

Abstract
In arterial smooth muscle, single or small clusters of Ca2+ channels operate in a high probability mode, creating sites of nearly continual Ca2+ influx (called “persistent Ca2+ sparklet” sites). Persistent Ca2+ sparklet activity varies regionally within any given cell. At present, the molecular identity of the Ca2+ channels underlying Ca2+ sparklets and the mechanisms that give rise to their spatial heterogeneity remain unclear. Here, we used total internal reflection fluorescence (TIRF) microscopy to directly investigate these issues. We found that tsA-201 cells expressing L-type Cavα1.2 channels recapitulated the general features of Ca2+ sparklets in cerebral arterial myocytes, including amplitude of quantal event, voltage dependencies, gating modalities, and pharmacology. Furthermore, PKCα activity was required for basal persistent Ca2+ sparklet activity in arterial myocytes and tsA-201 cells. In arterial myocytes, inhibition of protein phosphatase 2A (PP2A) and 2B (PP2B; calcineurin) increased Ca2+ influx by evoking new persistent Ca2+ sparklet sites and by increasing the activity of previously active sites. The actions of PP2A and PP2B inhibition on Ca2+ sparklets required PKC activity, indicating that these phosphatases opposed PKC-mediated phosphorylation. Together, these data unequivocally demonstrate that persistent Ca2+ sparklet activity is a fundamental property of L-type Ca2+ channels when associated with PKC. Our findings support a novel model in which the gating modality of L-type Ca2+ channels vary regionally within a cell depending on the relative activities of nearby PKCα, PP2A, and PP2B.
INTRODUCTION
During the myogenic response (Bayliss, 1902), smooth muscle lining the walls of resistance arteries respond to increased intravascular pressure by undergoing gradual depolarization, thus increasing the open probability of dihydropyridine-sensitive, voltage-gated L-type Ca2+ channels located in the sarcolemma of arterial smooth muscle cells (Harder et al., 1987; Fleischmann et al., 1994; Rubart et al., 1996; Knot and Nelson, 1998). Increased opening of L-type Ca2+ channels causes greater Ca2+ influx that culminates in a global rise in intracellular Ca2+ ([Ca2+]i) and arterial constriction. Accordingly, by regulating the activity of L-type Ca2+ channels, smooth muscle cells are able to modulate [Ca2+]i, arterial diameter, and therefore blood flow.
We recently observed single and small clusters of seemingly coupled Ca2+ channels (presumably L-type) operating in a high activity gating mode that created local areas of nearly continual Ca2+ influx termed “persistent Ca2+ sparklet” sites (Navedo et al., 2005). On the basis of these findings it was proposed that steady-state Ca2+ influx in arterial smooth muscle occurs through persistent Ca2+ sparklet sites in combination with random, infrequent openings of solitary L-type Ca2+ channels (Fleischmann et al., 1994; Rubart et al., 1996). At present, however, this “persistent Ca2+ sparklet model” remains largely untested.
The goal of this study was to address four fundamental, yet unresolved, issues raised by this provocative model. First, we investigated the molecular identity of the channels that underlie persistent Ca2+ sparklets. This is of particular importance because the conclusion that persistent Ca2+ sparklets are produced by the opening of single, or small clusters, of L-type Ca2+ channels was based largely on pharmacological evidence (i.e., sensitivity to dihydropyridines), which is equivocal. Second, we identified the minimal molecular components required for persistent Ca2+ sparklet activity. Third, we investigated the mechanisms underlying dynamic, regional variations in Ca2+ sparklet activity. Fourth, we examined the molecular identities of the signaling molecules involved in the regional modulation of persistent Ca2+ activity.
Our data indicate that expression of PKCα and L-type Ca2+ (Cavα1.2) channels was sufficient to reproduce the basic features of persistent Ca2+ sparklet activity in a heterologous expression system. This provides the first direct demonstration that Cavα1.2 channels underlie persistent Ca2+ sparklets in smooth muscle. Accordingly, our data indicate, for the first time, that persistent Ca2+ sparklet activity is a fundamental feature of L-type Ca2+ channels (with PKCα), which suggests the intriguing possibility that persistent Ca2+ sparklets may be a general mechanism underlying steady-state Ca2+ entry in excitable cells. Finally, our data support the novel concept that subcellular compartmentalization of Ca2+ influx via L-type Ca2+ channels is determined by the local balance between PKCα and opposing phosphatase (protein phosphate 2A and 2B) activities.
MATERIALS AND METHODS
Isolation of Arterial Myocytes
Rats (Sprague-Dawley; ≈250 g) as well as wild type and PKCα knockout mice (≈25 g) (Braz et al., 2004) were euthanized in strict accordance to the regulations of the University of Washington Institutional Animal Care and Use Committee using a lethal dose of sodium pentobarbital (100 mg/kg, intraperitoneally). Myocytes were dissociated from cerebral arteries using standard enzymatic techniques described in detail elsewhere (Amberg and Santana, 2003). After dissociation, cells were maintained in a nominally Ca2+-free Ringer's solution until used. Thapsigargin (1 μM) was included in all solutions to eliminate Ca2+ release from intracellular stores during experimentation.
Heterologous Expression of Cavα1.2 and PKCα in tsA-201 Cells
Cultures of tsA-201 cells were maintained in Dulbecco's modified essential Media supplemented with 10% fetal bovine serum, l-glutamine (2 mM), and a 1% streptomycin and penicillin solution. Cells were transiently transfected with the pcDNA clones of Cavα1.2, Cavβ3, Cavα2δ1 (a gift from D. Lipscombe, Brown University, Providence, RI), and the enhanced green fluorescent protein using Lipofectamine 2000. In some experiments, tsA-201 cells were transfected with Cavα1.2 and accessory subunits as well as PKCα tagged with the enhanced green fluorescent protein (provided by J. Exton, Vanderbilt University, Nashville, TN). Successfully transfected cells were identified on the basis of enhanced green fluorescent protein fluorescence.
Electrophysiology
We used the conventional whole-cell patch-clamp technique to control membrane voltage using an Axopatch 200B amplifier. During experiments, cells were continuously superfused with a solution with the following constituents (in mM): 140 NMDG, 5 CsCl, 1 MgCl2, 10 glucose, 10 HEPES, and 2 or 20 CaCl2 adjusted to pH 7.4. NMDG concentration was 120 mM when 20 mM CaCl2 was used. Pipettes were filled with a solution composed of (in mM) 87 Cs-aspartate, 20 CsCl, 1 MgCl2, 5 MgATP, 10 HEPES, 10 EGTA, and 0.2 Fluo-5F or Rhod-2 adjusted to pH 7.2 with CsOH. A voltage error of 10 mV attributable to the liquid junction potential was corrected for. In some experiments, Ca2+ currents were recorded and later analyzed using pCLAMP 9.0 software. In these experiments, currents were sampled at 20 kHz and low pass filtered at 2 kHz. All experiments were performed at room temperature (22–25°C).
Total Internal Reflection Fluorescence (TIRF) Microscopy
Ca2+ sparklets were recorded using a through-the-lens TIRF microscope built around an inverted Olympus IX-70 microscope equipped with an Olympus PlanApo (60X, numerical aperture = 1.45) oil-immersion lens and an XR Mega 10 intensified CCD camera (Solamere Technology Group). To monitor [Ca2+]i, cells were loaded with the calcium indicators Fluo-5F or Rhod-2. Rhod-2 was used in all experiments in which the enhanced green fluorescent protein was expressed. Excitation of Fluo-5F and Rhod-2 was achieved with the 488- or 568-nm line of an argon or krypton laser, respectively (Dynamic Lasers). Excitation and emission light was separated with the appropriate set of filters. Images were acquired at 30–90 Hz.
Background-subtracted fluorescence signals were converted to concentration units using the “Fmax” equation (Maravall et al., 2000):
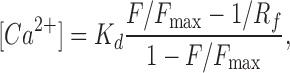 |
where F is fluorescence, Fmax is the fluorescence intensity of Fluo-5N or Rhod-2 in the presence of a saturating free Ca2+ concentration, Kd is the dissociation constant of the fluorescence indicator used (Fluo-5N = 1100 nM; Rhod-2 = 600 nM), and Rf (Fluo-5N = 210; Rhod-2 = 150) is this indicator's Fmax/Fmin. Fmin is the fluorescence intensity of Fluo-5N or Rhod-2 in a solution where the Ca2+ concentration is 0. Kd and Rf values for Fluo-5N and Rhod-2 were determined in vitro using standard methods (Woodruff et al., 2002). Fmax was determined at the end of the experiments by exposing cells to a solution to which the Ca2+ ionophore ionomycin (10 μM) and 20 mM external Ca2+ had been added.
Ca2+ sparklets were detected and defined for analysis using an automated algorithm written in IDL language. Ca2+ sparklets had an amplitude equal to or larger than the mean basal [Ca2+]i plus three times its standard deviation. For a [Ca2+]i elevation to be considered a sparklet, a grid of 3 × 3 contiguous pixels had to have a [Ca2+]i value at or above the amplitude threshold. These detection criteria for Ca2+ sparklets are similar to those used by other investigators (Cheng et al., 1999; Demuro and Parker, 2004, 2005).
By simultaneously recording single Ca2+ channel currents and Ca2+ sparklets in arterial myocytes, we recently reported that at −70 mV and with 20 mM external Ca2+, a single Ca2+ channel current of ≈0.5 pA produced a Ca2+ sparklet of ≈37 nM (Navedo et al., 2005). As shown in Fig. S1 (see online supplemental material, available at http://www.jgp.org/cgi/content/full/jgp200609519/DC1), an “all-points” histogram from representative [Ca2+]i records obtained from arterial myocytes had multiple, clearly separated peaks and could be fit with the following multi Gaussian function:
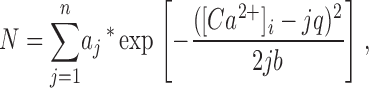 |
where a and b are constants and [Ca2+]i and q are intracellular Ca2+ and the quantal unit of Ca2+ influx, respectively. Using this analysis, and consistent with our previous study, we obtained a q value of 34 nM in the all-points histogram, a value that is similar to that obtained previously (see below). This analysis provides further support to the hypothesis that Ca2+ sparklets are quantal in nature and that the size of Ca2+ sparklet depends on the number of quanta activated.
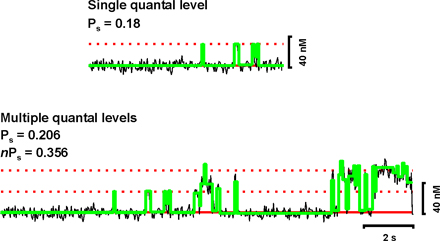
Analogous to single-channel data analysis, we determined the activity of Ca2+ sparklets by calculating the nP s of each sparklet site, where n is the number of quantal levels and P s is the probability that a quantal Ca2+ sparklet event is active. To do this, we used the single channel analysis module of pCLAMP 9.0. First, [Ca2+]i from previously identified sparklet sites were imported into this program and a baseline defined. To estimate nPs, Ca2+ sparklet events were detected using pCLAMP's “threshold detection analysis” using no duration constraints and a unitary Ca2+ elevation of 38 nM as a starting point for event detection (note that the amplitude of the unitary event was not fixed). Traces were then fitted with these initial parameters. Each one of the events detected with this analysis were then cross-referenced with the original image stack to verify that they met the amplitude and spatial criteria described above. Only Ca2+ influx events that met the spatial and amplitude criteria were used to estimate nPs for each experimental condition. An example of this type of analysis is shown in the online supplemental material (Fig. S2).
Amplitude histograms were constructed using the amplitudes of the detected Ca2+ sparklet events. The resulting histogram was fitted with the multicomponent Gaussian function described above, which allowed us to obtain an estimate of the amplitude of quantal Ca2+ sparklets under varied experimental conditions. It is important to note that the observation of multiple peaks and similar q values in our all-points and event histograms (e.g., Figs. 1–3 and Fig. S1) indicate that the use of the event histogram for our quantal analysis is appropriate.
Figure 1.

Ca2+ sparklets in a heterologous expression system. (A) Ca2+ current evoked by a depolarization from −70 to +20 mV in a representative nontransfected (control) cell and cell expressing Cavα1.2. (B) Image of a cell transfected with Cavα1.2 only. The traces below the images show the time course of [Ca2+]i in the sites marked by the green circle before and after the application of 500 nM Bay-K 8644. (C) Amplitude histogram of Ca2+ sparklets in tsA-201 cells expressing Cavα1.2 before and after Bay-K 8644 treatment. The black and red lines are the best fit to the control (q = 37 nM) and Bay-K 8644 (q = 37 nM) data, respectively, with the multicomponent Gaussian function described in the Materials and Methods section. (D) Bar plot of the mean ± SEM of the nPs before and after Bay-K 8644 application.
Figure 3.

Cavα1.2 and PKCα are required for persistent Ca2+ sparklet activity. (A) Image of tsA-201 cells expressing PKCα and Cavα1.2 with silent, low, and high activity Ca2+ sparklet sites. The traces below the images show the time course of [Ca2+]i in the sites marked by the green circle before and after the application of 500 nM Bay-K 8644. (B) Amplitude histogram of Ca2+ sparklets in tsA-201 cells expressing Cavα1.2 and PKCα before and after Bay-K 8644 treatment. The black and red lines are the best fit to the control (q = 38 nM) and Bay-K 8644 (q = 37 nM) data, respectively, with a multicomponent Gaussian function. (E) Bar plot of the mean ± SEM nPs under control conditions and after application of Bay-K 8644 or nifedipine.
Figure 2.

Ca2+ sparklets in tsA-201 cells expressing Cavα1.2 and PKCα. (A) Ca2+ current evoked by a depolarization from −70 to +20 mV in representative cells expressing PKCα or PKCα and Cavα1.2. (B) Time course of [Ca2+]i in Ca2+ sparklet site before and after the application of 10 μM nifedipine. (C) Time course of [Ca2+]i in Ca2+ sparklet site under control conditions and after perfusion with a solution without Ca2+. (D) Amplitude histogram of Ca2+ sparklets in tsA-201 cells expressing Cavα1.2 and PKCα. The black line is the best fit to the data (q = 36 nM) with the multicomponent Gaussian function described in the Materials and Methods section. (E) Voltage dependence of quantal Ca2+ sparklet amplitude in tsA-201 cells. Solid line is a linear fit to the data. The inset shows representative quantal Ca2+ sparklets recorded from tsA-201 cells expressing Cavα1.2 channels at −90 and −50 mV. The dotted lines mark the quantal level for Ca2+ sparklets at −90 and −50 mV.
As previously reported (Navedo et al., 2005), Ca2+ sparklet activity was bimodal in arterial myocytes and tsA-201 cells expressing Cavα1.2 and PKCα, with sites of low activity (nPs = 0.07 ± 0.01) and sites of high activity (nPs = 0.73 ± 0.7). Based on this behavior, we grouped Ca2+ sparklets into three categories; silent (by default has an nPs of 0), low (nPs between 0 and 0.2), and high (nPs higher than 0.2). Note that a silent Ca2+ sparklet site represents a site that is ordinarily inactive, but can be activated by an agonist.
Chemicals and Statistics
All PKC inhibitors were acquired from Calbiochem. Cell culture media and supplements were from Life Technologies. Lipofectamine 2000 was purchased from Invitrogen; all other chemicals were from Sigma-Aldrich. Data are presented as mean ± SEM. Two-sample comparisons were made using Student's t test. For datasets containing more than two groups, an ANOVA was used. A P value of <0.05 was considered significant. The asterisk (*) symbol used in the figures denotes a significant difference between groups.
Online Supplemental Material
The online supplemental material (Figs. S1 and S2, available at http://www.jgp.org/cgi/content/full/jgp.200609519/DC1) provides an example of our Ca2+ sparklet activity analysis.
RESULTS
Cavα1.2 Channels Produce Ca2+ Sparklets
All experiments in this study were performed in the presence of the SERCA pump inhibitor thapsigargin (1 μM) to eliminate Ca2+ release from intracellular stores. TIRF microscopy was used to image Ca2+ sparklet events with high spatial and temporal resolution. To increase the driving force for Ca2+ entry, experiments were performed in the presence of 20 mM external Ca2+ while cells were held at −70 mV. Analogous to single-channel data analysis, we determined the activity of Ca2+ sparklets by calculating the nP s of each sparklet site as described in the Materials and Methods.
First, we tested the hypothesis that L-type Ca2+ channels underlie Ca2+ sparklet activity in arterial myocytes. If L-type Ca2+ channels underlie Ca2+ sparklets, then Ca2+ sparklets should be observed in a heterologous expression system expressing these channels. Thus, we examined Ca2+ sparklet activity in tsA-201 cells expressing the L-type Ca2+ channel pore-forming Cavα1.2 and accessory Cavβ3 and Cavα2δ1 subunits. Cavα1.2 channels are the predominant L-type Ca2+ channels expressed in arterial smooth muscle (Koch et al., 1990; Sinnegger-Brauns et al., 2004). To prevent any potential effect of endogenous PKC activity on Cavα1.2 function, these experiments were performed using the PKC inhibitory peptide (100 μm) in the pipette solution.
To verify that tsA-201 cells transfected with Cavα1.2, Cavβ3, and Cavα2δ1 expressed functional channels, we depolarized these cells from −70 to +20 mV. As shown in Fig. 1 A, this protocol evoked robust Ca2+ currents in these cells. Ca2+ currents were not detected in nontransfected cells (n = 25). In addition, in tsA-201 cells expressing Cavα1.2, Ca2+ sparklets were observed at −70 mV (Fig. 1 B). Ca2+ sparklets were never observed in nontransfected cells (n = 25). As shown in the amplitude histogram in Fig. 1 C, the distribution of Ca2+ sparklet amplitudes in tsA-201 cells was modal. Indeed, the data could be fit with a multicomponent Gaussian function with a quantal unit of Ca2+ elevation of 37.9 nM (χ2 = 0.76). Interestingly, the amplitude of quantal Ca2+ sparklets in these cells is similar to that reported in arterial myocytes (≈38 nM) under identical experimental conditions (Navedo et al., 2005). This analysis suggests that, as with Ca2+ sparklets recorded in arterial myocytes, Ca2+ entry via heterologuosly expressed Cavα1.2 channels is quantal in nature and that the size of Ca2+ sparklet depends on the number of quanta activated.
Detailed analysis revealed that the activity of Ca2+ sparklets sites in these cells had a mean nPs value of 0.05 ± 0.02 (n = 9) (Fig. 1 D). This nPs value is similar (P > 0.05) to that of low activity Ca2+ sparklet sites reported in rat arterial myocytes (see also new data from rat and mouse myocytes presented below) (Navedo et al., 2005). Indeed, it is important to note that high activity, persistent Ca2+ sparklet sites (nPs > 0.2) were never observed in tsA-201 cells expressing Cavα1.2 only.
In rat arterial myocytes, Bay-K 8644 increases Ca2+ influx, at least in part, by increasing persistent Ca2+ sparklet activity (Navedo et al., 2005). However, the mechanisms by which Bay-K 8644 increases persistent Ca2+ sparklet activity are unclear. We investigated whether Bay-K 8644 could induce persistent Ca2+ sparklet activity in tsA-201 cells expressing Cavα1.2 (Fig. 1, B–D). Application of 500 nM Bay-K 8644 recruited new Ca2+ sparklet sites and increased the activity of previously active sites. Indeed, Bay-K 8644 increased the average Ca2+ sparklet activity (i.e., nPs) from 0.05 ± 0.03 to 0.13 ± 0.04 (n = 6, P < 0.05; Fig. 1 D) without increasing the amplitude of quantal Ca2+ sparklets (control = 37.0 nM vs. Bay-K 8644 = 37.6 nM; Fig. 1 C). Interestingly, even in the presence of 500 nM Bay-K 8644 we did not detect high nPs, persistent Ca2+ sparklet sites in tsA-201 cells expressing Cavα1.2 only. These data suggest that although Cavα1.2 channel can produce low activity Ca2+ sparklets sites, expression of these channels alone is not sufficient to produce persistent Ca2+ sparklet activity under control conditions or after Bay-K 8644 treatment.
Cavα1.2 and PKCα Are Required for Persistent Ca2+ Sparklet Activity
We recently observed that PKC activity is required for persistent Ca2+ sparklet activity in rat arterial myocytes (Navedo et al., 2005). Thus, we examined Ca2+ sparklet activity in tsA-201 cells expressing Cavα1.2 channels and PKCα (Fig. 2). We used PKCα in these experiments because this isoform is highly expressed in cerebral artery myocytes (but see below) (Pang et al., 2002; Wickman et al., 2003). Depolarization from the holding potential of −70 mV to +20 mV evoked large Ca2+ currents in tsA-201 expressing Cavα1.2 channels and PKCα (Fig. 2 A). Ca2+ currents were not observed in cells expressing PKCα alone (n = 28).
Accordingly, Ca2+ sparklet sites were frequently observed at −70 mV in tsA-201 cells expressing Cavα1.2 channels and PKCα, but never in cells expressing PKCα alone (n = 28). To provide further support to the hypothesis that Ca2+ sparklets are produced by Ca2+ influx via Cavα1.2 channels and not Ca2+ release from a thapsigargin-insensitive intracellular Ca2+ store in tsA-201 cells, we examined the effects of the L-type Ca2+ channel blocker nifedipine, which blocks sparklets in arterial myocytes (Navedo et al., 2005), and removing external Ca2+ on Ca2+ sparklets. As shown in Fig. 2 B, nifedipine completely eliminated Ca2+ sparklet activity (n = 15). Furthermore, we found that perfusion of an external solution with 0 Ca2+ (without nifedipine) rapidly eliminated Ca2+ sparklet activity in tsA-201 cells expressing Cavα1.2 channels (n = 10; Fig. 2 C). Together with our observation that Ca2+ sparklets are only detected in tsA-201 cells expressing Cavα1.2, these findings support the hypothesis that Ca2+ sparklets in tsA-201 cells are produced by Ca2+ influx events through plasma membrane Cavα1.2 channels.
An amplitude histogram of Ca2+ sparklets in tsA-201 cells expressing Cavα1.2 channels and PKCα is shown in Fig. 2 D. The histogram was fitted (χ2 = 0.84) with a multicomponent Gaussian function with a quantal unit of Ca2+ elevation of 36.0 nM, a value that is similar to that of cells expressing Cavα1.2 alone (see Fig. 1 C above). We also investigated the effects of membrane potential on the amplitude of quantal Ca2+ sparklets in tsA-201 cells expressing Cavα1.2 and PKCα. The amplitude of quantal Ca2+ sparklets at the voltages examined was obtained from the event amplitude histogram of all Ca2+ sparklets using the multi-Gaussian analysis described above. The inset in Fig. 2 E shows two [Ca2+]i records from a representative cell with quantal Ca2+ sparklets at −90 and −50 mV. Note that the amplitude of these Ca2+ sparklets decreased as the driving force for Ca2+ entry was decreased by membrane depolarization from −90 to −50 mV. Indeed, as previously reported in rat arterial myocytes (Navedo et al., 2005), the amplitude of quantal Ca2+ sparklets in tsA-201 cells decreased linearly over this range of potentials (Fig. 2 E), providing further support to the view that Ca2+ sparklets are produced by Ca2+ influx via L-type Cavα1.2 channels in the plasma membrane.
We analyzed the modalities of Ca2+ sparklet activity in tsA-201 cells expressing Cavα1.2 and PKCα (Fig. 3, A and C). Although most of the surface membrane in these cells did not show signs of Ca2+ influx at −70 mV (i.e., nPs = 0), there were sites of low and high Ca2+ sparklet activity (Fig. 2, C and D). Because cells expressing Cavα1.2 alone did not have high nPs sites, the combined nPs value of cells expressing Cavα1.2 and PKCα (0.16 ± 0.03, n = 12) was about threefold higher than in cells expressing Cavα1.2 alone (0.05 ± 0.02, n = 6, P < 0.05). These data suggest that Ca2+ sparklets in tsA-201 expressing Cavα1.2 and PKCα and rat arterial myocytes have similar gating modalities.
The effects of Bay-K 8644 on Ca2+ sparklets in tsA-201 cells expressing Cavα1.2 and PKCα were also investigated (Fig. 3). Application of 500 nM Bay-K 8644 increased the number of Ca2+ sparklet sites threefold (n = 8). Consistent with this, Bay-K 8644 activated quiescent Ca2+ channels in silent sites and increased Ca2+ sparklet activity in low nP s sites (P < 0.05), thus increasing the number of high nPs, persistent Ca2+ sparklets in tsA-201 cells (Fig. 3, A–C). Bay-K 8644 did not increase Ca2+ sparklet activity in high nP s sites (P > 0.05), suggesting maximal channel activity at these sites. Bay-K 8644 increased the number of Ca2+ sparklets of all amplitude levels without altering the value of the quantal event (38.4 nM; Fig. 3 B). Importantly, the actions of Bay-K 8644 on Ca2+ sparklets in these cells are similar to those reported in arterial myocytes (Navedo et al., 2005). Taken together, these data indicate that Ca2+ sparklets in tsA-201 cells expressing Cavα1.2 and PKCα have similar pharmacology, gating modalities, amplitude of quantal event, and voltage dependencies than sparklets in arterial myocytes. This is consistent with the view that Cavα1.2 and PKCα are the minimal molecular components required for persistent Ca2+ sparklet activity under control conditions and after Bay-K 8644 treatment.
Dynamic Modulation of Ca2+ Sparklet Activity Depends on the Relative Activities of PKCα and Opposing Phosphatases
Next, we investigated the mechanisms that underlie dynamic regional variations of persistent Ca2+ sparklet activity in arterial myocytes. We tested the hypothesis that regional differences in Ca2+ sparklet activity result from regional differences in the relative activities of PKC, which we have shown here to be essential for persistent Ca2+ sparklet activity in tsA-201 cells, and nearby opposing phosphatases. To begin, we investigated which of the PKC isoforms expressed in cerebral arterial smooth muscle are required for persistent Ca2+ sparklet activity. Although the experiments described above indicate that expression of PKCα and Cavα1.2 channels is sufficient to reproduce the basic features of Ca2+ sparklets in tsA-201 cells, two recent studies indicate that cerebral artery myocytes express three PKC isoforms: Ca2+-dependent PKCα and β and, to a lesser extent, the Ca2+-independent PKCɛ isoform (Pang et al., 2002; Wickman et al., 2003). We used isoform-specific PKC inhibitors to determine which of these three PKC isoforms influence persistent Ca2+ sparklet activity in cerebral arterial myocytes.
Like the experiments in tsA-201 cells, all experiments with arterial myocytes were performed with solutions containing the SERCA pump inhibitor thapsigargin (1 μM) to eliminate Ca2+ release from intracellular stores. To verify that Ca2+ sparklets were not produced by Ca2+ release from an intracellular store insensitive to thapsigargin, Ca2+ sparklets were recorded in rat arterial myocytes under control conditions (i.e., 20 mM external Ca2+ while the cells were held at −70 mV), in the presence of the dihydropyridine nifedipine and after perfusion of a solution without Ca2+. As shown in Fig. 4 A, Ca2+ sparklets in arterial myocytes were completely abolished by the application of the dihydropyridine nifedipine (10 μM; n = 7). Furthermore, note that Ca2+ sparklets were rapidly abolished by superfusion of a Ca2+-free solution (without nifedipine) (Fig. 4 B). Similar results were obtained in 10 independent experiments. Together, these findings indicate that Ca2+ sparklets are produced by Ca2+ influx via a sarcolemma L-type Ca2+ channel in arterial smooth muscle.
Figure 4.

Basal PKCβ and PKCɛ activity is not required for Ca2+ sparklet activity in arterial smooth muscle. (A) Time course of [Ca2+]i in a Ca2+ sparklet site before and after the application of 10 μM nifedipine. (B) Time course of [Ca2+]i in a Ca2+ sparklet under control conditions and after perfusion of a solution without Ca2+. Time course of [Ca2+]i in a Ca2+ sparklet site before and after the application of (C) 100 nM Gö6976 or (D) 50 nM PKCβi. (E) Bar plot of the mean ± SEM nPs under control conditions and after application of Gö6976 or PKCβi. *, significantly different from control.
Fig. 4 (A and B, top traces) shows the time course of [Ca2+]i in a Ca2+ sparklet site from a representative rat arterial myocyte under control conditions. An amplitude histogram of Ca2+ sparklets recorded under these conditions could be fit with a multi-Gaussian function with a quantal unit of Ca2+ increase of 37.5 nM (not depicted), which is similar to that in tsA-201 cells (see Fig. 1 C and Fig. 2 D above). We found that the majority of the sarcolemma of rat myocytes was optically silent (i.e., nPs = 0). However, we observed foci of low (nPs = 0.06 ± 0.01, n = 75) and high (nPs = 0.7 ± 0.1, n = 58) Ca2+ sparklet activity. The average nPs value of Ca2+ sparklet sites under control conditions was 0.17 ± 0.05 (n = 110). Interestingly, nPs values in rat arterial myocytes and tsA-201 cells (low nPs = 0.05 ± 0.01, high nPs = 0.6 ± 0.01, average nPs = 0.14 ± 0.04) expressing Cavα1.2 and PKCα were similar (P > 0.05).
Application of Gö6976 (100 nM), which selectively inhibits PKCα and PKCβ (Gschwendt et al., 1996), eliminated (i.e., nPs = 0, number of sparklet sites = 0) Ca2+ sparklet activity in rat arterial myocytes (Fig. 4, C–E). This suggests that the Ca2+-dependent PKCα and/or PKCβ isoforms underlie spontaneous persistent Ca2+ sparklet activity in rat arterial myocytes. To distinguish which of these two isoforms underlie spontaneous Ca2+ sparklet activity in these cells, we recorded Ca2+ sparklets before and after the application of a specific PKCβ inhibitor (PKCβi, 50 nM) (Tanaka et al., 2004). In contrast to Gö6976, the averaged Ca2+ sparklet activity did not change after PKCβi application (control nPs = 0.21 ± 0.04, n = 15 vs. PKCβi nPs = 0.29 ± 0.08, n = 15; P > 0.05) (Fig. 4, D and E). In addition, we found that Ca2+ sparklet activity and the number of Ca2+ sparklet sites per cell were not changed by dialysis with a specific PKCɛ inhibitory peptide (PKCɛi) (Johnson et al., 1996) (unpublished data, n = 5 cells, P > 0.05). These data indicate that neither PKCβ nor PKCɛ activity is required for spontaneous Ca2+ sparklet activity in arterial smooth muscle.
By excluding PKCβ and PKCɛ, our data suggest that PKCα is required for Ca2+ sparklet activity in arterial myocytes. To directly test this hypothesis, we examined Ca2+ sparklet activity in mouse wild-type (WT) and PKCα knockout (PKCα−/−) (Braz et al., 2004) arterial myocytes. Fig. 5 (A and C) shows that mouse arterial myocytes produce Ca2+ sparklets. Indeed, it is important to note that Ca2+ sparklets in WT mouse myocytes were similar (P > 0.05) to those in tsA-201 cells and rat arterial myocytes in all parameters examined (i.e., amplitude of the quantal events, 38 nM, and activity modalities). Control mouse-WT myocytes had silent (i.e., nPs = 0) as well as low (nPs = 0.08 ± 0.02, n = 35) and high activity (nPs = 0.80 ± 0.30, n = 15) Ca2+ sparklet sites. Like rat arterial myocytes (Navedo et al., 2005), application of the broad-spectrum PKC activator phorbol 12, 13-dibutyrate (PDBu, 200 nM) increased Ca2+ influx in mouse arterial myocytes by activating new Ca2+ sparklet sites and by increasing the activity of previously active sites (unpublished data). Indeed, PDBu increased the number of Ca2+ sparklet sites in mouse arterial myocytes twofold (n = 7). Thus, persistent Ca2+ sparklets and their modulation by PKC appears to be a conserved feature of arterial myocytes.
Figure 5.

PKCα is required for Ca2+ sparklets in arterial smooth muscle. Sample images of typical WT (A) and PKCα−/− (B and C) mouse arterial myocytes. The traces below the images (A and C) show the time course of [Ca2+]i in the sites marked by the green circle before and after the application of 200 nM PDBu. (D) Bar plot of the mean ± SEM nPs under control conditions and after application of PDBu. *, significantly different from control.
Consistent with our data from rat arterial myocytes and tsA-201 cells, we found that PKCα−/− myocytes were devoid of persistent Ca2+ sparklet activity under control conditions (n = 20 cells). Indeed, in only 1 out of 20 cells examined was a single Ca2+ sparklet event (amplitude = 38 nM) evident at −70 mV. Interestingly, application of 200 nM PDBu, which would activate other PKC isoforms expressed in PKCα−/− cells, had a small effect on Ca2+ sparklet activity in these cells, only activating a few, low nPs Ca2+ sparklet sites (nPs = 0.02 ± 0.01, n = 21; Fig. 5, B–D). In total, three Ca2+ sparklet sites were observed in PKCα−/− cells in the presence of PDBu. In PKCα−/− cells, Ca2+ sparklet amplitudes ranged from 34 to 41 nM. Note that this range of amplitudes is similar to the amplitude of quantal Ca2+ sparklets in WT mouse and rat myocytes and tsA-201 cells, indicating that the probability of coincidental openings of nearby L-type Ca2+ channels in PKCα−/− cells was low. Indeed, the effects of PDBu on Ca2+ sparklet activity in PKCα−/− cells (nPs ≈ 0.02 ± 0.01, n = 21) were 15-fold smaller than in WT cells (nPs = 0.30 ± 0.1, n = 15) (Fig. 5 D). Taken together, these data support the hypothesis that basal PKCα activity is necessary for spontaneous Ca2+ sparklet activity. Furthermore, our data suggest that regional variations in the activity of PKCα underlie heterogeneous Ca2+ sparklet activity in arterial myocyte.
Having established that basal PKCα activity is necessary for spontaneous persistent Ca2+ sparklet activity in arterial myocytes, we investigated the role of protein phosphatases in modulating the activity of these Ca2+ influx events. Recent studies have suggested that the serine/threonine phosphatases PP1, PP2A, and PP2B modulate L-type Ca2+ channel function (Santana et al., 2002; duBell and Rogers, 2004). Thus, we tested the hypothesis that local variations in the relative activities of PKCα and opposing protein phosphatases determine regional variations in Ca2+ sparklet activity in arterial myocytes.
First, we examined the role of PP2B (calcineurin) on Ca2+ sparklets (Fig. 6). Inhibition of PP2B with cyclosporine A (CsA; 500 nM) increased Ca2+ influx by activation of previously silent sites and by increasing the activity of low activity sites (Fig. 6, A and C). CsA did not increase the activity of high nPs sites, suggesting maximal activity at these sites (Fig. 6, A and C). CsA induced a twofold increase in the number of Ca2+ sparklet sites per cell (n = 12, P < 0.05). The quantal amplitude of Ca2+ sparklets was unchanged by CsA (Fig. 6 D). It is important to note that CsA failed to activate Ca2+ sparklets in cells dialyzed with the PKC inhibitor PKCi (Fig. 6 B). These results suggest that PP2B dampens Ca2+ sparklet activity by opposing PKC-mediated phosphorylation.
Figure 6.

PP2B opposes PKC-dependent activation of Ca2+ sparklets. (A) Images of representative cells with silent (left), low (center), and high (right) nPs sites. Traces below the images show the time course of [Ca2+]i in the green circle before (top) and after (bottom) application of 500 nM cyclosporine A (CsA). (B) Sample image from a cell dialyzed for 10 min with an internal solution containing PKCi (100 μM) before and after CsA. The time course of [Ca2+]i in the region marked by the green circle is shown below the image. (C) Mean ± SEM nP s of Ca2+ sparklet sites in control conditions and in the presence of CsA or PKCi and CsA. (D) Mean ± SEM amplitude of quantal Ca2+ sparklets before and after application of CsA. *, significantly different from control.
Next, we investigated the effects of the PP1 and PP2A inhibitor calyculin A (100 nM) (duBell et al., 2002) on Ca2+ sparklets. Like CsA, calyculin A increased Ca2+ influx by activating previously silent Ca2+ sparklet sites and by increasing the activity of low nPs sites (Fig. 7, A and C). Again, the activity of high nPs sites did not change upon application of calyculin A, suggesting maximal activity at these sites. Calyculin A induced a 2.3-fold increase in the number of Ca2+ sparklets sites per cell (n = 7, P < 0.05). As with CsA, calyculin A did not activate Ca2+ sparklets in cells dialyzed with PKCi (Fig. 7 B). These results suggest that PP1 and/or PP2A modulate Ca2+ sparklets by opposing PKC.
Figure 7.

Calyculin A increases Ca2+ sparklet activity. (A) Time course of [Ca2+]i in silent (left), low (center), and high (left) nPs sites from a representative cell before (top) and after (bottom) application of 100 nM calyculin A. (B) Sample image from a typical cell dialyzed for 10 min with an internal solution containing PKCi (100 μM). The traces below show the time course of [Ca2+]i in the region of the cell marked by the green circle before (top trace) and after (bottom trace) calyculin A. (C) Mean ± SEM nPs before and after application of calyculin A or PKCi plus calyculin A. (D) Mean ± SEM amplitude of quantal Ca2+ sparklets before and after application of calyculin A. *, significantly different from control.
We used different concentration of okadaic acid (OA) to determine the relative contribution of PP1 and PP2A to Ca2+ sparklet activity (Fig. 8). At concentrations of between 1 and 10 nM, OA inhibits PP2A, while at concentrations >100 nM, it blocks PP2A and PP1 (Bialojan and Takai, 1988; duBell and Rogers, 2004). Application of 1 nM OA increased Ca2+ sparklet activity nearly twofold (Fig. 8 A). It is important to note that the increase in Ca2+ sparklet activity produced by 1 nM OA was not significantly different from that observed with calyculin A (P > 0.05). At 1 nM, OA induced a 2.5-fold increase in the number of Ca2+ sparklets sites per cell (n = 9, P < 0.05). These data suggest that calyculin A increased Ca2+ sparklet activity by inhibiting PP2A. Consistent with this hypothesis, increasing OA in the bath from 1 to 100 nM, thus also inhibiting PP1, did not elicit any further increase in Ca2+ sparklet activity (nPs = 0.37 ± 0.1, n = 31, P > 0.05) or sparklet density (n = 9, P > 0.05). As with CsA and calyculin A, OA did not activate Ca2+ sparklets in cells dialyzed with PKCi and did not change the quantal level of the Ca2+ sparklets (Fig. 8). From these data we conclude that PP2A, PP2B, and PKCα form a signaling module that tunes local Ca2+ influx via L-type Ca2+ channels in arterial smooth muscle cells.
Figure 8.

PP2A inhibits Ca2+ sparklets by opposing PKC. (A) Mean ± SEM nPs before and after application of 1 or 100 nM okadaic acid (OA) in the absence or presence of PKCi in the patch pipette. (B) Mean ± SEM amplitude of quantal Ca2+ sparklets before and after application of 1 nM or 100 nM OA. *, significantly different from control.
DISCUSSION
We used TIRF microscopy to investigate with high resolution the spatiotemporal organization of functional Ca2+ channels. Using this approach, we provide the first direct demonstration that persistent Ca2+ sparklet activity is a fundamental property of L-type Ca2+ channels when associated with PKC. Furthermore, we describe a novel mechanism for the local, dynamic control of steady-state Ca2+ influx via L-type Ca2+ channels. We found that PKCα and the phosphatases PP2A and PP2B (calcineurin) have opposing effects on persistent Ca2+ sparklet activity. Our results also suggest that PP2A and PP2B oppose the actions of PKCα on L-type Ca2+ channels. Based on these findings we propose that the degree of steady-state Ca2+ influx in various regions of the cell via L-type Ca2+ channels is determined by a local balance between PKCα, PP2A, and PP2B activities.
Our data, in conjunction with earlier studies (Nelson et al., 1995), suggest that smooth muscle cells are capable of generating multiple types of local Ca2+ signals. For example, simultaneous activation of a small cluster of ryanodine-sensitive Ca2+ channels in the sarcoplasmic reticulum of these cells produces Ca2+ sparks (Nelson et al., 1995). Because Ca2+ sparks are produced by the release of Ca2+ from intracellular stores, they are insensitive to dihydropyridines or the removal of external Ca2+, their amplitude is independent of changes in membrane voltage, and they are abolished by depleting the SR of Ca2+ with thapsigargin (Cannell et al., 1995; López-López et al., 1995; Nelson et al., 1995). In sharp contrast to Ca2+ sparks, the Ca2+ sparklets described here meet all the criteria for a Ca2+ influx event via sarcolemmal Ca2+ channels. First, Ca2+ sparklets are insensitive to thapsigargin. Second, Ca2+ sparklets are rapidly eliminated by perfusion with a Ca2+-free solution. Third, the amplitude of quantal Ca2+ sparklets decreased with membrane depolarization (i.e., decreased driving force). Fourth, a dihydropyridine antagonist and agonist inhibited and activated Ca2+ sparklets, respectively. Taken together, these data provide compelling support to the hypothesis that Ca2+ sparklets are produced by Ca2+ influx via sarcolemma Ca2+ channels.
Our data unequivocally demonstrate that L-type Cavα1.2 channels have the ability to produce persistent Ca2+ sparklets even at hyperpolarized potentials (−70 mV). Interestingly, L-type Cavα1.2 channels require PKCα activity to operate in a persistent gating mode; in the absence of PKCα activity, Ca2+ sparklet activity in tsA-201 cells and arterial myocytes at −70 mV was very low. An important finding in our study is that heterologous expression of PKCα and Cavα1.2 channels was sufficient to reproduce all the basic features (i.e., amplitude of quantal event, voltage dependencies, gating modalities, and pharmacology) of persistent Ca2+ sparklet activity in arterial myocytes. These results strongly support the hypothesis that, as in heart (Wang et al., 2001), L-type Ca2+ channels underlie Ca2+ sparklets in arterial smooth muscle.
The data presented here, and in our previous study (Navedo et al., 2005), indicate that Ca2+ sparklets in arterial smooth muscle are produced by the opening of a single or a cluster of L-type Ca2+ channels. Accordingly, simultaneous recordings of single Ca2+ channel currents and Ca2+ sparklets under conditions similar to those used in the current study (i.e., −70 mV and 20 mM external Ca2+) demonstrated that an opening of a single Ca2+ channel (0.5 pA) could produce a Ca2+ sparklet of an amplitude of ≈37 nM (Navedo et al., 2005). Because Ca2+ sparklet amplitude is variable (between 38 and 300 nM), these data strongly suggest that Ca2+ sparklets could be produced by the opening of a single or a cluster of L-type Ca2+ channels. Consistent with this, the all-points (see Fig. S1, available at http://www.jgp.org/cgi/content/full/jgp.200609519/DC1) and event histograms (e.g., Fig. 1 C, Fig. 2 D, and Fig. 3 B) presented here clearly show well-defined peaks with a quantal unit of Ca2+ influx of ∼34–37 nM. Together, these data provide compelling support to the view that openings of a single or a cluster of L-type Ca2+ channels underlie Ca2+ sparklets in arterial smooth muscle.
Although Ca2+ sparklets in cardiac and smooth muscle are produced by the same molecular entity (hence the same name), there are important differences between Ca2+ sparklets in these cells. For example, as noted above, Ca2+ sparklets in arterial myocytes are produced by the opening of a single or a cluster of L-type Cavα1.2 channels. In contrast, Ca2+ sparklets in ventricular myocytes are produced by the opening of a single L-type Ca2+ channel (Wang et al., 2001). Furthermore, unlike smooth muscle, persistent Ca2+ sparklet activity has not been observed in ventricular myocytes. This is interesting because PKCα is expressed in ventricular myocytes (Braz et al., 2004) and Cavα1.2 is the predominant Cavα isoform in these cells. Although the reasons for these differences are presently unclear, it is intriguing to speculate that basal PKCα activity in ventricular myocytes is lower than in arterial myocytes, thus decreasing the probability of persistent Ca2+ sparklet activity in these cells. This hypothesis is supported by our data suggesting that Ca2+ sparklet activity in tsA-201 expressing Cavα1.2 only and PKCα−/− myocytes, as in ventricular myocytes, is low and mostly resulting from the activation of single Cavα1.2 channels. Future studies should examine the mechanisms modulating Ca2+ sparklet activity in heart.
A recent study (Yang et al., 2005) provides insight into the molecular mechanisms underlying PKC-induced modulation of Ca2+ sparklet activity. Yang and coworkers found that PKC could directly phosphorylate serine 1928 of Cavα1.2 channels. Thus, it is intriguing to speculate that direct phosphorylation of this L-type Ca2+ channel subunit by PKC may play a critical role in the induction of persistent Ca2+ sparklet activity. Consistent with this, our data indicate that coexpression of Cavα1.2 and PKCα is sufficient to produce persistent Ca2+ sparklet activity in tsA-201 cells. Future experiments should examine the molecular mechanisms by which PKCα promotes persistent L-type Ca2+ channel gating.
A particularly interesting finding in this study is that inhibition of PP2A or PP2B increases Ca2+ influx by recruiting new Ca2+ sparklet sites and increasing the activity of low nPs sites. Inhibition of these phosphatases did not increase the activity of high nPs sites, indicating that these sites were maximally activated under control conditions. These findings suggest that in silent Ca2+ sparklet sites, PP2A and/or PP2B activity is sufficiently high to exceed PKCα, thus favoring dephosphorylation of Ca2+ channels. Accordingly, in low nPs sites, the balance between PKCα and PP2A/PP2B favors submaximal PKCα-dependent phosphorylation, which induces low Ca2+ sparklet activity. In high nPs sites, however, PKCα activity exceeds PP2A/PP2B activity, thus favoring maximal phosphorylation of Ca2+ channels. Because silent, low, and high activity Ca2+ sparklet sites coexist in the same cell, these findings support a model in which Ca2+ influx is determined locally by the relative balance between PKCα-dependent phosphorylation and opposing phosphatases. Such a situation allows for dynamic, local modulation of Ca2+ channel gating modalities.
This model implies that when prevailing conditions favor dephosphorylation, Ca2+ influx is most likely dominated by random, sporadic openings of solitary L-type Ca2+ channels. A physiological stimulus that increases PKCα activity, or decreases the activity of PP2A and/or PP2B, would promote Ca2+ influx via persistent Ca2+ sparklet sites. In this case, Ca2+ influx would be determined by persistent Ca2+ channel activity in addition to rare, stochastic openings of these channels. Accordingly, vasoactive agents that activate PKC (e.g., angiotensin II and UTP) would increase Ca2+ influx and thereby constrict arterial smooth muscle, at least in part, by increasing persistent Ca2+ sparklet activity.
An issue that was not addressed by Navedo et al. (2005) is the mechanism by which Bay-K 8644 increases persistent Ca2+ sparklet activity in arterial myocytes. To our knowledge, Bay-K 8644 does not directly activate PKCα. Thus, how does Bay-K 8644 increase persistent Ca2+ sparklet activity in these cells? The experiments in this study provide insight into this issue. Note that Bay-K 8644 induced high nPs, persistent Ca2+ sparklet activity in tsA-201 cells expressing Cavα1.2 and PKCα; persistent Ca2+ sparklet activity was never observed after Bay-K 8644 in tsA-201 cells expressing Cavα1.2 only. Based on these data, we propose a positive feedback model to explain this apparent discrepancy between the effects of Bay-K 8644 on Ca2+ sparklets in tsA-201 cells and arterial myocytes. In this model, application of Bay-K 8644 (or any other Ca2+ channel opener) to arterial myocytes causes an increase in Ca2+ influx (i.e., by increasing the mean open time and/or open probability of L-type Ca2+ channels) that would activate nearby Ca2+-sensitive PKCα. Once activated, this kinase can induce L-type Ca2+ channels to operate in a persistent gating mode, thus causing further Ca2+ influx, which could presumably maintain PKCα activity and hence persistent Ca2+ sparklet activity. Because PKC expression in arterial myocytes is punctate (Maasch et al., 2000; Navedo et al., 2005), PKCα and persistent Ca2+ sparklet activity does not propagate throughout the cell. As noted above, protein phosphatases provide additional negative control to this system. An interesting implication of this model is that while Ca2+ sparklets could be evoked by voltage or pharmacological means wherever Cavα1.2 channels are expressed, persistent Ca2+ channel activity would only occur in regions of the cell membrane where both Cavα1.2 and PKCα are expressed. Future experiments should investigate the spatial and temporal relationship between Ca2+ sparklets and PKCα in arterial myocytes.
To conclude, we demonstrated that persistent Ca2+ sparklet activity is a fundamental feature of L-type Cavα1.2 channels in association with PKCα. Because PKC and Cavα1.2 are ubiquitously expressed, our findings support the concept that Ca2+ influx via persistent L-type Ca2+ channels may represent a general mechanism for the control of steady-state Ca2+ influx in excitable cells.
Furthermore, our observations support the concept that PKCα, PP2A, and PP2B form a signaling module that tunes the activity of Ca2+ channels, allowing for local, dynamic regulation of steady-state Ca2+ influx in cerebral arterial smooth muscle and perhaps excitable cells in general.
Supplemental Material
Acknowledgments
We thank Drs. Charles F. Rossow, Keith W. Dilly, Sharona E. Gordon, and Carmen A. Ufret-Vincenty for reading this manuscript.
This work was supported by National Institutes of Health grants HL077115, HL077115S1, HL07828, and HL07312.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: CsA, cyclosporine A; OA, okadaic acid; PDBu, phorbol 12, 13-dibutyrate; PKCβi, PKCβ inhibitor; PP2A, protein phosphatase 2A; TIRF, total internal reflection fluorescence; WT, wild-type.
References
- Amberg, G.C., and L.F. Santana. 2003. Downregulation of the BK channel β1 subunit in genetic hypertension. Circ. Res. 93:965–971. [DOI] [PubMed] [Google Scholar]
- Bayliss, W.M. 1902. On the local reaction of the arterial wall to changes in internal pressure. J. Physiol. 28:220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialojan, C., and A. Takai. 1988. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem. J. 256:283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz, J.C., K. Gregory, A. Pathak, W. Zhao, B. Sahin, R. Klevitsky, T.F. Kimball, J.N. Lorenz, A.C. Nairn, S.B. Liggett, et al. 2004. PKCα regulates cardiac contractility and propensity toward heart failure. Nat. Med. 10:248–254. [DOI] [PubMed] [Google Scholar]
- Cannell, M.B., H. Cheng, and W.J. Lederer. 1995. The control of calcium release in heart muscle. Science. 268:1045–1049. [DOI] [PubMed] [Google Scholar]
- Cheng, H., L.S. Song, N. Shirokova, A. Gonzalez, E.G. Lakatta, E. Rios, and M.D. Stern. 1999. Amplitude distribution of calcium sparks in confocal images: theory and studies with an automatic detection method. Biophys. J. 76:606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro, A., and I. Parker. 2004. Imaging the activity and localization of single voltage-gated Ca2+ channels by total internal reflection fluorescence microscopy. Biophys. J. 86:3250–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro, A., and I. Parker. 2005. “Optical patch-clamping”: single-channel recording by imaging Ca2+ flux through individual muscle acetylcholine receptor channels. J. Gen. Physiol. 126:179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- duBell, W.H., M.S. Gigena, S. Guatimosim, X. Long, W.J. Lederer, and T.B. Rogers. 2002. Effects of PP1/PP2A inhibitor calyculin A on the E-C coupling cascade in murine ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 282:H38–H48. [DOI] [PubMed] [Google Scholar]
- duBell, W.H., and T.B. Rogers. 2004. Protein phosphatase 1 and an opposing protein kinase regulate steady-state L-type Ca2+ current in mouse cardiac myocytes. J. Physiol. 556:79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann, B.K., R.K. Murray, and M.I. Kotlikoff. 1994. Voltage window for sustained elevation of cytosolic calcium in smooth muscle cells. Proc. Natl. Acad. Sci. USA. 91:11914–11918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwendt, M., S. Dieterich, J. Rennecke, W. Kittstein, H.J. Mueller, and F.J. Johannes. 1996. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase C isoenzymes. FEBS Lett. 392:77–80. [DOI] [PubMed] [Google Scholar]
- Harder, D.R., R. Gilbert, and J.H. Lombard. 1987. Vascular muscle cell depolarization and activation in renal arteries on elevation of transmural pressure. Am. J. Physiol. 253:F778–F781. [DOI] [PubMed] [Google Scholar]
- Johnson, J.A., M.O. Gray, C.H. Chen, and D. Mochly-Rosen. 1996. A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. J. Biol. Chem. 271:24962–24966. [DOI] [PubMed] [Google Scholar]
- Knot, H.J., and M.T. Nelson. 1998. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J. Physiol. 508:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, W.J., P.T. Ellinor, and A. Schwartz. 1990. cDNA cloning of a dihydropyridine-sensitive calcium channel from rat aorta. Evidence for the existence of alternatively spliced forms. J. Biol. Chem. 265:17786–17791. [PubMed] [Google Scholar]
- López-López, J.R., P.S. Shacklock, C.W. Balke, and W.G. Wier. 1995. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science. 268:1042–1045. [DOI] [PubMed] [Google Scholar]
- Maasch, C., S. Wagner, C. Lindschau, G. Alexander, K. Buchner, M. Gollasch, F.C. Luft, and H. Haller. 2000. Protein kinase C alpha targeting is regulated by temporal and spatial changes in intracellular free calcium concentration [Ca2+]i. FASEB J. 14:1653–1663. [DOI] [PubMed] [Google Scholar]
- Maravall, M., Z.F. Mainen, B.L. Sabatini, and K. Svoboda. 2000. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys. J. 78:2655–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navedo, M.F., G. Amberg, S.V. Votaw, and L.F. Santana. 2005. Constitutively active L-type Ca2+ channels. Proc. Natl. Acad. Sci. USA. 102:11112–11117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, M.T., H. Cheng, M. Rubart, L.F. Santana, A.D. Bonev, H.J. Knot, and W.J. Lederer. 1995. Relaxation of arterial smooth muscle by calcium sparks. Science. 270:633–637. [DOI] [PubMed] [Google Scholar]
- Pang, L., M. Nie, L. Corbett, R. Donnelly, S. Gray, and A.J. Knox. 2002. Protein kinase C-epsilon mediates bradykinin-induced cyclooxygenase-2 expression in human airway smooth muscle cells. FASEB J. 16:1435–1437. [DOI] [PubMed] [Google Scholar]
- Rubart, M., J.B. Patlak, and M.T. Nelson. 1996. Ca2+ currents in cerebral artery smooth muscle cells of rat at physiological Ca2+ concentrations. J. Gen. Physiol. 107:459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana, L.F., E.G. Chase, V.S. Votaw, M.T. Nelson, and R. Greven. 2002. Functional coupling of calcineurin and protein kinase A in mouse ventricular myocytes. J. Physiol. 544:57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnegger-Brauns, M.J., A. Hetzenauer, I.G. Huber, E. Renstrom, G. Wietzorrek, S. Berjukov, M. Cavalli, D. Walter, A. Koschak, R. Waldschutz, et al. 2004. Isoform-specific regulation of mood behavior and pancreatic β cell and cardiovascular function by L-type Ca2+ channels. J. Clin. Invest. 113:1430–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, M., S. Sagawa, J. Hoshi, F. Shimoma, I. Matsuda, K. Sakoda, T. Sasase, M. Shindo, and T. Inaba. 2004. Synthesis of anilino-monoindolylmaleimides as potent and selective PKCβ inhibitors. Bioorg. Med. Chem. Lett. 14:5171–5174. [DOI] [PubMed] [Google Scholar]
- Wang, S.Q., L.S. Song, E.G. Lakatta, and H. Cheng. 2001. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 410:592–596. [DOI] [PubMed] [Google Scholar]
- Wickman, G., C. Lan, and B. Vollrath. 2003. Functional roles of the rho/rho kinase pathway and protein kinase C in the regulation of cerebrovascular constriction mediated by hemoglobin: relevance to subarachnoid hemorrhage and vasospasm. Circ. Res. 92:809–816. [DOI] [PubMed] [Google Scholar]
- Woodruff, M.L., A.P. Sampath, H.R. Matthews, N.V. Krasnoperova, J. Lem, and G.L. Fain. 2002. Measurement of cytoplasmic calcium concentration in the rods of wild-type and transducin knock-out mice. J. Physiol. 542:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L., G. Liu, S.I. Zakharov, J.P. Morrow, V.O. Rybin, S.F. Steinberg, and S.O. Marx. 2005. Ser1928 is a common site for Cav1.2 phosphorylation by protein kinase C isoforms. J. Biol. Chem. 280:207–214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}