

Abstract
Electrophysiological events are of central importance during the phagocyte respiratory burst, because NADPH oxidase is electrogenic and voltage sensitive. We investigated the recent suggestion that large-conductance, calcium-activated K+ (BK) channels, rather than proton channels, play an essential role in innate immunity (Ahluwalia, J., A. Tinker, L.H. Clapp, M.R. Duchen, A.Y. Abramov, S. Page, M. Nobles, and A.W. Segal. 2004. Nature. 427:853–858). In PMA-stimulated human neutrophils or eosinophils, we did not detect BK currents, and neither of the BK channel inhibitors iberiotoxin or paxilline nor DPI inhibited any component of outward current. BK inhibitors did not inhibit the killing of bacteria, nor did they affect NADPH oxidase-dependent degradation of bacterial phospholipids by extracellular gIIA-PLA2 or the production of superoxide anion (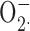 ). Moreover, an antibody against the BK channel did not detect immunoreactive protein in human neutrophils. A required role for voltage-gated proton channels is demonstrated by Zn2+ inhibition of NADPH oxidase activity assessed by H2O2 production, thus validating previous studies showing that Zn2+ inhibited
). Moreover, an antibody against the BK channel did not detect immunoreactive protein in human neutrophils. A required role for voltage-gated proton channels is demonstrated by Zn2+ inhibition of NADPH oxidase activity assessed by H2O2 production, thus validating previous studies showing that Zn2+ inhibited 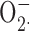 production when assessed by cytochrome c reduction. In conclusion, BK channels were not detected in human neutrophils or eosinophils, and BK inhibitors did not impair antimicrobial activity. In contrast, we present additional evidence that voltage-gated proton channels serve the essential role of charge compensation during the respiratory burst.
production when assessed by cytochrome c reduction. In conclusion, BK channels were not detected in human neutrophils or eosinophils, and BK inhibitors did not impair antimicrobial activity. In contrast, we present additional evidence that voltage-gated proton channels serve the essential role of charge compensation during the respiratory burst.
INTRODUCTION
A crucial component of innate immunity is the respiratory burst; the assembly and activation of the NADPH oxidase complex that produces superoxide anion (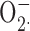 ), a precursor to an array of reactive oxygen species. NADPH oxidase is electrogenic (Henderson et al., 1987) because it transfers electrons from intracellular NADPH to extracellular (or intraphagosomal) oxygen. This charge movement must be compensated to prevent extreme depolarization that directly turns off NADPH oxidase (DeCoursey et al., 2003). Proton channels have been generally believed to compensate the obligate charge translocation that occurs during NADPH oxidase activity (Henderson et al., 1987; Cross and Jones, 1991; DeCoursey and Cherny, 1993; Schrenzel et al., 1998; Geiszt et al., 2001; Babior et al., 2002; Vignais, 2002; Lee et al., 2003; DeCoursey, 2003; Demaurex and Petheõ, 2005). However, it was reported recently that the main ionic conductance in PMA-stimulated human neutrophils and eosinophils is a K+ conductance, due to large-conductance, voltage-, and calcium-activated (BK) channels (Ahluwalia et al.., 2004). These channels are referred to as BK channels, because of their “big” conductance (compared with IK or SK, intermediate or small conductance Ca2+-activated K+ channels) and K+ selectivity. In that study, the BK channel inhibitors iberiotoxin (IbTX) and paxilline (PAX) inhibited the ability of neutrophils to kill Staphylococcus aureus, Serratia marcescens, and Candida albicans, and IbTX inhibited all outward currents in human neutrophils and eosinophils. To our knowledge PAX is specific for BK channels, but IbTX also inhibits IK channels (Schilling et al., 2002; 2004). The identification of BK channels in these cells was unexpected for several reasons. Although macrophages under certain conditions express BK channels (Gallin, 1984; DeCoursey et al., 1996), these channels had not been reported in any previous study (using appropriate ionic conditions) of neutrophils (von Tscharner et al., 1986; Krause and Welsh, 1990) or eosinophils (Gordienko et al., 1996; Saito et al., 1997; Tare et al., 1998; Schwingshackl et al., 2000; Morgan et al., 2003). Equally surprising was the failure to detect voltage-gated proton currents in these cells (Ahluwalia et al., 2004), the existence of which has been demonstrated in numerous previous studies (for review see DeCoursey, 2004). Previous pharmacological evidence that proton channels were required for sustained NADPH oxidase activity, based on inhibition by Zn2+ (Beswick et al., 1986; Henderson et al., 1988b; Simchowitz et al., 1990; Lowenthal and Levy, 1999; Bankers-Fulbright et al., 2001; DeCoursey et al., 2003; Rada et al., 2004) or Cd2+ (Henderson et al., 1988a; Simchowitz et al., 1990; Kapus et al., 1992) of cytochrome c reduction, was discounted as being artifactual (Ahluwalia et al., 2004).
), a precursor to an array of reactive oxygen species. NADPH oxidase is electrogenic (Henderson et al., 1987) because it transfers electrons from intracellular NADPH to extracellular (or intraphagosomal) oxygen. This charge movement must be compensated to prevent extreme depolarization that directly turns off NADPH oxidase (DeCoursey et al., 2003). Proton channels have been generally believed to compensate the obligate charge translocation that occurs during NADPH oxidase activity (Henderson et al., 1987; Cross and Jones, 1991; DeCoursey and Cherny, 1993; Schrenzel et al., 1998; Geiszt et al., 2001; Babior et al., 2002; Vignais, 2002; Lee et al., 2003; DeCoursey, 2003; Demaurex and Petheõ, 2005). However, it was reported recently that the main ionic conductance in PMA-stimulated human neutrophils and eosinophils is a K+ conductance, due to large-conductance, voltage-, and calcium-activated (BK) channels (Ahluwalia et al.., 2004). These channels are referred to as BK channels, because of their “big” conductance (compared with IK or SK, intermediate or small conductance Ca2+-activated K+ channels) and K+ selectivity. In that study, the BK channel inhibitors iberiotoxin (IbTX) and paxilline (PAX) inhibited the ability of neutrophils to kill Staphylococcus aureus, Serratia marcescens, and Candida albicans, and IbTX inhibited all outward currents in human neutrophils and eosinophils. To our knowledge PAX is specific for BK channels, but IbTX also inhibits IK channels (Schilling et al., 2002; 2004). The identification of BK channels in these cells was unexpected for several reasons. Although macrophages under certain conditions express BK channels (Gallin, 1984; DeCoursey et al., 1996), these channels had not been reported in any previous study (using appropriate ionic conditions) of neutrophils (von Tscharner et al., 1986; Krause and Welsh, 1990) or eosinophils (Gordienko et al., 1996; Saito et al., 1997; Tare et al., 1998; Schwingshackl et al., 2000; Morgan et al., 2003). Equally surprising was the failure to detect voltage-gated proton currents in these cells (Ahluwalia et al., 2004), the existence of which has been demonstrated in numerous previous studies (for review see DeCoursey, 2004). Previous pharmacological evidence that proton channels were required for sustained NADPH oxidase activity, based on inhibition by Zn2+ (Beswick et al., 1986; Henderson et al., 1988b; Simchowitz et al., 1990; Lowenthal and Levy, 1999; Bankers-Fulbright et al., 2001; DeCoursey et al., 2003; Rada et al., 2004) or Cd2+ (Henderson et al., 1988a; Simchowitz et al., 1990; Kapus et al., 1992) of cytochrome c reduction, was discounted as being artifactual (Ahluwalia et al., 2004).
The proposal that BK channels and not proton channels are essential for antibacterial activity of human neutrophils and eosinophils (Ahluwalia et al., 2004) represents a radical departure from prevailing thought about the electrophysiological events that occur during the respiratory burst. As logical extensions of this novel assignment of BK channel activity for phagocyte function, one would predict that BK channel inhibition would compromise NADPH oxidase-dependent events, including bacterial killing and staphylococcal phospholipid degradation. We therefore reexamined these issues using several complementary approaches. Our data fail to identify a contribution of BK channels to the respiratory burst or the antibacterial activity of human neutrophils and eosinophils, but support an essential role for voltage-gated proton channels.
MATERIALS AND METHODS
Eosinophils and Neutrophils
Venous blood was drawn from healthy adult volunteers under informed consent according to procedures approved by the Institutional Review Boards at the University of Iowa, Rush University, and Semmelweis University, respectively. Polymorphonuclear leukocytes (PMN) were purified as previously described (Boyum, 1968). For killing assays, PMN were used within 1 h of isolation. Patch-clamp studies were done on freshly isolated eosinophils and eosinophils incubated overnight at 37°C in RPMI 1640 medium containing 25 mM HEPES and L-glutamine (GIBCO BRL), supplemented with 10% FBS (Bio-Whittaker), 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich), and 1 ng/ml recombinant human GM-CSF (R&D Systems, Inc.). Eosinophils were isolated from PMN by negative selection using anti-CD16 immunomagnetic beads (DeCoursey et al., 2001) as described by the manufacturer (Miltenyi Biotec Inc.). The eosinophils or neutrophils were suspended in HEPES (10 mM)-buffered HBSS, pH 7.4, containing 1 mg/ml human serum albumin (HEPES-HBSS-HSA buffer) or in PBS with 2 mM EDTA and 0.5% BSA. Neutrophil purity was routinely 95% or greater. Eosinophil purity was routinely >98% as determined by counting Wright-stained cytospin preparations.
A549 Cells
The A549 cell line, established in 1972 from a human alveolar cell carcinoma (Lieber et al., 1976), was generously provided by Dr. Beverly L. Davidson, University of Iowa (Iowa City, IA). A549 cells were incubated at 37°C in Dulbecco's Modified Eagle's Medium (GIBCO BRL) supplemented with 10% FCS, 10 mM HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml Fungizone (amphotericin B) (GIBCO BRL).
THP-1 Cells
THP-1 cells were obtained from American Type Culture Collection. Cells were cultured in suspension at 1–2 × 106 cells/ml in RPMI medium supplemented with 0.29 mg/ml glutamine, 10% FBS, 100 U/ml of penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml Fungizone (GIBCO BRL). Cells were incubated at 37°C in a humidified atmosphere of 5% CO2 in air. Every 2–3 d, about half of the media was replaced with fresh media, and once per week the cells were removed, centrifuged at 1,800 rpm for 10 min at 4°C in a Napco 2028R refrigerated centrifuge. The cell pellet was resuspended in fresh media at 1–2 × 106 cells/ml. To induce differentiation, THP-1 cells were incubated with 10 ng/ml PMA (Sigma-Aldrich) for 3 d in 35-mm tissue culture dishes containing several small pieces of sterile glass coverslips. One manifestation of differentiation is adherence; cells adhering to the coverslip fragments were transferred to the recording chamber.
Transient Transfection of COS-7 Cells
COS-7 cells were maintained in 5% CO2 and 95% air in a humidified incubator at 37°C in Dulbecco's modified Eagle's medium containing 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. The cells were seeded in 35-mm tissue culture dishes (104 cells/dish) 3 d before transfection. To transfect the cells, the culture medium was replaced by 1.2-ml aliquots of Opti-MEM I (Invitrogen) containing 2 μl Lipofectamine 2000 reagent (Invitrogen), 0.5 μg pIRES-CD8, and 1 μg pcDNA3-BK-zero-HA plasmid DNA. After a 7-h incubation period, the cells were rinsed once and fresh culture medium was added. Electrophysiological measurements were performed 48–60 h after the initiation of the transfection procedure.
Bacterial Killing
Bacterial killing assays in Fig. 1 A were performed as described previously (Femling et al., 2005). In brief, subcultured S. aureus strain ALC1435 (Cheung et al., 1998) was opsonized in pooled human serum for 20 min before incubation with neutrophils that were pretreated with buffer alone, buffer with 10 μM DPI, or buffer with 100 nM IbTX. Final incubation mixtures contained 5 × 106 bacterial colony forming units/ml and 5 × 106 neutrophils/ml suspended in 20 mM HEPES-buffered HBSS supplemented with 1% human serum albumin and 10% pooled human serum. Incubations were performed in 5 ml round-bottom polypropylene tubes swirling at 120 rpm in a water bath at 37°C for 120 min. After incubations, a 10-μl aliquot of each cell suspension was added to 490 μl of 1% saponin in H2O. Samples were vortexed vigorously, incubated for 15 min at room temperature to lyse the PMN, and then serially diluted in PBS. Aliquots (10–20 μl) representing ∼200 CFU were added to a 50-mm Petri dish, followed by ∼5 ml of molten agar (50°C) and stirred briefly. The agar was then allowed to harden and CFU were counted after overnight incubation at 37°C.
Figure 1.

Bacterial killing by human neutrophils is not affected by IbTX. (A) Percent of S. aureus surviving, relative to the inoculum after 120 min of incubation with neutrophils (PMN) alone or in the presence of 10 μM DPI or 100 nM IbTX. Mean ± SEM of five observations using the method of Femling et al. (2005). (B) Percent of S. aureus remaining, relative to the initial number, after 30 min incubation in the presence of neutrophils and media alone, 5 μM DPI, or 100 nM IbTX. Mean ± SEM of four determinations using the method of Rada et al. (2004).
The bacterial killing assay in Fig. 1 B was performed using methods described previously (Rada et al., 2004), which differs from that in Fig. 1 A in several respects. Opsonization was for 5 min (B), rather than 20 min (A). After lysis of PMN by saponin, we add a freezing–thawing step for more complete disruption of PMN (B). As discussed in Results, the ratio of bacteria:PMN is 10:1 (B) vs. 1:1 (A).
Subcellular Fractionation of Human Neutrophils
Neutrophils disrupted by nitrogen cavitation, and subcellular fractions were isolated by Percoll density gradient centrifugation as described by Borregaard et al. (1983) and Clark et al. (1987). In brief, PMNs were incubated for 20 min at RT with diisopropylfluorophosphate (1 mM) and resuspended in relaxation buffer (10 mM PIPES, 100 mM KCl, 3 mM NaCl, 3.5 mM MgCl2) with ATP (1 mM ATP(Na)2) at 50 × 106 PMN/ml with added PMSF (1 mM). Cells were disrupted with N2 at 350 psi and collected in buffer containing EGTA (final 1.25 mM) and centrifuged 200 g for 20 min to remove nuclear debris and unbroken cells. Postnuclear supernatants were placed on top of two-layer Percoll gradients as previously described (Borregaard et al., 1983) and centrifuged to separate subcellular fractions. Percoll was removed by ultracentrifugation and fractions were washed with relaxation buffer.
Cell Lysate Preparation
Lysates from myometrial cells, transfected- and sham-transfected-HEK-293 cells were prepared as described previously (Brainard et al., 2005), grown to 70% confluence, pelletted, and resuspended in cell lysate buffer (250 mM sucrose, 5 mM MOPS, 2 mM EDTA, 2 mM EGTA, 1% Triton X-100, pH 7.4). Lysis was performed using three methanol:dry ice freeze–thaw cycles, and postnuclear material was collected after centrifugation at 15,000 g for 15 min at 4°C.
Protein Quantitation
Cell lysates and subcellular fractions were analyzed for protein content using the BCA assay microplate technique (Pierce Chemical Co.) and read at 562 nm with the Spectromax spectrometer and SoftMaxPro software. After protein quantitation, samples were resuspended in sample buffer (62.5 mM Tris-HCl, pH 6.8, 5% β-mercaptoethanol, 2.3% SDS) and heat denatured for immunoblotting at 100°C for 3 min.
Protein Electrophoresis and Immunoblotting
Samples were resolved by 10% SDS-PAGE (PAGE) and transferred to nitrocellulose. Immunoblots were probed for BK channels using mouse IgG1 monoclonal antibody specific for the COOH terminus of the human K+ channel α subunit (Maxi-K antibody, BD Biosciences) at 1:200 dilution in TBS buffer with 0.5% Tween-20 and 3% milk. Immunoblots were probed with horseradish peroxidase (HRP)-labeled goat anti-mouse secondary antibody at 1:1,500 dilution in buffer as above (Bio-Rad Laboratories) followed by enhanced chemiluminescence (ECL) detection (West Femto Super Signal Substrate; Pierce Chemical Co.). Immunoblots were scanned using the Typhoon imager (GE Healthcare) and Image Quant software for the phosphorimager.
Measurement of O2. − Production
Superoxide production of the cells was determined as the superoxide dismutase-inhibitable cytochrome c reduction. To measure 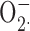 production in the extracellular medium with cytochrome c, 106 cells/ml were suspended in H-medium containing 100 μM cytochrome c. Control samples contained 12.5 μg/ml superoxide dismutase. Aliquots (200 μl) of the suspension were added into wells of a 96-well plate and prewarmed to 37°C for 5 min in a shaking ELISA reader (Labsystems iEMS Reader MF). The cells were stimulated with the appropriate stimuli by the addition of 5 μl of the stimulus solution. Changes in the absorption at 550 nm were recorded for 10 min, with two measurements/min at 37°C with gentle shaking. After subtracting the background values,
production in the extracellular medium with cytochrome c, 106 cells/ml were suspended in H-medium containing 100 μM cytochrome c. Control samples contained 12.5 μg/ml superoxide dismutase. Aliquots (200 μl) of the suspension were added into wells of a 96-well plate and prewarmed to 37°C for 5 min in a shaking ELISA reader (Labsystems iEMS Reader MF). The cells were stimulated with the appropriate stimuli by the addition of 5 μl of the stimulus solution. Changes in the absorption at 550 nm were recorded for 10 min, with two measurements/min at 37°C with gentle shaking. After subtracting the background values, 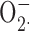 production was calculated with the use of an absorption coefficient of 21 mM−1cm−1 for cytochrome c.
production was calculated with the use of an absorption coefficient of 21 mM−1cm−1 for cytochrome c.
Assay of Metabolism of 14C-bacterial Phospholipids
The procedures used were described recently in detail (Femling et al., 2005). In brief, S. aureus were labeled with 14C-oleic acid in order to specifically follow the fate of bacterial lipids. Bacteria were then incubated as in the assay for bacterial viability. Samples at the end of incubation were extracted via a modified Bligh-Dyer method and chloroform-soluble lipids were resolved by thin-layer chromatography, and resolved 14C-labeled lipids were visualized.
Electrophysiology of Phagocytes, A549 Cells, and THP-1 Cells
The data collection setups and analysis software are unremarkable and have been described previously (Morgan et al., 2003). Seals were formed with Ringer's solution (in mM: 160 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 5 HEPES, pH 7.4) in the bath, and the potential zeroed after the pipette was in contact with the cell. For perforated patch recording, the pipette solution contained 80 mM KCH3SO3, 50 mM 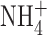 in the form of 25 mM (NH4)2SO4, 2 mM MgCl2, 5 mM BES buffer, 1 mM EGTA, and was titrated to pH 7.0 with TMAOH. We added ∼500 μg/ml solubilized amphotericin B (∼45% purity) (Sigma-Aldrich), after first dipping the pipette tip in amphotericin-free solution. The bath solution contained (in mM) 100 TMAMeSO3, 25 (NH4)2SO4, 2 MgCl2, 1.5 CaCl2, 1 EGTA, and 10 BES at pH 7.0. For whole-cell recording of BK currents, we used solutions buffered to various [Ca2+] by adding various amounts of CaCl2 to 10 mM EGTA, assuming apparent binding constants of 0.216 M for Mg2+ and 3.76 × 10−7 M for Ca2+ (Kim et al., 1996). Voltage-independent, IK channels in THP-1 cells (Kim et al., 1996) overwhelmed all other conductances at high [Ca2+]i, and thus BK currents were better distinguished at lower [Ca2+]i. All solutions were roughly 300 mOsm. Studies were done at room temperature (20–25°C).
in the form of 25 mM (NH4)2SO4, 2 mM MgCl2, 5 mM BES buffer, 1 mM EGTA, and was titrated to pH 7.0 with TMAOH. We added ∼500 μg/ml solubilized amphotericin B (∼45% purity) (Sigma-Aldrich), after first dipping the pipette tip in amphotericin-free solution. The bath solution contained (in mM) 100 TMAMeSO3, 25 (NH4)2SO4, 2 MgCl2, 1.5 CaCl2, 1 EGTA, and 10 BES at pH 7.0. For whole-cell recording of BK currents, we used solutions buffered to various [Ca2+] by adding various amounts of CaCl2 to 10 mM EGTA, assuming apparent binding constants of 0.216 M for Mg2+ and 3.76 × 10−7 M for Ca2+ (Kim et al., 1996). Voltage-independent, IK channels in THP-1 cells (Kim et al., 1996) overwhelmed all other conductances at high [Ca2+]i, and thus BK currents were better distinguished at lower [Ca2+]i. All solutions were roughly 300 mOsm. Studies were done at room temperature (20–25°C).
Electrophysiology of COS-7 Cells
Whole-cell currents in COS-7 cells were recorded with an RK-400 amplifier (Biologic) using microelectrodes made of thin-walled borosilicate glass (Clark Electromedical Instruments) with resistance of 7–11 MΩ when fire polished and filled with pipette solution, containing (in mM): KCl 76, MgCl2 1.6, EGTA 20, CaCl2 19.2, HEPES 20 (pH 7.3 with NaOH, calculated free [Ca2+] ≈ 5 μM). The bath solution contained (in mM): NaCl 136, KCl 4, CaCl2 1, MgCl2 4, HEPES 10 (pH 7.4 with NaOH). Experiments were performed at room temperature and the perfusing solutions were applied by a gravity-driven perfusion system. Transfected cells were visualized by Dynabeads M-450 anti-CD8 antibody-coated beads (Dynal). Typical seal resistance was >10 GΩ. Cutoff frequency of the low pass filter of the amplifier was adjusted to 0.3 kHz, and data were acquired at 1 kHz with Digidata Interface 1200 (Axon Instruments, Inc.). BK currents were measured with a voltage protocol consisting of a 200-ms step to −100 mV followed by a 600-ms ramp to +60 mV, repeated every 2 s. The holding potential was −50 mV. Recording and data analysis were performed using pCLAMP software 6.0.4 (Axon Instruments, Inc.).
Amplex Red Assay for H2O2
H2O2 release was measured using Amplex red kits purchased from Molecular Probes, according to the instructions. The basis for this assay is the HRP-catalyzed oxidation of A6550 to the highly fluorescent and stable resorufin (Mohanty et al., 1997). Measurements were done in a volume of 120 μl, with typically 2 × 104 cells/well. Because actual cell numbers varied, results are expressed per cell. Concentrations of H2O2 were quantified colorimetrically. Absolute concentrations of H2O2 were determined from a calibration curve, which was not affected by addition of up to 10 mM ZnCl2. The default buffer in the Amplex Red assay includes phosphate. Phosphate binds Zn2+ with somewhat higher affinity than it binds Ca2+ (Smith and Martel, 1976) and zinc phosphate is only sparingly soluble (Immerwahr, 1901). For the experiments shown in this paper, except those in Fig. 8 C, we substituted (phosphate-free) Ringer's solution for the buffer to avoid zinc phosphate formation.
Figure 8.

Inhibition of H2O2 release by ZnCl2 and impairment of ZnCl2 inhibition of H2O2 release by phosphate. (A) Time course of H2O2 release by neutrophils in the presence of PMA alone (▪) or various concentrations of ZnCl2 (as indicated in mM). Mean ± SEM of four batches of cells is plotted. Cells were stimulated with 60 nM PMA at time = 0. (B) The cumulative total H2O2 release after 30 min is plotted as a function of ZnCl2 concentration (▪). Also plotted (red ▾) are data from cells treated with PMA and ZnCl2 but with the addition of the protonophore, CCCP. We used 12 μM CCCP, a concentration that is fivefold lower than our previous study (DeCoursey et al., 2003), because 60 μM CCCP appeared to partially inhibit 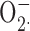 production. Curves show best-fitting Michaelis-Menten plots with Hill coefficient fixed at 1.0. (C) The data from B are replotted (▪) for comparison with identical measurements (n = 4) made using buffer that includes ∼0.4 mM phosphate (red •). Phosphate binds Zn2+ and consequently shifts the concentration–response relationship to the right (see text).
production. Curves show best-fitting Michaelis-Menten plots with Hill coefficient fixed at 1.0. (C) The data from B are replotted (▪) for comparison with identical measurements (n = 4) made using buffer that includes ∼0.4 mM phosphate (red •). Phosphate binds Zn2+ and consequently shifts the concentration–response relationship to the right (see text).
RESULTS
PAX and IbTX Do Not Inhibit Bacterial Killing by Human Neutrophils
If BK channels were required for the antibacterial potency of neutrophils, the BK inhibitors iberiotoxin (IbTX) and paxilline (PAX) should prevent this process. Recognizing that assays of bactericidal activity are not standardized and methods differ widely as to the inoculum size, relative concentrations of bacteria and phagocytes, opsonization conditions, and duration of incubation, we first used our established method of measuring the bactericidal activity of human neutrophils against S. aureus (Femling et al., 2005). In this assay, S. aureus opsonized with pooled human serum are promptly ingested by normal neutrophils, with phagocytosis complete within 10 min, and killed in an NADPH oxidase-dependent fashion (Femling et al., 2005). The antimicrobial activity of neutrophils was completely inhibited by treatment with diphenylene iodonium (DPI), a flavoprotein inhibitor that blocks the neutrophil NADPH oxidase (Fig. 1 A). In contrast, 100 nM IbTX did not prevent the killing of S. aureus by human neutrophils after 120 min incubation at 37°C. In five similar experiments, 300 nM PAX did not inhibit killing (unpublished data).
Given the observed discrepancy between the reported requirement for BK channels for neutrophil killing (Ahluwalia et al., 2004) and the lack of impact of IbTX or PAX, we conducted an independent set of experiments using a different killing assay in which bacterial survival was assessed by a recently described semi-automated method (Rada et al., 2004). Fig. 1 B illustrates survival of S. aureus at the end of 30 min incubation with PMN at 37°C compared with the initial number of bacteria. As in Fig. 1 A, DPI treatment prevented neutrophil-mediated bacterial killing, but IbTX had no effect. In these experiments, the ratio of bacteria:PMN was 10:1 in contrast with the 1:1 ratio used both in the study in Fig. 1 A and in that of Ahluwalia et al. (2004). Thus, although the neutrophils in the conditions of Fig. 1 B had a greater microbial challenge, they were equally effective in killing bacteria in the presence and absence of IbTX. Neither method of measuring bacterial killing indicated a role for BK channels in the antimicrobial action of human neutrophils against S. aureus. Both methods confirmed the requirement for NADPH oxidase activity. In summary, we were unable to identify conditions under which BK channel inhibitors had a detectable effect on the ability of human neutrophils to kill S. aureus.
IbTX Does Not Affect NADPH Oxidase-enhanced gIIA-PLA2-mediated Degradation of Bacterial Phospholipids
The importance of the NADPH oxidase to neutrophil antibacterial activity has been demonstrated repeatedly, primarily by measurements of its direct contribution to bacterial killing (e.g., Fig. 1). NADPH oxidase activity is also necessary for PMN to act in synergy with group IIA PLA2 (gIIA-PLA2) in the digestion of the principal S. aureus phospholipids phosphatidylglycerol (PG) and cardiolipin (CL) (Femling et al., 2005). To test whether BK channels are necessary for neutrophils to act in synergy with gIIA-PLA2, we compared the effects of DPI or IbTX on degradation of the phospholipids of S. aureus. Fig. 2 illustrates the effect of neutrophils and gIIA-PLA2 against S. aureus. As shown previously, purified gIIA-PLA2 degraded bacterial phospholipids. The addition of 150 ng/ml of gIIA-PLA2, a concentration at which the enzyme alone has negligible effects on phospholipids, resulted in significant degradation of the phospholipids of phagocytosed S. aureus. The addition of neutrophils increased phospholipid degradation >10-fold over that seen with the gIIA-PLA2 alone (Fig. 2), consistent with previous results (Femling et al., 2005). This synergy requires an intact and functional NADPH oxidase, as DPI completely abolished the enhanced phospholipid degradation. Were BK channels required for NADPH oxidase activity, their inhibition should likewise compromise phospholipid degradation. However, IbTX treatment did not affect this process, indicating that BK channels were not required for degradation of bacterial phospholipids.
Figure 2.

Synergistic degradation of bacterial phospholipids by gIIA-PLA2 and neutrophils is not affected by IbTX. The percent of the bacterial phospholipids phosphatidylglycerol (PG) and cardiolipin (CL) degraded in the presence of the indicated concentrations of exogenous gIIA-PLA2 in the presence or absence of neutrophils (PMN or None, respectively) is shown. DPI was added at 10 μM and IbTX at 100 nM. Mean ± SEM of three observations.
IbTX Does Not Inhibit NADPH Oxidase Activity
A possible role for BK channels in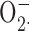 production by human neutrophils was excluded by measuring cytochrome c reduction stimulated with 100 nM PMA in the presence or absence of IbTX. Whereas 5 μM DPI practically abolished cytochrome c reduction to 4.3 ± 0.3% of control values (mean ± SEM, n = 3), 100 nM IbTX had no effect: 100.1 ± 1.6% of control. Thus, consistent with a previous study (Ahluwalia et al., 2004), we see no evidence that BK channels contribute to the respiratory burst of stimulated human neutrophils.
production by human neutrophils was excluded by measuring cytochrome c reduction stimulated with 100 nM PMA in the presence or absence of IbTX. Whereas 5 μM DPI practically abolished cytochrome c reduction to 4.3 ± 0.3% of control values (mean ± SEM, n = 3), 100 nM IbTX had no effect: 100.1 ± 1.6% of control. Thus, consistent with a previous study (Ahluwalia et al., 2004), we see no evidence that BK channels contribute to the respiratory burst of stimulated human neutrophils.
Absence of BK Channel Protein in Human Neutrophils
A prerequisite for BK channel involvement in innate immunity is the expression of this protein in neutrophils and eosinophils. In Fig. 3, unstimulated PMN were subjected to nitrogen cavitation and separation on a two layer Percoll gradient and the isolated subcellular fractions were probed, along with appropriate controls, by immunoblot for the maxi-K (BK) channel α subunit. The anti-BKα antibody, targeted against a conserved COOH-terminal epitope, detected endogenous BK protein (∼125 kD) in human myometrial smooth muscle cell lysate (lane 1) and BK channels transfected into HEK cells (lane 2). The protein was absent from the nontransfected HEK cell lysate (lane 3). The final three lanes include the neutrophil subcellular fractions, γ, β, and α, that contain the secretory vesicles/plasma membrane, secondary granules, and azurophilic granules, respectively (Borregaard et al., 1983). No BK channel protein was detected in any fraction from neutrophils. Analysis of relatively large quantities of neutrophil protein (100–200 μg of protein per fraction) did not result in detection of BK channel protein on immunoblots.
Figure 3.

BK channel protein is not detectable by immunoblot in human neutrophil fractions. All lanes contain an equal amount of protein (104 μg). From left to right, the lanes have (ML) human myometrial tissue lysate, (H+) lysate from HEK-293 cell transfected with BK channels, (H-) lysate from nontransfected HEK-293 cells, (γ) human neutrophil fraction that contains mainly secretory vesicles and plasma membrane, (β) human neutrophil fraction that contains mainly secondary granules, (α) human neutrophil fraction that contains mainly azurophilic granules.
Membrane Currents in Human Eosinophils Are Inhibited by Zn2+, but not by IbTX
If the level of BK channel expression were very low, protein might be functional yet undetectable by immunoblotting. However, a single BK channel could be detected by electrophysiological recording, due to the characteristically large single-channel currents and the sensitivity of this assay. Fig. 4 illustrates typical ionic currents in human eosinophils elicited by identical families of depolarizing pulses. This eosinophil was studied in perforated-patch configuration, which preserves intracellular constituents and many signaling pathways. In unstimulated cells (Fig. 4 A), H+ currents turned on slowly during depolarizing pulses to +40 mV and more positive voltages. There was no other detectable conductance in the voltage range examined, despite the high K+ concentration in the pipette solution, and hence in the cell.
Figure 4.

Currents in an eosinophil studied in perforated-patch configuration with K-containing pipette solution. Identical families of pulses (inset) were applied (A) before stimulation, (B) 5.6 min after PMA addition, (C) 2.3 min after addition of 100 nM IbTX, (D) and after addition of 3 mM ZnCl2. Arrows indicate zero current. From a holding potential of −60 mV, 8-s pulses were applied every 20 s in 20-mV increments from −40 to +80 mV. The total inward current (leak plus electron current) was −1.8 pA in A, −8.8 pA in B, −10.8 pA in C, and −8.5 pA in D. Taking the time the family in A was recorded as 0 min, the family in B was started at 11.0 min, C at 15.1 min, and D at 24.7 min. Filter, 1 kHz, bath solution pH 7.0 TMAMeSO3 and pipette solution KMeSO3; both solutions contain 50 mM 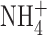 .
.
In a previous study (Ahluwalia et al., 2004), large currents identified pharmacologically as BK currents appeared after PMA stimulation of human neutrophils and eosinophils studied in perforated-patch configuration. In our studies, the only effect of PMA on eosinophils or neutrophils in perforated-patch configuration was to activate electron current and enhance proton currents, as illustrated in Fig. 4 B. Several minutes after PMA stimulation, the proton currents displayed properties typical of activated phagocytes (Bánfi et al., 1999; DeCoursey et al., 2000): they turned on more rapidly upon depolarization, turned off more slowly upon repolarization, and were larger in amplitude. In addition, the voltage at which H+ channels began to open, V threshold, was shifted ∼40 mV more negative, resulting in distinct H+ conductance at 0 mV. By comparison with zero current (indicated by the arrows), the “holding current” at −60 mV increased from −2 pA in Fig. 4 A to −9 pA in Fig. 4 B. The difference between these values reflects −7 pA of electron current generated by NADPH oxidase activity. No other discernable conductance was present.
To determine whether a small fraction of outward current might be BK current, we added 100 nM IbTX to the bath (Fig. 4 C). There was no effect on any current component. Both the proton and electron currents increased slightly, reflecting progression of PMA activation during the 7-min interval between the records in Fig. 4 (B and C). Currents during identical pulses immediately before and after addition or washout of 100 nM IbTX revealed no detectable effect in eosinophils or neutrophils either before (n = 5 and 2, respectively) or after PMA stimulation (n = 5 and 2, respectively). Addition of 3 mM ZnCl2 to the bath (Fig. 4 D) abolished the proton current and had little or no effect on the electron current. Proton currents are sensitive to Zn2+, but 3 mM Zn2+ had little effect on BK currents in A549 cells (not depicted) or on the putative BK currents reported in phagocytes (Ahluwalia et al.., 2004). After elimination of H+ current by Zn2+, no outward current remained in this cell. Finally, we added 6 μM DPI, which was reported to inhibit BK currents in eosinophils (Ahluwalia et al., 2004). DPI is a classical inhibitor of NADPH oxidase (Cross and Jones, 1986) and does not block proton channels in human neutrophils or eosinophils (Bánfi et al., 1999; DeCoursey et al., 2000; 2001). DPI abolished the electron current (not depicted), but had no other discernable effect. This result was anticipated, because after eliminating the proton current with Zn2+ and the electron current with DPI, no identifiable current remained. The whole cell membrane resistance measured between −60 and 0 mV was 86 GΩ in this cell in the presence of Zn2+, and averaged 53 ± 4 GΩ (mean ± SEM, n = 11). Examination of 140 eosinophils studied in perforated-patch configuration with K+-containing pipette solutions in which voltage-clamp families were recorded before and after PMA stimulation, including some experiments that were reported elsewhere (Morgan et al., 2003), revealed no outward K+ currents. We never detected BK currents in 20 neutrophils studied with K+-containing pipette solutions (perforated-patch and whole-cell), although other K+ conductances of modest amplitude were occasionally present. Voltage-gated proton currents were present in every viable cell studied. Some cells had or developed a large conductance with no distinguishing features (no reproducible voltage or time dependence), which we considered evidence of membrane damage, loss of seal resistance, or cell death. This large conductance was never affected by IbTX.
BK Channels Are Present in Other Cells, but not in Human Eosinophils
As a control, we examined BK currents in macrophage-related THP-1 cells. The characteristic appearance of individual BK channel openings can be seen in Fig. 5 A in a cell-attached patch on a THP-1 cell incubated for 3 d with PMA. This treatment resulted in the expression of inwardly rectifying K+ channels and BK channels (DeCoursey et al., 1996), both of which are evident in Fig. 5 A. At this gain, the numerous individual IR channels were not clearly distinguished and appeared as a macroscopic inward current. The BK channels had characteristically large unitary conductance, and five discrete levels can be seen as the channels open stochastically with depolarization.
Figure 5.

BK channels are present in macrophages but not eosinophils. (A) Appearance of BK channel currents in a cell-attached patch on a PMA-treated THP-1 cell. In this cell line, PMA induces the expression of inwardly rectifying K+ channels (IR) and large-conductance Ca2+-activated K+ channels (BK). The patch membrane was ramped from −40 to +100 mV relative to the resting potential of the cell. By the end of the ramp, five BK channels are open. The pipette contained 10 μM Ca2+ KMeSO3 and the bath contained Ringer's solution. Filter was 2 kHz. (B) Confirmation that at the mole fractions of 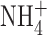 and K+ used in perforated-patch studies, BK channel currents remain large and readily detectable. A single leak-subtracted current record is shown, recorded during a voltage ramp in a cell-attached patch on a PMA-treated THP-1 cell. The leak current was obtained by averaging segments of currents in which no channels were open during several identical ramps. In cell-attached patches, the exact membrane potential is not known, because it depends on the resting potential of the cell. (C) Absence of BK channels in a PMA-activated human eosinophil in perforated-patch configuration. The voltage was ramped from −120 to +240 mV; currents during six consecutive ramps are superimposed. The bath included 5 mM ZnCl2 to inhibit proton current. Previously, addition of PMA had evoked −6 pA of electron current at −60 mV. During voltage ramps, the current may be offset (up or down) by an unknown constant amount (probably a few pA) due to residual uncompensated capacitance; therefore the horizontal lines do not necessarily represent zero current. The records were not corrected for leak current.
and K+ used in perforated-patch studies, BK channel currents remain large and readily detectable. A single leak-subtracted current record is shown, recorded during a voltage ramp in a cell-attached patch on a PMA-treated THP-1 cell. The leak current was obtained by averaging segments of currents in which no channels were open during several identical ramps. In cell-attached patches, the exact membrane potential is not known, because it depends on the resting potential of the cell. (C) Absence of BK channels in a PMA-activated human eosinophil in perforated-patch configuration. The voltage was ramped from −120 to +240 mV; currents during six consecutive ramps are superimposed. The bath included 5 mM ZnCl2 to inhibit proton current. Previously, addition of PMA had evoked −6 pA of electron current at −60 mV. During voltage ramps, the current may be offset (up or down) by an unknown constant amount (probably a few pA) due to residual uncompensated capacitance; therefore the horizontal lines do not necessarily represent zero current. The records were not corrected for leak current.
Because BK channels are gated by both elevated intracellular free calcium concentration, [Ca2+]i, and depolarization, the likelihood of their opening increases with depolarization. We did not observe BK channel currents in eosinophils, even during extreme depolarization. We reanalyzed data that were collected for another study (DeCoursey et al., 2003) in which the conditions serendipitously were ideal for revealing BK channels. The bath solution included 3–5 mM Zn2+ to eliminate the hundreds of picoamperes of H+ current that would otherwise be activated during extreme depolarization. Fig. 5 C illustrates six superimposed current records obtained during voltage ramps between −120 and +240 mV in an eosinophil in perforated-patch configuration with a pipette solution containing a high K+ concentration. This eosinophil was stimulated with PMA, activating 7–8 pA of electron current. After elimination of H+ currents by Zn2+, essentially no conductance remained in this cell. At this high gain, the opening of a single BK channel would be obvious; its amplitude would be ∼15 pA at +100 mV (Fig. 5 A). Although we occasionally observed small-conductance channel-like events, we never saw a credible example of a BK channel in 14 eosinophils studied in similar conditions, during repeated voltage ramps to extreme positive voltages (+180 to +240 mV). It should be noted that in perforated-patch configuration, current through the entire cell membrane is observed, in contrast with the experiment in Fig. 5 A, which shows channels in a small patch of cell membrane.
The perforated-patch experiments demonstrate that BK currents are absent from human neutrophils and eosinophils under physiological conditions in which both proton channels and NADPH oxidase respond vigorously to PMA. To determine whether BK currents can be detected under any conditions, in some perforated-patch experiments, we added Ca2+ ionophores, A23187 or ionomycin, to PMA-activated eosinophils. Little or no change was observed in the proton current, consistent with its being insensitive to [Ca2+]i in most studies (DeCoursey, 2003). No additional conductance appeared (not depicted). No BK channels were observed in cell-attached patches before or after stimulation with PMA, although BK channels are readily observed in cell-attached patch configuration in THP-1 cells (Fig. 5 B). We also studied neutrophils and eosinophils in whole-cell configuration with a pipette solution containing KMeSO3 with free Ca2+ buffered with EGTA to ∼1 or ∼10 μM. The bath contained either TMAMeSO3 or Ringer's solution. In these experiments, we observed voltage-gated proton currents of normal magnitude, voltage dependence, and kinetics, with proton selectivity confirmed by V rev measurements, and sometimes nonselective leak currents that reversed near 0 mV (not depicted). Despite using a variety of approaches that routinely reveal BK currents in other cells, we found no evidence that BK channels exist in human neutrophils or eosinophils.
Anomalous Mole-fraction Effects Do Not Account for the Absence of Detectable BK Currents in Human Eosinophils
One difference between our experimental conditions and those of Ahluwalia et al. (2004) is that in our perforated patch studies, we included 50 mM 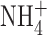 in both pipette and bath solutions to clamp pHi near pHo. Because
in both pipette and bath solutions to clamp pHi near pHo. Because 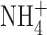 is permeant through BK channels (Blatz and Magleby, 1984; Hu et al., 1989), it was conceivable that anomalous mole-fraction effects (paradoxically smaller current in a mixture of permeant ions than in solutions of either ion alone) in a mixture of K+ and
is permeant through BK channels (Blatz and Magleby, 1984; Hu et al., 1989), it was conceivable that anomalous mole-fraction effects (paradoxically smaller current in a mixture of permeant ions than in solutions of either ion alone) in a mixture of K+ and 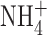 might attenuate the BK current unexpectedly. To test whether such an effect might account for the absence of BK current in our studies, we recorded BK currents in a cell-attached patch on a THP-1 cell, using 50
might attenuate the BK current unexpectedly. To test whether such an effect might account for the absence of BK current in our studies, we recorded BK currents in a cell-attached patch on a THP-1 cell, using 50 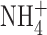 in bath and pipette solutions. Fig. 5 B illustrates a single leak-subtracted current record, obtained during a voltage ramp in which two BK channels opened. The amplitude of a single channel exceeds 10 pA. After the second BK channel opened, their combined amplitude decreased with further depolarization. This likely reflects voltage-dependent block by cytoplasmic Na+, a hallmark characteristic of BK channel currents in cell-attached patches (Marty, 1983). The presence of
in bath and pipette solutions. Fig. 5 B illustrates a single leak-subtracted current record, obtained during a voltage ramp in which two BK channels opened. The amplitude of a single channel exceeds 10 pA. After the second BK channel opened, their combined amplitude decreased with further depolarization. This likely reflects voltage-dependent block by cytoplasmic Na+, a hallmark characteristic of BK channel currents in cell-attached patches (Marty, 1983). The presence of 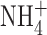 did not noticeably affect the BK current amplitude or the general appearance of openings. It is clear that under the ionic conditions used in this study, BK channels are readily detectable in cells that express them.
did not noticeably affect the BK current amplitude or the general appearance of openings. It is clear that under the ionic conditions used in this study, BK channels are readily detectable in cells that express them.
Confirmation that the IbTX and PAX Used in this Study Inhibit BK Current
IbTX is used routinely in studies of BK channel function (Galvez et al., 1990). Nevertheless, we confirmed that the toxins used in bacterial killing assays were active on BK currents. For this purpose, we used COS-7 cells transfected with BK channel DNA and two cell types that express native BK channels, the human alveolar epithelial cell line A549 (Jovanović et al., 2003), and the human monocytic leukemia cell line THP-1 after incubation with PMA (DeCoursey et al., 1996). The A549 cell shown in Fig. 6 was dialyzed with a pipette solution containing KMeSO3 with free Ca2+ buffered to nominally 10 μM, which activated large BK currents. The noisy outward currents activated rapidly, but not instantaneously, at voltages positive to 0 mV. Most of the outward current was inhibited by 100 nM IbTX (Fig. 6 B). Inhibition was not immediate, but progressed over a minute or two. Inhibition of macroscopic BK currents by 100 nM IbTX was observed in four PMA-treated THP-1 cells, in two A549 cells, and in six COS-7 cells transfected with BK channels. PAX (300 nM to 100 μM) inhibited BK currents in three THP-1 cells. Some of these studies were done using recently purchased blockers, other aliquots were identical to those used for the bacterial killing assays in Fig. 1 A. The blockers were effective under all conditions. The identical solutions used in Fig. 1 B inhibited BK currents in COS-7 cells transfected with BK channel DNA.
Figure 6.

Confirmation that the IbTX used in this study inhibits BK currents. (A) Whole-cell currents in an A549 cell (human alveolar epithelial cell line) with nominally 10 μM Ca2+ in a KMeSO3 pipette solution. The cell was held at −60 mV and pulses were applied in 20-mV increments from −80 to +100 mV. The bath contained Ringer's solution. (B) Currents in the same cell during the same pulses in the presence of 100 nM IbTX. Filter 2 kHz. Capacity 13 pF.
BK currents were studied in the A549 cell in Fig. 6 in Ringer's solution. To be sure that the ionic conditions of our measurements did not affect BK currents, we studied THP-1 cells using KMeSO3 in the pipette solution and TMAMeSO3 in the bath, with 50 mM 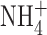 in both solutions. BK currents were observed during large depolarizations and were inhibited by PAX and IbTX. After the BK current was inhibited, slowly activating proton current remained (not depicted).
in both solutions. BK currents were observed during large depolarizations and were inhibited by PAX and IbTX. After the BK current was inhibited, slowly activating proton current remained (not depicted).
ZnCl2 Inhibits NADPH Oxidase Activity by Blocking Proton Channels
Numerous studies have supported the idea that Zn2+ inhibits NADPH oxidase activity (most often measured as the reduction of cytochrome c by 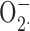 ), and that this inhibition is mediated by the blockade of charge compensation through the proton channel (see Discussion). After finding that Zn2+ could catalyze the conversion of
), and that this inhibition is mediated by the blockade of charge compensation through the proton channel (see Discussion). After finding that Zn2+ could catalyze the conversion of 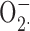 to H2O2, Ahluwalia et al. (2004) speculated that the inhibition of cytochrome c reduction by Zn2+ in previous studies was artifactual and that Zn2+ did not inhibit NADPH oxidase activity.
to H2O2, Ahluwalia et al. (2004) speculated that the inhibition of cytochrome c reduction by Zn2+ in previous studies was artifactual and that Zn2+ did not inhibit NADPH oxidase activity.
According to this interpretation (illustrated in the cartoon in Fig. 7), the previously observed results reflect the successful competition of Zn2+ with cytochrome c for interacting with 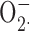 , rather than its inhibition of oxidase activity. To distinguish between these two activities of Zn2+, we measured ZnCl2 effects on H2O2 release using the Amplex Red assay. Fig. 8 A shows the average time course of H2O2 release from human neutrophils in the presence of a range of ZnCl2 concentrations. The corresponding ZnCl2 concentration–response relationship for inhibition of H2O2 release is plotted in Fig. 8 B (▪). To confirm that ZnCl2 did not interfere with the Amplex red assay, we repeated the H2O2 calibration curve in the presence of 3–10 mM ZnCl2, the highest concentrations used here, and saw no effect (not depicted). Thus, the inhibition of H2O2 release by ZnCl2 was due to reduction of NADPH oxidase activity via inhibition of proton current.
, rather than its inhibition of oxidase activity. To distinguish between these two activities of Zn2+, we measured ZnCl2 effects on H2O2 release using the Amplex Red assay. Fig. 8 A shows the average time course of H2O2 release from human neutrophils in the presence of a range of ZnCl2 concentrations. The corresponding ZnCl2 concentration–response relationship for inhibition of H2O2 release is plotted in Fig. 8 B (▪). To confirm that ZnCl2 did not interfere with the Amplex red assay, we repeated the H2O2 calibration curve in the presence of 3–10 mM ZnCl2, the highest concentrations used here, and saw no effect (not depicted). Thus, the inhibition of H2O2 release by ZnCl2 was due to reduction of NADPH oxidase activity via inhibition of proton current.
Figure 7.
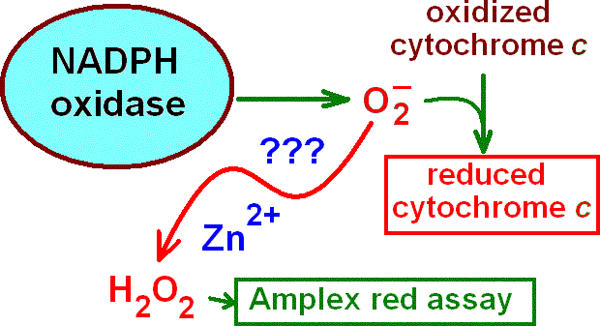
Zinc catalysis theory. A standard method of measuring 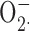 production is to assay the reduction of cytochrome c. It was proposed recently (Ahluwalia et al., 2004) that metals like Zn2+ or Cd2+ may rapidly convert
production is to assay the reduction of cytochrome c. It was proposed recently (Ahluwalia et al., 2004) that metals like Zn2+ or Cd2+ may rapidly convert 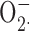 to H2O2 such that the observed cytochrome c reduction would be attenuated. If this were the case, then the H2O2 production rate would be unaffected by Zn2+. The data in Fig. 8 (Amplex red assay) disprove this theory.
to H2O2 such that the observed cytochrome c reduction would be attenuated. If this were the case, then the H2O2 production rate would be unaffected by Zn2+. The data in Fig. 8 (Amplex red assay) disprove this theory.
If Zn2+ acts by preventing H+ channel-mediated charge compensation, then its effects might be overcome by providing an alternative route. Fig. 8 B includes data (red ▾) in the presence of both ZnCl2 and the protonophore CCCP. As reported previously for cytochrome c measurements (Henderson et al., 1988b; DeCoursey et al., 2003), CCCP partially restored NADPH oxidase activity assessed by H2O2 release, suggesting that providing a proton pathway can overcome the loss of proton channels.
The Amplex red kit buffer contains phosphate, which binds Zn2+ avidly (Collier, 1979). Fig. 8 C illustrates the effect of including 0.4 mM phosphate in the assay solution. There is a rightward shift of the curve, with the midpoint shifting from 29 to 205 μM. Evidently, phosphate complexed with Zn2+ and reduced its effective concentration. Consequently, we used phosphate-free solutions for the H2O2 measurements in Fig. 8 (A and B).
DISCUSSION
BK Channels Are Absent and Play No Functional Role
The electrophysiology of the respiratory burst was reexamined in light of a recent study concluding that BK channels play an essential role in innate immunity (Ahluwalia et al., 2004). We found no evidence that BK channel inhibition affected superoxide anion production by stimulated neutrophils or compromised their ability to kill S. aureus. Killing assays were performed independently in two labs using different experimental methods and conditions. Under no experimental condition did we detect any effect of BK inhibitors on bacterial killing by human neutrophils. These observations contrast with those of Ahluwalia et al. (2004) and could reflect experimental conditions unique to their assays. To determine if IbTX or PAX had other effects on neutrophil antimicrobial activity, we tested the capacity of the NADPH oxidase to synergistically facilitate the degradation of bacterial phospholipids by gIIA-PLA2 (Femling et al., 2005). This activity was likewise unaffected by BK inhibitors but was abolished in the presence of DPI, demonstrating its absolute dependence on a functional NADPH oxidase. The capacity of the IbTX and PAX used in the killing and phospholipase degradation assays to inhibit BK channel currents was confirmed in cells expressing native BK channels and in COS-7 cells transfected with BK channels.
The lack of effect of BK channel inhibitors on these phagocyte antibacterial functions raises the question of whether BK channels are expressed in human neutrophils and eosinophils. Whereas an anti-BK channel antibody detected the 125-kD BK channel endogenously expressed by myometrial cells or present in HEK transfectants, no immunoreactive protein was detected in fractions isolated from human neutrophils. Despite examining >100 μg of protein per sample, we saw no immunoreactive protein in neutrophil fractions that might correspond with BK channel protein.
Even if the level of BK channel expression were too low to detect by immunoblot, BK channel currents should still be detected by electrophysiological recording because of their characteristically large single-channel currents. A single BK channel opening in the eosinophil plasma membrane would have been readily detected during voltage ramps using the perforated-patch configuration (Fig. 5 C). We observed no BK channel currents during voltage ramps up to +240 mV, roughly 200 mV beyond the depolarization that occurs during the respiratory burst in neutrophils (Geiszt et al., 1997; Jankowski and Grinstein, 1999; Rada et al., 2004) or eosinophils (Bánfi et al., 1999; Bankers-Fulbright et al., 2003). In the initial report of large BK currents in human eosinophils and neutrophils, the identification was based on inhibition of the current by IbTX and PAX (Ahluwalia et al., 2004). Neither K+ selectivity nor activation by increased [Ca2+]i was demonstrated. In addition, none of several previous studies of human eosinophils (Gordienko et al., 1996; Saito et al., 1997; Tare et al., 1998; Schwingshackl et al., 2000; Morgan et al., 2003) or neutrophils (von Tscharner et al., 1986; Krause and Welsh, 1990) reported BK currents. In the present study, we specifically searched for evidence of BK currents. In PMA-stimulated human eosinophils, only proton and electron currents were observed. Neither IbTX nor DPI inhibited any component of outward current.
Because our results differed so completely from those of Ahluwalia et al. (2004), we searched for experimental differences that might have interfered with detection of BK currents. One difference is that our pipette and bath solutions contained 50 mM 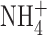 in order to clamp pHi near pHo. Although
in order to clamp pHi near pHo. Although 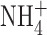 is permeant through many K+ channels (Hille, 2001), including BK channels (Blatz and Magleby, 1984; Hu et al., 1989), it was conceivable that anomalous mole-fraction effects might have attenuated the BK channel conductance. Anomalous mole-fraction effects in
is permeant through many K+ channels (Hille, 2001), including BK channels (Blatz and Magleby, 1984; Hu et al., 1989), it was conceivable that anomalous mole-fraction effects might have attenuated the BK channel conductance. Anomalous mole-fraction effects in 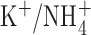 mixtures have been observed in several K+ channels (Eisenman et al., 1986; Wagoner and Oxford, 1987; Shapiro and DeCoursey, 1991). However, single-channel BK currents appeared normal in THP-1 cells exposed to the mole fractions of
mixtures have been observed in several K+ channels (Eisenman et al., 1986; Wagoner and Oxford, 1987; Shapiro and DeCoursey, 1991). However, single-channel BK currents appeared normal in THP-1 cells exposed to the mole fractions of 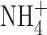 and K+ used in the perforated patch studies (Fig. 5 B). Another difference is that we controlled pHi using an
and K+ used in the perforated patch studies (Fig. 5 B). Another difference is that we controlled pHi using an 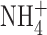 gradient, whereas pHi was uncontrolled in the cells studied by Ahluwalia et al. (2004). However, although BK channels are inhibited by low pHi (Brelidze and Magleby, 2004), their pH sensitivity is rather weak: BK current is reduced by <50% at pHi 5.0. Another factor, albeit one that did not differ in the two studies, is that in both studies, [Ca2+]i was uncontrolled. Although increasing [Ca2+]i promotes BK channel opening by shifting the voltage dependence of gating negatively, BK channels open above +100 mV even with nominally Ca2+-free intracellular solutions (Horrigan and Aldrich, 2002). BK channels were readily observed in cell-attached patches of THP-1 cells that contained only a few channels (Fig. 5 A), but were never observed when the entire eosinophil membrane was ramped to extreme positive voltages (Fig. 5 C).
gradient, whereas pHi was uncontrolled in the cells studied by Ahluwalia et al. (2004). However, although BK channels are inhibited by low pHi (Brelidze and Magleby, 2004), their pH sensitivity is rather weak: BK current is reduced by <50% at pHi 5.0. Another factor, albeit one that did not differ in the two studies, is that in both studies, [Ca2+]i was uncontrolled. Although increasing [Ca2+]i promotes BK channel opening by shifting the voltage dependence of gating negatively, BK channels open above +100 mV even with nominally Ca2+-free intracellular solutions (Horrigan and Aldrich, 2002). BK channels were readily observed in cell-attached patches of THP-1 cells that contained only a few channels (Fig. 5 A), but were never observed when the entire eosinophil membrane was ramped to extreme positive voltages (Fig. 5 C).
We searched for BK currents in whole-cell configuration in neutrophils and/or eosinophils using pipette solutions buffered to high [Ca2+] (1–10 μM). We also searched for single-channel BK currents in cell-attached patches, as was done in THP-1 cells in Fig. 5 A, before, during, and after stimulation of the cell with PMA. In neither configuration did we detect BK currents. However, Ahluwalia et al. (2004) reported that BK currents were observed mainly in perforated-patch configuration after stimulation with PMA. Therefore, most of our efforts were focused on this approach. A hybrid approach was used previously in human eosinophils, in which the pipette solution in whole-cell configuration (zero current mode) included NADPH, GTP-γs, and other factors to preserve NADPH oxidase function (Bánfi et al., 1999). In that study, despite high [K+] in the pipette solution, the membrane potential closely approached E H over a wide range of pH, indicating that the predominant conductance was H+ selective, not K+ selective.
It is astonishing that we consistently observe voltage-gated proton channels and have never seen evidence of BK channels in human neutrophils or eosinophils, whereas Ahluwalia et al. (2004) reported nanoamperes of BK current and no evidence of proton currents. It is extremely improbable that these apparent differences in ion channel expression reflect donor variability. Over the period that our studies were done, we used 50 different normal blood donors, and several donors with chronic granulomatous disease. We have never found a donor whose granulocytes lacked proton currents, and we have never found a donor whose granulocytes expressed BK channels. As discussed previously (DeCoursey, 2004), numerous other labs have studied human neutrophils and eosinophils under conditions of high cytoplasmic K+ concentration, and none has reported BK currents, whereas proton currents have been observed consistently. Our results are in agreement with the existing literature in this area.
It is well established that the electrogenic activity of NADPH oxidase results in membrane depolarization (Henderson et al., 1987). Several groups have reported massive depolarization (well positive to 0 mV) of the plasma membrane in human eosinophils or neutrophils stimulated with PMA (Geiszt et al., 1997; Bánfi et al., 1999; Jankowski and Grinstein, 1999; Bankers-Fulbright et al., 2003; Rada et al., 2004). Recently, the electrical events during the respiratory burst were modeled quantitatively, using empirical values for proton channel properties and expression, and for the electrogenic properties of NADPH oxidase (Murphy and DeCoursey, 2006). It was found that depolarization of the plasma membrane beyond 0 mV could occur only if any postulated K+ conductance was <240 pS, the conductance of a single BK channel. A large K+ conductance would simply clamp the membrane potential near E K, and depolarization could not occur.
Do Potassium Fluxes Play a Role?
Despite the absence of BK channels in human neutrophils and eosinophils, agonist-stimulated K+ and Rb+ fluxes have been measured during the respiratory burst (Reeves et al., 2002; Ahluwalia et al., 2004; Rada et al., 2004, 2005). The identity of the transport mechanism remains unknown; it could be a carrier, antiporter, symporter, pump, channel, or membrane leak. Indirect evidence that K+ (by analogy with Rb+) flux is electrogenic comes from a linear relationship between membrane depolarization and Rb+ flux (Rada et al., 2004; Rada et al., 2005). Rada et al. (2005) proposed that a g K sets the resting potential in neutrophils, and that as NADPH oxidase activity increases, proton fluxes increasingly dominate the charge compensation process. The Rb+ fluxes in these studies were measured across the plasma membrane, not the phagosome membrane. NADPH oxidase assembles in the plasma membrane in eosinophils and PMA-stimulated neutrophils, but the charge compensation mechanism may differ drastically from that which occurs in the phagosome, where bacteria are killed by neutrophils. As proposed by Reeves et al. (2002), any charge compensation by K+ flux rather than H+ flux from cytoplasm into the phagosome would tend to increase pHphagosome. Several factors (osmotic, electrical, biochemical, pH) constrain the possible mechanism of charge compensation in the phagosome (Murphy and DeCoursey, 2006). These constraints combine to limit the extent of charge compensation by any combination of K+ influx into the phagosome and Cl− efflux to ∼5% of the total (Murphy and DeCoursey, 2006), in agreement with the estimate by Reeves et al. (2002) that ∼6% is due to K+. Identification of the pathway that mediates K+ flux would facilitate understanding of this process.
Proton Channels Are Required for NADPH Oxidase Activity
Some of the most compelling data implicating proton channels as the major vehicle for charge compensation during NADPH oxidase activity comes from inhibition of NADPH activity, assessed by cytochrome c reduction assays, by divalent metal inhibitors of proton currents. Most such studies were done with Zn2+ (Beswick et al., 1986; Henderson et al., 1988b; Simchowitz et al., 1990; Lowenthal and Levy, 1999; Bankers-Fulbright et al., 2001; DeCoursey et al., 2003; Rada et al., 2004), but a few used Cd2+ (Henderson et al., 1988a; Simchowitz et al., 1990; Kapus et al., 1992). Both metals are classical inhibitors of voltage-gated proton channels (Thomas and Meech, 1982; Cherny and DeCoursey, 1999; DeCoursey, 2003). Ahluwalia et al. (2004) confirmed that 3 mM Zn2+ or Cd2+ inhibited cytochrome c reduction, but saw no inhibition of respiratory burst-associated O2 consumption. This result was surprising, because in four previous studies, respiratory burst-associated O2 consumption was inhibited by >60% by Zn2+ at 1 mM or less (Stankova et al., 1976; Chvapil et al., 1977; Yatsuyanagi and Ogiso, 1988; Ogino et al., 1994). We observed 53% inhibition of PMA-stimulated O2 consumption by 1 mM Zn2+ in measurements using an oxygen electrode. Ahluwalia et al. (2004) reported that Zn2+ or Cd2+ could induce dismutation of chemically generated 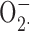 to H2O2 and thereby concluded that these metals do not inhibit NADPH oxidase activity but instead interfere with the cytochrome c assay, as illustrated in Fig. 7. Although several types of control measurements in previous studies indicated that this mechanism did not occur under conditions in which the cytochrome c reduction method has been used (see DeCoursey, 2004), we tested the theory directly by assessing H2O2 production. If Zn2+ did not inhibit NADPH oxidase function but instead caused rapid dismutation of
to H2O2 and thereby concluded that these metals do not inhibit NADPH oxidase activity but instead interfere with the cytochrome c assay, as illustrated in Fig. 7. Although several types of control measurements in previous studies indicated that this mechanism did not occur under conditions in which the cytochrome c reduction method has been used (see DeCoursey, 2004), we tested the theory directly by assessing H2O2 production. If Zn2+ did not inhibit NADPH oxidase function but instead caused rapid dismutation of 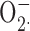 , thereby precluding cytochrome c reduction (Fig. 7), then Zn2+ should not decrease H2O2 production. Contrary to this prediction, we observed progressive and profound reduction of H2O2 production with increasing [Zn2+]. At 1 mM, Zn2+ inhibited H2O2 release by ∼90%, mirroring its inhibition of cytochrome c reduction (DeCoursey et al., 2003), thus supporting the obligatory link between proton channel activity and NADPH oxidase function.
, thereby precluding cytochrome c reduction (Fig. 7), then Zn2+ should not decrease H2O2 production. Contrary to this prediction, we observed progressive and profound reduction of H2O2 production with increasing [Zn2+]. At 1 mM, Zn2+ inhibited H2O2 release by ∼90%, mirroring its inhibition of cytochrome c reduction (DeCoursey et al., 2003), thus supporting the obligatory link between proton channel activity and NADPH oxidase function.
Zn2+ and Cd2+ act on proton channels, not on NADPH oxidase per se; Zn2+ has no effect in a cell-free system in which NADPH oxidase is reconstituted without proton channels (Yatsuyanagi and Ogiso, 1988). Three types of evidence indicate that the function of proton channels is charge compensation. First, providing an alternative proton efflux pathway by a protonophore (CCCP, Fig. 8 B) partially restores function (Henderson et al., 1988b; DeCoursey et al., 2003). Second, Zn2+ does not directly inhibit NADPH oxidase-generated electron current despite abolishing proton flux in voltage-clamped cells (Schrenzel et al., 1998; DeCoursey et al., 2003), because the voltage clamp amplifier automatically compensates charge. Finally, Zn2+ and Cd2+ exacerbate the depolarization that occurs during the respiratory burst (Henderson et al., 1987; Bánfi et al., 1999; Rada et al., 2004; Demaurex and Petheõ, 2005; Murphy and DeCoursey, 2006). We demonstrate here that proton channels are necessary for sustained NADPH oxidase function. We did not directly test whether proton channels are required for bacterial killing. However, we would expect this to be the case, to the extent that reactive oxygen species generated by NADPH oxidase are involved in killing. The recent identification of voltage-gated proton channel genes should facilitate testing this hypothesis (Ramsey et al., 2006; Sasaki et al., 2006).
In summary, we did not detect protein or functional evidence for BK channels in human neutrophils or eosinophils. Furthermore, BK channel inhibitors had no effect on the antibacterial activity of neutrophils as assessed by bacterial killing, 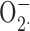 production, or bacterial phospholipid degradation, events that are dependent on a functional NADPH oxidase. In contrast, we have confirmed that voltage-gated proton channels in human eosinophils and neutrophils are required to sustain the respiratory burst, and probably do so by compensating charge.
production, or bacterial phospholipid degradation, events that are dependent on a functional NADPH oxidase. In contrast, we have confirmed that voltage-gated proton channels in human eosinophils and neutrophils are required to sustain the respiratory burst, and probably do so by compensating charge.
Acknowledgments
The authors thank Prof. M. Lazdunski and Dr. F. Lesage (CNRS-Universite de Nice-Sophia Antipolis, Valbonne, France) for pIRES-CD8 plasmid, Prof. F. Antoni (University of Edinburgh, Scotland, UK) for pcDNA3-BK-zero-HA construct coding for the HA-tagged high conductance calcium-activated K+ channel, and Drs. Tatiana Iastrebova and Jamie Schlomann for excellent technical assistance.
This work was supported in part by the Hungarian Research Fund (OTKA T37755, T46954, and Ts49851 to E. Ligeti), the Hungarian Ministry of Health (to E. Ligeti and P. Enyedi), National Institute of Allergy and Infectious Disease at the National Institutes of Health (AI34879 to W.M. Nauseef), and by the Heart, Lung, and Blood Institute of the National Institutes of Health (research grants HL52671 and HL61437 to T. DeCoursey).
David C. Gadsby served as editor.
Note added in proof. In agreement with the data presented in Fig. 1, Decleva et al. (Decleva, E., F. Defendi, R. Menegazzi, S. Busetto, P. Patriarca, and P. Dri. 2006. Eur. J. Clin. Invest. 36:41. Abstr.) recently reported that they also found no effect of IbTX or PAX on the ability of neutrophils to kill S. aureus or C. albicans.
Abbreviations used in this paper: BK channel, large-conductance, calcium-activated, potassium-selective channel; CCCP, carbonyl cyanide m-chlorophenylhydrazone; DPI, diphenylene iodonium; HRP, horseradish peroxidase; IbTX, iberiotoxin; IK, intermediate-conductance Ca2+-activated K+; KMeSO3, potassium methanesulfonate; PAX, paxilline; PMN, polymorphonuclear leukocytes.
References
- Ahluwalia, J., A. Tinker, L.H. Clapp, M.R. Duchen, A.Y. Abramov, S. Page, M. Nobles, and A.W. Segal. 2004. The large conductance Ca2+-activated K+ channel is essential for innate immunity. Nature. 427:853–858. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Babior, B.M., J.D. Lambeth, and W. Nauseef. 2002. The neutrophil NADPH oxidase. Arch. Biochem. Biophys. 397:342–344. [DOI] [PubMed] [Google Scholar]
- Bánfi, B., J. Schrenzel, O. Nüsse, D.P. Lew, E. Ligeti, K.-H. Krause, and N. Demaurex. 1999. A novel H+ conductance in eosinophils: unique characteristics and absence in chronic granulomatous disease. J. Exp. Med. 190:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankers-Fulbright, J.L., G.J. Gleich, G.M. Kephart, H. Kita, and S.M. O'Grady. 2003. Regulation of eosinophil membrane depolarization during NADPH oxidase activation. J. Cell Sci. 116:3221–3226. [DOI] [PubMed] [Google Scholar]
- Bankers-Fulbright, J.L., H. Kita, G.J. Gleich, and S.M. O'Grady. 2001. Regulation of human eosinophil NADPH oxidase activity: a central role for PKCδ. J. Cell. Physiol. 189:306–315. [DOI] [PubMed] [Google Scholar]
- Beswick, P.H., P.C. Brannen, and S.S. Hurles. 1986. The effects of smoking and zinc on the oxidative reactions of human neutrophils. J. Clin. Lab. Immunol. 21:71–75. [PubMed] [Google Scholar]
- Blatz, A.L., and K.L. Magleby. 1984. Ion conductance and selectivity of single calcium-activated potassium channels in cultured rat muscle. J. Gen. Physiol. 84:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borregaard, N., J.M. Heiple, E.R. Simons, and R.A. Clark. 1983. Subcellular localization of the b-cytochrome component of the human neutrophil microbicidal oxidase: translocation during activation. J. Cell Biol. 97:52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyum, A. 1968. Isolation of mononuclear cells and granulocytes from human blood. Isolation of monocular cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand. J. Clin. Lab. Invest. Suppl. 97:77–89. [PubMed] [Google Scholar]
- Brainard, A.M., A.J. Miller, J.R. Martens, and S.K. England. 2005. Maxi-K channels localize to caveolae in human myometrium: a role for an actin-channel-caveolin complex in the regulation of myometrial smooth muscle K+ current. Am. J. Physiol. Cell Physiol. 289:C49–C57. [DOI] [PubMed] [Google Scholar]
- Brelidze, T.I., and K.L. Magleby. 2004. Protons block BK channels by competitive inhibition with K+ and contribute to the limits of unitary currents at high voltages. J. Gen. Physiol. 123:305–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherny, V.V., and T.E. DeCoursey. 1999. pH-dependent inhibition of voltage-gated H+ currents in rat alveolar epithelial cells by Zn2+ and other divalent cations. J. Gen. Physiol. 114:819–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung, A.L., C.C. Nast, and A.S. Bayer. 1998. Selective activation of sar promoters with the use of green fluorescent protein transcriptional fusions as the detection system in the rabbit endocarditis model. Infect. Immun. 66:5988–5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chvapil, M., L. Stankova, D.S. Bernhard, P.L. Weldy, E.C. Carlson, and J.B. Campbell. 1977. Effect of zinc on peritoneal macrophages in vitro. Infect. Immun. 16:367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, R.A., K.G. Leidal, D.W. Pearson, and W.M. Nauseef. 1987. NADPH oxidase of human neutrophils. Subcellular localization and characterization of an arachidonate-activatable superoxide-generating system. J. Biol. Chem. 262:4065–4074. [PubMed] [Google Scholar]
- Collier, H.B. 1979. Binding of Zn2+ by buffers. Clin. Chem. 25:495–496. [PubMed] [Google Scholar]
- Cross, A.R., and O.T.G. Jones. 1986. The effect of the inhibitor diphenylene iodonium on the superoxide-generating system of neutrophils. Specific labelling of a component polypeptide of the oxidase. Biochem J. 237:111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross, A.R., and O.T.G. Jones. 1991. Enzymic mechanisms of superoxide production. Biochim. Biophys. Acta. 1057:281–298. [DOI] [PubMed] [Google Scholar]
- DeCoursey, T.E. 2003. Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 83:475–579. [DOI] [PubMed] [Google Scholar]
- DeCoursey, T.E. 2004. During the respiratory burst, do phagocytes need proton channels or potassium channels or both? Sci. STKE. 2004:pe21. [DOI] [PubMed]
- DeCoursey, T.E., and V.V. Cherny. 1993. Potential, pH, and arachidonate gate hydrogen ion currents in human neutrophils. Biophys. J. 65:1590–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey, T.E., V.V. Cherny, A.G. DeCoursey, W. Xu, and L.L. Thomas. 2001. Interactions between NADPH oxidase-related proton and electron currents in human eosinophils. J. Physiol. 535:767–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey, T.E., V.V. Cherny, W. Zhou, and L.L. Thomas. 2000. Simultaneous activation of NADPH oxidase-related proton and electron currents in human neutrophils. Proc. Natl. Acad. Sci. USA. 97:6885–6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey, T.E., S.Y. Kim, M.R. Silver, and F.N. Quandt. 1996. III. Ion channel expression in PMA-differentiated human THP-1 macrophages. J. Membr. Biol. 152:141–157. [DOI] [PubMed] [Google Scholar]
- DeCoursey, T.E., D. Morgan, and V.V. Cherny. 2003. The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature. 422:531–534. [DOI] [PubMed] [Google Scholar]
- Demaurex, N., and G.L. Petheõ. 2005. Electron and proton transport by NADPH oxidases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360:2315–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman, G., R. Latorre, and C. Miller. 1986. Multi-ion conduction and selectivity in the high-conductance Ca++-activated K+ channel from skeletal muscle. Biophys. J. 50:1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Femling, J.K., W.M. Nauseef, and J.P. Weiss. 2005. Synergy between extracellular Group IIA phospholipase A2 and phagocyte NADPH oxidase in digestion of phospholipids of Staphylococcus aureus ingested by human neutrophils. J. Immunol. 175:4653–4661. [DOI] [PubMed] [Google Scholar]
- Gallin, E.K. 1984. Calcium and voltage activated potassium channels in human macrophages. Biophys. J. 46:821–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez, A., G. Gimenez-Gallego, J.P. Reuben, L. Roy-Contancin, P. Feigenbaum, G.J. Kaczorowski, and M.L. Garcia. 1990. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J. Biol. Chem. 265:11083–11090. [PubMed] [Google Scholar]
- Geiszt, M., A. Kapus, and E. Ligeti. 2001. Chronic granulomatous disease: more than the lack of superoxide? J. Leukoc. Biol. 69:191–196. [PubMed] [Google Scholar]
- Geiszt, M., A. Kapus, K. Nemet, L. Farkas, and E. Ligeti. 1997. Regulation of capacitative Ca2+ influx in human neutrophil granulocytes. Alterations in chronic granulomatous disease. J. Biol. Chem. 272:26471–26478. [DOI] [PubMed] [Google Scholar]
- Gordienko, D.V., M. Tare, S. Parveen, C.J. Fenech, C. Robinson, and T.B. Bolton. 1996. Voltage-activated proton current in eosinophils from human blood. J. Physiol. 496:299–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, L.M., J.B. Chappell, and O.T.G. Jones. 1987. The superoxide-generating NADPH oxidase of human neutrophils is electrogenic and associated with an H+ channel. Biochem. J. 246:325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, L.M., J.B. Chappell, and O.T.G. Jones. 1988. a. Internal pH changes associated with the activity of NADPH oxidase of human neutrophils: further evidence for the presence of an H+ conducting channel. Biochem. J. 251:563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, L.M., J.B. Chappell, and O.T.G. Jones. 1988. b. Superoxide generation by the electrogenic NADPH oxidase of human neutrophils is limited by the movement of a compensating charge. Biochem. J. 255:285–290. [PMC free article] [PubMed] [Google Scholar]
- Hille, B. 2001. Ion Channels of Excitable Membranes. Third edition. Sinauer Associates, Inc., Sunderland, MA. 814 pp.
- Horrigan, F.T., and R.W. Aldrich. 2002. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J. Gen. Physiol. 120:267–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, S.L., Y. Yamamoto, and C.Y. Kao. 1989. Permeation, selectivity, and blockade of the Ca2+-activated potassium channel of the guinea pig Taenia coli myocyte. J. Gen. Physiol. 94:849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immerwahr, C. 1901. Beiträge zur Kenntnis der Löslichkeit von Schwermetall-Niederschlägen auf Elektrochemischem Wege. Zeitschrift für Elektrochemie. 7:477–483. [Google Scholar]
- Jankowski, A., and S. Grinstein. 1999. A noninvasive fluorometric procedure for measurement of membrane potential. J. Biol. Chem. 274:26098–26104. [DOI] [PubMed] [Google Scholar]
- Jovanović, S., R.M. Crawford, H.J. Ranki, and A. Jovanović. 2003. Large conductance Ca2+-activated K+ channels sense acute changes in oxygen tension in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 28:363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapus, A., K. Szászi, and E. Ligeti. 1992. Phorbol 12-myristate 13-acetate activates an electrogenic H+-conducting pathway in the membrane of neutrophils. Biochem. J. 281:697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.Y., M.R. Silver, and T.E. DeCoursey. 1996. I. Ion channels in human THP-1 monocytes. J. Membr. Biol. 152:117–130. [DOI] [PubMed] [Google Scholar]
- Krause, K.-H., and M.J. Welsh. 1990. Voltage-dependent and Ca2+-activated ion channels in human neutrophils. J. Clin. Invest. 85:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, W.L., R.E. Harrison, and S. Grinstein. 2003. Phagocytosis by neutrophils. Microbes Infect. 5:1299–1306. [DOI] [PubMed] [Google Scholar]
- Lieber, M., B. Smith, A. Szakal, W. Nelson-Rees, and G. Todaro. 1976. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int. J. Cancer. 17:62–70. [DOI] [PubMed] [Google Scholar]
- Lowenthal, A., and R. Levy. 1999. Essential requirement of cytosolic phospholipase A2 for activation of the H+ channel in phagocyte-like cells. J. Biol. Chem. 274:21603–21608. [DOI] [PubMed] [Google Scholar]
- Marty, A. 1983. Blocking of large unitary calcium-dependent potassium currents by internal sodium ions. Pflügers Arch. 396:179–181. [DOI] [PubMed] [Google Scholar]
- Mohanty, J.G., J.S. Jaffe, E.S. Schulman, and D.G. Raible. 1997. A highly sensitive fluorescent micro-assay of H2O2 release from activated human leukocytes using a dihydroxyphenoxazine derivative. J. Immunol. Methods. 202:133–141. [DOI] [PubMed] [Google Scholar]
- Morgan, D., V.V. Cherny, R. Murphy, W. Xu, L.L. Thomas, and T.E. DeCoursey. 2003. Temperature dependence of NADPH oxidase in human eosinophils. J. Physiol. 550:447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, R., and T.E. DeCoursey. 2006. Charge compensation in phagocytes. Biochim. Biophys. Acta: Bioenergetics. In press. [DOI] [PubMed]
- Ogino, K., Y. Izumi, H. Segawa, Y. Takeyama, H. Ishiyama, T. Houbara, T. Uda, and S. Yamashita. 1994. Zinc hydroxide induced respiratory burst in rat neutrophils. Eur. J. Pharmacol. 270:73–78. [DOI] [PubMed] [Google Scholar]
- Rada, B.K., M. Geiszt, K. Káldi, C. Tímár, and E. Ligeti. 2004. Dual role of phagocytic NADPH oxidase in bacterial killing. Blood. 104:2947–2953. [DOI] [PubMed] [Google Scholar]
- Rada, B.K., M. Geiszt, C. Hably, and E. Ligeti. 2005. Consequences of the electrogenic function of the phagocytic NADPH oxidase. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360:2293–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey, I.S., M.M. Moran, J.A. Chong, and D.E. Clapham. 2006. A voltage-gated proton-selective channel lacking the pore domain. Nature. 440:1213–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves, E.P., H. Lu, H.L. Jacobs, C.G.M. Messina, S. Bolsover, G. Gabella, E.O. Potma, A. Warley, J. Roes, and A.W. Segal. 2002. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 416:291–297. [DOI] [PubMed] [Google Scholar]
- Saito, M., R. Sato, N.M. Muňoz, A. Herrnreiter, M. Oyaizu, H. Kasugai, T. Narahashi, and A.R. Leff. 1997. Association of granular exocytosis with Ca2+-activated K+ channels in human eosinophils. Am. J. Physiol. 273:L16–L21. [DOI] [PubMed] [Google Scholar]
- Sasaki, M., M. Takagi, and Y. Okamura. 2006. A voltage sensor-domain protein is a voltage-gated proton channel. Science. 312:589–592. [DOI] [PubMed] [Google Scholar]
- Schilling, T., H. Repp, H. Richter, A. Koschinski, U. Heinemann, F. Dreyer, and C. Eder. 2002. Lysophospholipids induce membrane hyperpolarization in microglia by activation of IKCa1 Ca2+-dependent K+ channels. Neuroscience. 109:827–835. [DOI] [PubMed] [Google Scholar]
- Schilling, T., C. Stock, A. Schwab, and C. Eder. 2004. Functional importance of Ca2+-activated K+ channels for lysophosphatidic acid-induced microglial migration. Eur. J. Neurosci. 19:1469–1474. [DOI] [PubMed] [Google Scholar]
- Schrenzel, J., L. Serrander, B. Bánfi, O. Nüsse, R. Fouyouzi, D.P. Lew, N. Demaurex, and K.-H. Krause. 1998. Electron currents generated by the human phagocyte NADPH oxidase. Nature. 392:734–737. [DOI] [PubMed] [Google Scholar]
- Schwingshackl, A., R. Moqbel, and M. Duszyk. 2000. Involvement of ion channels in human eosinophil respiratory burst. J. Allergy Clin. Immunol. 106:272–279. [DOI] [PubMed] [Google Scholar]
- Shapiro, M.S., and T.E. DeCoursey. 1991. Selectivity and gating of the type L potassium channel in mouse lymphocytes. J. Gen. Physiol. 97:1227–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simchowitz, L., M.A. Foy, and E.J. Cragoe. 1990. A role for Na+/Ca2+ exchange in the generation of superoxide radicals by human neutrophils. J. Biol. Chem. 265:13449–13456. [PubMed] [Google Scholar]
- Smith, R.M., and A.E. Martel. 1976. Critical Stability Constants. Vol. 4: Inorganic Complexes. Plenum Press, New York. 257 pp.
- Stankova, L., G.W. Drach, T. Hicks, C.F. Zukoski, and M. Chvapil. 1976. Regulation of some functions of granulocytes by zinc of the prostatic fluid and prostate tissue. J. Lab. Clin. Med. 88:640–648. [PubMed] [Google Scholar]
- Tare, M., S.A. Prestwich, D.V. Gordienko, S. Parveen, J.E. Carver, C. Robinson, and T.B. Bolton. 1998. Inwardly rectifying whole cell potassium current in human blood eosinophils. J. Physiol. 506:303–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, R.C., and R.W. Meech. 1982. Hydrogen ion currents and intracellular pH in depolarized voltage-clamped snail neurones. Nature. 299:826–828. [DOI] [PubMed] [Google Scholar]
- Vignais, P.V. 2002. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell. Mol. Life Sci. 59:1428–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Tscharner, V., B. Prod'hom, M. Baggiolini, and H. Reuter. 1986. Ion channels in human neutrophils activated by a rise in free cytosolic calcium concentration. Nature. 324:369–372. [DOI] [PubMed] [Google Scholar]
- Wagoner, P.K., and G.S. Oxford. 1987. Cation permeation through the voltage-dependent potassium channel in the squid axon. Characteristics and mechanisms. J. Gen. Physiol. 90:261–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsuyanagi, J., and T. Ogiso. 1988. Zinc inhibition of respiratory burst in zymosan-stimulated neutrophils: a possible membrane action of zinc. Chem. Pharm. Bull. (Tokyo). 36:1035–1040. [DOI] [PubMed] [Google Scholar]