Fig. 4(C and F) with an n value of 4, a value suggested by the fourfold symmetry of these K channels. As can be seen in these figures (dashed lines), this simple model can readily and accurately account for the relationship between the IK1 and maxi-K channels.
It should be noted that this is perhaps the simplest of an entire class of models that would include various levels of reciprocal activity between these two channels. Future studies may uncover more detail and may require the consideration of more complex interaction schemes. However, for now, this simple model appears able to capture many of the important elements of the interaction between the maxi-K and IK1 channels.
Thus, the experimental results from this study and the agreement of the above model with these data strongly suggest that the IK1 channel regulates, either directly or through a closely associated intermediary, the activity of maxi-K channels. That the interaction occurred in a heterologous expression system suggests that it may be a general one and could occur in other cells in which both channels are expressed, which include vascular smooth muscle cells (Neylon et al., 1999), endothelial cells (Grgic et al., 2005), and macrophages (Hanley et al., 2004).
While there are many examples of protein–protein interactions in many physiological systems, there are not, to our knowledge, any other reported examples in which the activation of one ion channel inhibits another. The closet comparison might be in skeletal muscle where the activation of the voltage-gated Ca channel in the T-tubule membrane activates the ryanodine receptor in the sarcoplasmic reticulum. Perhaps the novel interaction between IK1 and maxi-K channels reported here is just the first of many similar interactions between other ion channel proteins.
Certainly, the inhibition of maxi-K activity by IK1 channels adds to the functional complexity of these classes of ion channels and raises questions about how these complexities may be used in their physiological roles. In fluid-secreting epithelia, the agonist-induced increase in intracellular Ca2+ activates Cl− channels and the resulting transepithelial Cl− ion movement is the primary driving force for fluid and electrolyte secretion. To maintain an electrical Cl− driving force, K channels need also to be activated. We have previously shown that the parotid Ca2+-activated Cl channels are voltage sensitive at low and moderate Ca2+ levels (Arreola et al., 1996). We suggest that fluid secretion at these low to moderate Ca2+ levels is sustained by a balance between these voltage-sensitive Cl channels and the voltage-sensitive maxi-K channels. At higher Ca2+ levels, the Cl channels lose their voltage sensitivity and we propose that the consequent activation of IK1 K channels will inhibit maxi-K channel activity. Thus, fluid secretion at these higher Ca2+ levels will involve a balance between the voltage-independent IK1 and the now voltage-independent Cl channels. If both maxi-K and IK1 channels were simultaneously active at high Ca2+ levels, we speculate the membrane potential would be more negative than with IK1 channels alone. This would lead to excessive Cl− loss and, perhaps, to excessive Ca2+ influx (driven by the negative membrane voltage), leading to unwanted Ca2+-signaling events, including apoptosis. One challenge of future studies will be to test this model of the role for IK1 inhibition of maxi-K channel activation.
Acknowledgments
We thank James E. Melvin for considerable discussion on this work and for critically reading the manuscript. We are grateful to Robert Dirksen for discussions, reading the manuscript, and for pointing out the skeletal muscle Ca channel/ryanodine receptor interaction. We thank Mark Wagner for preparing high quality parotid acinar cells.
This work was supported by National Institutes of Health grant DE-13539.
Angus C. Nairn served as editor.
Abbreviations used in this paper: CCh, carbachol; DCEBIO, 5,6-dichloro-1-ethyl-1,3-dihydro-2H-benzimidazol-2-one; SMEM, Eagle's minimum essential medium.
References
- Alvarez, J., M. Montero, and J. Garcia-Sancho. 1992. High affinity inhibition of Ca(2+)-dependent K+ channels by cytochrome P-450 inhibitors. J. Biol. Chem. 267:11789–11793. [PubMed] [Google Scholar]
- Arreola, J., J.E. Melvin, and T. Begenisich. 1996. Activation of calcium-dependent chloride channels in rat parotid acinar cells. J. Gen. Physiol. 108:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begenisich, T., T. Nakamoto, C.E. Ovitt, K. Nehrke, C. Brugnara, S.L. Alper, and J.E. Melvin. 2004. Physiological roles of the intermediate conductance, Ca2+-activated potassium channel Kcnn4. J. Biol. Chem. 279:47681–47687. [DOI] [PubMed] [Google Scholar]
- Behrens, R., A. Nolting, F. Reimann, M. Schwarz, R. Waldschutz, and O. Pongs. 2000. hKCNMB3 and hKCNMB4, cloning and characterization of two members of the large-conductance calcium-activated potassium channel β subunit family. FEBS Lett. 474:99–106. [DOI] [PubMed] [Google Scholar]
- Bers, D.M., C.W. Patton, and R. Nuccitelli. 1994. A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol. 40:3–29. [DOI] [PubMed] [Google Scholar]
- Brenner, R., T.J. Jegla, A. Wickenden, Y. Liu, and R.W. Aldrich. 2000. Cloning and functional characterization of novel large conductance calcium-activated potassium channel β subunits, hKCNMB3 and hKCNMB4. J. Biol. Chem. 275:6453–6461. [DOI] [PubMed] [Google Scholar]
- Bruce, J.I., T.J. Shuttleworth, D.R. Giovannucci, and D.I. Yule. 2002. Phosphorylation of inositol 1,4,5-trisphosphate receptors in parotid acinar cells. A mechanism for the synergistic effects of cAMP on Ca2+ signaling. J. Biol. Chem. 277:1340–1348. [DOI] [PubMed] [Google Scholar]
- Cui, J., D.H. Cox, and R.W. Aldrich. 1997. Intrinsic voltage dependence and Ca2+ regulation of mslo large conductance Ca-activated K+ channels. J. Gen. Physiol. 109:647–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworetzky, S.I., C.G. Boissard, J.T. Lum-Ragan, M.C. McKay, D.J. Post-Munson, J.T. Trojnacki, C.P. Chang, and V.K. Gribkoff. 1996. Phenotypic alteration of a human BK (hSlo) channel by hSlo beta subunit coexpression: changes in blocker sensitivity, activation/relaxation and inactivation kinetics, and protein kinase A modulation. J. Neurosci. 16:4543–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskett, J.K., and J.E. Melvin. 1989. Activation of salivary secretion: coupling of cell volume and [Ca2+]i in single cells. Science. 244:1582–1585. [DOI] [PubMed] [Google Scholar]
- Foskett, J.K., P.J. Gunter-Smith, J.E. Melvin, and R.J. Turner. 1989. Physiological localization of an agonist-sensitive pool of Ca2+ in parotid acinar cells. Proc. Natl. Acad. Sci. USA. 86:167–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardos, G. 1958. The function of calcium in the potassium permeability of human erythrocytes. Biochim. Biophys. Acta. 30:653–654. [DOI] [PubMed] [Google Scholar]
- Gray, P.T. 1988. Oscillations of free cytosolic calcium evoked by cholinergic and catecholaminergic agonists in rat parotid acinar cells. J. Physiol. 406:35–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grgic, I., I. Eichler, P. Heinau, H. Si, S. Brakemeier, J. Hoyer, and R. Kohler. 2005. Selective blockade of the intermediate-conductance Ca2+-activated K+ channel suppresses proliferation of microvascular and macrovascular endothelial cells and angiogenesis in vivo. Arterioscler. Thromb. Vasc. Biol. 25:704–709. [DOI] [PubMed] [Google Scholar]
- Gutman, G.A., K.G. Chandy, J.P. Adelman, J. Aiyar, D.A. Bayliss, D.E. Clapham, M. Covarriubias, G.V. Desir, K. Furuichi, B. Ganetzky, et al. 2003. International union of pharmacology. XLI. Compendium of voltage-gated ion channels: potassium channels. Pharmacol. Rev. 55:583–586. [DOI] [PubMed] [Google Scholar]
- Hanley, P.J., B. Musset, V. Renigunta, S.H. Limberg, A.H. Dalpke, R. Sus, K.M. Heeg, R. Preisig-Muller, and J. Daut. 2004. Extracellular ATP induces oscillations of intracellular Ca2+ and membrane potential and promotes transcription of IL-6 in macrophages. Proc. Natl. Acad. Sci. USA. 101:9479–9484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanner, M., W.A. Schmalhofer, P. Munujos, H.G. Knaus, G.J. Kaczorowski, and M.L. Garcia. 1997. The β subunit of the high-conductance calcium-activated potassium channel contributes to the high-affinity receptor for charybdotoxin. Proc. Natl. Acad. Sci. USA. 94:2853–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer, A.R., P.M. Smith, and D.V. Gallacher. 2004. Local and global calcium signals and fluid and electrolyte secretion in mouse submandibular acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 288:G118–G124. [DOI] [PubMed] [Google Scholar]
- Horn, R. 1991. Estimating the number of channels in patch recordings. Biophys. J. 60:433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii, T.M., C. Silvia, B. Hirschberg, C.T. Bond, J.P. Adelman, and J. Maylie. 1997. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. USA. 94:11651–11656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, B.S., D. Strobaek, P. Christophersen, T.D. Jorgensen, C. Hansen, A. Silahtaroglu, S.P. Olesen, and P.K. Ahring. 1998. Characterization of the cloned human intermediate-conductance Ca2+-activated K+ channel. Am. J. Physiol. 275:C848–C856. [DOI] [PubMed] [Google Scholar]
- Joiner, W.J., L.Y. Wang, M.D. Tang, and L.K. Kaczmarek. 1997. hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc. Natl. Acad. Sci. USA. 94:11013–11018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus, H.G., O.B. McManus, S.H. Lee, W.A. Schmalhofer, M. Garcia-Calvo, L.M. Helms, M. Sanchez, K. Giangiacomo, J.P. Reuben, A.B. Smith III, et al. 1994. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry. 33:5819–5828. [DOI] [PubMed] [Google Scholar]
- Latorre, R., A. Oberhauser, P. Labarca, and O. Alvarez. 1989. Varieties of calcium-activated potassium channels. Annu. Rev. Physiol. 51:385–399. [DOI] [PubMed] [Google Scholar]
- Logsdon, N.J., J. Kang, J.A. Togo, E.P. Christian, and J. Aiyar. 1997. A novel gene, hKCa4, encodes the calcium-activated potassium channel in human T lymphocytes. J. Biol. Chem. 272:32723–32726. [DOI] [PubMed] [Google Scholar]
- Marty, A., and E. Neher. 1995. Tight-seal whole-cell recording. In Single-channel Recording. B. Sakmann, and E. Neher, editors. Plenum Press, New York. 31-52.
- Maruyama, Y., D.V. Gallacher, and O.H. Petersen. 1983. Voltage and Ca2+-activated K+ channel in baso-lateral acinar cell membranes of mammalian salivary glands. Nature. 302:827–829. [DOI] [PubMed] [Google Scholar]
- Meera, P., M. Wallner, and L. Toro. 2000. A neuronal β subunit (KCNMB4) makes the large conductance, voltage- and Ca2+-activated K+ channel resistant to charybdotoxin and iberiotoxin. Proc. Natl. Acad. Sci. USA. 97:5562–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melvin, J.E., D. Yule, T. Shuttleworth, and T. Begenisich. 2005. Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu. Rev. Physiol. 67:445–469. [DOI] [PubMed] [Google Scholar]
- Nehrke, K., C.C. Quinn, and T. Begenisich. 2003. Molecular identification of Ca2+-activated K+ channels in parotid acinar cells. Am. J. Physiol. Cell Physiol. 284:C535–C546. [DOI] [PubMed] [Google Scholar]
- Neylon, C.B., R.J. Lang, Y. Fu, A. Bobik, and P.H. Reinhart. 1999. Molecular cloning and characterization of the intermediate-conductance Ca2+-activated K+ channel in vascular smooth muscle: relationship between KCa channel diversity and smooth muscle cell function. Circ. Res. 85:e33–e43. [DOI] [PubMed] [Google Scholar]
- Petersen, O.H., and Y. Maruyama. 1984. Calcium-activated potassium channels and their role in secretion. Nature. 307:693–696. [DOI] [PubMed] [Google Scholar]
- Rittenhouse, A.R., C. Parker, C. Brugnara, K.G. Morgan, and S.L. Alper. 1997. Inhibition of maxi-K currents in ferret portal vein smooth muscle cells by the antifungal clotrimazole. Am. J. Physiol. 273:C45–C56. [DOI] [PubMed] [Google Scholar]
- Santarelli, L.C., J. Chen, S.H. Heinemann, and T. Hoshi. 2004. The β1 subunit enhances oxidative regulation of large-conductance calcium-activated K+ channels. J. Gen. Physiol. 124:357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh, C.C., M. Coghlan, J.P. Sullivan, and M. Gopalakrishnan. 2000. Potassium channels: molecular defects, diseases, and therapeutic opportunities. Pharmacol. Rev. 52:557–594. [PubMed] [Google Scholar]
- Singh, S., C.A. Syme, A.K. Singh, D.C. Devor, and R.J. Bridges. 2001. Benzimidazolone activators of chloride secretion: potential therapeutics for cystic fibrosis and chronic obstructive pulmonary disease. J. Pharmacol. Exp. Ther. 296:600–611. [PubMed] [Google Scholar]
- Stocker, M. 2004. Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat. Rev. Neurosci. 5:758–770. [DOI] [PubMed] [Google Scholar]
- Takahata, T., M. Hayashi, and T. Ishikawa. 2003. SK4/IK1-like channels mediate TEA-insensitive, Ca2+-activated K+ currents in bovine parotid acinar cells. Am. J. Physiol. Cell Physiol. 284:C127–C144. [DOI] [PubMed] [Google Scholar]
- Takeda, M., T. Tanimoto, M. Ikeda, J. Ka, and S. Matsumoto. 2004. Activaton of GABAB receptor inhibits the excitability of rat small diameter trigeminal root ganglion neurons. Neuroscience. 123:491–505. [DOI] [PubMed] [Google Scholar]
- Tojyo, Y., A. Tanimura, A. Nezu, and Y. Matsumoto. 1998. Activation of β-adrenoceptors does not cause any change in cytosolic Ca2+ distribution in rat parotid acinar cells. Eur. J. Pharmacol. 360:73–79. [DOI] [PubMed] [Google Scholar]
- Wulff, H., M.J. Miller, W. Hansel, S. Grissmer, M.D. Cahalan, and K.G. Chandy. 2000. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc. Natl. Acad. Sci. USA. 97:8151–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]