

Abstract
The interaction of palytoxin with the Na,K-ATPase was studied by the electrochromic styryl dye RH421, which monitors the amount of ions in the membrane domain of the pump. The toxin affected the pump function in the state P-E2, independently of the type of phosphorylation (ATP or inorganic phosphate). The palytoxin-induced modification of the protein consisted of two steps: toxin binding and a subsequent conformational change into a transmembrane ion channel. At 20°C, the rate-limiting reaction had a forward rate constant of 105 M−1s−1 and a backward rate constant of about 10−3 s−1. In the palytoxin-modified state, the binding affinity for Na+ and H+ was increased and reached values between those obtained in the E1 and P-E2 conformation under physiological conditions. Even under saturating palytoxin concentrations, the ATPase activity was not completely inhibited. In the Na/K mode, ∼50% of the enzyme remained active in the average, and in the Na-only mode 25%. The experimental findings indicate that an additional exit from the inhibited state exists. An obvious reaction pathway is a slow dephosphorylation of the palytoxin-inhibited state with a time constant of ∼100 s. Analysis of the effect of blockers of the extracellular and cytoplasmic access channels, TPA+ and Br2-Titu3+, respectively, showed that both access channels are part of the ion pathway in the palytoxin-modified protein. All experiments can be explained by an extension of the Post-Albers cycle, in which three additional states were added that branch off in the P-E2 state and lead to states in which the open-channel conformation is introduced and returns into the pump cycle in the occluded E2 state. The previously suggested molecular model for the channel state of the Na,K-ATPase as a conformation in which both gates between binding sites and aqueous phases are simultaneously in their open state is supported by this study.
INTRODUCTION
The lethal marine toxin palytoxin (PTX) can be extracted from polyps of the genus Palythoa (Moore and Scheuer, 1971). It was found to depolarize mammalian cells by causing cation conductance with relatively low ion selectivity (Weidmann, 1977). Small unselective cation channels with a single-channel conductance on the order of 10 pS were identified as origin of the electric currents (Ikeda et al., 1988; Muramatsu et al., 1988; Tosteson et al., 1991; Hirsh and Wu, 1997), and finally clear evidence was shown that these cation channels were formed upon interaction of PTX with the Na,K-ATPase (Ozaki et al., 1985; Habermann, 1989; Wang and Horisberger, 1997). A large series of experimental studies identified some details of the mechanism (Grell et al., 1988; Ikeda et al., 1988; Muramatsu et al., 1988; Scheiner-Bobis and Schneider, 1997; Guennoun and Horisberger, 2000, 2002; Farley et al., 2001; Wu et al., 2003; Horisberger et al., 2004), nevertheless a molecular concept of the action of the toxin could not be formulated. A significant step forward in the understanding of the PTX action on the Na,K-ATPase was obtained by recent electrophysiological experiments and their interpretation on the basis of the pump cycle of the Na,K-ATPase (Artigas and Gadsby, 2003b; Artigas and Gadsby, 2004).
The Na,K-ATPase is a housekeeping enzyme of almost all animal cells and belongs to the family of P-type ATPases, which share common features of the ion transport mechanism. Stimulated by the presentation of the structure with atomic resolution of the Ca-ATPase of the sarcoplasmic reticulum (Toyoshima et al., 2000; Toyoshima and Inesi, 2004), the discussion of structure–function relations of the Na,K-ATPase led to a consistent model of the transport on the basis of the generally accepted Post-Albers cycle of the P-type ATPases (Apell, 2004). The main features are as follows: (a) a ping-pong mechanism, i.e., both transported ion species are transferred successively and in opposite direction across the membrane; (b) the transport process for each ion species consists of a sequence of reaction steps, which are ion binding, ion occlusion, conformational transition of the protein, successive deocclusion of the ions, and release to the other side of the membrane; and (c) recent experimental evidence showed that the ion binding sites are placed inside the transmembrane section of the proteins and that ion movements occur preferentially during the ion binding and release processes.
Because the binding sites are located in the middle of the membrane domain, the ions have to migrate through so-called access channels before they bind to, or after they dissociate from, their sites. And convincing evidence has provided support for this charge movement (de Weer et al., 2000; Apell, 2003). To prevent a channel-like, electric short circuiting by the Na,K-ATPase under physiological conditions, conformations of the protein have to be strictly prohibited in which the binding sites have simultaneous access to both aqueous compartments. This is achieved by the existence of gates between the central binding sites and the protein surface, and by a rigorous control of the gates during the progress of the pump process in a way that at least one gate is always closed. An obvious consequence of this constraint is the existence of “occluded states” in which both gates are closed simultaneously, and the bound ions are trapped inside the membrane domain (Forbush, 1988; Glynn and Karlish, 1990).
On the basis of this mechanistic concept, Artigas and Gadsby (2003a,b) proposed that the effect of PTX is a modification of the gating condition of the Na,K-ATPase in the P-E2 conformation. In the P-E2 state, the outside gate (i.e., the gate between binding sites and extracellular aqueous phase) is opened and allows the exchange of the three Na+ ions against two K+ ions, according to the physiological task of the pump. The toxin interacts with this state in a way that allows the cytoplasmic gate (i.e., the gate between binding sites and cytoplasm) to also be opened, and thus establishes a relatively nonselective cation channel. This channel is the reason for the toxicity of PTX. By patch-clamp experiments, single-channel properties of the Na,K-pump channels were analyzed, and their conductance and voltage dependence determined under various conditions (Artigas and Gadsby, 2003b; Artigas and Gadsby, 2004). Single-channel recordings also showed burst-like behavior in which single channels were opened for several seconds and showed conductance flickering by short closure events on the order of 50 ms (Artigas and Gadsby, 2004).
An experimental method alternative to electrophysiological studies to investigate partial reactions of the Na,K-ATPase is the application of electrochromic styryl dyes, such as RH421, which are a convenient approach to study the presence and movements of cations in the membrane domain of P-type ATPases (Heyse et al., 1994; Pedersen et al., 2002). Due to the mechanism of the dye molecules, which are dissolved in the lipid phase of the membrane bilayer and detect changes of the local electric fields, entry and binding of cations can be monitored by a fluorescence decrease. The time course reveals the kinetics and the fluorescence level reveals the amount of charge in the binding sites. This method was already applied to determine the occupancy of the binding sites by ions in the case of the ouabain-inhibited sodium pump (Stürmer and Apell, 1992), and to explain the apparent electroneutrality of K+ binding in the E1 conformation of the Na,K-ATPase (Apell and Diller, 2002). Therefore, it could be expected that the properties of the styryl dye RH421 may be used to investigate the effects of PTX on the ion transport pathway of the Na,K-ATPase.
MATERIALS AND METHODS
Materials
Phosphoenolpyruvate (PEP), pyruvate kinase (PK), lactate dehydrogenase (LDH), NADH, BSA, and ATP (disodium salt, special quality) were from Boehringer. PTX was bought from Calbiochem (Palythoa toxica, lot B32897) and Sigma-Aldrich (Palythoa tuberculosa, lot 22K1357; Palythoa caribaeorum, lot 61K1637). The electrochromic styryl dye RH421 was ordered from MoBiTec. All other reagents were purchased from Merck or Sigma-Aldrich at the highest quality available. 1,3-dibromo-2,4,6-tris(methylisothiouronium)benzene (Br2-Titu3+) was a gift from S.D. Karlish (Weizmann Institute of Science, Rehovot, Israel).
Membrane Preparations
Purified membrane preparations with a high concentration of Na,K-ATPase (∼5.000 pumps per μm2) were prepared from the outer medulla of rabbit kidneys using the procedure C of Jørgensen (1974). The enzyme activity of the Na,K-ATPase (Schwartz et al., 1971) was determined in buffer containing 25 mM imidazole (pH 7.2), 100 mM NaCl, 10 mM KCl, 5 mM MgCl2, 1.5 mM Na2ATP, 2 mM PEP, 450 U/ml of pyruvate kinase (PK) and lactate dehydrogenase (LDH), and initially 80 μM NADH. The specific ATPase activity was in the range of 2,000–2,400 μmol Pi per hour and mg protein at 37°C, ouabain-insensitive activity was <1%. The enzyme activity could be completely blocked in the presence of 1 μM ouabain. For comparison, some experiments were performed with a purified membrane preparation from duck salt gland. The crude microsomal preparation from duck was provided by O.D. Lopina (Moscow State University, Moscow, Russia).
Fluorescence Experiments with Styryl Dye RH421
RH421 is an amphiphilic styryl dye that dissolves in lipid membranes with a partition coefficient of 2.5 × 105 (Bühler et al., 1991), with its negatively charged sulfonyl residue directed toward the aqueous phase. By light absorption at the red edge of the absorption spectrum, the dye is excited with its delocalized positive charge shifted from the pyridine unit of the chromophore toward the more interior placed aniline unit (Pedersen et al., 2002). The spectral changes of the styryl dye predominantly result from an electrochromic effect, i.e., a shift of the absorption band occurs when the energy difference between ground state and excited state depends on electric field strength. The electric field may also affect the fluorescence quantum yield.
The local electric field strength is modified by charge translocations in the course of the pump cycle of the Na,K-ATPase, and the styryl dye responds with shifts of the emission spectra to longer (red) or shorter (blue) wavelength corresponding to changes of the (local) electric potential inside the membrane to more negative or more positive values, respectively (Bühler et al., 1991).
The fluorescence measurements in equilibrium-titration experiments were performed with a self-constructed setup using a HeNe laser with a wavelength of 594 nm (Laser 2000) to excite the fluorescence of RH421. The emitted light was collected perpendicularly to the incident light, filtered by a narrow-band interference filter (λmax = 660 nm, half width 15 nm) and detected by a head-on photo multiplier (R2066, Hamamatsu). The photo current was amplified by a Keithley current amplifier 427 (Keithley Instruments) and collected by a data acquisition board of a PC (PCI-T112, Imtec) with sampling frequencies between 1 and 10 Hz. The experimental data were displayed on the monitor, stored, and analyzed on the PC. The temperature in the permanently stirred cuvette (2 ml) was maintained by a thermostat at 20°C, if not mentioned otherwise.
The raw data obtained from a fluorescence experiment stored in arbitrary units were normalized according to the function ΔF/F 0 = (F − F 0)/F 0, with respect to the initial fluorescence level, F 0, so that different experiments can be easily compared.
RESULTS
Detection of ion movements in the Na,K-ATPase by the electrochromic styryl dye RH421 was applied successfully to study the effect of inhibitors of the ion pump and identify the affected reaction steps of the pump cycle and to analyze the mechanism of cardiac glycosides (Stürmer and Apell, 1992), and recently that of the so-called MCS derivatives (Stimac et al., 2005). Accordingly, the technique was applied to learn new details of the mechanism of PTX when interacting with the Na,K-ATPase.
Identification of Affected Partial Reactions
The fluorescent dye RH421 monitors the amount of charge (proportional to the number of ions) present in the binding sites of the Na,K-ATPase, which are located inside the membrane domain of the ion pump (Stimac et al., 2005). Well-defined enzyme states can be stabilized by an appropriate choice of buffer compositions (Stürmer et al., 1991), as shown in Fig. 1 A. When Na,K-ATPase (9 μg/ml) of a purified membrane preparation is equilibrated in standard buffer (25 mM histidine, 0.1 mM EDTA, 1 mM MgCl2, 0.1 mM H3BO3, 28 μg/ml BSA, pH 7.2) and 200 nM RH421, the resulting fluorescence level, F 0, corresponds to the state H2E1 (Apell and Diller, 2002). When successive solutions of NaCl (50 mM), ATP (500 μM), and KCl (20 mM) are added, transition into the states Na3E1, P-E2, and (ATP·E2[K2] + Na3E1) are induced, respectively. Addition of KCl generates the buffer composition in which the Na,K-ATPase works under turnover condition. Therefore, the ion pumps are distributed mainly between the two states, (Na3E1-P and E2[K2]), which precede the rate-limiting steps of the cycle. The state Na3E1-P is expected to be preferentially populated at the chosen high ATP concentration. This sequence of additions we call our “standard experiment.” A typical standard experiment is shown in Fig. 1 B. It can be seen that the ion pumps react within a short time, controlled by the mixing time after addition of microliter aliquots to the buffer. When 100 nM PTX was added under turnover conditions, no significant fluorescence change was observed (Fig. 1 B).
Figure 1.

Standard experiments with addition of 100 nM PTX at four defined states of the Na,K-ATPase. (A) Post-Albers cycle of the Na,K-ATPase under physiological conditions. E1 and E2 are conformations of the ion pump with ion binding sites facing the cytoplasm and extracellular medium, respectively. Three Na+ and two K+ are transported out of and into the cytoplasm of the cell, respectively. (Na3)E1-P, E2(K2), and ATP·E2(K2) are occluded states in which the ions bound are unable to exchange with either aqueous phases. In the absence of Na+ and K+, the E1 state is actually a H2E1 state (Apell and Diller, 2002). The pump states numbered 1–4 in the scheme of A can be stabilized by appropriate substrate additions, as shown in B except that no ATP was present in the beginning of the experiments shown in B–E and that, therefore, states 1 and 2 did not carry ATP. When all substrates are present, the pump will run through the cycle and most of the pumps are accumulated in the states before the two rate-limiting steps, labeled by “4.” In B–E, the initial state is always H2E1, with the normalized fluorescence level ΔF/F0 = 0. Subsequently, 50 mM NaCl, 0.5 mM ATP, and 20 mM KCl were added, which stabilize the states listed in A. PTX was added at different states of the protein, as indicated by an arrow: (B) Na3E1-P + ATP·E2(K2), (C) H2E1, (D) Na3E1, (E) P-E2.
Such standard experiments were repeated with modifications. PTX was added in defined protein states. In Fig. 1, the effects of the toxin on the pump function can be seen when added in states E1 (Fig. 1 C), Na3E1 (Fig. 1 D), and P-E2 (Fig. 1 E). When 100 nM PTX was added in state E1 (Fig. 1 C), no significant modifications of the fluorescence signal in states E1 and Na3E1 were detected. But in both cases, after addition of ATP, the fluorescence level corresponding to P-E2 was no longer stable but decayed exponentially with a time constant of ∼100 s to a level that matched the condition of a state believed to contain two monovalent cations bound to the binding sites. When KCl was added subsequently, the first third of the fluorescence change happened rapidly (τ < 5 s) while the rest decayed with a single exponential function with a time constant of ∼100 s. The final fluorescence level corresponded to the level obtained under turnover condition in the absence of PTX. When PTX was added in state Na3E1 (Fig. 1 D), no significant fluorescence change was observed in this state and the behavior upon addition of ATP was identical to the results in Fig. 1 C. In the last version of the standard experiment, PTX was added in state P-E2 (Fig. 1 E). Again, an exponential fluorescence decay with a time constant of 100 s was found, and the K+-induced change was the same as in Fig. 1 (C and D).
The first and obvious result from this set of experiments is that PTX modifies the Na,K-ATPase–induced fluorescence signal only in state P-E2. This modification is a rather slow reaction when compared with time constants of the unaffected pump cycle, and, in the absence of K+, the new steady state obtained upon interaction of pump and toxin was always with the level corresponding to two positive charges inside the membrane domain of the Na,K-ATPase, probably two Na+ ions. Addition of K+ ions led to a fluorescence level that corresponded to that observed with ion pumps under turnover conditions.
Rate Constants of the PTX-induced Effect
The fluorescence decay of the state P-E2 upon addition of PTX was investigated in two different sets of experiments. In one series, the Na,K-ATPases were transferred from the E1 state in standard buffer to predominantly the P-E2 state by addition of 50 mM NaCl and 500 μM ATP, then PTX was added (as shown in Fig. 1 E) in concentrations between 50 and 500 nM. The fluorescence decay could be well fitted by a single exponential function, F(f) = ΔF·exp(−k·t), and the rate constant, k, determined. Such experiments were performed with PTX commercially available from three different species of Palythoa and with various purified Na,K-ATPase preparations, including those from duck salt gland. The rate constants k were plotted as function of the PTX concentration (Fig. 2). The observed stationary fluorescence level after the toxin-induced transition was independent of the PTX concentration and corresponded in all cases to the level with two monovalent cations bound in the binding sites.
Figure 2.

Kinetics of the PTX-induced transition from P-E2 into the toxin-modified state,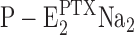 . The fluorescence decrease upon addition of PTX as shown in Fig. 1 E could be perfectly fitted by a single exponential function, ΔF·exp(−kt). The rate constant k was determined for PTX concentrations between 100 and 600 nM for different Na,K-ATPase preparations and PTX sources. (A) PTX from P. caribaeorum (1 and 2) and two different preparations from rabbit kidney, (3) PTX from P. tuberculosa and the same enzyme as in trace 1. (B) PTX from P. caribaeorum, (1) rabbit kidney Na,K-ATPase and (2) duck salt gland Na,K-ATPase.
. The fluorescence decrease upon addition of PTX as shown in Fig. 1 E could be perfectly fitted by a single exponential function, ΔF·exp(−kt). The rate constant k was determined for PTX concentrations between 100 and 600 nM for different Na,K-ATPase preparations and PTX sources. (A) PTX from P. caribaeorum (1 and 2) and two different preparations from rabbit kidney, (3) PTX from P. tuberculosa and the same enzyme as in trace 1. (B) PTX from P. caribaeorum, (1) rabbit kidney Na,K-ATPase and (2) duck salt gland Na,K-ATPase.
In all experiments, the PTX concentration dependence of the rate constant resulted in a linear relation that is consistent with a chemical reaction of first order,
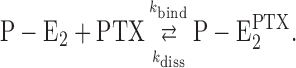 |
(1) |
The reaction is controlled by two rate constants, k bind and k diss, which characterize the forward and backward reaction step, respectively, of toxin and ion-pump interaction. According to the principles of chemical kinetics, the slope of the line through the data points in Fig. 2 represents the value of k bind, and the intercept with the y axis ([PTX] = 0) provides the value of k diss. The quantitative analysis is summarized in Table I, and the results of the experiments with the different preparations of Na,K-ATPase showed that the binding constant, k bind, differed by about a factor of 2 (between 0.9 × 105 s−1M−1 for rabbit enzyme and 1.9 × 105 s−1M−1 for salt gland enzyme). The deviation in the case of the experiments with PTX from P. tuberculosa with k bind = 0.57 × 105 s−1M−1 (Fig. 2 A) may be caused by an inaccurate filling of the delivered sample. We had no means to verify the accuracy of the “25 μg” printed on the label. The dissociation rate constant, k diss, varied between 9.0 × 10−4 s−1 (rabbit kidney) and 19.5 × 10−4 s−1 (salt gland). This value corresponds to a time constant for the backward reaction in the order of 1,000 s for the renal enzyme in a buffer with 50 mM NaCl and pH 7.2.
TABLE I.
Overview of the Kinetic Parameters of the PTB Binding Kinetics
| Toxin, experimental condition | k bind/s−1M−1 | k diss/s−1 | K m/nM |
|---|---|---|---|
| P. carib., renal enz., 50 NaCl | (0.90 ± 0.05) 105 | (9.0 ± 3) 10−4 | 10 ± 3 |
| P. toxica, renal enz., 50 NaCl | (0.80 ± 0.04) 105 | (13.0 ± 4) 10−4 | 16 ± 4 |
| P. tuberc., renal enz., 50 NaCl | (0.58 ± 0.01) 105 | (12.6 ± 5) 10−4 | 22 ± 5 |
| P. carib., salt gland, 50 NaCl | (1.87 ± 0.13) 105 | (19.5 ± 8) 10−4 | 10 ± 8 |
| P. carib., renal enz., 5 NaCl | ND | ND | 34.5 ± 6 |
| P. carib., renal enz., 50 NaCl | ND | ND | 31.8 ± 6 |
| P. carib., renal enz, 200 NaCl | ND | ND | 25.0 ± 4 |
| P. carib., renal enz, 0 NaCl, Pi | (0.208 ± 0.02) 105 | (29.6 ± 6) 10−4 | 142 ± 6 |
Three sources of PTX were used: from Palythoa caribaeorum (P. carib.), Palythoa toxica (P. toxica), and Palythoa tuberculosa (P. tuberc.). In the time-solved experiments, k bind and k diss were obtained from experiments and the equilibrium dissociation constant calculated as K m = k diss/k bind. The Na+ concentration dependence of PTX binding was performed as equilibrium titration experiments.
The values of k bind and k diss determined from the experiments can be used to calculate the half-saturating PTX concentration for the Na,K-ATPase, K m = k diss/ k bind, to be 10 ± 3 nM (50 mM NaCl) and 13.4 nM (5 mM NaCl). In addition, equilibrium titration experiments were performed in the presence of 5, 50, and 200 mM NaCl, in which the enzyme was phosphorylated by 500 μM ATP. After a steady-state fluorescence level was obtained, PTX was added in small aliquots up to 300 nM and the toxin-induced fluorescence decrease was detected (unpublished data). The respective fluorescence levels were plotted against the PTX concentrations, and the resulting concentration dependence could be fitted by a binding isotherm with K m = 34.5 ± 6 nM (5 mM NaCl), K m = 31.8 ± 6 nM (50 mM NaCl), and K m = 25 ± 4 nM (200 mM NaCl).
Reversibility of PTX-induced Modification of the Na,K-ATPase
To study the effect of PTX on the turnover rate of the Na,K-ATPase in its Na-only mode, the following series of experiments was performed. The Na-only mode is a noncanonical mode of the pump in which Na+ ions are pumped in the absence of K+ ions. A standard RH421 experiment was started as shown in Fig. 1 B, up to the point where ATP was added. This time, however, only 1 μM ATP was added. In these experiments, the cuvette volume was 2 ml, and thus n ATP = 2 × 10−9 mol. The amount of protein was 9 μg (or n prot = 6 × 10−11 mol). Therefore, such a small amount of ATP provided substrate for n ATP/n prot = ∼33 turnovers per ATPase as an average. Under this condition the buffer was depleted of ATP after ∼100 s (Fig. 3); the Na,K-ATPases stopped cycling and were trapped in the Na3E1, the proximate state of the pump cycle before enzyme phosphorylation. The experiment was then continued in the manner of Fig. 1 D, when 50 nM PTX was added, followed ∼1 min later by another aliquot of 1 μM ATP. After the ATP was completely hydrolyzed and the pumps returned again to Na3E1, another 50 nM PTX was added followed by 1 μM ATP ∼1 min later. This procedure was repeated three more times. The resulting fluorescence trace is shown in Fig. 3. The modification of the pump function by PTX can be seen best at the highest (here 200 nM) PTX concentration. After addition of 1 μM ATP, all pumps proceeded to the P-E2 state (represented by the highest fluorescence level) in which the inhibitor interacts with the protein by transforming it into a channel with higher ion-binding affinities, and thereupon Na+ ions bound and caused the observed exponential fluorescence decrease. The subsequent plateau phase represents the condition when the Na,K-ATPase molecules were preferentially in the PTX-modified state in which they nevertheless were able to hydrolyze ATP, as will be shown below. With increasing depletion of ATP in the buffer, more and more pumps that left the PTX-modified state were trapped in the state before enzyme phosphorylation, Na3E1. This transition can be seen in the third phase of the fluorescence signal when it returned to the low level before addition of ATP.
Figure 3.

Effect of PTX on the P-E2 state of the Na,K-ATPase. After the Na,K-ATPase was equilibrated in standard buffer at 20°C, the fluorescence level, F0, represents the state H2E1. Addition of NaCl (50 mM) induced the transition to Na3E1 and the subsequent addition of ATP (1 μM) led to enzyme phosphorylation, transition into the P-E2 conformation, and release of the Na+ ions bound. Due to the fact that the amount of ATP present was so small, within ∼100 s, all ATP was hydrolyzed and all pumps returned into the equilibrium state, Na3E1, under this buffer condition. Then 50 nM PTX was added as well as another 1 μM ATP. In the presence of PTX, the time course of the fluorescence signal was slightly modified. Repetitive additions of 50 nM PTX and 1 μM ATP up to a final PTX concentration of 200 nM led to a distinct fluorescence pattern with an intermediate fluorescence level, which corresponded approximately to the level of the ion pump with two monovalent cations bound. After each reaction sequence, however, the final equilibrium state was Na3E1.
Three significant modifications in the time course of the fluorescence signals were found in the presence of PTX: (1) with increasing PTX concentrations, the time taken for all pumps to return to the Na3E1 state became longer, (2) with increasing PTX concentrations, an intermediate state developed with a fluorescence level corresponding to two cations bound in the membrane domain, and (3) as already shown (Fig. 2), the transition from the initial P-E2 state immediately following phosphorylation by ATP is also dependent on PTX, its time constant being shorter the higher the toxin concentration. The latter fact is not strikingly evident in Fig. 3, but exponential fits to the initial parts of the fluorescence decay curves showed that the dependence of their rate constants on PTX concentration was indistinguishable from that shown in Fig. 2.
After the last addition of PTX, the total concentration was 200 nM. A corresponding return into the state Na3E1 after ∼1,100 s was also observed in the presence of 700 nM (not depicted). This fact indicates that even at a saturating concentration of the toxin, all pumps eventually end up in a state in which PTX does alter fluorescence. Since the final fluorescence level following ATP exhaustion is identical to that of Na3E1 before PTX addition, we can discard the hypothesis that only the enzyme fraction unaffected by PTX consumed the ATP. If that were the case, the higher fluorescence of the PTX-modified enzyme, with two cations bound, would have raised the overall fluorescence above the Na3E1 level in the steady state. In consequence, the conclusion is that PTX modifications of the Na,K-ATPase are reversible.
In the presence of 50 mM NaCl and 200 nM PTX, a steady state occurs in which ∼86% (assuming K m = 31.8 nM; Table I) to 95% (assuming K m = 10 nM) of the enzyme molecules to be in the PTX-bound state. As discussed above, an average of 33 turnovers per pump must occur to consume the 1 μM ATP added at the start of the experiment. Given a PTX “off” time constant in the order of 1,000 s (Table I), this complete consumption of ATP, requiring 33 turnovers, could not have taken place within 1,000 s (Fig. 2) unless the PTX-modified enzyme, in a state with two bound cations as inferred from its fluorescence, still participates in ATP hydrolysis. To check this, the effect of the Na+ concentration in the buffer on Na,K-ATPase modification by PTX was investigated. Experiments were performed in which the ion pump was phosphorylated by 1 μM ATP in the presence of various concentrations of NaCl (5–50 mM) and 100 nM PTX. One series of experiments is shown in Fig. 4. One interesting result was that in the investigated Na+ concentration range, the transition kinetics into the PTX-modified state at a concentration of 100 nM PTX was not significantly affected. The average rate constant of the fitted exponential decays was 0.015 ± 0.0004 s−1. The second result was that duration of the PTX-modified state until the pumps became dephosphorylated and returned to the Na3E1 state became shorter with increasing Na+ concentration, i.e., ATP was consumed faster, or in other words, Na+ destabilized the toxin-induced state. The overall reduced fluorescence amplitude in the presence of 5 mM NaCl (Fig. 4, trace a) can easily be explained by the fact that at this concentration below half-saturating cytoplasmic Na+ binding affinity, part of the enzyme did not participate in ion pumping, thus reducing the fluorescence amplitude.
Figure 4.

Dependence of the PTX-modified state of the Na,K-ATPase on the Na+ concentration in the buffer. The NaCl concentrations were 5 mM (a), 10 mM (b), 15 mM (c), 20 mM (d), 25 mM (e). The final fluorescence level represents state Na3E1 in all experiments and it was achieved faster the higher the Na+ concentration was. The time, Δt r, at which the fluorescence level returned halfway from the intermediate PTX-inhibited level to the final level is indicated by diamonds. The reciprocal value, 1/Δt r, is plotted in the inset against the respective Na+ concentration in the buffer. The data points were fitted with Eq. 2 as described in the text.
The duration between PTX addition (at t = 0) and the return to the “resting” state, Na3E1, was defined as the time period, Δt
r, until the fluorescence amplitude returns halfway from the state 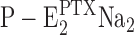 to Na3E1 (indicated by diamonds on the fluorescence traces in Fig. 4). It is obvious that Na+ binding speeds up the turnover rate and thus ATP consumption. To quantify the Na+ concentration dependence, the following estimate was performed. The reciprocal time period, 1/Δt
r, was plotted against the Na+ concentration in the inset of Fig. 4. Since the pump rate may be defined as v = n
ATP/Δt, in a first rough approximation we introduced a mean turnover rate v
r = n
ATP/Δt
r. Such a treatment may be justified by the fact that in all experiments the same amount of ATP and ATPases was used. Given that the PTX-modified enzyme hydrolyzes ATP with the rate v
0 when no Na+ ions are in the binding sites, and with the rate v
∞ when both sites are occupied by Na+, then a Na+ concentration–dependent turnover rate can be assumed to be
to Na3E1 (indicated by diamonds on the fluorescence traces in Fig. 4). It is obvious that Na+ binding speeds up the turnover rate and thus ATP consumption. To quantify the Na+ concentration dependence, the following estimate was performed. The reciprocal time period, 1/Δt
r, was plotted against the Na+ concentration in the inset of Fig. 4. Since the pump rate may be defined as v = n
ATP/Δt, in a first rough approximation we introduced a mean turnover rate v
r = n
ATP/Δt
r. Such a treatment may be justified by the fact that in all experiments the same amount of ATP and ATPases was used. Given that the PTX-modified enzyme hydrolyzes ATP with the rate v
0 when no Na+ ions are in the binding sites, and with the rate v
∞ when both sites are occupied by Na+, then a Na+ concentration–dependent turnover rate can be assumed to be
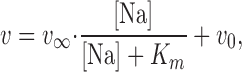 |
(2) |
where [Na] is the Na+ concentration and K m is the Na+ concentration for half-maximal activation. With this approximation, and under the assumption that the calculated v is proportional to v r, the Na+ concentration dependence of v r was fitted (Fig. 4, inset). The fit of the data was obtained with v ∞/v 0 = 10.1, which means that Na+ ions in the binding sites reduce the stability of the PTX-modified state by more than a factor of 10. K m was estimated to be 20.1 mM, which is significantly smaller than the value obtained for the native enzyme, ∼400 mM (Heyse et al., 1994). This difference in apparent binding affinity explains well why the RH421 fluorescence decreased upon addition of PTX: Na+ ions are more readily able to bind to the toxin-modified Na,K-ATPase.
When Na+ and K+ titration experiments were performed in the P-E2 state in the absence and presence of 100 nM PTX, the effect of the toxin on the affinity of the ion-binding sites could be determined. In these experiments, Na,K-ATPase was equilibrated in standard buffer and 200 nM RH421. Then the following were added: 100 nM PTX (or left out in the control experiments), 5 mM NaCl, and 0.5 mM ATP (similar to Fig. 1 C). After a steady-state fluorescence level was reached, small aliquots of NaCl and KCl were added and the fluorescence change recorded. When the normalized fluorescence levels were plotted against the respective cation concentration, a fluorescence decrease corresponding to the occupation of the binding sites was obtained, which was fitted by a binding isotherm (unpublished data). In the case of Na+ additions, the half-saturation concentrations were 18.1 ± 2.0 mM (≥100 nM PTX) and ∼400 mM (0 PTX). When the binding sites were titrated with K+, the obtained K m values were 0.14 ± 0.02 mM, the same in the absence and presence of 100 nM PTX.
Temperature Dependence of the PTX-induced Effect
Na,K-ATPase was incubated in standard buffer at various temperatures between 5°C and 38°C. 50 mM NaCl and 0.5 mM ATP were added to transfer the ion pumps preferentially into state P-E2. Then 100 nM PTX was added and the fluorescence decay was recorded (similar to the experiment in Fig. 1 E). The rate constant of the monoexponential fit of the fluorescence signal was derived from the experiments and plotted as Arrhenius diagram (Fig. 5). In this plot, two regions with a linear relation of ln(k) vs. 1/T were found. On the basis of the underlying theory, the linear slope, m, of an Arrhenius plot is proportional to the activation energy, E A, of the rate-limiting reaction step E A = m·R. R is the gas constant. The two activation energies determined by linear regression from the data points were E A,1 = 28.5 ± 8.2 kJ/mol above 23°C, and E A,2 = 71.0 ± 3.1 kJ/mol below this temperature. Such an abrupt change of the activation energy suggests a sharp cooperative phase change such as the lipid phase of the purified microsomal preparation of the Na,K-ATPase used in these experiments.
Figure 5.

Temperature dependence of the PTX-induced transition from P-E2 into the toxin-modified state. The fluorescence decrease upon PTX addition was measured at various temperatures between 5°C and 38°C, and fitted by a single exponential function, ΔF·exp(−kt). The rate constant, k, was plotted against the temperature in form of an Arrhenius plot. The data show different temperature dependences below and above 23°C (1/T = 3.378 × 10−3 K−1). The dashed lines through the data represent (1) 71 kJ/mol and (2) 28.5 kJ/mol.
Due to the high activation energy at low temperatures, this finding indicates that at least two separate reaction steps have to exist in the reaction 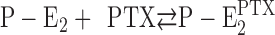 , since a Q
10 of ∼2.5 corresponds in the case of the Na,K-ATPase to a conformation transition rather than to substrate binding. This observation leads to the following mechanistic proposal:
, since a Q
10 of ∼2.5 corresponds in the case of the Na,K-ATPase to a conformation transition rather than to substrate binding. This observation leads to the following mechanistic proposal:
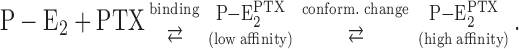 |
(3) |
According to the well-accepted concept of the pump mechanism, the extracellular gate is able to open in the P-E2 conformation. Upon binding of PTX, the toxin-induced conformational modification enhanced the affinity of the ion-binding sites for cations. According to electrophysiological studies, this conformational rearrangement may also allow the second, cytoplasmic gate to open, which creates the ion channel as reported recently (Artigas and Gadsby, 2003b; Artigas and Gadsby, 2004).
Effect of PTX on the Enzyme Activity
The enzyme activity was determined with the standard PK/LDH assay as described in Materials and Methods. The activity was determined in the absence and in the presence of various concentrations of PTX up to 1500 nM. The specific activity, A E([PTX]), was normalized to the value in the absence of PTX, A E (0). These experiments were repeated with three different preparations of rabbit kidney enzyme and one preparation from duck salt gland enzyme. The results are shown in Fig. 6. When the experiments were performed in buffer containing 10 mM KCl (curve a), PTX inhibited the enzyme activity significantly less than in buffer without KCl (curve b). When the data were fitted by a binding isotherm, the PTX concentration for half-maximal inhibition, Km, was found to be 812 nM in the Na/K mode and 283 nM in the Na-only mode. Maximum inhibition of the Na,K-ATPase at saturating PTX concentrations was extrapolated to be 52% in the Na/K mode and 75% in the Na-only mode. This result agrees well with the PTX inhibition of detergent-activated kidney microsomes with a Km of 800 nM as obtained by Böttinger and Habermann (1984). In previously published work, the Km of guinea pig heart enzyme was found to be ∼3 μM in the Na/K mode and an estimated maximum inhibition of >80% was found (Ishida et al., 1983).
Figure 6.

Effect of PTX on the enzyme activity of the Na,K-ATPase. The ATP-hydrolyzing activity was measured with the standard PK/LDH assay and set to 100% in the absence of PTX. The inhibiting action of PTX was studied in buffer containing 100 mM Na+ and 10 mM K+ (a, Na,K-mode) or in 110 mM Na+ (b, Na-only mode). In the Na,K mode, no difference was found in enzyme from rabbit kidney (solid circles) and duck salt gland (open squares). In the presence of 10 mM K+, a significantly lower level of inhibition was observed. When the data were fitted by a single binding isotherm (solid lines), the fraction of inhibition at saturating PTX was 52% in the Na,K mode and 75% in the Na-only mode.
The observed, limited inhibition of the Na,K-ATPase by PTX is another indicator that the PTX-modified state of the ion pump can be drained by a reaction pathway different from the back reaction of Eq. 1. If the only exit from state 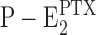 would be the back reaction to P-E2, then a high enough concentration of PTX would have to inhibit enzyme activity completely, as is known for cardiac glycosides. The second interesting result was that in the presence of KCl, even less of the ion pump can be kept in the inhibited state and that an approximately three-times higher concentration of PTX is necessary to reach a half-maximal inhibition of the enzyme activity. An obvious explanation is that binding of K+ in the membrane domain of the pump destabilizes the PTX-modified state even stronger than Na+ ions. Possible suggestions for such a destabilizing reaction sequence would be either the dephosphorylation step or the reversal of the PTX modification first (see Eq. 4).
would be the back reaction to P-E2, then a high enough concentration of PTX would have to inhibit enzyme activity completely, as is known for cardiac glycosides. The second interesting result was that in the presence of KCl, even less of the ion pump can be kept in the inhibited state and that an approximately three-times higher concentration of PTX is necessary to reach a half-maximal inhibition of the enzyme activity. An obvious explanation is that binding of K+ in the membrane domain of the pump destabilizes the PTX-modified state even stronger than Na+ ions. Possible suggestions for such a destabilizing reaction sequence would be either the dephosphorylation step or the reversal of the PTX modification first (see Eq. 4).
After two monovalent cations (X+ = Na+, K+, H+) are bound, two different pathways are plausible. The ions in the binding site either trigger a dephosphorylation, which causes a closing of the occlusion gate on the extracellular side (indicated by the unpaired “)” in Eq. 4). This step is followed by a reversal of the PTX-induced conformation change and a closing of the cytoplasmic gate. An alternative reaction sequence could be the reverse order of the above described sequence. Binding of the two cations causes first a reversal of the PTX-induced conformation change and a closing of the cytoplasmic gate (indicated by the unpaired “(” in Eq. 4), then followed by dephosphorylation of the enzyme with a complete occlusion of the ions. The third step could be accompanied in both cases by a complete dissociation of the PTX molecule or only by dissociation from its direct reaction partner on the protein while remaining attached to the pump, as assumed in Eq. 4. In both cases, the last step would be the conformation transition back to E1 resuming the normal pump cycle. The data of Tosteson et al. (2003) support the possibility that PTX remains attached to the protein.
Backdoor Phosphorylation and Effects of PTX
So far, all PTX-induced reactions were performed in the presence of at least Na+, one of the physiologically transported cations. To study the interaction of PTX in the absence of Na+ and K+ ions, experiments were performed in standard buffer without monovalent cations other than protons (pH 7.2). Addition of Tris phosphate (0.5 mM Pi) produced an RH421 fluorescence increase corresponding to a release of the two positive elementary charges, i.e., two H+, which are bound to the Na,K-ATPase in E1 when no Na+ or K+ are present (Apell et al., 1996; Apell and Diller, 2002). As shown in the inset of Fig. 7 A, subsequent addition of 200 nM PTX induced an exponential decrease of the fluorescence with a time constant on the order of 120 s and reached an equilibrium state at a fluorescence level corresponding to the initial level before addition of Pi. Experiments of this kind were repeated with PTX concentrations between 25 and 350 nM. The observed Pi-induced fluorescence rise was the same in the absence and presence of PTX (unpublished data), indicating that the same partial reaction E2(H2)→P-E2H2→P-E2 was triggered by addition of Pi.
Figure 7.

Modification of the Na,K-ATPase by PTX under the condition of backdoor phosphorylation. When 0.5 mM inorganic phosphate (Pi) is added to the Na,K-ATPase in the absence of Na+ and K+ ions, the following reaction sequence is triggered: (H2E1→)E2(H2)→P-E2H2→P-E2 (Apell and Diller, 2002), and the dissociation of the two protons leads to an increase of the RH421 fluorescence as shown in the inset of A. Addition of 100 nM PTX produced an exponential decay of the fluorescence intensity whose rate constant, k, and fluorescence amplitude, ΔFmax, was determined. When plotted as function of the applied PTX concentration (A), a linear dependence of k on PTX concentration was found from which k bind = 0.22 × 105 M−1s−1 and k diss = 35 × 10−4 s−1 were determined. (B) The concentration dependence of ΔFmax could be fitted by a binding isotherm with a half-saturating binding concentration of K m = 25.8 nM.
The analysis of the PTX-induced fluorescence decrease is shown in Fig. 7. The linear dependence of the relaxation rate, k, is consistent with a first-order binding process of PTX (Eq. 1), and the rate constants obtained from Fig. 7 A were k bind = 0.22 × 105 M−1s−1 and k diss = 35 × 10−4 s−1. The binding constant, k bind, is a factor of four smaller than in the case of the ATP-phosphorylated rabbit kidney enzyme, while the k diss is larger by a factor of four. The equilibrium dissociation constant for PTX in the case of backdoor-phosphorylated pumps, K m, is 160 nM. The steady-state amplitude of the fluorescence decrease was also analyzed as a function of the PTX concentration (Fig. 7 B). Fitting the data with a binding isotherm, a half-saturating PTX concentration, K m = 25.8 ± 5.8 nM was obtained, and a match of the final fluorescence level and the initial (before Pi addition) was found. Comparable experiments were also performed at pH 6.0 and pH 8.0. While at low pH the binding rate constant, k bind = 0.20 × 105 M−1s−1, was the same as in buffer of pH 7.2, it was significantly increased at high pH, k bind = 0.80 × 105 M−1s−1. The rate of the reverse reaction step, k diss, varied between 15·10−4 s−1 (pH 6) and 60 × 10−4 s−1 (pH 8). In summary, these results demonstrate by the low steady-state fluorescence level that the PTX-modified Na,K-ATPase binds two H+ ions in the absence of other monovalent cations, and the rate constant of the transformation into the open channel is slowed down in the presence of H+, while the reversal is accelerated.
 |
(4) |
Role of the Access Channels on the PTX-modified State
The concept of PTX modification of the Na,K-ATPase is that both occlusion gates may be open at the same time and, therefore, that the pump may behave as an ion channel. The role of both the cytoplasmic and extracellular access channels may be tested by specific blockers of each half channel. In the literature, those blockers were introduced before. To block the extracellular access channel, tetrapropylammonium (TPA+) was used (Gatto et al., 2005; Kropp and Sachs, 1977), and on the cytoplasmic side, access to the binding sites was prevented effectively by the large organic cation Br2-Titu3+ (Hoving et al., 1995).
When RH421 experiments were performed to investigate the effect of TPA+ on the extracellular access channel in the absence of PTX, it was found (unpublished data) that (a) up to 20 mM TPA+, the RH421 fluorescence in standard experiments was not affected by TPA+, (b) the half-saturating K+ concentration increased from 0.13 mM (0 TPA+) to 1.77 mM (20 mM TPA+), and (c) the enzyme activity in the Na/K mode of the Na,K-ATPase was reduced to <5% in the presence of 20 mM TPA+. At concentrations of 30 mM and above, the property of TPA+ as a hydrophobic cation (similar to TPP+) came into play; therefore, no data were collected above 30 mM. The fact that TPA+ did not modify the RH421 fluorescence at a concentration at which >95% of the pumps were inhibited shows that the organic cation does not penetrate significantly into the membrane domain of the protein but acts on the extracellular outside of the Na,K-ATPase more like a bottle cap.
The action of PTX on the Na,K-ATPase in the presence of TPA+ was studied in the kind of experiments shown in Fig. 3. Na,K-ATPase was equilibrated in standard buffer, then 50 mM Na+, 200 nM PTX, and 1 μM ATP were added. The experiments were repeated with 10 or 20 mM TPA+ added to the standard buffer before addition of Na+ (Fig. 8 A). The time course of the PTX-induced fluorescence change in P-E2 was similar at all TPA+ concentrations. The initial, exponential fluorescence decay had the same rate constant, k, of 0.031 ± 0.002 s−1 irrespective of the TPA+ concentration. The time period until 1 μM ATP was hydrolyzed was increased by ∼15% when TPA+ was increased from 0 to 20 mM. The most significant change was, however, the elevated intermediate fluorescence level in the presence of TPA+, which with 20 mM TPA+ corresponded to an average occupation of one elementary charge in the binding sites.
Figure 8.

Effect of access-channel blockers on the PTX-induced action of the Na,K-ATPase. Na,K-ATPase was equilibrated in standard buffer: 200 nM RH421, 50 mM NaCl, and PTX. Upon addition of 1 μM ATP, the enzyme turned over into the P-E2 state to allow PTX (200 nM in [A] and 100 nM [B]) to modify the pump. (A) In the absence (a) or in the presence of 10 mM (b) and 20 mM (c) TPA+, the main difference of the ATP-induced fluorescence signal was an enhanced intermediate fluorescence level in the presence of the channel blocker. (B) In the absence (a) or in the presence of 5 μM (b), 10 μM (c), 15 μM (d), and 20 μM (e) Br2-Titu3+, the only significant difference was the duration of the intermediate, PTX-dependent state, which was elongated by the presence of Br2-Titu3+.
Assuming that TPA+ produces an all-or-none effect on the extracellular access channel, the higher fluorescence level in the presence of 20 mM TPA+ (Fig. 8 A) would account for a condition in which access to the binding sites was blocked by TPA+ on average in 50% of the pumps. If in the presence of PTX the cytoplasmic occlusion gate of the Na,K-ATPase is open while access from the external side is blocked by TPA+, and under this condition still a lesser occupation of the binding sites is found as shown in Fig. 8 A, this fact indicates that the access to the binding sites through the cytoplasmic channel must be somehow restricted, possibly due to the repelling Coulomb force of TPA+ in the entrance of the extracellular access channel.
To study the properties of the cytoplasmic access channel, experiments similar to that shown in Fig. 8 A were performed; however, Br2-Titu3+ was added instead of TPA+. Results are shown in Fig. 8 B. The main findings are that Br2-Titu3+ in concentrations up to 20 μM did not affect the binding rate constant of PTX, k, nor the fluorescence level of the intermediate state. Solely the overall time until all ATP was hydrolyzed was extended by the presence of Br2-Titu3+.
These results are in agreement with the concept of a blockade of the cytoplasmic access channel since all processes occurring from the extracellular side were unmodified, while the restricted connection of binding sites and cytoplasm decelerated the overall turnover of the pumps, and therefore, the duration until all ATP was hydrolyzed is elongated.
To study the action of the access channel blockers in the absence of Na+ and K+ ions, the Na,K-ATPase was backdoor phosphorylated by 0.5 mM Tris phosphate in the absence or presence of the blockers and then exposed to 150 nM PTX. The results of these experiments in Fig. 9 reveal several interesting properties. (a) Backdoor phosphorylation is significantly slower in the presence of 15 μM of the trivalent cation Br2-Titu3+ (τ ∼42 s), probably by a slowed-down formation of the transient E2(H2) state, which is needed for phosphorylation. In contrast, backdoor phosphorylation was not affected by the presence of TPA+, and the phosphorylation kinetics was limited only by the mixing time upon Pi addition, as in the case of the control experiment without blockers. (b) When PTX was added to the phosphorylated state, there was no significant difference in time course and amplitude of the fluorescence decay when the control experiment is compared with the experiment in the presence of 30 mM TPA+. (c) In the presence of Br2-Titu3+, however, the amplitude of the PTX-induced fluorescence decrease was reduced to ∼20% of the control. This effect was independent of the presence of TPA+. This observation is also in contrast to the “normal” fluorescence decrease when Na+ (50 mM) was present (Fig. 8 B). (d) In all four traces, the fluorescence decrease could be fitted by a single exponential and the rate constant was the same, k = 0.010 ± 0.0002 s−1.
Figure 9.

Effect of access-channel blockers on the PTX-induced action of the Na,K-ATPase in the absence of monovalent cations other than H+. Na,K-ATPase was phosphorylated in standard buffer by 0.5 mM Tris phosphate in the absence or presence of 15 μM Br2-Titu3+ and/or 30 mM TPA+. After reaching a steady-state level of the fluorescence, corresponding to state P-E2, 150 nM PTX was added. Only Br2-Titu3+, the blocker of the cytoplasmic access channel, produced a striking difference by reducing the amplitude of the fluorescence decrease by about a factor of five. TPA+ had no significant effect. In all four cases, the rate constant of the exponential decay was 0.01 ± 0.0002 s−1.
The observed significant reduction of the PTX-induced fluorescence decrease in the presence of Br2-Titu3+ indicates that on average, only one or less H+ are bound to the Na,K-ATPase. A possible explanation of this finding may be the electrostatic repulsion produced by the trivalent cation Br2-Titu3+ bound at the entrance of the cytoplasmic access channel that diminishes the apparent proton binding affinity of the binding sites.
According to Fig. 9, H+ binding was not affected by the presence of TPA+ in the PTX-induced state. Therefore, the pH dependence of the RH421 fluorescence was studied in the absence and presence of 15 μM Br2-Titu3+. The Na,K-ATPase was equilibrated in standard buffer and 200 nM RH421, the enzyme was phosphorylated by 0.5 mM Tris phosphate in the absence or presence of 15 μM Br2-Titu3+ (initial pH 7.0), and then 150 nM PTX was added, and a stationary state was allowed to be reached. The respective pH upon each addition of HCl was measured with a pH electrode. The resulting fluorescence decrease reflects H+ binding to the binding sites. The results are shown in Fig. 10 A. The pK of the fluorescence decrease represents the half-saturating H+ concentration of the site that was initially unoccupied, and it was found to be 6.45 ± 0.05 in the absence and 5.98 ± 0.04 in the presence of Br2-Titu3+ when the data were fitted with a binding isotherm. At saturating H+ concentrations (pH < 4.5), the fluorescence reached the same level for both conditions, which corresponded to occupancy of the binding sites by two H+. The shift of pK (and the reduced fluorescence level at pH 7) is again compatible with a repulsive electrostatic potential generated by the positively charged Br2-Titu3+ bound at/in the entrance of the cytoplasmic access channel. The ion-binding experiments were repeated with K+, which is known to bind with a high affinity. The results are shown in Fig. 10 B. The experiments were performed as described above with the only difference that aliquots of KCl were added (instead of HCl). The concentration dependence was fitted by a binding isotherm, and the half-saturating K+ concentrations, K m, were found to be independent of the addition of the channel blocker, 0.28 ± 0.04 mM (0 Br2-Titu3+) and 0.31 ± 0.03 mM (15 μM Br2-Titu3+). Again, at saturating K+ concentrations, the fluorescence levels would be identical, corresponding to two K+ bound. The initial difference at low K+ is caused by initially less H+ bound (at pH 7) in the presence of Br2-Titu3+ (compare to Fig. 10 A).
Figure 10.

Effect of Br2-Titu3+ on H+ and K+ binding to the Na,K-ATPase in the PTX-modified state (150 nM PTX). Titration of the binding sites was studied in the absence (open circles) and presence of 15 μM Br2-Titu3+ (solid circles). (A) The pH titration showed that in the presence of Br2-Titu3+, the pK was shifted by 0.5 pH units to a lower value, indicating a repulsive effect of the positively charged channel blocker on H+ binding. Merging of the data at low pH represents a saturation of ion binding with both binding sites in the Na,K-ATPase to be saturated. (B) In the case of high-affinity K+ binding, the half-saturating K+ concentration was identical under both conditions (∼300 μM), and saturation at high concentration was also found for both conditions to be the same state with two ions bound. The difference of the initial fluorescence level was caused by the varying H+ binding at pH 7.
DISCUSSION
The most detailed analysis of the molecular mechanism of PTX/sodium pump interaction by now was recently contributed by Artigas and Gadsby who studied the effect of the toxin on the Na,K-ATPase–mediated electric currents in outside-out and inside-out patches of HEK293 cells and in whole-cell current recordings of guinea pig ventricular myocytes (Artigas and Gadsby, 2003b; Artigas and Gadsby, 2004). From their results it could be clearly derived that the Na,K-ATPase is transformed by the toxin into a cation-selective ion channel with a suggested (minimal) width of 7.5 Å. PTX interacts preferentially with the P-E2 state, stabilizes the ion pump in a state different from the states of the Post-Albers pump cycle, and preserves enzyme phosphorylation. The obvious and reasonable proposal for the molecular mechanism is that PTX provokes a conformational rearrangement of the α helices in the protein's membrane domain, which allows the cytoplasmic gate of the ion pump to become open (in addition to the extracellular gate that can already open in the P-E2 state). These findings and detailed proposals for PTX-dependent partial reactions could be gained from measurements of the electric currents enabled by the open-channel state(s) of the Na,K-ATPase and by their substrate kinetics.
The results presented in this study are complementary to the previous investigations. Fluorescence experiments with the electrochromic styryl dye RH421 monitor states of the Na,K-ATPase according to the number of ions bound in the membrane domain of the ion pump as well as transitions between states (Heyse et al., 1994; Pedersen et al., 2002; Stimac et al., 2005). By appropriate choice of the buffer composition, a single (or a few) well-defined state(s) of the Post-Albers cycle (Fig. 11) can be emphasized, and identified by the number of ions bound. Instead of detecting the amount of charge passing through the open Na,K-ATPase channel per time, the fluorescence monitors the amount of charge that is present (on average) inside the membrane domain of the protein.
Figure 11.

Proposal for an extended Post-Albers pump cycle of the Na,K-ATPase induced by the effect PTX. Due to the weak ion selectivity of the PTX-modified ion pump, the ions bound can be Na+, K+, and H+. Therefore, “X” denotes that different ions are transported under appropriate buffer composition from the extracellular side to the cytoplasm. (In the case of H+, the indicated exchange of two X+ against two H+ in the E1 state is unnecessary.) The underlined states, 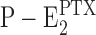 and
and 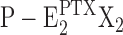 , represent the channel-like states of the Na,K-ATPase. The missing left parenthesis in the state
, represent the channel-like states of the Na,K-ATPase. The missing left parenthesis in the state 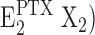 shall indicate that the cytoplasmic gate of the Na,K-ATPase is still held in its open position.
shall indicate that the cytoplasmic gate of the Na,K-ATPase is still held in its open position.
The following discussion is based on two mechanistic proposals that so far could be successfully applied to explain experiments on structure–function relations of the Na,K-ATPase: (1) the ion pump consists of binding sites for two or three ions in the middle of the membrane domain, two access channels that connect the binding sites with both aqueous phases of the membrane, and two gates that disconnect the binding sites (alternately) from either phase when closed (Apell, 2004), and (2) PTX reacts reversibly with the Na,K-ATPase and opens an ion channel without a major rearrangement of the transmembrane helices but in a conformation that consists of mainly the “normal” ion pathway with occasionally simultaneously open gates on the cytoplasmic and extracellular side of the binding sites. These two conditions imply that the ions migrate (diffusion controlled) through the access channels and interact with the protein preferentially in the binding sites, which are preserved overall in the PTX-modified state.
The PTX-affected State of the Na,K-ATPase
As demonstrated by the experiments in Fig. 1, PTX induces a state of the Na,K-ATPase that can be distinguished by its specific fluorescence level from the precedent state, P-E2, from which it develops, and from subsequent states to which it may exit. The amplitude of the fluorescence is in agreement with a state that on average contains two monovalent cations, like, e.g., the initial state H2E1, to which the fluorescence amplitude is normalized in the presented figures. The amount of charge in the PTX-modified enzyme was also the same when the pump was backdoor phosphorylated by inorganic phosphate.
The sequence of standard experiments in Fig. 1 shows that addition of PTX did not affect the substrate-dependent partial reactions in a detectable manner before the pump reached the P-E2 state. The Na,K-ATPase had to reach inevitably P-E2 (without ions in the binding sites) to allow a PTX–pump interaction that led to an uptake (and binding) of ions in the membrane domain. There was no difference in the kinetics of the PTX-induced protein transformation when PTX was present before enzyme phosphorylation or after the enzyme was in the P-E2 state (Figs. 1 and 2), as well as in both methods of phosphorylation, by ATP or Tris phosphate (Fig. 7). Therefore, it can be proposed that a functional interaction occurs in the purified membrane preparation only in the P-E2 state, even if PTX may be already attached to the extracellular part of the Na,K-ATPase. The fact that PTX interaction with the Na,K-ATPase could not be detected in other states of the enzyme is in contrast to the results of Artigas and Gadsby (2004) who proposed an interaction of PTX with all states of the ion pump. This difference may be due to so far disguised toxin–enzyme interactions, which are also reflected by the significantly lower toxin affinity of our enzyme preparation and under our experimental conditions. Artigas and Gadsby report a half-maximum PTX-induced current through the Na,K-ATPase at a concentration of 33 pM in Na+ buffer and high ATP concentrations in HEK239 whole-cell patches. In our microsomal enzyme preparation, the apparent half-saturating PTX concentration varied between 8 and 280 nM, depending on the experiments and the experimental condition.
The Inhibition Kinetics
After the Na,K-ATPase reached the state P-E2, which is characterized by the highest fluorescence level in the pump cycle, the transition into the PTX-modified state can be monitored in a time-resolved manner, and the PTX concentration dependence of the monoexponential decay of the fluorescence indicates a bimolecular reaction, 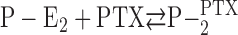 (Fig. 2 and Fig. 7 A). Due to mechanistic reasons, this reaction sequence should be split into two consecutive reaction steps (compare Eq. 3) of which the rate-limiting process has below 23°C such a high activation energy (71 kJ/mol) that it can be assigned to the conformational modification that opens the cytoplasmic gate. This step follows a preceding PTX binding to its site as trigger process. This proposed reaction sequence of PTX binding and transition into the open “channel” molecule is in agreement with the interpretation of the investigation of the channel behavior as published by Artigas and Gadsby (2004).
(Fig. 2 and Fig. 7 A). Due to mechanistic reasons, this reaction sequence should be split into two consecutive reaction steps (compare Eq. 3) of which the rate-limiting process has below 23°C such a high activation energy (71 kJ/mol) that it can be assigned to the conformational modification that opens the cytoplasmic gate. This step follows a preceding PTX binding to its site as trigger process. This proposed reaction sequence of PTX binding and transition into the open “channel” molecule is in agreement with the interpretation of the investigation of the channel behavior as published by Artigas and Gadsby (2004).
When from time-resolved kinetics and from equilibrium titration experiments the PTX binding affinity was determined under otherwise the same experimental conditions, half-saturating PTX concentrations varied by about a factor of three (Table I). In addition, the Na+ concentration in the buffer played a modulatory role with an increased overall turnover rate of the modified pump at higher cation concentrations. These observations are a clue that the effect of the reaction of the protein with PTX, which produces the open channel, is not a state that is the end point of the reaction sequence but allows subsequent steps draining the 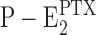 state. In both types of experiments it was observed that higher Na+ concentrations increased the apparent PTX affinity, most probably caused by the reaction
state. In both types of experiments it was observed that higher Na+ concentrations increased the apparent PTX affinity, most probably caused by the reaction 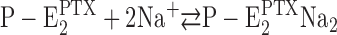 . Higher Na+ produced a shift of the equilibrium to the right side and thus reduced the probability of the back reaction
. Higher Na+ produced a shift of the equilibrium to the right side and thus reduced the probability of the back reaction 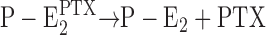 .
.
Properties of the PTX-modified Membrane Domain of the Na,K-ATPase
The most prominent modification of the protein in the presented experiments is the uptake of cations in the membrane domain, as indicated by the fluorescence decrease of ∼30–40%. This effect was observed in the presence of Na+ (Fig. 4) and H+ ions (Fig. 7, inset). Since no electrochemical potential gradient is applied across the membrane, the state after the transformation into an ion channel is a steady state, and therefore, it can be assumed that the ions are located in binding sites. This concept is supported by the result that in ion titration experiments, binding isotherms were found (e.g., Fig. 10). A consequence is that PTX not only opens the cytoplasmic gate but possibly also affects the binding affinity of the sites. This was clearly found for Na+ binding. Binding affinities of the unmodified Na,K-ATPase for Na+ in the P-E2 state were determined to be 90 mM for the first, 1.5 M for the second, and 100 mM for the third ion for the rabbit kidney preparation (Wuddel and Apell, 1995). In the PTX-modified protein, two binding sites are already completely occupied at 50 mM NaCl (Fig. 1). From Fig. 4, a half-saturating concentration of 12 mM was estimated. From direct Na+ titration experiments in the 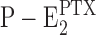 state, a K
m of 18.1 ± 2.0 mM was determined. These values are closer to the half-saturating Na+ concentration in the E1 conformation of the Na,K-ATPase, which is 2 mM in the presence of 1 mM Mg2+ (Schneeberger and Apell, 1999), but still significantly different. Therefore, it has to be proposed that the binding sites are neither in the E1 nor P-E2 form.
state, a K
m of 18.1 ± 2.0 mM was determined. These values are closer to the half-saturating Na+ concentration in the E1 conformation of the Na,K-ATPase, which is 2 mM in the presence of 1 mM Mg2+ (Schneeberger and Apell, 1999), but still significantly different. Therefore, it has to be proposed that the binding sites are neither in the E1 nor P-E2 form.
In the case of H+ binding, the affinity for protons in the native P-E2 state is low enough that at pH 7.2, no H+ are bound (Apell and Diller, 2002), while in 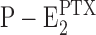 , more than one H+ could be detected in the membrane domain (Fig. 9), similar to what was found in E1. In the case of K+ ions, no significant differences were observed in K+ binding between P-E2 and
, more than one H+ could be detected in the membrane domain (Fig. 9), similar to what was found in E1. In the case of K+ ions, no significant differences were observed in K+ binding between P-E2 and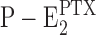 (Fig. 10 B) as well as E1 (Schneeberger and Apell, 2001).
(Fig. 10 B) as well as E1 (Schneeberger and Apell, 2001).
Properties of the Access Channels
The experiments presented above show that TPA+ affects the extracellular access channel of the Na,K-ATPase in both the native and PTX-modified protein. The mechanism did not lead to a complete block of the access channel but generated a behavior more like a flickering gate that prevents a full occupation of the binding sites and thus slowed down dephosphorylation (and overall enzyme activity). A competition of K+ binding with TPA+ has been shown before (Gatto et al., 2005) and was reproduced here. TPA+ does not bind inside the membrane domain close to the binding sites since it did not affect the RH421 fluorescence, nevertheless it reduced the enzyme activity by >95% when present in a concentration of 20 mM. All TPA+-induced effects, in the presence and absence of PTX can be explained by a (reversible) blockade in the extracellular access channel produced by TPA+ as a steric hindrance.
In a similar way Br2-Titu3+ blocked the cytoplasmic access channel rather effectively in the absence as well as in the presence of PTX. It is known that this channel blocker binds on or close to the surface and reduced significantly the movement of Na+ or H+ ions through the access channel from or to the binding sites. This mechanism is in agreement with the observations that the time period is prolonged until 1 μM ATP is hydrolyzed by the Na,K-ATPase (Fig. 8 B) and that backdoor phosphorylation is decelerated by more than a factor of 10 (Fig. 9) in the presence of Br2-Titu3+.
Since the observed effects produced by TPA+ and Br2-Titu3+ can be assigned to the interactions with the extracellular and cytoplasmic access channel, respectively, and since no new quality of the effects of action of these “channel blockers” were observed in the PTX-modified Na,K-ATPase, the findings support the concept that the ion channel through the membrane domain of the protein consists of both access (half) channels and the moiety of the binding sites and that no additional pathway for the ions was opened.
Proposal of a simple reaction scheme for the PTX-modified Na,K-ATPase
To describe the observed kinetical behavior of the ion pump in a comprehensive manner, the Post-Albers cycle (Fig. 11) has to be extended to account for the toxin-induced effects. The simplest extension requires three additional states, two of which may be assigned as “open channel” (Fig. 11). As will be discussed below, in this model, PTX modifies the Na,K-ATPase exclusively in the P-E2 state. The toxin binds in a first step to a specific site on the extracellular domain of the ion pump and provokes a modification in the membrane domain that opens the channel and modifies the ion-binding sites slightly so that the binding affinity, especially for Na+ and H+, is increased. A possible mechanism could be a higher mobility of the amino acid side chains, which are the coordination partners of the ions bound.
In the open-channel conformation, ions pass through the channel as soon as a driving force is present in the form of an electrochemical potential gradient. Ion transport may be described as diffusion-controlled movement through the access channels and a transient binding to the two binding sites present in the P-E2 conformation, similar to what is known to happen in the selectivity filter of a classical ion channel. In the short period when two ions are bound to their sites, the state 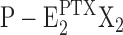 , the enzyme can be dephosphorylated, according to what is happening under physiological conditions in the state P-E2K2 of the Post-Albers cycle. The higher the ion concentration, X+, the higher is the probability that both sites are occupied simultaneously, and this fact would explain the faster escape from the inhibited state and faster ATP hydrolyzation rate at higher Na+ concentrations (Fig. 4). Under the assumption that PTX opens the cytoplasmic gate in the membrane domain, dephosphorylation could close the extracellular gate, similar to the situation under physiological conditions, while the cytoplasmic gate is still open. Then the PTX-modified state is destabilized, and after reversal of the PTX action, the pump returns to the genuine, occluded state E2(X2) of the Post-Albers cycle.
, the enzyme can be dephosphorylated, according to what is happening under physiological conditions in the state P-E2K2 of the Post-Albers cycle. The higher the ion concentration, X+, the higher is the probability that both sites are occupied simultaneously, and this fact would explain the faster escape from the inhibited state and faster ATP hydrolyzation rate at higher Na+ concentrations (Fig. 4). Under the assumption that PTX opens the cytoplasmic gate in the membrane domain, dephosphorylation could close the extracellular gate, similar to the situation under physiological conditions, while the cytoplasmic gate is still open. Then the PTX-modified state is destabilized, and after reversal of the PTX action, the pump returns to the genuine, occluded state E2(X2) of the Post-Albers cycle.
In the presence of ATP, the ion pump continues through the cycle and can, or will, be affected again by PTX when it gets to the P-E2 state. This conception is in agreement with observations on the level of the single-channel experiments by Artigas and Gadsby (2004), who observed long open bursts with brief closures of 20–30 ms duration. Since the rate-limiting reaction steps in the Post-Albers cycle are all included in the sequence from E2(K2) to P-E2, this period of a closed channel of 20–30 ms can be related to a turnover rate of 30–50 s−1, which is to be expected at the temperature of 22–25°C in these experiments. The observed burst durations on the order of a minute with several of these short closures indicate that the PTX remains attached to the pump.
From the kinetical behavior of the Na,K-ATPase in the presence of PTX, some details of reaction steps in the reaction scheme of Fig. 11 may be revealed. The fact that experiments on backdoor phosphorylation of the Na,K-ATPase, which was preincubated with PTX, always show a transition through the (highest) fluorescence level of the P-E2 state, provides a strong indication for the constraints that (a) the backward-oriented reaction sequence, E2X2→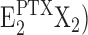 Z →
Z →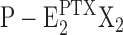 Z (with X = H), in Fig. 11 is kinetically inhibited, and (b) the PTX-dependent reaction P-E2X2→
Z (with X = H), in Fig. 11 is kinetically inhibited, and (b) the PTX-dependent reaction P-E2X2→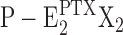 Z does not occur to a detectable extent. The open-channel state is only reached through the P-E2 state.
Z does not occur to a detectable extent. The open-channel state is only reached through the P-E2 state.
Enzyme dephosphorylation in the PTX-modified state, 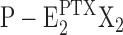 →
→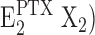 Z is significantly slower than under physiological conditions without toxin. This can be seen from comparison of the fluorescence response on KCl addition in Fig. 1 B (0 PTX) and Fig. 1 (C–E) (100 nM PTX). While in the absence of toxin the transition to state E2K2 cannot be resolved in the time resolution of the experiment (∼2 s), in the PTX-modified ion pump, a response with two components is obvious. The fast initial component is assigned to the fraction of ion pumps uninhibited at that moment (∼25%) while the slow component with a time constant of ∼100 s (∼75%) may be assigned to the slow dephosphorylation. Such an impaired dephosphorylation was also reported from electrophysiological studies (Artigas and Gadsby, 2004).
Z is significantly slower than under physiological conditions without toxin. This can be seen from comparison of the fluorescence response on KCl addition in Fig. 1 B (0 PTX) and Fig. 1 (C–E) (100 nM PTX). While in the absence of toxin the transition to state E2K2 cannot be resolved in the time resolution of the experiment (∼2 s), in the PTX-modified ion pump, a response with two components is obvious. The fast initial component is assigned to the fraction of ion pumps uninhibited at that moment (∼25%) while the slow component with a time constant of ∼100 s (∼75%) may be assigned to the slow dephosphorylation. Such an impaired dephosphorylation was also reported from electrophysiological studies (Artigas and Gadsby, 2004).
Based on the presented data, the action of PTX on the Na,K-ATPase can be described by the concise mechanism as depicted in Fig. 11. Although PTX can be attached to the extracellular side of the Na,K-ATPase in all states of the protein, as discussed above, in this scheme, “PTX” is introduced as superscript to the P-E2 states only to indicate the toxin-modified states. When the ion pump enters into the state P-E2 in the presence of the toxin, binding to its specific site triggers a conformational modification of the membrane domain that opens the cytoplasmic gate in addition to the already open extracellular gate. This process depends on the PTX concentration and occurs with a time constant of ∼40 s at 200 nM PTX (Fig. 2). Under physiological buffer conditions, i.e., in the presence of K+, this step is in competition with ion binding, P-E2 + 2 K+→P-E2K2, which is reflected by an only 50% inhibition of the enzyme activity even at a saturating PTX concentration (Fig. 6). Once in the “channel state,” the binding affinity of the ion binding sites is somehow increased so that the average occupation is higher than in state P-E2; however, binding is not so tight that it would prevent significant ion flux through the channel in the presence of an electrochemical potential gradient for the cations. In the state with two cations bound, 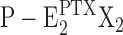 , enzyme dephosphorylation is possible, this process occurs only rather slowly (τ ≈ 100 s). So far it is not clear whether this behavior is a consequence of the short fraction of time in which both binding sites are occupied or a small rate constant of the dephosphorylation step. The dephosphorylated state with PTX bound is proposed to no longer be an open-channel state and has the extracellular gate closed. Finally, the return into the canonical Post-Albers cycle occurs by a release of the membrane domain from the PTX modification; the cytoplasmic gate is closed so that the occluded state is regained, E2(X2). This step may be fast, so far no kinetical information is available; however, on all accounts, the equilibrium of the reaction,
, enzyme dephosphorylation is possible, this process occurs only rather slowly (τ ≈ 100 s). So far it is not clear whether this behavior is a consequence of the short fraction of time in which both binding sites are occupied or a small rate constant of the dephosphorylation step. The dephosphorylated state with PTX bound is proposed to no longer be an open-channel state and has the extracellular gate closed. Finally, the return into the canonical Post-Albers cycle occurs by a release of the membrane domain from the PTX modification; the cytoplasmic gate is closed so that the occluded state is regained, E2(X2). This step may be fast, so far no kinetical information is available; however, on all accounts, the equilibrium of the reaction, 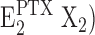 ↔E2(X2), has to be shifted strictly to the right side to prevent the direct transition into the PTX-modified conformation.
↔E2(X2), has to be shifted strictly to the right side to prevent the direct transition into the PTX-modified conformation.
Acknowledgments
We thank Milena Roudna and Christian Mayer for excellent technical assistance. We thank Dr. D.C. Gadsby and Dr. P. Artigas for stimulating discussions and critical reading of the manuscript.
This work was financially supported by the Deutsche Forschungsgemeinschaft (Ap 45/4) and INTAS (Project 011-0224).
Olaf S. Andersen served as editor.
Abbreviations used in this paper: LDH, lactate dehydrogenase; PK, pyruvate kinase; PTX, palytoxin; TPA+, tetrapropylammonium.
References
- Apell, H.J. 2004. How do P-type ATPases transport ions? Bioelectrochemistry. 63:149–156. [DOI] [PubMed] [Google Scholar]
- Apell, H.-J. 2003. Toward an understanding of ion transport through the Na,K-ATPase. Ann. N. Y. Acad. Sci. 986:133–140. [DOI] [PubMed] [Google Scholar]
- Apell, H.-J., and A. Diller. 2002. Do H+ ions obscure electrogenic Na+ and K+ binding in the E1 state of the Na,K-ATPase? FEBS Lett. 532:198–202. [DOI] [PubMed] [Google Scholar]
- Apell, H.-J., M. Roudna, J.E. Corrie, and D.R. Trentham. 1996. Kinetics of the phosphorylation of Na,K-ATPase by inorganic phosphate detected by a fluorescence method. Biochemistry. 35:10922–10930. [DOI] [PubMed] [Google Scholar]
- Artigas, P., and D.C. Gadsby. 2003. a. Ion occlusion/deocclusion partial reactions in individual palytoxin-modified Na/K pumps. Ann. N. Y. Acad. Sci. 986:116–126. [DOI] [PubMed] [Google Scholar]
- Artigas, P., and D.C. Gadsby. 2003. b. Na+/K+-pump ligands modulate gating of palytoxin-induced ion channels. Proc. Natl. Acad. Sci. USA. 100:501–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artigas, P., and D.C. Gadsby. 2004. Large diameter of palytoxin-induced Na/K pump channels and modulation of palytoxin interaction by Na/K pump ligands. J. Gen. Physiol. 123:357–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttinger, H., and E. Habermann. 1984. Palytoxin binds to and inhibits kidney and erythrocyte Na+, K+-ATPase. Naunyn Schmiedebergs Arch. Pharmacol. 325:85–87. [DOI] [PubMed] [Google Scholar]
- Bühler, R., W. Stürmer, H.-J. Apell, and P. Läuger. 1991. Charge translocation by the Na,K-pump: I. Kinetics of local field changes studied by time-resolved fluorescence measurements. J. Membr. Biol. 121:141–161. [DOI] [PubMed] [Google Scholar]
- de Weer, P., D.C. Gadsby, and R.F. Rakowski. 2000. The Na/K-ATPase: a current-generating enzyme. In The Na/K Pump and Related ATPases. K. Taniguchi and S. Kaya, editors. Elsevier Science B.V., Amsterdam. 27–34.
- Farley, R.A., S. Schreiber, S.G. Wang, and G. Scheiner-Bobis. 2001. A hybrid between Na+,K+-ATPase and H+,K+-ATPase is sensitive to palytoxin, ouabain, and SCH 28080. J. Biol. Chem. 276:2608–2615. [DOI] [PubMed] [Google Scholar]
- Forbush, B., III. 1988. Occluded ions and Na, K-ATPase. Prog. Clin. Biol. Res. 268A:229–248. [PubMed] [Google Scholar]
- Gatto, C., J.B. Helms, M.C. Prasse, K.L. Arnett, and M.A. Milanick. 2005. Kinetic characterization of tetrapropylammonium inhibition reveals how ATP and Pi alter access to the Na+-K+-ATPase transport site. Am. J. Physiol. Cell Physiol. 289:C302–C311. [DOI] [PubMed] [Google Scholar]
- Glynn, I.M., and S.J. Karlish. 1990. Occluded cations in active transport. Annu. Rev. Biochem. 59:171–205. [DOI] [PubMed] [Google Scholar]
- Grell, E., E. Lewitzki, and D. Uemura. 1988. Interaction between palytoxin and purified Na, K-ATPase. Prog. Clin. Biol. Res. 268B:393–400. [PubMed] [Google Scholar]
- Guennoun, S., and J.D. Horisberger. 2000. Structure of the 5th transmembrane segment of the Na,K-ATPase alpha subunit: a cysteine-scanning mutagenesis study. FEBS Lett. 482:144–148. [DOI] [PubMed] [Google Scholar]
- Guennoun, S., and J.D. Horisberger. 2002. Cysteine-scanning mutagenesis study of the sixth transmembrane segment of the Na,K-ATPase alpha subunit. FEBS Lett. 513:277–281. [DOI] [PubMed] [Google Scholar]
- Habermann, E. 1989. Palytoxin acts through Na+,K+-ATPase. Toxicon. 27:1171–1187. [DOI] [PubMed] [Google Scholar]
- Heyse, S., I. Wuddel, H.-J. Apell, and W. Stürmer. 1994. Partial reactions of the Na,K-ATPase: determination of rate constants. J. Gen. Physiol. 104:197–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsh, J.K., and C.H. Wu. 1997. Palytoxin-induced single-channel currents from the sodium pump synthesized by in vitro expression. Toxicon. 35:169–176. [DOI] [PubMed] [Google Scholar]
- Horisberger, J.D., S. Kharoubi-Hess, S. Guennoun, and O. Michielin. 2004. The fourth transmembrane segment of the Na,K-ATPase α subunit: a systematic mutagenesis study. J. Biol. Chem. 279:29542–29550. [DOI] [PubMed] [Google Scholar]
- Hoving, S., M. Bar-Shimon, J.J. Tijmes, R. Goldshleger, D.M. Tal, and S.J. Karlish. 1995. Novel aromatic isothiouronium derivatives which act as high affinity competitive antagonists of alkali metal cations on Na/K-ATPase. J. Biol. Chem. 270:29788–29793. [DOI] [PubMed] [Google Scholar]
- Ikeda, M., K. Mitani, and K. Ito. 1988. Palytoxin induces a nonselective cation channel in single ventricular cells of rat. Naunyn Schmiedebergs Arch. Pharmacol. 337:591–593. [DOI] [PubMed] [Google Scholar]
- Ishida, Y., K. Takagi, M. Takahashi, N. Satake, and S. Shibata. 1983. Palytoxin isolated from marine coelenterates. The inhibitory action on (Na,K)-ATPase. J. Biol. Chem. 258:7900–7902. [PubMed] [Google Scholar]
- Jørgensen, P.L. 1974. Isolation of (Na++K+)-ATPase. Methods Enzymol. 32:277–290. [PubMed] [Google Scholar]
- Kropp, D.L., and J.R. Sachs. 1977. Kinetics of the inhibition of the Na-K pump by tetrapropylammonium chloride. J. Physiol. 264:471–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, R.E., and P.J. Scheuer. 1971. Palytoxin: a new marine toxin from a coelenterate. Science. 172:495–498. [DOI] [PubMed] [Google Scholar]
- Muramatsu, I., M. Nishio, S. Kigoshi, and D. Uemura. 1988. Single ionic channels induced by palytoxin in guinea-pig ventricular myocytes. Br. J. Pharmacol. 93:811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki, H., H. Nagase, and N. Urakawa. 1985. Interaction of palytoxin and cardiac glycosides on erythrocyte membrane and (Na+ + K+) ATPase. Eur. J. Biochem. 152:475–480. [DOI] [PubMed] [Google Scholar]
- Pedersen, M., M. Roudna, S. Beutner, M. Birmes, B. Reifers, H.-D. Martin, and H.-J. Apell. 2002. Detection of charge movements in ion pumps by a family of styryl dyes. J. Membr. Biol. 185:221–236. [DOI] [PubMed] [Google Scholar]
- Scheiner-Bobis, G., and H. Schneider. 1997. Palytoxin-induced channel formation within the Na+/K+-ATPase does not require a catalytically active enzyme. Eur. J. Biochem. 248:717–723. [DOI] [PubMed] [Google Scholar]
- Schneeberger, A., and H.-J. Apell. 1999. Ion selectivity of the cytoplasmic binding sites of the Na,K-ATPase: I. Sodium binding is associated with a conformational rearrangement. J. Membr. Biol. 168:221–228. [DOI] [PubMed] [Google Scholar]
- Schneeberger, A., and H.-J. Apell. 2001. Ion selectivity of the cytoplasmic binding sites of the Na,K-ATPase: II. Competition of various cations. J. Membr. Biol. 179:263–273. [DOI] [PubMed] [Google Scholar]
- Schwartz, A.K., M. Nagano, M. Nakao, G.E. Lindenmayer, and J.C. Allen. 1971. The sodium- and potassium-activated adenosinetriphosphatase system. Meth. Pharmacol. 1:361–388. [Google Scholar]
- Stimac, R., F. Kerek, and H.-J. Apell. 2005. Mechanism of the Na,K-ATPase inhibition by MCS derivatives. J. Membr. Biol. 205:89–101. [DOI] [PubMed] [Google Scholar]
- Stürmer, W., and H.-J. Apell. 1992. Fluorescence study on cardiac glycoside binding to the Na,K-pump. Ouabain binding is associated with movement of electrical charge. FEBS Lett. 300:1–4. [DOI] [PubMed] [Google Scholar]
- Stürmer, W., R. Bühler, H.-J. Apell, and P. Läuger. 1991. Charge translocation by the Na,K-pump: II. Ion binding and release at the extracellular face. J. Membr. Biol. 121:163–176. [DOI] [PubMed] [Google Scholar]
- Tosteson, M.T., J.A. Halperin, Y. Kishi, and D.C. Tosteson. 1991. Palytoxin induces an increase in the cation conductance of red cells. J. Gen. Physiol. 98:969–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosteson, M.T., J. Thomas, J. Arnadottir, and D.C. Tosteson. 2003. Effects of palytoxin on cation occlusion and phosphorylation of the (Na+,K+)-ATPase. J. Membr. Biol. 192:181–189. [DOI] [PubMed] [Google Scholar]
- Toyoshima, C., and G. Inesi. 2004. Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 73:269–292. [DOI] [PubMed] [Google Scholar]
- Toyoshima, C., M. Nakasako, H. Nomura, and H. Ogawa. 2000. Crystal structure of the calcium pump of sarcoplasmatic reticulum at 2.6 Å resolution. Nature. 405:647–655. [DOI] [PubMed] [Google Scholar]
- Wang, X., and J.-D. Horisberger. 1997. Palytoxin effects through interaction with the Na,K-ATPase in Xenopus oocyte. FEBS Lett. 409:391–395. [DOI] [PubMed] [Google Scholar]
- Weidmann, S. 1977. Effects of palytoxin on the electrical activity of dog and rabbit heart. Experientia. 33:1487–1489. [DOI] [PubMed] [Google Scholar]
- Wu, C.H., L.A. Vasilets, K. Takeda, M. Kawamura, and W. Schwarz. 2003. Functional role of the N-terminus of Na+,K+-ATPase alpha- subunit as an inactivation gate of palytoxin-induced pump channel. Biochim. Biophys. Acta. 1609:55–62. [DOI] [PubMed] [Google Scholar]
- Wuddel, I., and H.-J. Apell. 1995. Electrogenicity of the sodium transport pathway in the Na,K-ATPase probed by charge-pulse experiments. Biophys. J. 69:909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]