

Abstract
Acid-sensing ion channels (ASICs) are Na+ channels gated by extracellular H+. Six ASIC subunits that are expressed in neurons have been characterized. The tarantula toxin psalmotoxin 1 has been reported to potently and specifically inhibit homomeric ASIC1a and has been useful to characterize ASICs in neurons. Recently we have shown that psalmotoxin 1 inhibits ASIC1a by increasing its apparent affinity for H+. However, the mechanism by which PcTx1 increases the apparent H+ affinity remained unclear. Here we show that PcTx1 also interacts with ASIC1b, a splice variant of ASIC1a. However, PcTx1 does not inhibit ASIC1b but promotes its opening; under slightly acidic conditions, PcTx1 behaves like an agonist for ASIC1b. Our results are most easily explained by binding of PcTx1 with different affinities to different states (closed, open, and desensitized) of the channel. For ASIC1b, PcTx1 binds most tightly to the open state, promoting opening, whereas for ASIC1a, it binds most tightly to the open and the desensitized state, promoting desensitization.
INTRODUCTION
Acid sensing ion channels (ASICs) are Na+-selective ion channels that are activated by extracellular H+ (Waldmann and Lazdunski, 1998; Krishtal, 2003). They are abundantly expressed in the central and the peripheral nervous system and participate in higher brain functions, such as learning and memory (Wemmie et al., 2002), and in perception of pain (Sutherland et al., 2001; Voilley et al., 2001; Chen et al., 2002; Mamet et al., 2002), taste (Ugawa et al., 2003), and mechanical stimuli (Price et al., 2000). ASIC subunits have a simple topology with two transmembrane domains, short intracellular termini, and the bulk of the protein in the extracellular space (Saugstad et al., 2004). In the genome of mammals there are four asic genes. ASIC1a and ASIC1b are splice variants of the asic1 gene, which differ in the first third of their amino acid sequence, including the first transmembrane domain TM1, whereas the remaining two thirds of the proteins are identical (Chen et al., 1998; Bässler et al., 2001). ASIC1a is highly expressed in the small neurons of the dorsal root ganglia and many regions, mostly those with excitatory input, in the brain (Waldmann et al., 1997; Wemmie et al., 2003). In contrast, ASIC1b is specifically expressed in sensory neurons (Chen et al., 1998). Native ASICs are homo- and heteromeric assemblies of probably four subunits (Sutherland et al., 2001; Baron et al., 2002; Benson et al., 2002; Xie et al., 2002).
Like the related epithelial Na channel, ENaC, ASICs are blocked by the diuretic amiloride, with an EC50 of ∼20 μM (Waldmann et al., 1997; Paukert et al., 2004). The first potent and specific blocker of ASICs to be identified was the tarantula toxin psalmotoxin 1, PcTx1 (Escoubas et al., 2000). It was reported that PcTx1 specifically inhibits ASIC1a with an EC50 of ∼1 nM (Escoubas et al., 2000). No other ASIC and also no heteromeric ASICs, even those containing the ASIC1a subunit, were inhibited (Escoubas et al., 2000). Recently, we reported that PcTx1 inhibits ASIC1a by increasing its apparent H+ affinity (Chen et al., 2005). This increase in apparent affinity for their ligand, H+, is sufficient to shift ASIC1a channels into the desensitized state at a resting pH of 7.4. In addition, PcTx1 promotes the opening of ASIC1a (Chen et al., 2005).
Apparent H+ affinity, however, is an unspecific description that does not provide much insight into the underlying mechanism (Colquhoun, 1998). According to the basic kinetic schemeASICs bind H+ in the closed state C, and from this H+-bound closed state they either reach the open state O or the desensitized state D. H+-bound states would be cyclically connected so that channels could reach the desensitized state D also from the open state O. As we previously proposed (Chen et al., 2005), the increase in apparent H+ affinity by PcTx1 could be explained in two different ways. First, PcTx1 could increase the true affinity of H+ to ASIC1a, modifying the energetics of the binding step. Second, it could modify the energetics of the gating step, shifting the equilibrium between the closed state with H+ bound and the open and desensitized state. Such a shift of the equilibrium between different states would be expected if PcTx1 would have a higher affinity to the open and/or the desensitized state than to the closed state. Since PcTx1 promoted steady-state desensitization as well as opening of ASIC1a, we were unable to decide if PcTx1 directly affects the H+ affinity of ASICs or if it indirectly modulates gating by state-dependent binding.
In this study, we further addressed the mechanism of inhibition of ASIC1 by PcTx1. We found that PcTx1 also interacts with ASIC1b. However, in contrast to ASIC1a, ASIC1b was almost not inhibited by PcTx1 but its opening was greatly facilitated and its desensitization slowed down by PcTx1. These results show that facilitated binding of H+ cannot explain the effects of PcTx1. They rather support a model in which PcTx1 binds with different affinity to different states of the channel, allosterically modifying gating.
MATERIALS AND METHODS
Electrophysiology
cDNAs for rat ASIC1a, ASIC1b, and chimeras C43, C61, C92, and C98 had been previously described (Bässler et al., 2001; Babini et al., 2002). Chimera C166 was constructed as described before (Babini et al., 2002). Capped cRNA was synthesized by SP6 RNA polymerase from linearized cDNA, using the mMessage mMachine kit (Ambion).
We injected 0.01–10 ng cRNA into defolliculated stage V–VI oocytes of Xenopus laevis and kept oocytes in OR-2 medium for 2–4 d before measurements. OR-2 (oocyte ringer-2) solution contained (in mM) NaCl 82.5, KCl 2.5, Na2HPO4 1.0, MgCl2 1.0, CaCl2 1.0, HEPES 5.0, PVP 0.5 g/liter, 1,000 U/liter penicillin, and 10 mg/liter streptomycin; pH was adjusted to 7.3 with NaOH. Whole cell currents were recorded with a TurboTec 03X amplifier (npi electronic) using an automated, pump-driven solution exchange system together with the oocyte testing carousel controlled by the interface OTC-20 (Madeja et al., 1995). Data acquisition and solution exchange were managed using the software CellWorks 5.1.1 (npi electronic). Data were filtered at 20 Hz and acquired at 0.1–1 kHz. Bath solution for whole oocyte current measurements contained (in mM) NaCl 140, HEPES 10, CaCl2 1.8, MgCl2 1.0; pH was adjusted with NaOH. For acidic test solutions, HEPES was replaced by MES. Glass electrodes filled with 3 M KCl were used; they had a resistance of 0.3–1.5 MΩ. The membrane potential was clamped at −70 mV.
PcTx1 synthesis and refolding was previously reported (Chen et al., 2005). Solutions containing PcTx1 were supplemented with 0.05% BSA (Sigma-Aldrich) in order to avoid absorption by the tubing. 0.05% BSA itself did not significantly affect ASICs currents. In addition, we obtained results that were qualitatively similar to the results with PcTx1, using solutions containing the Psalmopoeus cambridgei venom (SpiderPharm) (not depicted). These solutions were not supplemented with BSA.
Data Analysis
Data were analyzed with the software IgorPro (Wave metrics, Lake Oswego, OR USA). pH response curves were fit with a Hill function:
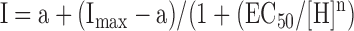 |
(1) |
where Imax is the maximal current, a is the residual current, [H] is the concentration of the H+, EC50 is the concentration at which half-maximal response occurs, and n is the Hill coefficient. The pH of half-maximal activation, pH50, was calculated as −log(EC50). The activation and desensitization curves of ASICs currents were fit with a mono-exponential function. Before fitting, currents from each measurement were normalized to the maximal value measured. Results are reported, in the text, as means ± SD or, on the figures, as means ± SEM. They represent the mean of n individual measurements on different oocytes. Statistical analysis was done with the paired or unpaired t test, as appropriate.
RESULTS
PcTx1 Only Weakly Affects Steady-state Desensitization of ASIC1b but Strongly Promotes H+ Activation
It has been reported that PcTx1 does not inhibit ASIC1b at pH 7.4 (Escoubas et al., 2000). However, since ASIC1b has a lower H+ affinity than ASIC1a and its steady-state desensitization curve is shifted by 0.25 pH units compared with ASIC1a (Babini et al., 2002), we reevaluated inhibition at a conditioning pH of 7.1. Fig. 1 shows that also at pH 7.1 ASIC1b currents were not inhibited, even by a high concentration (500 nM) of synthetic PcTx1 (n = 5). At a more acidic conditioning pH of 6.9, 100 nM PcTx1 slightly inhibited ASIC1b currents (n = 5). To more systematically address the interaction of PcTx1 with ASIC1b, we determined the steady-state desensitization curve of ASIC1b in the absence and in the presence of 100 nM PcTx1. As is shown in Fig. 1, the toxin slightly (P = 0.04) shifted the pH50 of steady-state desensitization from pH 6.95 ± 0.03 (n = 6) without PcTx1 to pH 6.99 ± 0.03 (n = 6) with PcTx1. This shift is tiny compared with ASIC1a, for which steady-state desensitization is shifted by 0.27 pH units by 30 nM PcTx1 (Chen et al., 2005), consistent with the finding that PcTx1 does not inhibit ASIC1b as clearly as it inhibits ASIC1a.
Figure 1.

PcTx1 did not strongly inhibit ASIC1b. (A) ASIC1b currents were repeatedly activated by pH 5.0 for 10 s. Even 500 nM PcTx1 did not inhibit the current, when it was applied at pH 7.1 (n = 5). When applied at pH 6.9, 100 nM PcTx1 slightly inhibited the current (n = 6). (B) H+ dependence of steady-state desensitization of ASIC1b currents in the absence (n = 6, open circles) or presence (n = 6, filled circles) of 100 nM PcTx1. PcTx1 was applied for 120 s in the conditioning period; available channels were assessed with pH 5.0. Solid lines are fits of the mean values of each data point to a Hill function (Eq. 1). PcTx1 slightly but significantly (P = 0.04) shifted the steady-state desensitization curve of ASIC1b leftwards.
Next we examined the apparent H+ affinity for activation of ASIC1b in the absence and presence of 100 nM PcTx1. As shown in Fig. 2 B, and similar to a previous reports (Babini et al., 2002), ASIC1b currents were half-maximally activated at a pH of 5.70 ± 0.04 (n = 7) in the absence of PcTx1. In the presence of 100 nM PcTx1, the pH50 of activation was significantly (P < 0.01) leftward shifted by 0.38 pH units to pH 6.08 ± 0.06 (n = 6). Hence PcTx1 robustly increased the apparent H+ affinity of ASIC1b for activation. The increased apparent H+ affinity of ASIC1b is also illustrated by a potentiation of ASIC1b currents by PcTx1 as shown in Fig. 3 A. Application of 100 nM PcTx1 in the conditioning period increased ASIC1b currents elicited by pH 6.0 by 1.9-fold (1.9 ± 0.8, n = 9). This result clearly shows that PcTx1 can bind to and interact with ASIC1b. It promotes the opening of ASIC1b but does not induce strong desensitization at steady state.
Figure 2.

PcTx1 facilitated opening of ASIC1b. (A) Representative current traces elicited by pH ranging from 4.8 to 6.6 with conditioning pH 7.5 (applied for 60 s). Top, without PcTx1; bottom, with 100 nM PcTx1. (B) H+ dependence of ASIC1b activation in the absence (n = 7, open squares) or in the presence (n = 6, filled squares) of 100 nM PcTx1. Solid lines are fits of the mean values of each data point to a Hill function (Eq. 1). PcTx1 significantly (P < 0.01) shifted the pH activation curve to lower H+ concentrations. The arrow illustrates how PcTx1 potentiates currents at pH 6.0, as shown in Fig. 3 A.
Figure 3.

(A) PcTx1 potentiated ASIC1b currents elicited by pH 6.0. Top, representative current traces. Conditioning pH was 7.5. PcTx1 was applied in the conditioning period for 120 s. Bottom, concentration–response relationship (n = 5–9). Fit to the Hill equation revealed half-maximal potentiation at 101 nM (dashed line). Since a maximal response was not reached, this EC50 value provides a lower limit for the apparent affinity. (B) PcTx1 directly and robustly opened ASIC1b. Top, different concentrations of PcTx1 were coapplied with pH 6.6. pH 6.6 alone did not open ASIC1b. Bottom, concentration–response relationship. Solid line represents a fit to the Hill equation and revealed half-maximal potentiation at 139 nM (n = 4–8). (C) PcTx1 directly opened ASIC1a. Top, different concentrations of PcTx1 were coapplied with pH 7.1 (n = 4–10). pH 7.1 alone did not open ASIC1a. Bottom, concentration–response relationship. Fit to the Hill equation revealed half-maximal potentiation at 156 nM (dashed line), providing a lower limit for the apparent affinity.
PcTx1 Binds with Different Affinities to Different States of the Channel
PcTx1 directly opens ASIC1a when either the H+ concentration is simultaneously increased or the Ca2+ concentration is simultaneously lowered (Chen et al., 2005). We, therefore, examined whether PcTx1 opens ASIC1b in a similar way. pH 6.6 by itself does not open ASIC1b (Fig. 3 B). However, as illustrated in Fig. 3 B, coapplication of 200 nM PcTx1 and pH 6.6 indeed opened ASIC1b. We estimated the apparent affinity of ASIC1b for PcTx1 by coapplying different concentrations of the toxin with pH 6.6. This analysis yielded an EC50 of ∼100 nM (Fig. 3 B). From a similar analysis with ASIC1a we estimated a lower limit for the EC50 of 150 nM when PcTx1 was coapplied with pH 7.1 (Fig. 3 C). Due to desensitization of the channel, these EC50 values were not measured at steady state and, therefore, provide only a rough estimate for the apparent affinity of the open state of the channels. They suggest that apparent affinities of PcTx1 for open ASIC1a and open ASIC1b channels are on the same order of magnitude.
We previously estimated the apparent IC50 of PcTx1 for ASIC1a to be ∼3 nM at pH 7.4 (Chen et al., 2005). This value was obtained by measuring the inhibition of the ASIC1a current and thus provides an estimate of the apparent affinity of PcTx1 for the desensitized state of ASIC1a. As shown above, the apparent IC50 of PcTx1 for ASIC1b was much larger than for ASIC1a (>500 nM at pH 7.1). It is not possible to directly compare the estimates of the IC50 with the estimates of the EC50, because IC50 values have been measured at steady state, whereas, as already mentioned, EC50 values cannot be measured at steady state.
PcTx1 Slows the Desensitization of ASIC1b Channels
As can be seen from the traces in Fig. 1 A and Fig. 2 A, PcTx1 appeared to slow down the desensitization of ASIC1b. Analyzing the decay time constant from experiments as the one shown in Fig. 2 A, we found that the decay time constant was indeed increased by a factor of ∼1.4, independent of the pH used (Fig. 4 A). Opening of ASIC1b channels with pH 6.0 and preincubating the channels with different concentrations of the toxin revealed that the increase of the desensitization time constant was concentration dependent (Fig. 4 B). The concentration dependence could be reasonably well fit with a Hill function assuming the above-determined EC50 (100 nM) for the toxin (Fig. 4 B).
Figure 4.

PcTx1 slowed down desensitization of ASIC1b. (A) Diagram showing the time constant of desensitization with and without 100 nM PcTx1 at different pH values (squares). Data are from measurements like the one shown in Fig. 2 A. The ratio of both time constants is shown in the same diagram (filled circles). It was not pH dependent. (B) Diagram showing the dependence of the ratio of the time constant of desensitization with and without PcTx1 at different concentrations of PcTx1. pH used to activate ASIC1b channels was always pH 6.0. Data are from measurements like the one shown in Fig. 3 A. The dashed line represents a fit to the Hill equation assuming an EC50 of 100 nM, as determined in Fig. 3, A and B.
At pH 5.0, ASIC1b currents decayed with a time constant τ of 0.6 ± 0.1 s. Preincubation with 500 nM PcTx1 robustly increased the decay time constant τ to 1.5 ± 0.1 s (n = 6; P < 0.01; Fig. 5). In contrast, the desensitization of ASIC1a was only slightly affected under the same conditions (τ = 1.2 ± 0.2 s without toxin and τ = 1.3 ± 0.2 s with 500 nM PcTx1; P < 0.01, n = 5; Fig. 5). Similarly, PcTx1 opened ASIC1b more persistently than ASIC1a (Fig. 6 A). ASIC1b currents elicited by 100 nM PcTx1 at pH 6.6 decayed significantly (P < 0.01) slower than ASIC1a currents elicited by 100 nM PcTx1 at pH 7.1. The decay time constant was τ = 17.6 ± 3.8 s (n = 12) for ASIC1b and τ = 4.3 ± 0.7 s (n = 11) for ASIC1a currents. The ASIC1b currents were sensitive to amiloride block (100 μM), several seconds after washing off PcTx1 (Fig. 6 A), indicating that PcTx1 dissociated slowly from the channel with a time constant on the order of several seconds. In contrast, the rising phase of ASIC1b currents was similar to that of ASIC1a currents under these conditions (P = 0.27). For both channels, the time constant of the rising phase was around 2 s, much larger than for opening by high H+ concentrations. This slow onset of current activation may be explained by the slow association of PcTx1. Slow on and off rates have recently also been shown for the interaction of hanatoxin, which is structurally related to PcTx1 (Escoubas et al., 2003), with Kv2.1 channels (Phillips et al., 2005). Both the time constant describing the rising phase and the time constant describing the decay phase of the PcTx1-induced ASIC1b current decreased with increasing toxin concentrations up to 200 nM (Fig. 6). However, increasing the toxin concentration >200 nM did not further decrease the decay time constant (τ ∼12 s). Such slowing of the desensitization by PcTx1 is expected when PcTx1 stabilized the open state of ASIC1b with respect to the desensitized state.
Figure 5.

PcTx1 significantly prolonged the desensitization of ASIC1 currents. 500 nM PcTx1 slightly but significantly (P < 0.01, n = 5) slowed down desensitization of ASIC1a currents. Desensitization of ASIC1b currents was more strongly slowed down (P ≪ 0.01, n = 6). Conditioning pH was pH 7.9 for ASIC1a and pH 8.0 for ASIC1b, respectively. Acidic test pH was always pH 5.0. Top, examples of current traces. Scaled overlaid traces before and after PcTx1 application are shown for better comparison. Bottom, bars representing desensitization time constants of ASIC1 currents before and after PcTx1 application.
Figure 6.

PcTx1 opens ASIC1b more persistently than ASIC1a. (A) When coapplied with pH 7.1, 100 nM PcTx1 induced relatively transient ASIC1a currents (left, n = 7). ASIC1b currents that were induced under comparable conditions were of greater relative amplitude and decayed more slowly. They could be blocked by 100 μM amiloride (right, n = 6). (B) Diagram showing the dependence on the PcTx1 concentration of the time constant describing the rising and the decay phase of the PcTx1-elicited currents, determined in the continuous presence of PcTx1 (n = 4–11 for ASIC1a; n = 4–12 for ASIC1b). The time constants of the rising phase were similar for ASIC1a and ASIC1b currents, while for the decay phase, they were significantly different. For ASIC1b, the decay time constant initially decreased with increasing concentrations of PcTx1 but remained constant around 12 s for high concentrations of PcTx1. Part of the data for ASIC1a have already been published (Chen et al., 2005). **, P < 0.01.
A Small Region in the Extracellular Loop Determines High PcTx1 Affinity of the Desensitized State
To gain insights, which structural differences between ASIC1a and ASIC1b determine their differential response to PcTx1, we used a chimeric approach that was based on a series of chimeras that exchange different parts between the two splice variants (Fig. 7 A). Some of these chimeras had already been previously described (Bässler et al., 2001; Babini et al., 2002). As shown in Fig. 7, in these chimeras increasing fragments from ASIC1b replaced the corresponding fragments of ASIC1a, leading to chimeras C43, C61, C92, C98, and C166, with the number denoting the number of amino acids replaced. For example, C166 contained 166 amino acids from ASIC1b at its NH2 terminus, followed by 19 amino acids from ASIC1a, and finally the common COOH terminus (Fig. 7).
Figure 7.

PcTx1 inhibits chimeras between ASIC1a and 1b. Left, chimeras are schematically drawn. NH2-terminal sequences from ASIC1a are shown as open bars, those from ASIC1b as gray bars. The first transmembrane domain TM1 is indicated as a black box and the common COOH terminus is shown as a black bar; only its first part is shown. The chimeras did have a shorter NH2 terminus than ASIC1b; this shorter NH2 terminus corresponds to M3 in Bässler et al. (2001). However, since the missing part of the NH2 terminus of ASIC1b is supposed to be located in the cytoplasm and since chimera C43 interacted with PcTx1 like ASIC1a, this NH2-terminal part does not seem to have any strong influence on the interaction with PcTx1. Right, PcTx1 strongly inhibited the currents of ASIC1a and all chimeras, but not of ASIC1b. Conditioning pH was chosen according to the pH50 of steady-state desensitization of each channel. Acidic test pH was always pH 5.0. PcTx1 (50 nM) was applied in the conditioning period for 120 s. Note the opening of chimeras C61, C92, C98, and C166 by application of PcTx1. The transient decrease in current amplitude with C92, after washout of PcTx1 and application of pH 5.0, is enlarged. Scale bars correspond to 5 μA and 60 s, respectively. Peak current amplitudes (mean ± SD) were −11.6 ± 6.1 μA for ASIC1a (n = 7), −35.9 ± 18.2 μA for C43 (n = 6), −8.4 ± 9.9 μA for C61 (n = 6), −12.3 ± 4.5 μA for C91 (n = 5), −12.0 ± 4.0 μA for C98 (n = 3), −11.4 ± 3.1 μA for C166 (n = 5), and −11.1 ± 6.0 μA for ASIC1b (n = 9).
First, we looked for inhibition of the chimeras by PcTx1. We determined for each chimera the pH50 of steady-state desensitization (Table I) and applied PcTx1 at a conditioning pH that was ∼0.2 pH units above this pH50. Fig. 7 illustrates that all five chimeras C43, C62, C92, C98, and C166, like ASIC1a wild type, were almost completely inhibited by 50 nM PcTx1, while no inhibition was observed on ASIC1b currents under similar conditions. We determined a complete curve for steady-state desensitization in the presence of PcTx1 only for chimera C166, which retained the smallest part of ASIC1a. As expected from the inhibition by PcTx1 of this chimera, 100 nM PcTx1 shifted the curve for steady-state desensitization of chimera C166 significantly (P < 0.01) by 0.27 pH units, from pH 6.90 ± 0.01 (n = 5) in the absence of PcTx1 to pH 7.17 ± 0.01 (n = 5) in the presence of 100 nM PcTx1 (Fig. 8 A). This robust shift of the steady-state desensitization curve is in strong contrast to the small shift of the steady-state desensitization curve of ASIC1b wild type (Fig. 1) and suggests that the 19 amino acids of ASIC1a that are retained in C166 are sufficient to confer a high PcTx1 affinity to the desensitized state of the channel. Analyzing inhibition by different concentrations of PcTx1 at pH 7.1 indeed revealed that C166 had an apparent PcTx1 affinity similar to ASIC1a (IC50 = 3.4 nM, n = 8–10; Fig. 8 B). Similar to the steady-state desensitization curve, the activation curve of C166 was shifted by 0.32 pH units by PcTx1 (Fig. 8 A).
TABLE I.
pH50 of Steady-state Desensitization and Activation
| ASIC1a | C43 | C62 | C92 | C98 | C166 | ASIC1b | |
|---|---|---|---|---|---|---|---|
| pH50 (SSD) | 7.19 ± 0.01 (8) | 7.26 ± 0.01 (6) | 7.23 ± 0.09 (8) | 7.17 ± 0.13 (8) | 7.02 ± 0.02 (9) | 6.90 ± 0.01 (5) | 6.95 ± 0.03 (6) |
| pH50 (Act) | 6.56 ± 0.04 (7) | 6.00 ± 0.11 (6) | 6.78 ± 0.06 (9) | 6.77 ± 0.08 (6) | 6.32 ± 0.07 (10) | 6.07 ± 0.05 (7) | 5.70 ± 0.04 (7) |
Data are mean ± SD for the number n of individual oocytes indicated in brackets. Data for ASIC1a are from Chen et al. (2005) and data for C62, C92, and C98 are from Babini et al. (2002). SSD, steady-state desensitization; Act, activation.
Figure 8.

(A) PcTx1 robustly shifted the steady-state desensitization and activation curves of C166. pH50 of activation was pH 6.07 ± 0.05 (n = 7) in the absence of PcTx1 (open squares) and pH 6.40 ± 0.06 n = 5, in the presence of 100 nM PcTx1 (closed squares). pH50 of steady-state desensitization was pH 6.90 ± 0.01 (n = 5) in the absence of PcTx1 (open circles) and pH 7.17 ± 0.01 (n = 5) in the presence of 100 nM PcTx1 (closed circles). Solid lines are fits of the mean values of each data point to the Hill function (Eq. 1).(B) Concentration–response relationship for inhibition of C166 currents by PcTx1. Different concentrations of the toxin were applied for 120 s during the conditioning period with pH 7.1. The line represents a fit to the Hill equation (IC50 = 3.4 nM; n = 8–10).
Chimeric Channels Reveal Partially Liganded Open Channels
Interestingly, PcTx1 clearly also opened chimeras C62, C92, C98, and C166 at the conditioning pH in this set of experiments, in addition to inhibit currents evoked by the acidic pulse (Fig. 7). This result suggests an unusually high PcTx1 affinity for the open state of chimeras C62, C92, C98, and C166. We tested this for chimera C92, which was most strongly opened by PcTx1. The apparent affinity for PcTx1, determined by coapplying different PcTx1 concentrations with pH 7.05, was indeed about 10-fold higher (EC50 = 11 nM; Fig. 9 A) than for ASIC1a or ASIC1b. As can be seen from Fig. 7, about 20 s after PcTx1 application, channels in the open state were in equilibrium with channels in the desensitized state, with up to 50% of the channels in the open state. Most likely, PcTx1 transferred part of the channels to the open and part of them to the desensitized state. Application of pH 5.0 immediately after PcTx1 did not open further channels, indicating that indeed no significant fraction of channels remained in the closed state. As mentioned before, such stabilization by PcTx1 of the open state with respect to the desensitized state would predict a slowing of the desensitization by PcTx1. Indeed, C92 currents elicited by coapplication of pH 7.05 with 100 nM PcTx1 decayed slowly with a time constant of 29.9 ± 8.4 s (n = 6; Fig. 9 B).
Figure 9.

(A) Concentration–response relationship for activation of chimera C92 by PcTx1. Different concentrations of the toxin were coapplied with pH 7.05 for 40 s (n = 5–6). The line represents a fit to the Hill equation (EC50 = 11 nM). (B) PcTx1 robustly opened chimera C92. Left, 100 nM PcTx1 was coapplied with pH 7.05 (n = 6). pH 7.05 alone did not open C92. Note the rebound of the current amplitude after returning to pH 7.4. Right, bars representing time constants of the rising and of the decay phase of the current.
Interestingly, as illustrated in the inset of Fig. 7, pH 5.0 quickly reduced the current amplitude and after returning to pH 7.4, current amplitude increased again. Such an unusual rebound of the current amplitude can also be seen in Fig. 9 B after returning to pH 7.4. This decrease of the current amplitude by low pH cannot be explained by the reaction Scheme 1 (see INTRODUCTION) and the fact that more than one H+ is needed for full opening of ASICs (Immke and McCleskey, 2003) has to be taken into account. Extending Scheme 1 to accommodate multiple H+ binding steps leads to the following scheme:According to Scheme 2, which is similar to a Monod-Wyman-Changeux model for cooperativity in allosteric proteins (Monod et al., 1965), there are five closed and open states, with different amounts of the ligand, H+, bound. From the fully liganded states, channels could reach also the desensitized state. Binding of H+ would be highly cooperative, and transition from partially liganded closed states to open and desensitized states would be very infrequent. Thus, usually, channels would open almost exclusively from the fully liganded closed state (black lines). Under these conditions, the more complex Scheme 2 can be condensed to the more simple Scheme 1 (see INTRODUCTION). Our finding that PcTx1 has an unusually high affinity to the open state of chimera C92 predicts that for this chimera, a significant fraction of channels opens also from partially liganded closed states. The decrease of the current amplitude by application of pH 5.0 to open C92 channel is then readily explained by the binding of additional H+ to these partially liganded open channels. From the fully liganded open state, these channels would then quickly desensitize. After washout of pH 5.0, with some PcTx1 still present, channels would again equilibrate between open states and the desensitized state, leading to the rebound of current. Thus, the effect of PxTc1 on chimera C92 reveals the existence of partially liganded open states, supporting an allosteric reaction scheme, where ligand binding and channel opening are coupled but distinct. Although we cannot exclude more complicated reaction schemes that take into account desensitization from partially liganded open or closed states, Scheme 2 most easily explains our data.
(SCHEME 1).

(SCHEME 2).

DISCUSSION
Our data show that PcTx1 does not interact specifically with ASIC1a but that it also interacts with ASIC1b. However, ASIC1b is not strongly inhibited by PcTx1 but rather its opening is promoted. Therefore, interaction with ASIC1b shows that facilitated binding of H+, an increase in the “true” affinity for H+, cannot account for the effect of PcTx1 on ASIC1. Otherwise, according to Scheme 1, one would expect a similar shift of the steady-state desensitization curve and the activation curve. This indicates that PcTx1 binds with different affinities to different states of the channel; PcTx1 binding is state dependent. Although our results do not allow us to directly compare the affinity of PcTx1 for different states of the channels, they suggest that, in the case of ASIC1b, the toxin binds with highest affinity to the open state, and in the case of ASIC1a, it binds more tightly to the desensitized and open states. The rationale behind this conclusion is as follows. First, the apparent affinity of PcTx1 for the open state is comparable for ASIC1a and ASIC1b; second, the apparent affinity for the desensitized state is much higher for ASIC1a than for ASIC1b; third, desensitization of ASIC1a was not much affected by PcTx1, suggesting that the equilibrium between these two states was not strongly affected; and fourth, desensitization of ASIC1b was slowed down, suggesting that PcTx1 stabilized the open state of ASIC1b relative to the desensitized state. The last interpretation is supported by the strong shift of the equilibrium between open and desensitized states for chimera C92 (Fig. 6), which was characterized by the highest apparent affinity of PcTx1 for the open state.
State dependence of binding of a modifier of channel gating is not surprising because usually channels undergo quite extensive conformational changes upon gating. To turn this argument around, state dependence of PcTx1 binding indicates that ASICs indeed undergo conformational changes upon gating, a view that is not easily compatible with more simple models of ASIC gating (Immke and McCleskey, 2003).
Similar to the inhibition of ASIC1 by PcTx1, tight binding to the inactivated state of voltage-gated Na+ channels is responsible for the inhibition of these channels by local anesthetics, according to the modulated receptor model (Hille, 1977). Block by local anesthetics is relieved by hyperpolarization (Hille, 1977), much like inhibition by PcTx1 is relieved by increasing the pH. Although local anesthetics bind within and occlude the ion pore, kinetic modeling has shown that their effect is best explained assuming that they also affect channel gating allosterically (Balser et al., 1996). Similarly, inhibition of voltage-gated Cl− channels by 2-(p-chlorophenoxy)butyric acid (CPB), is state dependent (Pusch et al., 2001). CPB binds more tightly to the closed state of the channel and inhibition vanishes at depolarized potentials (Pusch et al., 2001). Inhibition of voltage-gated Ca2+ channels by dihydropyridines (DHPs) also has many parallels to the inhibition of ASICs by PcTx1. Nitrendipine, for example, binds more tightly to the inactive state than to the closed state of the channel (Bean, 1984). Moreover, the inhibition by DHPs is antagonized by extracellular Ca2+ (Kass and Krafte, 1987), much as the inhibition by PcTx1 is antagonized by extracellular Ca2+ (Chen et al., 2005). Strikingly, some DHPs (e.g., BAY K 8644) act as agonists rather than antagonists of Ca2+ channels (Hess et al., 1984). Similarly, PcTx1 acts on ASIC1b also like an agonist rather than an antagonist. DHPs potentiate the Ca2+ current and shift the activation curve to more negative potentials (Sanguinetti et al., 1986), much like PcTx1 shifts the ASIC1 activation curve to higher pH values. Again, kinetic modeling has shown that DHP agonists affect channel gating allosterically (Marks and Jones, 1992). Moreover, it has been proposed that DHP agonists allow open states when not all the voltage sensors have moved upon depolarization (Marks and Jones, 1992), similar to our interpretation that, for some chimeras, PcTx1 allows open states when not all subunits have ligand bound. Given the high similarity to the interaction of local anesthetics with Na+ channels and of DHPs with Ca2+ channels, we think that PcTx1 most likely is an allosteric modifier of ASIC gating that binds with different affinities to different states of the channel. In this context, it is worthwhile to remember that PcTx1 is structurally related to other spider toxins (Escoubas et al., 2003) that act as gating modifiers of voltage-gated channels (Swartz and MacKinnon, 1997).
The rates of onset of PcTx1 inhibition and of recovery from PcTx1 inhibition are rather large (Chen et al., 2005). We have previously explained this slow process by a membrane-access mechanism for PcTx1 (Chen et al., 2005). Therefore, even after washout of PcTx1, PcTx1 is probably still present in rather high concentrations within the membrane. We can therefore not exclude that the on- and off-reactions of PcTx1 occur preferentially from the open or, in the case of ASIC1a, desensitized state of the channel, even though PcTx1 was applied to the closed state of the channel.
We used a series of chimeras between ASIC1a and ASIC1b to identify regions of the two splice variants that determine their differential behavior to PcTx1. We identified a small region of only 19 amino acids from ASIC1a (amino acids 167–185) that determines a high affinity of PcTx1 for the desensitized state of ASIC1. This result is in agreement with a very recent study identifying amino acids 157–185 of ASIC1a as conferring a high affinity for PcTx1 (Salinas et al., 2006). In this study, affinity had been measured with a binding assay using a radiolabeled toxin. Binding was performed at pH 7.25 and, therefore, the affinity that was determined most likely corresponds to the affinity for the desensitized state. Although the authors of this study proposed that amino acids 157–185 constitute part of the PcTx1 binding site (Salinas et al., 2006), it cannot be excluded that this region allosterically determines the affinity of the desensitized state for PcTx1.
Acknowledgments
We thank M. Pusch for critical reading of the manuscript.
This work was supported by a grant of the Deutsche Forschungsgemeinschaft to S. Gründer (GR1771/3-3).
Olaf S. Andersen served as editor.
Abbreviations used in this paper: ASIC, acid-sensing ion channel; DHP, dihydropyridine; PcTx1, psalmotoxin 1.
References
- Babini, E., M. Paukert, H.S. Geisler, and S. Gründer. 2002. Alternative splicing and interaction with di- and polyvalent cations control the dynamic range of acid-sensing ion channel 1 (ASIC1). J. Biol. Chem. 277:41597–41603. [DOI] [PubMed] [Google Scholar]
- Balser, J.R., H.B. Nuss, D.W. Orias, D.C. Johns, E. Marban, G.F. Tomaselli, and J.H. Lawrence. 1996. Local anesthetics as effectors of allosteric gating. Lidocaine effects on inactivation- deficient rat skeletal muscle Na channels. J. Clin. Invest. 98:2874–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron, A., R. Waldmann, and M. Lazdunski. 2002. ASIC-like, proton- activated currents in rat hippocampal neurons. J. Physiol. 539:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean, B.P. 1984. Nitrendipine block of cardiac calcium channels: high-affinity binding to the inactivated state. Proc. Natl. Acad. Sci. USA. 81:6388–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, C.J., J. Xie, J.A. Wemmie, M.P. Price, J.M. Henss, M.J. Welsh, and P.M. Snyder. 2002. Heteromultimers of DEG/ENaC subunits form H+-gated channels in mouse sensory neurons. Proc. Natl. Acad. Sci. USA. 99:2338–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bässler, E.L., T.J. Ngo-Anh, H.S. Geisler, J.P. Ruppersberg, and S. Gründer. 2001. Molecular and functional characterization of acid-sensing ion channel (ASIC) 1b. J. Biol. Chem. 276:33782–33787. [DOI] [PubMed] [Google Scholar]
- Chen, C.C., S. England, A.N. Akopian, and J.N. Wood. 1998. A sensory neuron-specific, proton-gated ion channel. Proc. Natl. Acad. Sci. USA. 95:10240–10245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.C., A. Zimmer, W.H. Sun, J. Hall, and M.J. Brownstein. 2002. A role for ASIC3 in the modulation of high-intensity pain stimuli. Proc. Natl. Acad. Sci. USA. 99:8992–8997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X., H. Kalbacher, and S. Gründer. 2005. The tarantula toxin psalmotoxin 1 inhibits acid-sensing ion channel (ASIC) 1a by increasing its apparent H+ affinity. J. Gen. Physiol. 126:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun, D. 1998. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacol. 125:924–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escoubas, P., J.R. De Weille, A. Lecoq, S. Diochot, R. Waldmann, G. Champigny, D. Moinier, A. Menez, and M. Lazdunski. 2000. Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. J. Biol. Chem. 275:25116–25121. [DOI] [PubMed] [Google Scholar]
- Escoubas, P., C. Bernard, G. Lambeau, M. Lazdunski, and H. Darbon. 2003. Recombinant production and solution structure of PcTx1, the specific peptide inhibitor of ASIC1a proton-gated cation channels. Protein Sci. 12:1332–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, P., J.B. Lansman, and R.W. Tsien. 1984. Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature. 311:538–544. [DOI] [PubMed] [Google Scholar]
- Hille, B. 1977. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 69:497–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immke, D.C., and E.W. McCleskey. 2003. Protons open acid-sensing ion channels by catalyzing relief of Ca2+ blockade. Neuron. 37:75–84. [DOI] [PubMed] [Google Scholar]
- Kass, R.S., and D.S. Krafte. 1987. Negative surface charge density near heart calcium channels. Relevance to block by dihydropyridines. J. Gen. Physiol. 89:629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishtal, O. 2003. The ASICs: signaling molecules? Modulators? Trends Neurosci. 26:477–483. [DOI] [PubMed] [Google Scholar]
- Madeja, M., U. Musshoff, and E.J. Speckmann. 1995. Improvement and testing of a concentration-clamp system for oocytes of Xenopus laevis. J. Neurosci. Methods. 63:211–213. [DOI] [PubMed] [Google Scholar]
- Mamet, J., A. Baron, M. Lazdunski, and N. Voilley. 2002. Proinflammatory mediators, stimulators of sensory neuron excitability via the expression of acid-sensing ion channels. J. Neurosci. 22:10662–10670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks, T.N., and S.W. Jones. 1992. Calcium currents in the A7r5 smooth muscle-derived cell line. An allosteric model for calcium channel activation and dihydropyridine agonist action. J. Gen. Physiol. 99:367–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod, J., J. Wyman, and J.P. Changeux. 1965. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12:88–118. [DOI] [PubMed] [Google Scholar]
- Paukert, M., S. Sidi, C. Russell, M. Siba, S.W. Wilson, T. Nicolson, and S. Gründer. 2004. A family of acid-sensing ion channels from the zebrafish: widespread expression in the central nervous system suggests a conserved role in neuronal communication. J. Biol. Chem. 279:18783–18791. [DOI] [PubMed] [Google Scholar]
- Phillips, L.R., M. Milescu, Y. Li-Smerin, J.A. Mindell, J.I. Kim, and K.J. Swartz. 2005. Voltage-sensor activation with a tarantula toxin as cargo. Nature. 436:857–860. [DOI] [PubMed] [Google Scholar]
- Price, M.P., G.R. Lewin, S.L. McIlwrath, C. Cheng, J. Xie, P.A. Heppenstall, C.L. Stucky, A.G. Mannsfeldt, T.J. Brennan, H.A. Drummond, et al. 2000. The mammalian sodium channel BNC1 is required for normal touch sensation. Nature. 407:1007–1011. [DOI] [PubMed] [Google Scholar]
- Pusch, M., A. Accardi, A. Liantonio, L. Ferrera, A. De Luca, D.C. Camerino, and F. Conti. 2001. Mechanism of block of single protopores of the Torpedo chloride channel ClC-0 by 2-(p-chlorophenoxy) butyric acid (CPB). J. Gen. Physiol. 118:45–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas, M., L.D. Rash, A. Baron, G. Lambeau, P. Escoubas, and M. Lazdunski. 2006. The receptor site of the spider toxin PcTx1 on the proton-gated cation channel ASIC1a. J. Physiol. 570:339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti, M.C., D.S. Krafte, and R.S. Kass. 1986. Voltage-dependent modulation of Ca channel current in heart cells by Bay K8644. J. Gen. Physiol. 88:369–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saugstad, J.A., J.A. Roberts, J. Dong, S. Zeitouni, and R.J. Evans. 2004. Analysis of the membrane topology of the acid-sensing ion channel 2a. J. Biol. Chem. 279:55514–55519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland, S.P., C.J. Benson, J.P. Adelman, and E.W. McCleskey. 2001. Acid-sensing ion channel 3 matches the acid-gated current in cardiac ischemia-sensing neurons. Proc. Natl. Acad. Sci. USA. 98:711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz, K.J., and R. MacKinnon. 1997. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron. 18:665–673. [DOI] [PubMed] [Google Scholar]
- Ugawa, S., T. Yamamoto, T. Ueda, Y. Ishida, A. Inagaki, M. Nishigaki, S. Shimada, Y. Minami, W. Guo, Y. Saishin, et al. 2003. Amiloride-insensitive currents of the acid-sensing ion channel-2a (ASIC2a)/ASIC2b heteromeric sour-taste receptor channel. J. Neurosci. 23:3616–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voilley, N., J. de Weille, J. Mamet, and M. Lazdunski. 2001. Nonsteroid anti-inflammatory drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors. J. Neurosci. 21:8026–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann, R., and M. Lazdunski. 1998. H+-gated cation channels: neuronal acid sensors in the NaC/DEG family of ion channels. Curr. Opin. Neurobiol. 8:418–424. [DOI] [PubMed] [Google Scholar]
- Waldmann, R., F. Bassilana, J. de Weille, G. Champigny, C. Heurteaux, and M. Lazdunski. 1997. Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J. Biol. Chem. 272:20975–20978. [DOI] [PubMed] [Google Scholar]
- Wemmie, J.A., J. Chen, C.C. Askwith, A.M. Hruska-Hageman, M.P. Price, B.C. Nolan, P.G. Yoder, E. Lamani, T. Hoshi, J.H. Freeman Jr., and M.J. Welsh. 2002. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 34:463–477. [DOI] [PubMed] [Google Scholar]
- Wemmie, J.A., C.C. Askwith, E. Lamani, M.D. Cassell, J.H. Freeman Jr., M.J. Welsh, A.S. Leonard, O. Yermolaieva, A. Hruska-Hageman, M.P. Price, et al. 2003. Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J. Neurosci. 23:5496–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, J., M.P. Price, A.L. Berger, and M.J. Welsh. 2002. DRASIC contributes to pH-gated currents in large dorsal root ganglion sensory neurons by forming heteromultimeric channels. J. Neurophysiol. 87:2835–2843. [DOI] [PubMed] [Google Scholar]