

Abstract
β subunits (Cavβ) increase macroscopic currents of voltage-dependent Ca2+ channels (VDCC) by increasing surface expression and modulating their gating, causing a leftward shift in conductance–voltage (G-V) curve and increasing the maximal open probability, Po,max. In L-type Cav1.2 channels, the Cavβ-induced increase in macroscopic current crucially depends on the initial segment of the cytosolic NH2 terminus (NT) of the Cav1.2α (α1C) subunit. This segment, which we term the “NT inhibitory (NTI) module,” potently inhibits long-NT (cardiac) isoform of α1C that features an initial segment of 46 amino acid residues (aa); removal of NTI module greatly increases macroscopic currents. It is not known whether an NTI module exists in the short-NT (smooth muscle/brain type) α1C isoform with a 16-aa initial segment. We addressed this question, and the molecular mechanism of NTI module action, by expressing subunits of Cav1.2 in Xenopus oocytes. NT deletions and chimeras identified aa 1–20 of the long-NT as necessary and sufficient to perform NTI module functions. Coexpression of β2b subunit reproducibly modulated function and surface expression of α1C, despite the presence of measurable amounts of an endogenous Cavβ in Xenopus oocytes. Coexpressed β2b increased surface expression of α1C approximately twofold (as demonstrated by two independent immunohistochemical methods), shifted the G-V curve by ∼14 mV, and increased Po,max 2.8–3.8-fold. Neither the surface expression of the channel without Cavβ nor β2b-induced increase in surface expression or the shift in G-V curve depended on the presence of the NTI module. In contrast, the increase in Po,max was completely absent in the short-NT isoform and in mutants of long-NT α1C lacking the NTI module. We conclude that regulation of Po,max is a discrete, separable function of Cavβ. In Cav1.2, this action of Cavβ depends on NT of α1C and is α1C isoform specific.
INTRODUCTION
Voltage-dependent Ca2+ channels are grouped into three families, Cav1–Cav3 (Ertel et al., 2000). The main structural component of all Cav channels is the α1 subunit that bears the archetypal features of a voltage-dependent channel, with four membrane-spanning domains and a large cytosolic domain comprising the NH2- and COOH-terminal parts of the protein (NT and CT, respectively), and three large intracellular loops, L1–L3, connecting the membrane-spanning domains (Fig. 1 A). In addition, members of Cav1 and Cav2 families also contain at least two auxiliary subunits, β (Cavβ1–Cavβ4) and α2δ (Isom et al., 1994; Varadi et al., 1995; De Waard et al., 1996; Birnbaumer et al., 1998; Walker and De Waard, 1998; Striessnig, 1999; Catterall, 2000). The α2δ subunit regulates channel expression and trafficking to the plasma membrane (PM) (Shistik et al., 1995; Yasuda et al., 2004; Canti et al., 2005), increases the open probability (Po) of α1C (Shistik et al., 1995), and regulates some pharmacological properties of the channel (De Waard et al., 1996). Cavβ subunits are modular MAGUK-type proteins with an SH3-like and a guanylate kinase (GK)-like domain (Chen et al., 2004; McGee et al., 2004; Opatowsky et al., 2004; Van Petegem et al., 2004). The latter binds with high affinity to a conserved AID (α-interaction domain) motif within the first intracellular loop (L1) of α1 (Pragnell et al., 1994). The β subunits profoundly modulate the properties of voltage-dependent Ca2+ channels. The most prominent effect is a great increase in the magnitude of macroscopic Ca2+ currents, caused by the expression of Cavβ on top of α1 or α1+α2δ in most heterologous expression systems (Mori et al., 1991; Singer et al., 1991; Varadi et al., 1991; Williams et al., 1992; Castellano et al., 1993; Lory et al., 1993), or by the expression of Cavβ in cardiac cells (Wei et al., 2000; Colecraft et al., 2002). Accordingly, depletion or elimination of endogenous Cavβ subunits by knockdown/knockout strategies greatly reduces voltage-dependent Ca2+ currents in various excitable cells (Gregg et al., 1996; Strube et al., 1996; Leuranguer et al., 1998; Namkung et al., 1998; Chu et al., 2004).
Figure 1.

The structure of the α1C subunit and its NH2 terminus. (A) Schematic presentation of the α1C subunit of the L-type Ca2+ channel, Cav1.2. Numbering is shown according to rabbit long-NT α1C-wt (Mikami et al., 1989). The principal NH2-terminal deletion mutants used in this work are indicated by short lines. (B) Comparison of protein sequences of NH2 termini of rabbit and human long-NT and short-NT isoforms. Dots stand for full sequence homology, dashes show gaps, and shaded areas show the conserved motif TxxYxP. Arrows indicate the location of the inserted methionine at the beginning of each NH2-terminal deletion mutant. The initial segments of long-NT and short-NT isoforms are encoded by exons 1a and exon 1, accordingly; the beginning of the transmembrane segment IS1 corresponds to the beginning of exon 3.
Extensive studies in heterologous expression systems revealed a multitude of effects of β subunits on biosynthesis and gating of VDCCs. First, Cavβ increases the surface expression of α1, by improving its trafficking from the ER to the PM (Chien et al., 1995; Brice et al., 1997; Tareilus et al., 1997; Gao et al., 1999), probably by relieving an ER retention signal (Bichet et al., 2000). Second, Cavβ regulates several gating properties of VDCCs. The extent of regulation varies among subtypes and isoforms of Cavβ and Cavα1. The most prominent changes in macroscopic currents include hyperpolarizing (leftward) shifts in current–voltage (I-V) (or conductance–voltage, G-V) and steady-state inactivation curves, an increase in the rate of activation, and changes in the kinetics of voltage-dependent inactivation (for reviews see Birnbaumer et al., 1998; Stotz and Zamponi, 2001; Dolphin, 2003). The shift in G-V curve reflects an improved coupling between gating charge movement and channel opening (Neely et al., 1993, 2004). On the single channel level, coexpression of Cavβ increases the open probability, Po, without changing the single channel conductance (Wakamori et al., 1993; Shistik et al., 1995; Costantin et al., 1998).
The overall increase in macroscopic Ca2+ channel currents caused by heterologous expression of Cavβ results both from increased surface expression, and from changes in gating that lead to increased Po. Regulation of trafficking by Cavβ is clearly separable from modulation of gating; changes in trafficking and gating occur on distinct time and Cavβ concentration scales, and mutations that disrupt the high-affinity interaction between AID and Cavβ disrupt colocalization of α1 and β in the PM and membrane targeting of α1, but spare some or all of the β-induced changes in gating parameters. Thus, the high-affinity binding of Cavβ to AID is obligatory for the regulation of trafficking but not gating (Yamaguchi et al., 1998; Canti et al., 1999, 2001; Gerster et al., 1999; Hullin et al., 2003; McGee et al., 2004; Leroy et al., 2005; Maltez et al., 2005). The SH3–GK domain interaction may also play an important role in regulating trafficking (Takahashi et al., 2005). Among gating effects of Cavβ, at least one, the regulation of kinetics of voltage-dependent inactivation (VDI), is separable from the others. A palmitoylated isoform of β2a (usually designated simply as β2a) decelerates the VDI of several Cavα, whereas all other β subunits, including a nonpalmitoylated isoform of β2a (np-β2a of rabbit, and its human orthologue β2b), accelerate VDI. At the same time, the enhanced trafficking of α1 and the hyperpolarizing shift in G-V curve are independent of palmitoylation of Cavβ (Olcese et al., 1994; Chien et al., 1996; Qin et al., 1996, 1998; Gao et al., 1999). The separability of different effects of Cavβ implies that they are determined by distinct molecular interactions between different parts of β and/or α1. Some actions of β may rely upon low-affinity interactions of β (the SH3 domain in particular) with regions outside the AID in α1, either in L1 or in the CT in some types of α1 (Tareilus et al., 1997; Walker et al., 1999; Takahashi et al., 2004; Maltez et al., 2005). At present, it is unclear whether gating effects of Cavβ other than change in kinetics are also separable, and what is the molecular basis of separate effects of Cavβ on different gating parameters.
It is notable that cells most widely used for heterologous expression of Ca2+ channels, Xenopus oocytes and human embryonic kidney (HEK) cells, contain small but measurable amounts of an endogenous Cavβ protein that undoubtedly aids the “β-less” channels to reach the PM (Tareilus et al., 1997; Canti et al., 2001; Leroy et al., 2005). Arguably, the presence of a minimal amount of endogenous Cavβ may be obligatory for surface expression of at least some subtypes of Cav1 and Cav2 channels (Tareilus et al., 1997; Leroy et al., 2005). An absence of such endogenous Cavβ may explain the reports that in some cell lines transfected with α1C alone or even with α1C+ α2δ, no functional Ca2+ channel expression is observed (Gao et al., 1999; Harry et al., 2004; Kobrinsky et al., 2004). The presence of the endogenous Cavβ, which is permissive for trafficking of α1 to PM, does not impair the ability of coexpressed or exogenously added Cavβ protein to modulate the biophysical properties of the channel (Tareilus et al., 1997; Yamaguchi et al., 1998; Garcia et al., 2002). Therefore, it has been proposed that the endogenous β only “chaperones” α1, helping it to leave the ER without staying with it in the PM; the added exogenous β then binds to α1 and modulates the gating (the single Cavβ-binding model). An alternative multiple Cavβ-binding model contends that the endogenous β remains irreversibly bound to AID, and additional β subunit(s) modulate channel gating by interacting with other parts of α1 (discussed by Birnbaumer et al., 1998; Jones, 2002; Dolphin, 2003). A recent study that used a β2b subunit tethered to the end of α1C strongly supports a functional 1:1 α1-β stoichiometry (Dalton et al., 2005); unfortunately, not all functions of β were fully recovered by the tethered β, leaving this fundamental issue open for argument.
Unfortunately, the exact extent of regulation of different Cav channels by various Cavβ is still debated (discussed in Yasuda et al., 2004), and the contribution of different mechanisms to the increase in whole-cell current has not yet been precisely assessed. The variations are aggravated by apparent inconsistencies between results obtained in different cells and the use of different isoforms of Cavα1 and Cavβ. To properly understand the molecular principles underlying the different actions of Cavβ, one needs to reliably monitor both the surface expression of α1 and a defined set of gating parameters, in a well characterized system. In this report, we implemented such an approach to analyze the mechanism of regulation of Cav1.2 (α1C), the L-type channel present in most excitable tissues, by β2b. The study focused on the parameters of channel activity that lead to changes in the magnitude of the macroscopic Ba2+ current (IBa), leaving the regulation of inactivation out of scope. We have previously found that in the cardiac (long-NT) isoform of α1C, the first 46 aa of the cytosolic NT constitute an NH2-terminal inhibitory (NTI) module whose removal greatly increases the macroscopic current and the Po. The presence of the NTI module is also crucial for β2b-induced increase in IBa via the long-NT isoform α1C in Xenopus oocytes. We therefore proposed that the β subunit acts, in part, by functionally counteracting the inhibitory effect of this module (Shistik et al., 1998; Ivanina et al., 2000). Here we demonstrate that the long-NT initial segment selectively regulates a single action of β2b: the elevation of Po,max. This modulation is absent in deletion mutants lacking the initial NT segment, and in a short-NT isoform of α1C (smooth muscle/brain subtype), in which the initial 46 aa encoded by exon 1a are replaced by a partially homologous stretch of 16 aa encoded by exon 1. Other effects of β (trafficking, G-V curve shift) are preserved. This is the first report on a discrete regulation by Cavβ of Po,max in Cav1.2. These findings bear upon the isoform-specific physiological properties of the L-type Ca2+ channel and upon the physiological role of Cavβ in different tissues.
MATERIALS AND METHODS
DNA Constructs and mRNA
cDNAs of rabbit heart α1C (X15539), rabbit heart np-β2a (L06110) (here termed β2b), skeletal muscle α2δ-1 (P13806) subunits, and the α1C NH2-terminal truncation mutants α1CΔ5, α1CΔ20, α1CΔ46, and α1CΔ139 were prepared and used as described previously (Shistik et al., 1998). The mutants Δ6-20, Δ21-46, T10A/Y13F/P15A, NTSL, and NTLS chimeras were constructed by standard PCR methods. To create α1C-HA, the hemagglutinin (HA) tag (SRYPYDVPDYA; the first two amino acids SR were added in order to create an XbaI restriction site in the corresponding cDNA sequence) has been inserted into the extracellular loop after the S5 transmembrane segment of α1C-wt, between amino acids Q713 and T714, by two consecutive PCRs. The Δ21-46-HA and NTLS-HA were then produced by standard subcloning procedures. All mutations and PCR products were verified by nucleotide sequencing at the Tel Aviv University Sequencing Facility.
All cDNA constructs of α1C and mutants were inserted into the same vector, pGEM-HE-GSB (Shistik et al., 1998), which is a derivative of pGEM-HE (Liman et al., 1992). This vector provides the necessary 5′ and 3′ untranslated regions (UTR) from Xenopus α-globin. Therefore, only the coding sequences of all α1C derivatives, without any residual original UTRs, were inserted into the vector. To minimize any variability that may be caused by variations in quality and quantity of RNAs, in each series of experiments all tested RNAs (all mutants of α1C under study and the wt α1C) were synthesized anew on the same day.
Oocyte Culture and Electrophysiology
All the experiments were performed in accordance with the Tel Aviv University Institutional Animal Care and Use Committee (permits no. 11–99-47 and 11–05-064). Xenopus laevis frogs were maintained and operated, and oocytes were collected, defolliculated, and injected with RNA as previously described (Dascal and Lotan, 1992). In brief, female frogs, maintained at 20 ± 2°C on an 11-h light/13-h dark cycle, were anaesthetized in a 0.15% solution of procaine methanesulfonate (MS222), and portions of ovary were removed through a small incision on the abdomen. The incision was sutured, and the animal was returned to a separate tank until it had fully recovered from the anesthesia, and afterwards was returned to a large tank where, together with the other postoperational animals, it was allowed to recover for at least 4 wk until the next surgery. The animals did not show any signs of postoperational distress. The oocytes were injected with the mRNAs of α1C or its mutants, α2δ, and β2b, according to the design of experiment (0.3–5 ng for electrophysiology and 2.5–5 ng for imaging). Unless indicated otherwise, equal amounts (by weight) of different RNAs were injected. The oocytes were incubated for 3–5 d at 20–22°C in ND96 solution (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 5 mM HEPES, pH 7.5) supplemented with 2.5 mM Na-pyruvate and 50 μg/ml gentamycin. In some batches with high endogenous chloride currents, oocytes were injected with 25–30 nl/oocyte of the Ca2+ chelator EGTA (50–100 mM), giving a final concentration of 2–5 mM within the oocyte (assuming oocyte's free water volume of 0.5 μl).
Whole cell currents were recorded using the Gene Clamp 500 amplifier (Axon Instruments) using the two-electrode voltage clamp technique, in a solution containing 40 mM Ba(OH)2, 50 mM NaOH, 2 mM KOH, and 5 mM HEPES, titrated to pH 7.5 with methanesulfonic acid (Shistik et al., 1995). Stimulation, data acquisition, and analysis were performed using pCLAMP software (Axon Instruments). Current–voltage (I-V) relation of Ba2+ currents was measured by 60-ms, 10-mV steps given every 10 s from a holding potential of −80 mV. In each cell, the net IBa was obtained by subtraction of the residual currents recorded with the same protocols after applying 200 μM Cd2+.
Immunocytochemistry and Confocal Imaging
Immunocytochemistry in giant PM patches was done essentially as previously described (Singer-Lahat et al., 2000; Peleg et al., 2002), as illustrated in Fig. 3. Oocytes were devitellinized by peeling off the vitelline membrane in ND96, and placed on plastic coverslips (Thermanox plastic coverslip; Nunc). After sticking to the coverslip, the oocyte was removed mechanically and/or by washing with a strong jet of solution. Pieces of membranes strongly attached to the coverslip were continuously washed until the membrane patch became transparent without any visible cytosolic content and pigment granules. After fixation for 10 min in 1% formaldehyde, the membranes were washed three times with 1% BSA dissolved in TBS solution (135 mM NaCl, 10 mM Tris-HCl, pH 7.4). Blocking of nonspecific binding sites was done with donkey immunoglobulin G (IgG, whole molecule, 1/200, Jackson ImmunoResearch Laboratories) for 30 min. Each coverslip was incubated for 1 h with CT4 antibody against α1C COOH terminus (1:500; provided by M. Hosey, Northwestern University, Chicago, IL; see Gao et al., 2001) or against L2 (1:500; Alomone Labs.). Residual antibody was washed out with 1% BSA three times, 5 min each. This was followed by a 30-min incubation with secondary antibody (Cy3 donkey anti–rabbit IgG, 1:400; Jackson ImmunoResearch Laboratories). Free secondary antibody was then washed out with 1% BSA three times, 5 min each in darkness and the coverslips were mounted on a glass slide. The fluorescent labeling was examined by a confocal laser scanning microscope (LSM 410 or LSM 510, Zeiss, Germany). 40× NA/1.2 C-apochromat water-immersion lens (Axiovert 135 M, Zeiss) was used for imaging. The Cy3-conjugated secondary antibody was excited at 488 nm and the emitted light at >568 nm was collected. The fluorescent signals were analyzed by measuring total luminosity (optical density) of the whole image using the Tina 2.1 (Raytest Isotopenmelgerilte GmbH) or Carl Zeiss MicroImaging, Inc. LSM5 software. In all confocal imaging procedures, care was taken to completely avoid saturation of the signal. The gain of the photomultiplier was kept <75% of maximum. In each experiment, all oocytes from the different groups were studied using a constant set of imaging parameters. The normalized intensities were always calculated relative to the control group of the same experiment. Net fluorescence intensity per unit area was obtained by subtracting an averaged background signal measured in the same way in membranes of native (uninjected) oocytes from the same batch.
Figure 3.

NT mutations and deletions do not alter the surface expression of α1C. (A) A simplified presentation of the method used to measure the surface expression of α1C in giant membrane patches (see Materials and Methods for details). A devitellinized intact oocyte is placed for 10–20 min on coverslip with its animal (dark) hemisphere facing the coverslip (a). After attachment, the oocyte is swept away and patches of membrane (usually >100 μm in diameter) remain stuck to the coverslip (b). The cytosolic surface of the PM, facing the external solution, is thoroughly washed until the membrane appears transparent (c) and then stained with an antibody directed against a cytosolic part of the channel. (B) Examples of confocal images of α1C-wt, α1CΔ46, α1CΔ20, and α1CTYP in giant patches of oocytes of the same batch. (C) NT mutations do not alter the surface expression of α1C (a), despite the typical differences in IBa (measured at +20 mV) in oocytes of one of these batches (b). The amount of injected RNA was 5 ng/oocyte.
Immunocytochemistry and imaging of whole oocytes has been done as follows. 3–4 d after the injection of RNA, the oocytes were fixated in 4% formaldehyde (37%) in Ca-free ND96 solution for 15 min. Blocking of nonspecific binding sites was done by 5% skim milk for 1 h. Then the oocytes were incubated for 1 h with the mouse monoclonal IgG2a antibody against HA (Santa Cruz Biotechnology), diluted 1:400 in 2.5% skim milk. Residual antibody was washed out with 2.5% skim milk three times, 5 min each. This was followed by 1 h incubation with the secondary antibody (Alexa-conjugated anti–mouse IgG, 1:400; Jackson ImmunoResearch Laboratories) in dark. Free secondary antibody was then washed out with Ca-free ND96. Oocytes were placed in a chamber with a transparent bottom, and fluorescence imaging of optical slices was performed with LSM 510 (×20 objective, zoom = 1, pinhole 3 Airy units). Alexa was excited at 594 nm and the emitted light was collected using long-pass (LP) 615-nm filter. The fluorescent signals were usually analyzed as described in Fig. 4 D. Alternatively, the intensity of fluorescence in the PM was measured by averaging the signal obtained from six standard circular regions of interest. Net fluorescence intensity per unit area was obtained by subtracting the background signal measured in native oocytes.
Figure 4.

Coexpression of β2b increases the surface expression of α1C-wt and of NT mutants. (A) Examples of confocal images of α1C-wt, α1CΔ46, α1CΔ20, and α1CTYP in giant PM patches, in absence and presence of β2b subunit. (B) Surface expression of the mutant proteins in α1α2δ subunit composition, without β2b, normalized to α1C-wt. This series of experiments included two oocyte batches and was separate from that shown in Fig. 3. Injected RNA was 5 ng/oocyte. (C) Summary of the effects of β2b on the surface expression of α1C mutants. For each construct, the surface expression in the presence of β2b was normalized to the average expression measured without β2b. (D) The effect of β2b on surface expression of α1C-HA; panel a shows representative confocal images of whole oocytes, either native (uninjected) or expressing α1C-HA + α2/δ (“α1C-HA”) or α1C-HA + α2/δ + β2b (“α1C-HA+β2b”). Injected RNA was 5 ng/oocyte. The dimensions of the image are 0.45 × 0.45 mm. The rightmost image exemplifies the method used to analyze the intensity of fluorescent labeling. A rectangle of a fixed width was superimposed on the image, and a profile of optical density (shown in b) along the axis approximately perpendicular to the PM (arrow) was obtained using the TINA software. (Optical density and distance were measured in arbitrary units inherent to the software). (b) Quantitative analysis of optical density profiles. The intensity was defined as the integral of the shaded area. The width of this area was defined as constant for all images taken in an experiment. The net intensity in each α1C-HA–expressing oocyte was calculated by subtracting the average intensity measured in uninjected oocytes of the same experiment, and then normalized to the average net intensity of [α1C-HA+α2/δ]-expressing oocytes of this experiment. The normalized values, shown in c, were summarized across all experiments (N = 3).
Data Analysis
Current–voltage (I-V) curve was fitted to the Boltzmann equation in the form
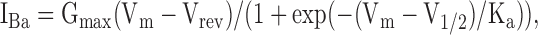 |
(1) |
where Ka is the slope factor, V1/2 is the voltage that causes half maximal activation, Gmax is the maximal macroscopic conductance, Vm is membrane voltage, IBa is the current measured at the same voltage, and Vrev is the reversal potential of IBa. The obtained parameters of Gmax and Vrev were then used to calculate fractional conductance at each Vm, G/Gmax, using the equation
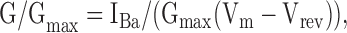 |
(2) |
where G is the total macroscopic conductance at Vm. The conductance–voltage (G-V) curves were plotted with the values of V1/2 and Ka obtained from the fit of the I-V curves, using the following form of the Boltzmann equation:
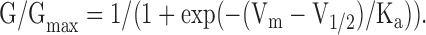 |
(3) |
The results were summarized from many groups of oocytes from different donors (“batches”). To avoid series resistance artifacts associated with very large currents (Schreibmayer et al., 1994), or inaccuracies resulting from a contribution from the small endogenous oocyte's Ca2+, Cl−, or K+ channel currents when the macroscopic IBa was low (mainly in oocytes expressing α1C+ α2δ without β2b; Dascal, 1987; Dascal et al., 1992), these quantitative analyses were performed only in experiments in which, at the peak of the I-V curve, 0.1 μA ≤ IBa ≤ 6 μA.
The calculation of changes in Po,max was based on the following considerations. The total macroscopic conductance, estimated by measuring peak currents at different voltages, is a linear function of Po:
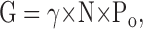 |
(4) |
where γ is the single channel conductance, and N is the total number of functional channels in the PM (Hille, 2002). In Cav channels, Po is voltage dependent whereas γ and N are not. Po reaches a maximal value, Po,max, at positive voltages where a maximal macroscopic conductance, Gmax, is attained. Both Gmax and Po,max are empirical parameters that are considered voltage independent and are interrelated as follows:
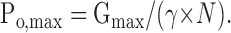 |
(5) |
The fold change in N caused by a treatment, RN, is defined as Ntreatment/Ncontrol. From here, if γ is constant, the change in Po,max caused by a treatment is given by
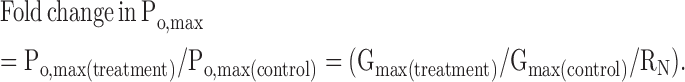 |
(6) |
In this study, RN was monitored by imaging measurements in giant PM patches or in whole oocytes. Eq. 6 is similar to those used previously (Wei et al., 1994; Takahashi et al., 2004) to assess changes in Po; in these studies, RN was estimated from the measurement of Qmax (the maximal gating charge).
To compare parameters (I40, surface α1C labeling, or Gmax) observed or calculated in oocytes of different batches, data from individual cells were averaged across batches using the following normalization procedure (Sharon et al., 1997). The value of the parameter in each cell was normalized to the mean value of this parameter in the control group of the same batch. This procedure was also applied to the cells of the control group, thus providing a useful measure of variability in this group and enabling an accurate statistical analysis. Normalized values were then averaged from all cells across all batches. The results are presented as means ± SEM Multiple group comparisons were done by one-way analysis of variance (ANOVA) test followed by Tukey's test. Two-group comparisons were done using Student's t test.
Online Supplemental Material
Table S1 (available at http://www.jgp.org/cgi/content/full/jgp.200609485/DC1) shows the mean values of Gmax and Vrev obtained in Boltzmann equation fits for some of the α1C constructs used in this study.
RESULTS
Mapping the Part of NT Crucial for the Regulation of Gmax
Deletions of initial segment of long-NT α1C increase the amplitude of IBa but attenuate the current enhancement caused by Cavβ. It is not known if the same structural elements of NT alter both macroscopic currents and modulation by Cavβ. To address this problem, we sought to delineate the regions that are important for these modulations. Fig. 1 B shows the protein sequences of the NH2-terminal part of long-NT and short-NT isoforms of rabbit and human α1C (the corresponding sequences in rat and mouse are also highly homologous; see Blumenstein et al., 2002). The cytosolic NT consists of 124 (short-NT) or 154 aa (long-NT), which are the products of transcription of alternative initial exons 1 or 1a, respectively, and an invariable exon 2 encoding 108 aa. Amino acids 6–20 of the long-NT show partial homology with the first 16 aa of the short-NT (Shistik et al., 1998); in particular, the amino acids T10, Y13, and P15 (which we call TxxYxP motif; numbering by long-NT) are conserved in both isoforms (Fig. 1 B, gray boxes). In the previous studies, we have created several NT deletion mutants of rabbit cardiac long-NT α1C. The logic of these deletions was dictated by the comparison of nucleotide sequences of long- and short-NT isoforms. Initially, we deleted amino acid stretches from the first methionine up to the positions corresponding to the boundaries of the region of partial homology (mutants α1CΔ5 and α1CΔ20), and then up to the beginning of the high homology region (α1CΔ46). We also deleted almost all of the cytosolic part of the NT (α1CΔ139). In the previous studies, only α1CΔ46 and α1CΔ139 have been characterized in detail; voltage-dependent characteristics, protein expression levels, and the effect of β subunit have not been compared systematically in most mutants. Such a comparative study has been done in the experiments described below. In addition, we have deleted the region of partial homology with short-NT α1C, aa 6–20, from long-NT α1C (mutant α1CΔ6-20). We also mutated the TxxYxP motif, creating the T10A/Y13F/P15A (α1CTYP) mutant.
To delineate the region of long-NT that regulates the magnitude of IBa, either long-NT wild-type α1C (α1C-wt) or the various NT deletion mutants were expressed without Cavβ. The expression of α1C-wt alone in Xenopus oocytes results in very small (a few nA) Ba2+ currents that are difficult to reliably resolve and analyze, and often difficult to separate from currents via oocyte's endogenous Cav channels, which are non–L type (Singer et al., 1991; Dascal et al., 1992; Singer-Lahat et al., 1994). Therefore, α2δ was coexpressed in all experiments. The latter greatly increases IBa compared with α1C-wt alone, but does not increase the current via the endogenous channels. α2δ aids trafficking α1C to the PM (Shistik et al., 1995; Yasuda et al., 2004; Canti et al., 2005) and produces certain synergistic (more than additive) effects with some β subunits (Singer et al., 1991; Yamaguchi et al., 2000), but most of the actions of β2b are similar in the presence or absence of α2δ (see Table IV). In oocytes expressing α1C+α2δ or α1C+α2δ+β2b, the contribution of endogenous channels to IBa was negligible; IBa was reduced by >95% by 10 μM of the dihydropyridine L-type Ca2+ channel blockers nitrendipine and nifedipine (Singer-Lahat et al., 1994; unpublished data).
Table IV.
A Summary of Published Quantitative Analyses of the Effects of Coexpression of Cavβ Subunits on Cav1.2 Channels in Heterologous Expression Systems

Surface expression, I-V shift, and Po are usually presented for α1c+α2δ+β subunit combination vs. α1c+α2δ, unless indicated otherwise. Mammalian cells were derivatives of HEK, COS, or similar cell lines lacking significant amounts of an endogenous α1C. In one study (Colecraft et al., 2002), β subunits were overexpressed in cardiac myocytes.
aChanges in N were measured from Qmax. We have calculated Po,max using Eq. 6 and the presented data.
bNo α2δ present.
cEstimated from current increase at peak, by comparing early (1 h) and late times after the injection of the β3 protein; the shift in I-V is already complete by 1 h, and the difference in peak current is only due to surface expression (see Fig. 4 in Yamaguchi et al., 1998).
dPurified β protein was injected into oocytes.
eBy imaging an intracellular HA tag added to α1C at the NT. Oocytes were permeabilized with Triton-X100 and stained with an anti-HA antibody.
fIn the presence of a dihydropyridine agonist.
gFrom total membrane [3H]PN200-110 binding. This method does not exclude the labeling of α1C located in ER or Golgi.
hBoth by immunoprecipitation of α1C from separated PM and by counting channels in cell-attached patches.
iBy counting channels in patches. Measurement by surface biotinylation confirmed the increase in N, but there is no quantitative estimate.
jβ2a was overexpressed in cardiomyocytes on the background of existing cardiac channels, probably the long-NT α1C type (Blumenstein et al., 2002; Pang et al., 2003). Po,max was measured in single channel records at +40 mV.
kA deletion mutant of the rabbit long-NT α1C missing the first 60 aa.
lThe types of α1C are defined as follows: Mikami (Mikami et al., 1989) is the first cloned long-NT. This is the rabbit cardiac isoform, used also in the present study (termed α1C-wt). Identical or nearly identical cDNAs were later cloned by Wei et al. (1991), Biel et al. (1991) and Lory et al. (1993).m Schultz (Schultz et al., 1993) is a human α1C bearing 93% similarity to Mikami's clone but missing the first 59 aa at the NT (the protein starts with Met60 encoded by the first in-frame ATG of exon 2). Biel (Biel et al., 1990) is a rabbit lung short-NT α1C with the 16-aa initial segment, and variations in a few downstream exons compared to Mikami's clone. Snutch (Snutch et al., 1991) is a short-NT (16 aa initial segment) rat cDNA; the most widely used is the rbII clone.
mThe cDNA is identical to that of Mikami et al. (1989) except for an alternative exon encoding for a part of IVS3.
Fig. 2 A shows the routine experimental protocol used to analyze the I-V characteristic of the expressed channels. Ca2+ channel currents were elicited by 60-ms depolarizing pulses from a holding potential of −80 mV, in 10-mV steps, in a solution containing 40 mM Ba2+ using the two-electrode voltage clamp technique. Net IBa was obtained by subtracting currents elicited by the same voltage protocol in the presence of 200 μM CdCl2 (Fig. 2 A, left). The absolute amplitude of IBa depended on the amount of RNA injected, period of incubation, and also varied among oocyte batches. The amount of the injected RNA (of both α1C-wt and α2δ) varied in our experiments between 1 and 5 ng/oocyte, depending on the experimental design. In this range, greater amounts of RNA always resulted in larger IBa. The right panel of Fig. 2 A shows a typical I-V curve of net IBa, averaged from six oocytes of the same batch (donor frog). Without Cavβ, IBa was maximal at ∼30 mV. In the following, to compare IBa across different groups and treatments, we chose to use the value of IBa measured at +40 mV (I40) rather than at the peak of the I-V curve, because the latter varied between different treatments. Also, at +40 mV, the whole-cell Ba2+ conductance is close to maximum under most conditions (Figs. 2 and 4), therefore changes in I40 should approximately reflect changes in Gmax.
Figure 2.

Effects of NH2-terminal deletions and T10A/Y13F/P15A mutation on IBa in the absence of Cavβ (the α1Cα2δ subunit composition). (A) The standard procedure used to monitor IBa, and an averaged I-V curve. See explanations in the text. (B) Comparison of IBa in α1C-wt and α1CΔ5 (2.5 ng RNA/oocyte of each subunit). Panel a shows representative current traces recorded at +40 mV in oocytes of one batch (donor). A current trace obtained in an oocyte expressing the α1Δ46 mutant is shown for comparison. Panel b shows averaged I-V curves from α1C-wt and α1Δ5 groups (n = 9 oocytes, N = 2 batches). The normalized values of I40 are shown in panel c. (C) Panel a shows the summary of relative I40 in the various mutants. I40 in each oocyte was normalized to the average I40 of α1C-wt of the same batch, as explained in Materials and Methods. Panels b and c show normalized I-V and G-V curves, respectively, of the indicated channel constructs, averaged from oocytes of two representative batches (n = 8–14, N = 2). Amount of injected RNA was varied from 0.3 ng/oocyte in NT mutants to 5 ng/oocyte in α1-wt to reach comparable IBa amplitudes in order to minimize the fitting artifacts. Note that Boltzmann fits have been performed separately in each oocyte. For illustration purposes, in averaged I-V and G-V curves shown in C and in the following figures, the solid lines through averaged experimental points were drawn using Boltzmann equation with values of V1/2 and Ka from Table I. In this and the following figures, the numbers above bars indicate the number of cells tested, and asterisks indicate statistically significant differences, as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
As reported previously (Shistik et al., 1999), in the α1CΔ5 mutant, I40 increased about twofold compared with α1C-wt, without any significant change in the voltage dependence of activation (Fig. 2 B). The calculated increase in Gmax in the α1CΔ5 mutant was 66% (Table II). In comparison, the deletion of 20 or more aa of the long-NT initial segment caused a robust seven to ninefold increase in I40 and in the calculated Gmax (Fig. 2, Ba and Ca, and Table II). For a summary of the absolute values of Gmax in the different groups, injected with the same RNA amount of 1 ng/oocyte, see Table S1 (available at http://www.jgp.org/cgi/content/full/jgp.200609485/DC1).
TABLE II.
Relative Changes in Surface Expression, Gmax, I40, and Po in NT Mutants in α1C+α2/δ Subunit Composition in the Absence of Cavβ
| Fold increase in PM α1C
labeling |
Fold increase in Po
|
||||
|---|---|---|---|---|---|
| Construct | Fold increase in Gmax | Fold increase in I40 | from Gmax | from I40 | |
| α1C-wt | 1 ± 0.05 (40; 7) | 1 ± 0.04 (56; 11) | 1 ± 0.07 (56; 11) | ||
| α1CΔ5 | 1.21 ± 0.18 (6; 2) | 1.66 ± 0.08c (9; 2)a | 1.76 ± 0.24b (9; 2)a | 1.37 | 1.45 |
| α1CΔ20 | 1.22 ± 0.12 (24; 3) | 7.64 ± 0.98d (30; 6) | 7.68 ± 0.34d (30; 6) | 6.26 | 6.3 |
| α1CΔ46 | 0.99 ± 0.12 (16; 3) | 7.63 ± 0.81d (30; 6) | 7.75 ± 0.95d (30; 6) | 7.7 | 7.82 |
| α1CΔ139 | Not examined | 6.81 ± 0.93d (19; 5) | 6.88 ± 0.94d (19; 5) | ||
| α1CTYP | 0.86 ± 0.1 (17; 3) | 5.71 ± 0.93d (23; 3) | 5.95 ± 0.95d (23; 3) | 6.63 | 6.91 |
| α1CΔ6-20 | Not examined | 8.22 ± 1.01d (14; 3) | 8.6 ± 1.23d (14; 3) | ||
| α1C-short | 1.16 ± 0.14 (9; 2) | 8.56 ± 0.98d (12; 2) | 8.71 ± 1.71d (12; 2) | 7.38 | 7.5 |
| NTSL | 1.13 ± 0.19 (11; 2) | 5.77 ± 0.56d (16; 3) | 5.98 ± 0.74d (16; 3) | 5.1 | 5.3 |
| HA-labeled constructs | |||||
| α1C-HA | 1 ± 0.12 (10; 1) | 1 ± 0.11 (9; 3) | 1 ± 0.31 (9; 3) | ||
| NTLS-HA | 1.15 ± 0.09 (11; 1) | 13.6 ± 1.85d (15; 3) | 14.08 ± 2.0d (15; 3) | 11.8 | 12.2 |
| α1CΔ21-46-HA | 0.88 ± 0.09 (11; 1) | 1.41 ± 0.29d (9; 3) | 1.47 ± 0.29d (9; 3) | 1.6 | 1.67 |
Surface expression was measured by confocal imaging of immunostained α1C in giant membrane patches or, for HA-labeled constructs, in intact oocytes. Numbers in parentheses indicate n; N (the total number of oocytes and the number of oocyte batches tested). To calculate fold change in a parameter, normalization procedure was performed as described in Materials and Methods. Statistical comparisons were performed using one way ANOVA (multiple comparisons versus control group, α1C-wt or α1C-HA, in oocytes injected with the same amount of RNA, followed by Tukey's test).
In two batches where α1C-wt and α1CΔ5 were compared, the statistical analysis has been done separately from other batches, since the amount of injected RNA was 2.5 ng per oocyte rather than 1 ng. Averaged normalized fold increase in Gmax and I40 for α1C-wt in these two batches was 1 ± 0.16 (n = 9, N = 2) and 1 ± 0.2 (n = 9, N = 2), respectively. The statistical analysis was done using t test.
P < 0.05.
P < 0.01.
P < 0.001.
As reported earlier (Wei et al., 1996; Shistik et al., 1998), the I-V curves in α1CΔ46 and α1CΔ139 appeared similar to α1C-wt. However, an exhaustive comparison of a large amount of records revealed a small hyperpolarizing shift caused by the NT deletions, as illustrated in normalized averaged I-V curves (Fig. 2 Cb). To assure that the shift was not an artifact resulting form a larger current amplitude in the NT deletion mutants, in two batches (14–16 oocytes) oocytes were injected with amounts of RNA adjusted to produce IBa of similar amplitude in α1C-wt, α1CΔ46, and α1CΔ20. The I-V curve shift was observed in all cases. Fitting I-V curves to Boltzmann equation (solid lines in Fig. 2 Cb) indicated a statistically significant 5–7 mV shift in the half activation voltage, V1/2, in all NT deletion mutants studied except α1CΔ5 (Table I). The leftward shift in the G-V curves is more clearly seen in conductance–voltage (G-V) curves that were drawn through the data points using the Boltzmann equation parameters obtained in I-V curve fits (Fig. 2 Cc). In addition, the slope appears to be increased by the NT deletions. Indeed, the slope factor Ka was slightly but statistically significantly reduced, from 9.6 mV in α1C-wt to ∼7.9 mV in most mutants (Table I). None of the mutations caused any changes in the reversal potential of IBa (see Table S1).
TABLE I.
Effects of NT Deletions and Mutations of α1C on the Gating Parameters of Ba2+ Current Activation
| V1/2, mV | Ka, mV | |||||||
|---|---|---|---|---|---|---|---|---|
| Construct | No β2b | With β2b | No β2b | With β2b | ||||
| α1C-wt | 15.66 ± 1.23 (56) | 1.49 ± 0.79 (56)d | 9.62 ± 0.31 (56) | 7.2 ± 0.13 (56)d | ||||
| α1Δ5 | 15.83 ± 3.17 (6) | 0.91 ± 0.37 (9)d | 9.48 ± 0.92 (6) | 5.59 ± 0.08 (9) b d | ||||
| α1Δ20 | 9.28 ± 0.98 (19)b | −1.14 ± 1.14 (19)d | 7.87 ± 0.18 (19)b | 6.41 ± 0.23 (19)b d | ||||
| α1Δ46 | 9.49 ± 0.99 (30)b | −1.40 ± 0.80 (20)d | 7.88 ± 0.24 (30)b | 5.87 ± 0.18 (20)b d | ||||
| α1Δ139 | 11.02 ± 1.00 (16)a | −1.83 ± 3.58 (13)d | 7.93 ± 0.15 (16)b | 5.38 ± 0.35 (13)b d | ||||
| α1Δ6-20 | 8.61 ± 1.56 (8) | −1.28 ± 2.21 (14)c | 7.83 ± 0.23 (8) | 6.29 ± 0.23 (14)b d | ||||
| α1TYP | 10.28 ± 0.94 (23)b | −2.36 ± 0.57 (12)d | 8.88 ± 0.24 (23) | 6.13 ± 0.27 (12)b d | ||||
| α1C-short | 9.83 ± 1.28 (22)b | −2.19 ± 1.20 (26)d | 7.15 ± 0.28 (22)b | 6.06 ± 0.19 (26)b c | ||||
| α1Δ21-46 | 20.75 ± 1.73 (9) | 5.68 ± 1.93 (12)d | 9.15 ± 0.33 (9) | 6.94 ± 0.11 (12)d | ||||
| NTSL | 9.77 ± 1.40 (13)a | −1.89 ± 2.59 (18)d | 8.03 ± 0.17 (13)b | 5.95 ± 0.3 (18)b d | ||||
| NTLS | 13.86 ± 2.18 (13) | −3.59 ± 2.9 (9)d | 6.58 ± 0.97 (13)b | 4.45 ± 0.62 (9)b c | ||||
| HA-labeled constructs | ||||||||
| α1C-HA | 17.88 ± 0.59 (6) | 1.43 ± 1.38 (6)d | 8.53 ± 0.21 (6) | 6.55 ± 0.27 (6)d | ||||
| α1CΔ21-46-HA | 18.37 ± 1.08 (7) | 3.6 ± 1.16 (6)d | 8.34 ± 0.39 (7) | 6.8 ± 0.17 (6)d | ||||
| NTSL-HA | 7.12 ± 1.67 (6)b | −3.06 ± 1.54 (6)d | 6.76 ± 0.4 (6)b | 4.89 ± 0.49 (6)a d | ||||
The values of V1/2 and Ka were obtained in each cell by fitting the I-V curve to the Boltzmann equation (Eq. 1 in Materials and Methods). Mean ± SEM are shown, with numbers of cells in parentheses.
Statistically significant difference in V1/2 or Ka as compared with α1C-wt (or α1C-HA) by one-way ANOVA followed by the Dunnett's test. P < 0.05.
Statistically significant difference in V1/2 or Ka as compared with α1C-wt (or α1C-HA) by one-way ANOVA followed by the Dunnett's test. P < 0.01.
Statistically significant difference compared to no β2b group by t test. P < 0.01.
Statistically significant difference compared to no β2b group by t test. P < 0.001.
The robust increase in IBa and the shift in G-V curve were also observed in the α1CΔ6-20 mutant lacking the 16 aa of the partial homology region. Mutation of the conserved TxxYxP motif produced similar but milder changes (Fig. 2 and Table I). The changes in Gmax (calculated from the Boltzmann fits) were similar to changes in the measured I40 (Table II). Taken together, these data imply that amino acids 6–20 of the long-NT isoform constitute a crucial part of the NTI module. The increase in IBa and in Gmax is accompanied by a moderate hyperpolarizing shift in the G-V curve; this effect is also fully dependent on the presence of the same 16 aa. The T10A/Y13F/P15A mutation interferes with the function of the NTI module by a mechanism yet to be determined.
Previously, controversial results have been reported regarding the surface expression of α1C with NT deletions of 40 aa or more from a long-NT α1C, expressed in Xenopus oocytes. Wei et al. (1996) reported an approximately sixfold increase in surface expression on the basis of measurements of Qmax in cut-open oocytes. In contrast, we did not observe any changes in surface expression in α1CΔ46 either by counting channels in cell-attached patches, or by immunoprecipitating α1C from manually separated plasma membranes (Shistik et al., 1998). Here we confirm these results by an independent imaging method (Singer-Lahat et al., 2000; Peleg et al., 2002). Proteins expressed in the PM were visualized by immunostaining in giant membrane patches in which the cytosolic surface of the PM is exposed to external solution (Fig. 3 A). The membrane protein in the patch is labeled with an antibody against a cytosolic segment, then with a fluorescently labeled secondary antibody, and visualized using a confocal microscope (see Materials and Methods for details). The image captures a randomly selected 105 × 105 μm area within a (larger) patch. The large size of the imaged area ensures a fair averaging of channel density even if the channels are clustered. Patches from many tens of oocytes can be screened during a 1-d experiment, providing statistically reliable data. This is a big advantage over the previously employed method of immunoprecipitation of α1C from manually separated PM of metabolically labeled oocytes (Shistik et al., 1995, 1998), which produced one experimental point for 20–30 whole-oocyte membranes.
Fig. 3 B shows images of representative membrane patches from oocytes of one batch expressing α1C-wt or various NT mutants of α1C without the β subunit. The antibody labeling was clearly detected in all channel constructs, and no significant labeling was observed in uninjected oocytes at these imaging parameters. The data from two oocyte batches are summarized in Fig. 3 Ca, showing that there were no significant differences in the expression of any of the channel variants tested. This result was reproduced later in a separate series of experiments (see Fig. 4, A and B) and also using an alternative imaging method (see Fig. 7 C). At the same time, IBa measured in oocytes of one of these two batches was enhanced by the NT mutations, as usual (Fig. 3 Cb). We conclude that physical or functional removal of the NTI module of the long NH2 terminus does not alter the surface expression of α1C. Since the single channel conductance is not changed either (Shistik et al., 1998), all of the increase in Gmax caused by these mutations must result from an increase in Po,max. Indeed, calculations using Eq. 6 show that all these mutations cause a greater than sixfold increase in Po,max (Table II), except α1CΔ5, which shows only a 37% increase. The increase in I40 was essentially identical (Table II).
Figure 7.

Design and surface expression of α1C-short, short/long NT chimeras, and α1CΔ21-46. (A) The design of initial NT segments of new constructs. Dashes show gaps. The homologous 15-aa sequence in long- and short-NT is highlighted in yellow and magenta, respectively. (B and C) Surface expression of the NT chimeric and deletion constructs in the absence and presence of β2b monitored in giant membrane patches (B) or in intact oocyte by imaging the external HA tag (C). The amount of injected RNA was 2.5 ng/oocyte in B and 5 ng in C. Examples of confocal images are shown in panel a, and the summary of the surface expression in the absence of β subunit (relative to α1C-wt) is shown in b. The effect of coexpression of β2b for each one of the constructs tested in these experiments is summarized in c. Numbers above bars indicate the number of patches (B) or oocytes (C) imaged.
Regulation by β2b Subunit of Po,max, But Not of Other Parameters, Depends on NT of α1C
We used the nonpalmitoylated np-β2a (Hullin et al., 1992), which is the rabbit orthologue of human β2b (96% identity), the most abundant cardiac β subunit in rat and humans (Colecraft et al., 2002; Hullin et al., 2003). To avoid confusion between palmitoylated and nonpalmitoylated forms of β2a, we refer to np-β2a as β2b. Unlike the palmitoylated splice variant isoform (β2a) of the same gene, β2b does not slow down the VDI and does not reside in PM in the absence of α1C (Olcese et al., 1994; Chien et al., 1998). When expressed in Xenopus oocytes, β2b accelerates the VDI of Cav1.2, and in addition causes a robust increase in the whole-cell current and affects all gating parameters (Hullin et al., 1992; Shistik et al., 1995). However, it is not clear whether coexpression of either β2a or β2b improves the trafficking of α1C to the PM in Xenopus oocytes. No change (Neely et al., 1993, 2004) or a mild ∼50% increase (Shistik et al., 1995) have been reported (see Table IV and Discussion).
Using the imaging method shown in Fig. 3, we have examined the effect of coexpression of β2b on the amount of α1C-wt, α1CΔ20, α1CΔ46, and α1CTYP in two to four batches of oocytes; α2δ was always coexpressed (Fig. 4). As before, in the absence of Cavβ, the surface expression of NT mutants was similar to that of α1C-wt (Fig. 4, A and B). In contrast, β2b induced a mild but reproducible and statistically significant ∼70% increase (P < 0.001) in the surface expression in all α1C constructs (Fig. 4, A and C).
The effect of β2b on surface expression of α1C was additionally verified using an independent method, using a modified α1C (termed α1C-HA) with an extracellular HA tag inserted in the extracellular loop following the S5 segment of domain I. A similar construct was previously used to study the trafficking of α1C in mammalian cells (Altier et al., 2002). Ba2+ currents via channels formed by α1C-HA, coexpressed with α2/δ with or without β2b, did not differ in amplitude or voltage dependency from α1C-wt (unpublished data; Table I). Surface expression of the HA label was measured using a confocal microscope in whole intact oocytes fixated with formaldehyde and treated with an anti-HA antibody and a secondary antibody conjugated to a fluorescent dye (Fig. 4 D). As shown in Fig. 4 Da, the expression of β2b caused a clear increase in surface expression of α1C-HA. The results were quantified as explained in Fig. 4 legend, and showed a 2.09 ± 0.34-fold increase in the intensity of fluorescence caused by β2b (Fig. 4 Dc). This estimate, though somewhat higher than by measuring the effect of β2b in giant PM patches, was not significantly different when compared in the same oocyte batch (not depicted). These results unequivocally demonstrate that, despite the theoretical possibility that some of the fluorescent signal in the giant membrane patches arises from α1C found in submembrane ER, the latter method provides a realistic estimate of α1C levels in the PM. We conclude that, despite the presence of an endogenous Cavβ in the oocytes, the expressed β2b further increases the surface expression of Cav1.2 channels about twofold (in the presence of α2δ). The ability of β2b to do so is not affected by the elimination of the NTI module.
Coexpression of β2b also increased the macroscopic currents in wt and all NT mutants. Examples are shown in Fig. 5 A for α1C-wt and α1CΔ46. Note that β2b accelerated the inactivation kinetics in both cases, despite the fact that without Cavβ, they were already faster in α1CΔ46 than in α1C-wt. This was a recurrent result in most mutants (unpublished data). Thus, although VDI is out of the scope of this paper, and although it is clear that the NH2 terminus itself plays a role in VDI (see also Shistik et al., 1998; Kobrinsky et al., 2004), we construe that the elimination of the NTI module does not impair the ability of β2b to speed up the VDI.
Figure 5.

Effects of mutations in NT, and of the β2b subunit, on voltage dependence of activation of IBa. (A) Representative net IBa of α1C-wt and α1CΔ46 constructs in the absence and presence of coexpressed β2b subunit, recorded at +40 mV. (B) I-V curves of channels in α1α2δ and α1α2δβ2b subunit compositions (n = 6–8, N = 1). Representative averaged I-V curves are shown for α1C-wt, α1CΔ5, α1CΔ6-20, and α1CΔ46. (C and D) Normalized averaged I-V curves (C) and G-V curves (D) from all experiments and all mutants tested, in absence (gray symbols) and presence (pink symbols) of the β2b subunit. The solid lines were drawn for illustration using the Boltzmann equation with the averaged parameters of V1/2 and Ka from Table I and Vrev from Table S1. Data are from 2–11 experiments, 14–56 cells.
The I-V curves of all α1C mutants, and of the α1C-wt, were shifted to the left by coexpression of β2b. Examples of averaged I-V curves of oocytes from representative batches are shown in Fig. 5 B, and a full summary of normalized I-V and G-V curves averaged from all oocytes is presented in Fig. 5 (C and D). The hyperpolarizing shift produced by β2b is observed both in α1C-wt and in all mutants lacking the NTI module. A closer examination of the results of Boltzmann fits in Table I reveals that the net change in V1/2 and Ka parameters caused by β2b in α1C-wt (14 and 2.4 mV, respectively) was somewhat greater than in some of the NT mutants (11–13 and 1.5–2 mV). However, these differences are small and may be within the experimental or fitting error. In all, we conclude that elimination of NT inhibitory module does not impair the ability of β2b to cause a hyperpolarizing shift in the voltage dependency of channel activation.
In contrast to parameters considered so far, the extent of increase in IBa and in Gmax caused by coexpression of β2b was altered dramatically by NT mutations (Figs. 5 B, Fig. 6, and Table III). In agreement with previous reports regarding α1CΔ20, α1CΔ46, and α1CΔ139 (Shistik et al., 1998, 1999), all mutations that impaired the function of the NTI module also greatly reduced the ability of β2b to increase IBa (Fig. 6, A and B). The differences were more pronounced at less positive potentials, because at least part of the increase in IBa at these potentials is due to the leftward shift in the I-V curve, as illustrated in Fig. 6 A for some of the mutants. However, even at +40 mV, the difference between α1C-wt and the various mutants is still very substantial (Fig. 6 B). The β2b-induced increase in the calculated Gmax, which in α1C-wt was 4.65 ± 0.39-fold, was diminished to only 1.92 ± 0.23-fold in α1CΔ46 (P < 0.01; Table III). A similar, though slightly milder, reduction was observed in α1CΔ20 and α1CTYP (Table III). The only outlier is α1CΔ5. The β2b-induced change in I40 was even greater in this mutant than in α1C-wt, as measured in two batches of oocytes (Fig. 6 C). This is at odds with a previous observation (Fig. 2 F in Shistik et al., 1999), but a comprehensive investigation into the reasons of controversy is not possible, because in the experiment reported in 1999, the effect of β2b was studied only marginally (Shistik et al., 1999); IBa was measured only at +10 mV, and no analysis of voltage dependency has been performed. Thus, dose-dependent or batch-dependent effects of β2b on α1CΔ5 cannot be excluded at present.
Figure 6.

The NTI module regulates the effect of β2b subunit. (A) The increase in IBa caused by coexpression of β2b (relative to currents observed in the same α1C constructs without β2b) is much smaller in NT deletion mutants than in α1C-wt, at all voltages. Shown are results of two representative experiments (n = 10–18). (B) A general summary of the effect of coexpression of β2b. Shown is the fold increase in relative I40 in the various constructs; numbers of assayed cells are indicated above the bars. N = 2–7 experiments. (C) The increase in IBa caused by coexpression of β2b is potentiated in the α1CΔ5 mutant. N = 2.
Table III.
Relative Changes in Surface Expression, Gmax, I40, and Po Caused by Coexpression of β2b
| Fold increase in Po
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Group | Fold increase in PM α1C labeling | Fold increase in Gmax | Fold increase in I40 | from Gmax | from I40 | |||||
| α1C-wt | 1.69 ± 0.19 (20; 4) | 4.65 ± 0.39 (45; 7) | 4.9 ± 0.37 (45; 7) | 2.75 | 2.9 | |||||
| α1CΔ20 | 1.65 ± 0.11 (11; 2) | 2.48 ± 0.58a (19; 4) | 2.36 ± 0.26a (19; 4) | 1.5 | 1.43 | |||||
| α1CΔ46 | 1.63 ± 0.08 (20; 3) | 2.01 ± 0.22a (28; 5) | 1.92 ± 0.23a (28; 5) | 1.23 | 1.18 | |||||
| α1CTYP | 1.79 ± 0.04 (9; 2) | 2.49 ± 0.21a (20; 3) | 2.24 ± 0.23a (20; 3) | 1.39 | 1.25 | |||||
| α1C-short | 1.64 ± 0.13 (12; 3) | 1.25 ± 0.14a (11; 2) | 1.31 ± 0.15a (11; 2) | 0.76 | 0.8 | |||||
| NTSL | 1.58 ± 0.17 (9; 2) | 4.12 ± 0.41 (16; 3) | 4.07 ± 0.49 (16; 3) | 2.6 | 2.57 | |||||
| HA-labeled constructs | ||||||||||
| α1C-HA | 2.22 ± 0.21 (13; 1) | 8.04 ± 0.93 (13; 3) | 8.49 ± 1 (13; 3) | 3.6 | 3.82 | |||||
| NTLS-HA | 1.99 ± 0.22 (7; 1) | 2.36 ± 0.31a (9; 3) | 2.29 ± 0.28a (9; 3) | 1.18 | 1.15 | |||||
| α1CΔ21-46-HA | 2.86 ± 0.38 (11; 1) | 10.9 ± 1.38 (12; 3) | 11.03 ± 1.54 (12; 3) | 3.8 | 3.85 | |||||
All comparisons and normalization procedures are as in Table II.
P < 0.01, compared with α1C-wt.
To summarize, the main conclusion from these experiments is that the ability of β2b to increase Gmax is greatly reduced by the removal of a crucial part of the NT inhibitory module, aa 6–20. The unique aa 2–5 of the long-NT channel are a part of the NTI module, since their removal already causes a mild increase in Po,max; however, it also potentiates the enhancing effect of β2b on Po,max, instead of reducing it. This peculiar effect deserves further attention in the future.
The β2b-induced increase in Gmax still remaining after the elimination of the NTI module is comparable to the increase in the surface expression caused by β2b. This indicates that in these mutants, the increase of the macroscopic Ca2+ channel conductance caused by β2b may be the result of an increase in the amount of the functional channels, N, whereas Po,max is not substantially changed. This is illustrated by comparing the values of β-induced change in Po,max calculated using Eq. 6 (Table III).
The Initial Segment of the Short-NT Isoform of α1C Is Not an NTI Module
Are aa 6–20 of long-NT sufficient to act as an NTI module? Does the initial segment of the short-NT isoform (aa 2–16) act as an inhibitory module, similarly to the partially homologous aa 6–20 segment in the long-NT? To answer these questions, we created a cDNA encoding for a short-NT isoform, α1C-short, in which exon 1a of the long-NT was replaced by the exon 1 of short-NT α1C (Fig. 7 A). The rest of the molecule was left as in the cardiac, long-NT α1C. This design was intended to single out the effects of NT and to avoid additional changes in channel's properties that can arise if one is using one of the several cloned short-NT isoforms, with additional variations in the other parts of the protein (Abernethy and Soldatov, 2002). We also created cDNAs encoding two chimeric proteins in which parts of short- and long-NT initial segments were switched (Fig. 7 A): (1) NTSL, which is a short-NT α1C, in which aa 2–16 of the short-NT α1C are replaced by aa 6–20 of the long-NT α1C; (2) NTLS, which is a long-NT α1C in which aa 6–20 are replaced by aa 2–16 of the short-NT. Two additional groups tested in these experiments included the standard α1CΔ46 mutant (which misses the entire initial segment), and a new internal deletion of long-NT α1C, α1CΔ21-46. External HA-tagged constructs were also made for all new mutants except short-NT α1C; they exhibited G-V curves and macroscopic IBa very similar to the untagged counterparts (Table I; unpublished data). Immunohistochemical measurements in giant membrane patches or in intact oocytes (for HA-tagged constructs) showed that all new constructs expressed similarly to α1C-wt in the PM, and coexpression of β2b increased the amount of all constructs in the PM similarly to α1C-wt (Fig. 7, B and C; Tables II and III).
Fig. 8 (A–C) reports on the results of two series of experiments (a and b) in which the macroscopic properties of IBa in the new constructs were tested, in different combinations and at two RNA doses. Without the β subunit, IBa in α1C-short, NTSL, and NTLS was substantially greater than in α1C-wt, and a small but significant leftward shift in V1/2 and a decrease in Ka were also observed. All these parameters were comparable to those of α1CΔ46 (Fig. 8; Tables I and II). I40 tended to be somewhat smaller in NTSL than in α1C-short or α1CΔ46, but the difference did not reach statistical significance. In contrast, both macroscopic IBa and all G-V curve parameters of α1CΔ21-46 were very similar to α1C-wt.
Figure 8.

Chimeras of long- and short-NT help demarcate the NTI module. Two separate series of experiments, that used different doses of RNA, are shown in A–C. In Aa, B, and Ca, all RNAs were injected at 1 ng/oocyte; in Ab and Cb, at 2.5 ng/oocyte. (A) Comparison of normalized I40 in the absence of coexpressed β subunit. N = 2–3. (B) Effect of β2b on the voltage dependency of IBa in the same experiments as in Aa. Normalized averaged G-V curves are shown, in the absence (black symbols) and presence (red symbols) of the β2b subunit. (C) Summary of the effect of coexpression of β2b on I40. (D) The voltage dependency of the relative effects of β2b on IBa in selected NT mutants. The results were summarized from experiments shown in Figs. 6 B and 8. In each experiment, the increase in IBa caused by the coexpression of β2b was normalized to the average increase observed in α1C-wt group of oocytes at the same voltage, and the results were averaged across all experiments. The linear regression (dotted lines) is shown for illustrative purposes.
Coexpression of β2b shifted the G-V curve to the left by 12–17 mV in all constructs (Fig. 8 B; Table I), whereas the effects on IBa amplitude were again diverse. The increase in I40 caused by β2b was poor (less than twofold) and comparable to α1CΔ46 in α1C-short and NTLS, which lack the crucial 16 aa of the long-NT. In NTSL, I40 increased much more than in α1CΔ46 but less than in α1C-wt, whereas α1CΔ21-46 again behaved almost exactly as α1C-wt (Fig. 8 C). The increase in Po,max caused by β2b almost disappeared in α1CΔ46 and NTLS mutants; in the α1C-short, the calculation even showed a small decrease in Po,max by β2b (Table III). In contrast, in NTSL, the calculated Po,max was increased by β2b almost like in the long-NT α1C-wt. α1CΔ21-46 behaved exactly like α1C-wt (Fig. 8 Cb). Taken together, these results strongly suggest that aa 1–20 of long-NT fully restore the function of the NTI module, whereas aa 6–20 of long-NT alone are insufficient. aa 2–16 of short-NT cannot replace the homologous 15 aa of the long-NT as a crucial component of NTI module.
The difference in effects of β2b on α1C-wt and the various NT deletion mutants is further explored in Fig. 8 D, where the β2b-induced increase in IBa at each voltage was normalized to that observed in α1C-wt. The rationale for this analysis is as follows. If the NTI module regulates the effect of β2b on Po,max (a nominally voltage-independent parameter) and not on the voltage dependence of activation, then the removal of the NTI module should alter the effect of β2b to a similar extent at all voltages. In general, this was indeed the case (Fig. 8 D), although the relative effect of β2b showed a slight voltage sensitivity; the difference between α1C-wt and NT mutants was somewhat greater at more negative voltages. This, however, is a predictable consequence of the mild hyperpolarizing shift in the G-V curve caused by the removal of the NTI module (see Fig. 2 C and Fig. 8 B). The latter leads to a slightly stronger voltage dependency of β2b-induced increase in IBa in α1C-wt than in the mutants. Fig. 8 D also illustrates the fact that, of all constructs tested, the increase in IBa by β2b was the greatest in α1C-wt and the smallest in α1C-short (even compared with α1CΔ46). The substantial recovery of the effect of β2b in NTSL and the full recovery in α1CΔ21-46 are also clearly visualized.
DISCUSSION
Overview of Findings and Conclusions
Despite the importance of regulation of Ca2+ channels by β subunits and the wide interest in structure, function, and interactions of Cavα and Cavβ, controversies still abound regarding the mechanisms and even the phenomenology of the effects of Cavβ. In this study we attempt to sort out some of the discrepancies, and to better understand the mechanism by which Cavβ increases Ca2+ channel currents, focusing on a single type of Cavα1 and Cavβ.
(a) We show that Cavβ-induced increase in maximal open probability (Po,max), at least in Cav1.2, is separable from the other mechanisms (shift in G-V curve and change in PM expression) that underlie the increase in macroscopic Ca2+ currents by Cavβ. This effect is crucially dependent on the presence of an NTI module. This separability, and the separability of trafficking from all gating effects, implies that Cavβ acts on α1C to increase Po,max via molecular determinants (in β and/or in α1) different from those used to change trafficking or to shift the G-V curve.
(b) Using deletion mutagenesis and chimeric constructs, we map the necessary and sufficient component of the NTI module to aa 1–20 of the long-NT and demonstrate that the NTI reduces Po,max, thus restraining the activation of the channel at all voltages. Loss of NTI module enhances IBa by increasing Po,max ∼6–10-fold and, in parallel, specifically eliminates the effect of Cavβ on Po,max; addition of the module restores the small amplitude of IBa and the enhancement of Po by Cavβ. This correlation supports the hypothesis (Shistik et al., 1998) that part of the effect of β on α1C is due to a suppression of the inhibitory action of the NTI module. In the absence of the NTI module, Cavβ cannot further increase Po,max, which is already high.
(c) We establish that the NTI module is completely missing from a short-NT Cav1.2, which represents a class of Cav1.2α isoforms abundant in the smooth muscle and brain. This is a striking example of a physiologically meaningful disparate regulation of two very similar isoforms of an ion channel by an auxiliary subunit.
(d) Using two independent methods, we unequivocally demonstrate that coexpression of Cavβ increases the surface expression of α1C in Xenopus oocytes approximately twofold, independently of the presence of the NTI module. By a critical examination of literature and separation of studies that used different NT isoforms, we resolve apparent controversies and single out remaining problems, regarding the effects of Cavβ on α1C.
A Summary of the Effects of Cavβ on α1C
It is known that the effects of β subunits differ among the various α1 subunits (Dolphin, 2003). However, even for a single species of Cavα1, the details and magnitude of effects of coexpressed Cavβ often remain controversial. For instance, estimates of the increase in macroscopic maximal IBa of Cav1.2 (α1C) in mammalian cells range between 15 and 20-fold (Lory et al., 1993; Takahashi et al., 2004) to almost no effect (Yasuda et al., 2004). The comparison between the different reports is thwarted by the unavoidable difference in recording conditions and often by an absence of quantitative estimates of some of the electrophysiological parameters. To understand the origins of the controversies, we summarized the data on Cavβ effects on Cav1.2 from reports that contain an explicit quantitative estimation of parameters that affect the macroscopic Ca2+ channel currents (Table IV). Data collected from short-NT (or mutants of α1C missing the initial NT segment) and the long-NT isoforms of α1C are separated. Macroscopic inactivation is unimportant in determining the whole-cell current when Ba2+ is used as the charge carrier (see below), and is left out of scope. Reports in which effects of different Cavβ have been compared but there are no data on channels expressed without Cavβ are not included. If several β subunits have been compared in the same study, we usually show β2a or np-β2a (β2b), which were the most widely used. While the summary in Table IV may not be exhaustive, it is quite revealing.
A Distinction between Po and Po,max
Whole-cell Ca2+ channel currents and/or conductance (G) depend both on the amount of functional channels (N) and on the open probability, Po, such that G = γ × N × Po, and Gmax = γ × N × Po,max (see Materials and Methods, Eqs. 4 and 5). Treatments used in our experiments (NT deletions or coexpression of β2b) do not affect γ (Shistik et al., 1995, 1998; Hullin et al., 2003), thus only N and Po were changing. N is voltage independent, and assessing the impact of changes in N (once monitored) on G is straightforward.
In comparison, the analysis of Po and the assessment of its effect on G are not trivial. First, in voltage-dependent channels, Po is voltage dependent, being described by the same Boltzmann equation as G/Gmax (Hille, 2002) (Eq. 3). Po ranges from zero at negative potentials to Po,max at “saturating” membrane voltages where the G-V curve reaches a plateau. Note that in this classical treatment Gmax and Po,max are voltage independent. An agent that improves the coupling between gating charge movement and pore opening, such as Cavβ (Neely et al., 1993), and shifts the G-V curve to the left will increase Po (and macroscopic currents) in a range of “nonsaturating” voltages, whether Po,max is changed or not. In contrast, an increase in Po,max leads to an increase in Ca2+ conductance at all voltages except those at which the channel is fully shut. Therefore, to understand the molecular mechanism of β's actions, it is important to distinguish between changes in Po caused by a G-V curve shift vs. an increase in Po,max.
In single-channel recordings it is possible to directly determine Po, which is the fraction of time that a channel spends in open state(s), out of total observation time (Colquhoun and Sigworth, 1995). Although estimation of Po can be distorted by the presence of inactivated states (Colquhoun and Sigworth, 1995), in Cav1.2 with Ba2+ as charge carrier, the inactivation is slow, and such distortion can be avoided by using short analyzed time segments (a few tens of milliseconds). Unfortunately, the estimation of changes in Po,max caused by Cavβ is almost impossible in single-channel recording of Cav channels. Most publications report single channel currents recorded between −10 and +10 mV in high-Ba2+ solutions, to ensure high signal-to-noise ratio; a full I-V curve is usually not constructed. (An exception is a study in cardiomyocytes [Colecraft et al., 2002], where a full I-V relation was explored; see Table IV.) Under these conditions, G (and thus Po) is very far from reaching a maximum, as evident from typical G-V curves shown in Figs. 2, 5, and 8, and the measured Po does not provide any estimate of Po,max.
In comparison, measurements of changes in macroscopic Gmax in conjunction with a reliable estimation of N should provide a good measure of relative changes in Po,max (Wei et al., 1994; Takahashi et al., 2004). Here, G/Gmax is calculated using the values of peak macroscopic current that are independent of the duration of voltage step, therefore the latter does not affect the calculated relative change in Po,max. Our estimates of Po,max were not affected by changes in the steady-state inactivation curve in the NT deletion mutants, because the availability of the channels for opening at the holding potential of −80 mV remained 100% (Shistik et al., 1998). There are two possible sources of inaccuracy. First, acceleration of the inactivation process by β2b may decrease peak IBa, if the rate constants of activation and inactivation are comparable (Aldrich et al., 1983; Hille, 2002). However, this artifact is probably negligible in Cav1.2. Here, the time constant of the faster of the two components of macroscopic VDI of IBa is ∼75 ms for a short-NT isoform (Shi and Soldatov, 2002) and >150 ms with the long-NT α1C (unpublished data). This is at least twofold slower than first latency time constant observed in single channel recordings (∼25–35 ms, see Hullin et al., 2003). Another artifact may arise when Gmax is estimated by fitting experimental I-V curves to Boltzmann equation. In this case, G usually appears to reach its maximum at +40 to +50 mV. G-V curves based on the measurement of tail currents, which are devoid of certain inaccuracies inherent to I-V curves (such as very small IBa at Vm close to the reversal potential), often imply that the G-V curve has two components and does not reach a plateau at +40 to +50 mV, but close to 100 mV (e.g., Takahashi et al., 2004). Assuming that this is the case and using the data from Takahashi et al. (2004), we calculated that we might have overestimated the changes in Gmax, but by no more than 30% (unpublished data). Since in α1C-wt the changes in Gmax caused by NT deletions or by β2b are between 260 and 770% (Tables II and III), such an error would not affect our main conclusions.
Cavβ-induced Changes in Voltage Dependency (G-V curve) and in Po,max Are Separable
(a) Only the Cavβ-induced increase in Po,max, but not the shift in G-V curve, depends on the presence of the NTI module. In all mutants lacking the crucial part of the NTI module (aa 6–20 of long NT), G-V curve shifts are like in α1C-wt, but Po,max is not changed by β2b (Table III). The mild approximately twofold increase in Gmax (or I40) caused by β2b in these mutants can be almost fully accounted for by the increase in N, which is 1.6–2.2-fold. Strikingly, in the short-NT isoform, the coexpression of β2b even causes an ∼20% decrease in calculated Po, despite a well-pronounced threefold increase in IBa at 0 mV (a Vm relevant for comparison with single-channel records). This increase is undoubtedly governed by the increase in N (see below) and the leftward shift in the G-V curve.
Published results (Table IV) fully support our data; separation of long-NT and short-NT isoforms eliminates controversies and allows solid conclusions. Coexpression of β subunits increases the Po measured in single-channel patches at −10 to +10 mV in high-Ba2+, in oocytes and in mammalian cells. However, the increase is consistently greater in the long-NT isoform (range: six to eightfold) than in the short-NT α1C forms (range: two to fivefold). Data necessary for the calculation of the relative change in Po,max are available from fewer works, but also fit our results; coexpression of Cavβ increases Po,max of the long-NT α1C two to fourfold, whereas a mild ∼20% decrease is observed in a short-NT α1C isoform.
(b) Changes in Po,max caused by coexpression of Cavβ are similar in Xenopus oocytes and mammalian cells (Table IV). In contrast, there is a consistent and striking difference in the reported shifts in the G-V curve: changes in ΔV1/2 range from 12 to 22 mV in oocytes but are always <10 mV in mammalian cells. This intriguing difference calls for study and may provide new clues to understanding VDCC gating; but it also supports the disparity of mechanisms of the effects of Cavβ on Po,max and on voltage dependence of activation.
On the basis of these arguments, we conclude that, most intriguingly, the two gating actions of Cavβ that are most important in determining the amplitude of whole-cell Ca2+ currents are separable, and therefore may rely on distinct molecular mechanisms. It is remarkable that one of the most prominent actions of Cavβ can be selectively eliminated by removing a short segment of α1C that does not even bind β, whereas other functions of Cavβ remain unchanged. This finding provides a hint at how a single Cavβ may regulate several different functions of α1, without the need to assume the involvement of multiple β subunits.
Changes in Surface Expression Caused by β2b
Surface expression of voltage-dependent ion channels can be gauged by quantitative immunohistochemistry of various kinds, or by measuring the maximal nonlinear gating charge movement, Qmax. The immunohistochemical methods that use monitoring of external surface-exposed parts of channel with or without genetically introduced epitopes reliably monitor the total number of channels in PM (e.g., Zerangue et al., 1999; Altier et al., 2002; Canti et al., 2005; Leroy et al., 2005) but do not guarantee that all detected channels are conducting (functional). Similarly, Qmax reflects the displacement of the voltage sensors in all channels in the PM, whether conducting or not. Relative changes in number of channel (N) caused by a regulatory factor (e.g., Cavβ) are faithfully reflected in changes of either surface labeling or Qmax; however, the latter is also altered by factors that alter the charge or movement of the voltage sensors (Aggarwal and MacKinnon, 1996; Barrett et al., 2005).
The prevailing view is that the Cavβ-induced increase in the amount of α1C in the PM, reproducibly observed in mammalian cells including even cardiomyocytes (Table IV), does not occur in Xenopus oocytes. This notion is based on the reported absence of a significant Cavβ-induced increase in Qmax measured in the oocytes using the cut-open voltage clamp, in which gating and ionic currents are measured from a large part of an internally perfused cell (Neely et al., 1993, 2004). In contrast, Yamaguchi et al. (1998, 2000) reported a threefold increase in α1C caused in Xenopus oocytes by either injecting a purified β3 subunit or by coexpressing β2a. Although imaging of an intracellular HA tag in permeabilized oocytes, used in these works to monitor surface expression of α1C, does not exclude the labeling of α1C located in submembrane ER, the approximately threefold increase is also supported by observing the enhancement of IBa at different times after the injection of the β3 protein (Yamaguchi et al., 1998; see Table IV legend for details). We have previously detected a β2b-induced, 20–55% increase in N measured by counting channels in cell-attached patches and by immunoprecipitating α1C from manually separated PM (Shistik et al., 1995), but it did not reach statistical significance. Interestingly, in the absence of α2δ, even this small increase was not observed (Table IV).
To address the controversy and to measure changes in surface expression of α1C in oocytes as accurately as possible, in this work we employed two additional, independent methods: immunolabeling of α1C, tagged by an extracellular HA epitope, in whole oocytes; and imaging of immunolabeled α1C in giant membrane patches of oocytes. Both methods clearly demonstrated a reproducible, highly statistically significant, isoform-independent increase in PM expression of α1C induced by coexpression of β2b. The estimates obtained by the widely accepted external tag measurement were slightly but usually insignificantly greater than by imaging in giant patches, when measured with the same construct of the channel (2.2-fold vs. 1.7-fold increase in α1C-wt, respectively). We conclude that β2b causes an approximately twofold increase in the PM content of α1C, in the presence of α2δ. The reason for the discrepancy with Neely et al. (1993, 2004) remains unclear; it would be interesting to see whether a change in Qmax occurs when α2δ is present. The increase in surface expression caused by coexpression Cavβ is absolutely independent of NTI module, in sharp contrast with Po,max. These results corroborate the previously demonstrated separability of the effects of Cavβ on surface expression and gating (Canti et al., 1999, 2001; Gerster et al., 1999).
The NTI Module and the Effect of Cavβ
In this report we demonstrated that α1C variants lacking the NTI module (the various mutants and the short NT isoform) express in the PM at the same level as the long-NT isoform, though they give much larger macroscopic currents. These and our previous results (Shistik et al., 1998) firmly establish the role of the initial segment of long-NT as a regulator of gating, rather than expression, of α1C. In support, Agler et al. (2005) recently found that the NT also constitutes an inhibitory module in the N-type (Cav2.2) channel; in contrast to Cav1.2, in Cav2.2 the inhibitory effect of the NT is G protein gated.
Since the main effect of NTI is to regulate the voltage-independent parameter Po,max, it is possible that the removal of the NTI module causes a modal shift in gating, like Cavβ (Costantin et al., 1998; Colecraft et al., 2002). If NTI module “tonically” restrains the opening of the channel's gate at all voltages (Shistik et al., 1998), its removal would cause a voltage-independent transition from a low-Po mode to a high-Po mode, in which the voltage independent transition from the last closed to the open state is more favorable than in the low-Po mode. The slight change in the voltage dependence of activation in NT mutants lacking the NTI module may be due to a reequilibration among closed states, the transitions between which are voltage dependent. Further studies including single channel recordings will be required to test this conjecture.
We find that aa 6–20 constitute a crucial and necessary part of the inhibitory module; its deletion causes a maximal enhancement of Po,max and an almost complete disappearance of the enhancing effect of β2b on Po,max. Nevertheless, aa 6–20 alone only partially recover the function of the NTI when added at the NH2 terminus of a α1CΔ46 mutant, which lacks all 46 initial amino acid residues (NTSL chimera). Full recovery of both functions of NTI (reduction in Po,max and enhancement of β2b-induced increase in Po,max) is achieved by the addition of aa 1–20 of the long-NT, which is thus established as a necessary and sufficient structural determinant of the NTI module. The function of aa 21–46, which link between the NTI module and the beginning of the highly conserved part of NT encoded by exon 2, is unknown at present. Our data conclusively demonstrate that the initial 16-aa NT segment of the short α1C isoform is not an inhibitory module. These 16 aa were unable to restore the NTI functions even in the context of a full 46-aa initial segment, i.e., in the presence of aa 2–5 and 21–46 of the long NT (NTLS chimera).
The interacting partners of NT in Cav1.2 are unknown. The regulation of inactivation kinetics of α1C (Cav1.2α) by β2a involves voltage-dependent rearrangements of NT regions of α1C and β2a, as deduced from fluorescent resonance energy transfer (FRET) experiments (Kobrinsky et al., 2004). However, no physical interaction between NT of α1C and Cavβ could be detected by direct biochemical measurements (Pragnell et al., 1994; Shistik et al., 1998), and the exact molecular interaction underlying the proximity between the NT of α1C and β2a indicated by FRET is yet to be determined. No direct interaction of NT with L1 (which would explain the intensive interplay with Cavβ function) could be detected in α1C (Agler et al., 2005). Another possible interactor is the CT of α1C. Similarly to NT, CT acts to prevent channel opening (Wei et al., 1994; Klockner et al., 1995; Gao et al., 2001; Mikala et al., 2003). Joint removal of NT and distal CT causes a more-than-additive increase in IBa (Ivanina et al., 2000), suggesting that the relaxed state of the channel is a high-Po one, and the termini are vital to prevent uncontrolled entry of Ca2+.
Using Xenopus Oocytes vs. Mammalian Cells to Study the Effects of Cavβ on Ca2+ Channels
The validity of conclusions regarding Cavβ effects, especially changes in surface expression, obtained in Xenopus oocytes has often been met with reservations, because of the presence of an endogenous β subunit. However, HEK cells also contain endogenous β (Leroy et al., 2005). In this work we demonstrate that all major effects of Cavβ can be reproducibly observed and reliably studied in Xenopus oocytes. The Cavβ-induced increase in surface α1C in oocytes is in qualitative agreement with that in mammalian systems, although quantitatively the increase reported in mammalian cells is usually greater, more than fivefold (Table IV; but see Yasuda et al., 2004). The variability among different cell types may reflect variable levels of endogenous Cavβ. The changes in gating parameters are also at least qualitatively (shift in G-V curve; see above) and quantitatively (change in Po,max) similar.
Physiological Significance
Cav1.2 channels are crucial for contraction of cardiac and smooth muscles, hormone secretion, and regulation of gene expression in the brain (Reuter, 1983; Finkbeiner and Greenberg, 1998; Catterall, 2000; West et al., 2001; Lipscombe et al., 2004). In humans, the Cav1.2 gene, CACNA1C (Soldatov, 1994) contains 55 exons, of which 19 are subject to alternative splicing, giving rise to a very large number of isoforms (Tang et al., 2004). The exact exon combinations underlying the isoforms of α1C in different tissues are yet to be catalogued (Abernethy and Soldatov, 2002; Jurkat-Rott and Lehmann-Horn, 2004; Tang et al., 2004). Long-NT isoform (isoforms?) is predominant in the heart but is also present in the brain, short-NT isoforms prevail in smooth muscle and brain (Slish et al., 1989; Koch et al., 1990; Snutch et al., 1991; Shistik et al., 1999; Blumenstein et al., 2002; Dai et al., 2002; Saada et al., 2003). At present we cannot exclude the possibility that in some of the short-NT isoforms of α1C the function of the NT initial segment is different than in the isoform that we used here, due to an interaction with some alternative distant parts of α1C; this remains to be investigated.
The isoforms may substantially differ in their physiological and pharmacological properties. For instance, splice variations in the COOH-terminal region regulate the inactivation of the channel and its sensitivity to certain pharmacological agents (Abernethy and Soldatov, 2002). The present work reveals a strikingly disparate regulation of two very similar isoforms of a Cav channel by a Cavβ subunit. It is notable that, in addition to the two isoforms of α1C corresponding to splice variants with the alternative first exons 1 and 1a, another short-NT isoform lacking either of these exons and starting from exon 2 may exist in human tissues (Schultz et al., 1993; Saada et al., 2003). This would correspond to the truncation mutant α1CΔ46 or, if the protein starts with the first methionine of exon 2, to α1CΔ59 (see Fig. 1). The differences in function of NH2 termini of these isoforms, discovered in this paper, are expected to have significant physiological implications. They will result in substantial differences in amplitudes of macroscopic Ca2+ currents and in the effects of Cavβ (whose association with α1 may change in development and growth; see Spafford et al., 2004) or regulators that act in a Cavβ-dependent manner, such as protein kinase A (Bunemann et al., 1999).
Supplemental Material
Acknowledgments
The authors want to thank Etay Artzy for help with confocal imaging, and Amy Lee and Bernard Attali for the critical reading of the manuscript.
This paper was supported by Israel Science Foundation grants 596/04 and 1396/05 and a grant from the Israel Ministry of Health.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: aa, amino acid; AID, α-interaction domain; CT, COOH terminus; HA, hemagglutinin; HEK, human embryonic kidney; NT, NH2 terminus; NTI, NT inhibitory; PM, plasma membrane; Po, open probability; VDCC, voltage-dependent calcium channel; VDI, voltage-dependent inactivation.
References
- Abernethy, D.R., and N.M. Soldatov. 2002. Structure-functional diversity of human L-type Ca2+ channel: perspectives for new pharmacological targets. J. Pharmacol. Exp. Ther. 300:724–728. [DOI] [PubMed] [Google Scholar]
- Aggarwal, S.K., and R. MacKinnon. 1996. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 16:1169–1177. [DOI] [PubMed] [Google Scholar]
- Agler, H.L., J. Evans, L.H. Tay, M.J. Anderson, H.M. Colecraft, and D.T. Yue. 2005. G protein-gated inhibitory module of N-type (Cav2.2) Ca2+ channels. Neuron. 46:891–904. [DOI] [PubMed] [Google Scholar]
- Aldrich, R.W., D.P. Corey, and C.F. Stevens. 1983. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 306:436–441. [DOI] [PubMed] [Google Scholar]
- Altier, C., S.J. Dubel, C. Barrere, S.E. Jarvis, S.C. Stotz, R.L. Spaetgens, J.D. Scott, V. Cornet, M. DeWaard, G.W. Zamponi, et al. 2002. Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J. Biol. Chem. 277:33598–33603. [DOI] [PubMed] [Google Scholar]
- Barrett, C.F., Y.Q. Cao, and R.W. Tsien. 2005. Gating deficiency in a familial hemiplegic migraine type 1 mutant P/Q-type calcium channel. J. Biol. Chem. 280:24064–24071. [DOI] [PubMed] [Google Scholar]
- Bichet, D., V. Cornet, S. Geib, E. Carlier, S. Volsen, T. Hoshi, Y. Mori, and M. De Waard. 2000. The I-II loop of the Ca2+ channel α1 subunit contains an endoplasmic reticulum retention signal antagonized by the β subunit. Neuron. 25:177–190. [DOI] [PubMed] [Google Scholar]
- Biel, M., R. Hullin, S. Freundner, D. Singer, N. Dascal, V. Flockerzi, and F. Hofmann. 1991. Tissue-specific expression of high-voltage-activated dihydropyridine-sensitive L-type calcium channels. Eur. J. Biochem. 200:81–88. [DOI] [PubMed] [Google Scholar]
- Biel, M., P. Ruth, E. Bosse, R. Hullin, W. Stuhmer, V. Flockerzi, and F. Hofmann. 1990. Primary structure and functional expression of a high voltage activated calcium channel from rabbit lung. FEBS Lett. 269:409–412. [DOI] [PubMed] [Google Scholar]
- Birnbaumer, L., N. Qin, R. Olcese, E. Tareilus, D. Platano, J. Costantin, and E. Stefani. 1998. Structures and functions of calcium channel β subunits. J. Bioenerg. Biomembr. 30:357–375. [DOI] [PubMed] [Google Scholar]
- Blumenstein, Y., N. Kanevsky, G. Sahar, R. Barzilai, T. Ivanina, and N. Dascal. 2002. A novel long-N-terminus isoform of human L-type Ca2+ channel is up-regulated by protein kinase C. J. Biol. Chem. 277:3419–3423. [DOI] [PubMed] [Google Scholar]
- Brice, N.L., N.S. Berrow, V. Campbell, K.M. Page, K. Brickley, I. Tedder, and A.C. Dolphin. 1997. Importance of the different β subunits in the membrane expression of the α1A and α2 calcium channel subunits: studies using a depolarization-sensitive α1A antibody. Eur. J. Neurosci. 9:749–759. [DOI] [PubMed] [Google Scholar]
- Bunemann, M., B.L. Gerhardstein, T. Gao, and M.M. Hosey. 1999. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the β2 subunit. J. Biol. Chem. 274:33851–33854. [DOI] [PubMed] [Google Scholar]
- Canti, C., A. Davies, N.S. Berrow, A.J. Butcher, K.M. Page, and A.C. Dolphin. 2001. Evidence for two concentration-dependent processes for β-subunit effects on α1B calcium channels. Biophys. J. 81:1439–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canti, C., M. Nieto-Rostro, I. Foucault, F. Heblich, J. Wratten, M.W. Richards, J. Hendrich, L. Douglas, K.M. Page, A. Davies, and A.C. Dolphin. 2005. The metal-ion-dependent adhesion site in the Von Willebrand factor-A domain of α2δ subunits is key to trafficking voltage-gated Ca2+ channels. Proc. Natl. Acad. Sci. USA. 102:11230–11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canti, C., K.M. Page, G.J. Stephens, and A.C. Dolphin. 1999. Identification of residues in the N terminus of α1B critical for inhibition of the voltage-dependent calcium channel by Gβγ. J. Neurosci. 19:6855–6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano, A., X. Wei, L. Birnbaumer, and E. Perez Reyes. 1993. Cloning and expression of a neuronal calcium channel β subunit. J. Biol. Chem. 268:12359–12366. [PubMed] [Google Scholar]
- Catterall, W.A. 2000. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16:521–555. [DOI] [PubMed] [Google Scholar]
- Chen, Y.H., M.H. Li, Y. Zhang, L.L. He, Y. Yamada, A. Fitzmaurice, Y. Shen, H. Zhang, L. Tong, and J. Yang. 2004. Structural basis of the α1-β subunit interaction of voltage-gated Ca2+ channels. Nature. 429:675–680. [DOI] [PubMed] [Google Scholar]
- Chien, A.J., K.M. Carr, R.E. Shirokov, E. Rios, and M.M. Hosey. 1996. Identification of palmitoylation sites within the L-type calcium channel β2a subunit and effects on channel function. J. Biol. Chem. 271:26465–26468. [DOI] [PubMed] [Google Scholar]
- Chien, A.J., T. Gao, E. Perez Reyes, and M.M. Hosey. 1998. Membrane targeting of L-type calcium channels. Role of palmitoylation in the subcellular localization of the β2a subunit. J. Biol. Chem. 273:23590–23597. [DOI] [PubMed] [Google Scholar]
- Chien, A.J., X. Zhao, R.E. Shirokov, T.S. Puri, C.F. Chang, D. Sun, E. Rios, and M.M. Hosey. 1995. Roles of a membrane-localized β subunit in the formation and targeting of functional L-type Ca2+ channels. J. Biol. Chem. 270:30036–30044. [DOI] [PubMed] [Google Scholar]
- Chu, P.-J., J.K. Larsen, C.-C. Chen, and P.M. Best. 2004. Distribution and relative expression levels of calcium channel β subunits within the chambers of the rat heart. J. Mol. Cell. Cardiol. 36:423–434. [DOI] [PubMed] [Google Scholar]
- Cohen, R.M., J.D. Foell, R.C. Balijepalli, V. Shah, J.W. Hell, and T.J. Kamp. 2005. Unique modulation of L-type Ca2+ channels by short auxiliary β1d subunit present in cardiac muscle. Am. J. Physiol. 288:H2363–H2374. [DOI] [PubMed] [Google Scholar]
- Colecraft, H.M., B. Alseikhan, S.X. Takahashi, D. Chaudhuri, S. Mittman, V. Yegnasubramanian, R.S. Alvania, D.C. Johns, E. Marban, and D.T. Yue. 2002. Novel functional properties of Ca2+ channel β subunits revealed by their expression in adult rat heart cells. J. Physiol. 541:435–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun, D., and F.J. Sigworth. 1995. Fitting and statistical analysis of single-channel records. In Single-channel Recording. B. Sakmann and E. Neher, editors. Plenum Press, New York. pp. 483–587.
- Costantin, J., F. Noceti, N. Qin, X. Wei, L. Birnbaumer, and E. Stefani. 1998. Facilitation by the β2a subunit of pore openings in cardiac Ca2+ channels. J. Physiol. 507:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, B., N. Saada, C. Echetebu, C. Dettbarn, and P. Palade. 2002. A new promoter for α1C subunit of human L-type cardiac calcium channel Cav1.2. Biochem. Biophys. Res. Commun. 296:429–433. [DOI] [PubMed] [Google Scholar]
- Dalton, S., S.X. Takahashi, J. Miriyala, and H.M. Colecraft. 2005. A single CaVβ can reconstitute both trafficking and macroscopic conductance of voltage-dependent calcium channels. J. Physiol. 567:757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dascal, N. 1987. The use of Xenopus oocytes for the study of ion channels. CRC Crit. Rev. Biochem. 22:317–387. [DOI] [PubMed] [Google Scholar]
- Dascal, N., and I. Lotan. 1992. Expression of exogenous ion channels and neurotransmitter receptors in RNA-injected Xenopus oocytes. In Protocols in Molecular Neurobiology. Volume 13. A. Longstaff and P. Revest, editors. Humana Press, Totowa, NJ. pp. 205–225.
- Dascal, N., I. Lotan, E. Karni, and A. Gigi. 1992. Calcium channel currents in Xenopus oocytes injected with rat skeletal muscle RNA. J. Physiol. 450:469–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Waard, M., C.A. Gurnett, and K.P. Campbell. 1996. Structural and functional diversity of voltage-activated calcium channels. Ion Channels. 4:41–87. [DOI] [PubMed] [Google Scholar]
- Dolphin, A.C. 2003. β subunits of voltage-gated calcium channels. J. Bioenerg. Biomembr. 35:599–620. [DOI] [PubMed] [Google Scholar]
- Ertel, E.A., K.P. Campbell, M.M. Harpold, F. Hofmann, Y. Mori, E. Perez-Reyes, A. Schwartz, T.P. Snutch, T. Tanabe, L. Birnbaumer, et al. 2000. Nomenclature of voltage-gated calcium channels. Neuron. 25:533–535. [DOI] [PubMed] [Google Scholar]
- Finkbeiner, S., and M.E. Greenberg. 1998. Ca2+ channel-regulated neuronal gene expression. J. Neurobiol. 37:171–189. [PubMed] [Google Scholar]
- Gao, T., A.J. Chien, and M.M. Hosey. 1999. Complexes of the α1C and β subunits generate the necessary signal for membrane targeting of class C L-type calcium channels. J. Biol. Chem. 274:2137–2144. [DOI] [PubMed] [Google Scholar]
- Gao, T., A.E. Cuadra, H. Ma, M. Bunemann, B.L. Gerhardstein, T. Cheng, R. Ten Eick, and M.M. Hosey. 2001. C-terminal fragments of the α1C (Cav1.2) subunit associate with and regulate L-type calcium channels containing C-terminally truncated α1C subunits. J. Biol. Chem. 276:21089–21097. [DOI] [PubMed] [Google Scholar]
- Garcia, R., E. Carrillo, S. Rebolledo, M.C. Garcia, and J.A. Sanchez. 2002. The β1a subunit regulates the functional properties of adult frog and mouse L-type Ca2+ channels of skeletal muscle. J. Physiol. 545:407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerster, U., B. Neuhuber, K. Groschner, J. Striessnig, and B.E. Flucher. 1999. Current modulation and membrane targeting of the calcium channel α1C subunit are independent functions of the β subunit. J. Physiol. 517:353–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg, R.G., A. Messing, C. Strube, M. Beurg, R. Moss, M. Behan, M. Sukhareva, S. Haynes, J.A. Powell, R. Coronado, and P.A. Powers. 1996. Absence of the β subunit (cchb1) of the skeletal muscle dihydropyridine receptor alters expression of the α1 subunit and eliminates excitation-contraction coupling. Proc. Natl. Acad. Sci. USA. 93:13961–13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harry, J.B., E. Kobrinsky, D.R. Abernethy, and N.M. Soldatov. 2004. New short splice variants of the human cardiac Cavβ2 subunit: redefining the major functional motifs implemented in modulation of the Cav1.2 channel. J. Biol. Chem. 279:46367–46372. [DOI] [PubMed] [Google Scholar]
- Hille, B. 2002. Ion Channels of Excitable Membranes. Sinauer, Sunderland, MA. 722 pp.
- Hohaus, A., M. Poteser, C. Romanin, N. Klugbauer, F. Hofmann, I. Morano, H. Haase, and K. Groschner. 2000. Modulation of the smooth-muscle L-type Ca2+ channel α1 subunit (α1C-b) by the β2a subunit: a peptide which inhibits binding of β to the I-II linker of α1 induces functional uncoupling. Biochem. J. 348(Pt 3): 657–665. [PMC free article] [PubMed] [Google Scholar]
- Hullin, R., I.F. Khan, S. Wirtz, P. Mohacsi, G. Varadi, A. Schwartz, and S. Herzig. 2003. Cardiac L-type calcium channel β-subunits expressed in human heart have differential effects on single channel characteristics. J. Biol. Chem. 278:21623–21630. [DOI] [PubMed] [Google Scholar]
- Hullin, R., D. Singer-Lahat, M. Freichel, M. Biel, N. Dascal, F. Hofmann, and V. Flockerzi. 1992. Calcium channel β subunit heterogeneity: functional expression of cloned cDNA from heart, aorta and brain. EMBO J. 11:885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom, L.L., K.S. De Jongh, and W.A. Catterall. 1994. Auxiliary subunits of voltage-gated ion channels. Neuron. 12:1183–1194. [DOI] [PubMed] [Google Scholar]
- Ivanina, T., Y. Blumenstein, E. Shistik, R. Barzilai, and N. Dascal. 2000. Modulation of L-type Ca2+ channels by Gβγ and calmodulin via interactions with N- and C-termini of α1C. J. Biol. Chem. 275:39846–39854. [DOI] [PubMed] [Google Scholar]
- Jones, S.W. 2002. Calcium channels: when is a subunit not a subunit? J. Physiol. 545:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephson, I.R., and G. Varadi. 1996. The β subunit increases Ca2+ currents and gating charge movements of human cardiac L-type Ca2+ channels. Biophys. J. 70:1285–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkat-Rott, K., and F. Lehmann-Horn. 2004. The impact of splice isoforms on voltage-gated calcium channel α1 subunits. J. Physiol. 554:609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp, T.J., M.T. Perez Garcia, and E. Marban. 1996. Enhancement of ionic current and charge movement by coexpression of calcium channel β1A subunit with α1C subunit in a human embryonic kidney cell line. J. Physiol. 492:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockner, U., G. Mikala, M. Varadi, G. Varadi, and A. Schwartz. 1995. Involvement of the carboxyl-terminal region of the α1 subunit in voltage-dependent inactivation of cardiac calcium channels. J. Biol. Chem. 270:17306–17310. [DOI] [PubMed] [Google Scholar]
- Kobrinsky, E., K.J. Kepplinger, A. Yu, J.B. Harry, H. Kahr, C. Romanin, D.R. Abernethy, and N.M. Soldatov. 2004. Voltage-gated rearrangements associated with differential β-subunit modulation of the L-type Ca2+ channel inactivation. Biophys. J. 87:844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, W.J., P.T. Ellinor, and A. Schwartz. 1990. cDNA cloning of a dihydropyridine-sensitive calcium channel from rat aorta. Evidence for the existence of alternatively spliced forms. J. Biol. Chem. 265:17786–17791. [PubMed] [Google Scholar]
- Lacinova, L., A. Ludwig, E. Bosse, V. Flockerzi, and F. Hofmann. 1995. The block of the expressed L-type calcium channel is modulated by the β3 subunit. FEBS Lett. 373:103–107. [DOI] [PubMed] [Google Scholar]
- Leroy, J., M.S. Richards, A.J. Butcher, M. Nieto-Rostro, W.S. Pratt, A. Davies, and A.C. Dolphin. 2005. Interaction via a key tryptophan in the I-II linker of N-type calcium channels is required for β1 but not for palmitoylated β2, implicating an additional binding site in the regulation of channel voltage-dependent properties. J. Neurosci. 25:6984–6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuranguer, V., E. Bourinet, P. Lory, and J. Nargeot. 1998. Antisense depletion of β subunits fails to affect T-type calcium channels properties in a neuroblastoma cell line. Neuropharmacology. 37:701–708. [DOI] [PubMed] [Google Scholar]
- Liman, E.R., J. Tytgat, and P. Hess. 1992. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 9:861–871. [DOI] [PubMed] [Google Scholar]
- Lipscombe, D., T.D. Helton, and W. Xu. 2004. L-type calcium channels: the low down. J. Neurophysiol. 92:2633–2641. [DOI] [PubMed] [Google Scholar]
- Lory, P., G. Varadi, D.F. Slish, M. Varadi, and A. Schwartz. 1993. Characterization of β subunit modulation of a rabbit cardiac L-type Ca2+ channel α1 subunit as expressed in mouse L cells. FEBS Lett. 315:167–172. [DOI] [PubMed] [Google Scholar]
- Maltez, J.M., D.A. Nunziato, J. Kim, and G.S. Pitt. 2005. Essential Cavβ modulatory properties are AID-independent. Nat. Struct. Mol. Biol. 12:372–377. [DOI] [PubMed] [Google Scholar]
- McGee, A.W., D.A. Nunziato, J.M. Maltez, K.E. Prehoda, G.S. Pitt, and D.S. Bredt. 2004. Calcium channel function regulated by the SH3-GK module in β subunits. Neuron. 42:89–99. [DOI] [PubMed] [Google Scholar]
- Mikala, G., I. Bodi, U. Klockner, M. Varadi, G. Varadi, S.E. Koch, and A. Schwartz. 2003. Characterization of auto-regulation of the human cardiac α1 subunit of the L-type calcium channel: importance of the C-terminus. Mol. Cell. Biochem. 250:81–89. [DOI] [PubMed] [Google Scholar]
- Mikami, A., K. Imoto, T. Tanabe, T. Niidome, Y. Mori, H. Takeshima, S. Narumiya, and S. Numa. 1989. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature. 340:230–233. [DOI] [PubMed] [Google Scholar]
- Mori, Y., T. Friedrich, M.S. Kim, A. Mikami, J. Nakai, P. Ruth, E. Bosse, F. Hofmann, V. Flockerzi, T. Furuichi, et al. 1991. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 350:398–402. [DOI] [PubMed] [Google Scholar]
- Namkung, Y., S.M. Smith, S.B. Lee, N.V. Skrypnyk, H.L. Kim, H. Chin, R.H. Scheller, R.W. Tsien, and H.S. Shin. 1998. Targeted disruption of the Ca2+ channel β3 subunit reduces N- and L-type Ca2+ channel activity and alters the voltage-dependent activation of P/Q-type Ca2+ channels in neurons. Proc. Natl. Acad. Sci. USA. 95:12010–12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely, A., J. Garcia-Olivares, S. Voswinkel, H. Horstkott, and P. Hidalgo. 2004. Folding of active calcium channel β1b subunit by size-exclusion chromatography and its role on channel function. J. Biol. Chem. 279:21689–21694. [DOI] [PubMed] [Google Scholar]
- Neely, A., R. Olcese, X. Wei, L. Birnbaumer, and E. Stefani. 1994. Ca2+-dependent inactivation of a cloned cardiac Ca2+ channel α1 subunit (α1C) expressed in Xenopus oocytes. Biophys. J. 66:1895–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely, A., X. Wei, R. Olcese, L. Birnbaumer, and E. Stefani. 1993. Potentiation by the β subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science. 262:575–578. [DOI] [PubMed] [Google Scholar]
- Olcese, R., N. Qin, T. Schneider, A. Neely, X. Wei, E. Stefani, and L. Birnbaumer. 1994. The amino terminus of a calcium channel β subunit sets rates of channel inactivation independently of the subunit's effect on activation. Neuron. 13:1433–1438. [DOI] [PubMed] [Google Scholar]
- Opatowsky, Y., C.C. Chen, K.P. Campbell, and J.A. Hirsch. 2004. Structural analysis of the voltage-dependent calcium channel β subunit functional core and its complex with the α1 interaction domain. Neuron. 42:387–399. [DOI] [PubMed] [Google Scholar]
- Pang, L., G. Koren, Z. Wang, and S. Nattel. 2003. Tissue-specific expression of two human Cav1.2 isoforms under the control of distinct 5′ flanking regulatory elements. FEBS Lett. 546:349–354. [DOI] [PubMed] [Google Scholar]
- Peleg, S., D. Varon, T. Ivanina, C.W. Dessauer, and N. Dascal. 2002. Gαi controls the gating of the G-protein-activated K+ channel, GIRK. Neuron. 33:87–99. [DOI] [PubMed] [Google Scholar]
- Perez Garcia, M.T., T.J. Kamp, and E. Marban. 1995. Functional properties of cardiac L-type calcium channels transiently expressed in HEK293 cells. Roles of α1 and β subunits. J. Gen. Physiol. 105:289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Reyes, E., A. Castellano, H.S. Kim, P. Bertrand, E. Baggstrom, A.E. Lacerda, X.Y. Wei, and L. Birnbaumer. 1992. Cloning and expression of a cardiac/brain β subunit of the L-type calcium channel. J. Biol. Chem. 267:1792–1797. [PubMed] [Google Scholar]
- Pragnell, M., M. De Waard, Y. Mori, T. Tanabe, T.P. Snutch, and K.P. Campbell. 1994. Calcium channel β subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1 subunit. Nature. 368:67–70. [DOI] [PubMed] [Google Scholar]
- Qin, N., R. Olcese, J. Zhou, O.A. Cabello, L. Birnbaumer, and E. Stefani. 1996. Identification of a second region of the beta-subunit involved in regulation of calcium channel inactivation. Am. J. Physiol. 271:C1539–C1545. [DOI] [PubMed] [Google Scholar]
- Qin, N., D. Platano, R. Olcese, J.L. Costantin, E. Stefani, and L. Birnbaumer. 1998. Unique regulatory properties of the type 2a Ca2+ channel β subunit caused by palmitoylation. Proc. Natl. Acad. Sci. USA. 95:4690–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter, H. 1983. Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature. 301:569–574. [DOI] [PubMed] [Google Scholar]
- Saada, N., B. Dai, C. Echetebu, S.K. Sarna, and P. Palade. 2003. Smooth muscle uses another promoter to express primarily a form of human CaV1.2 L-type calcium channel different from the principal heart form. Biochem. Biophys. Res. Commun. 302:23–28. [DOI] [PubMed] [Google Scholar]
- Schreibmayer, W., H.A. Lester, and N. Dascal. 1994. Voltage clamp of Xenopus laevis oocytes utilizing agarose cushion electrodes. Pflugers Arch. 426:453–458. [DOI] [PubMed] [Google Scholar]
- Schultz, D., G. Mikala, A. Yatani, D.B. Engle, D.E. Iles, B. Segers, R.J. Sinke, D.O. Weghuis, U. Klockner, M. Wakamori, et al. 1993. Cloning, chromosomal localization, and functional expression of the α1 subunit of the L-type voltage-dependent calcium channel from normal human heart. Proc. Natl. Acad. Sci. USA. 90:6228–6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon, D., D. Vorobiov, and N. Dascal. 1997. Positive and negative coupling of the metabotropic glutamate receptors to a G protein-activated K+ channel, GIRK, in Xenopus oocytes. J. Gen. Physiol. 109:477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, C., and N.M. Soldatov. 2002. Molecular determinants of voltage-dependent slow inactivation of the Ca2+ channel. J. Biol. Chem. 277:6813–6821. [DOI] [PubMed] [Google Scholar]
- Shistik, E., T. Ivanina, Y. Blumenstein, and N. Dascal. 1998. Crucial role of N terminus in function of cardiac L-type Ca2+ channel and its modulation by protein kinase C. J. Biol. Chem. 273:17901–17909. [DOI] [PubMed] [Google Scholar]
- Shistik, E., T. Ivanina, T. Puri, M. Hosey, and N. Dascal. 1995. Ca2+ current enhancement by α2/δ and β subunits in Xenopus oocytes: contribution of changes in channel gating and α1 protein level. J. Physiol. 489:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shistik, E., T. Keren-Raifman, G.H. Idelson, N. Dascal, and T. Ivanina. 1999. The N-terminus of the cardiac L-type Ca2+ channel α1C subunit: the initial segment is ubiquitous and crucial for protein kinase C modulation, but it is not directly phosphorylated. J. Biol. Chem. 274:31145–31149. [DOI] [PubMed] [Google Scholar]
- Singer, D., M. Biel, I. Lotan, V. Flockerzi, F. Hofmann, and N. Dascal. 1991. The roles of the subunits in the function of the calcium channel. Science. 253:1553–1557. [DOI] [PubMed] [Google Scholar]
- Singer-Lahat, D., N. Dascal, L. Mittelman, S. Peleg, and I. Lotan. 2000. Imaging plasma membrane proteins in large membrane patches of Xenopus oocytes. Pflugers Arch. 440:627–633. [DOI] [PubMed] [Google Scholar]
- Singer-Lahat, D., I. Lotan, M. Biel, V. Flockerzi, F. Hofmann, and N. Dascal. 1994. Cardiac calcium channels expressed in Xenopus oocytes are modulated by dephosphorylation but not by cAMP-dependent phosphorylation. Receptors Channels. 2:215–226. [PubMed] [Google Scholar]
- Slish, D.F., D.B. Engle, G. Varadi, I. Lotan, D. Singer, N. Dascal, and A. Schwartz. 1989. Evidence for the existence of a cardiac specific isoform of the α1 subunit of the voltage dependent calcium channel. FEBS Lett. 250:509–514. [DOI] [PubMed] [Google Scholar]
- Snutch, T.P., W.J. Tomlinson, J.P. Leonard, and M.M. Gilbert. 1991. Distinct calcium channels are generated by alternative splicing and are differentially expressed in the mammalian CNS. Neuron. 7:45–57. [DOI] [PubMed] [Google Scholar]
- Soldatov, N.M. 1994. Genomic structure of human L-type Ca2+ channel. Genomics. 22:77–87. [DOI] [PubMed] [Google Scholar]
- Spafford, J.D., J. Van Minnen, P. Larsen, A.B. Smit, N.I. Syed, and G.W. Zamponi. 2004. Uncoupling of calcium channel α1 and β subunits in developing neurons. J. Biol. Chem. 279:41157–41167. [DOI] [PubMed] [Google Scholar]
- Stotz, S.C., and G.W. Zamponi. 2001. Structural determinants of fast inactivation of high voltage-activated Ca2+ channels. Trends Neurosci. 24:176–182. [DOI] [PubMed] [Google Scholar]
- Striessnig, J. 1999. Pharmacology, structure and function of cardiac L-type Ca2+ channels. Cell. Physiol. Biochem. 9:242–269. [DOI] [PubMed] [Google Scholar]
- Strube, C., M. Beurg, P.A. Powers, R.G. Gregg, and R. Coronado. 1996. Reduced Ca2+ current, charge movement, and absence of Ca2+ transients in skeletal muscle deficient in dihydropyridine receptor β1 subunit. Biophys. J. 71:2531–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, S.X., J. Miriyala, and H.M. Colecraft. 2004. Membrane-associated guanylate kinase-like properties of β-subunits required for modulation of voltage-dependent Ca2+ channels. Proc. Natl. Acad. Sci. USA. 101:7193–7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, S.X., J. Miriyala, L.H. Tay, D.T. Yue, and H.M. Colecraft. 2005. A Cavβ SH3/guanylate kinase domain interaction regulates multiple properties of voltage-gated Ca2+ channels. J. Gen. Physiol. 126:365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z.Z., M.C. Liang, S. Lu, D. Yu, C.Y. Yu, D.T. Yue, and T.W. Soong. 2004. Transcript scanning reveals novel and extensive splice variations in human L-type voltage-gated calcium channel, Cav1.2 α1 subunit. J. Biol. Chem. 279:44335–44343. [DOI] [PubMed] [Google Scholar]
- Tareilus, E., M. Roux, N. Qin, R. Olcese, J. Zhou, E. Stefani, and L. Birnbaumer. 1997. A Xenopus oocyte β subunit: evidence for a role in the assembly/expression of voltage-gated calcium channels that is separate from its role as a regulatory subunit. Proc. Natl. Acad. Sci. USA. 94:1703–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem, F., K.A. Clark, F.C. Chatelain, and D.L. Minor Jr. 2004. Structure of a complex between a voltage-gated calcium channel β subunit and an α subunit domain. Nature. 429:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi, G., P. Lory, D. Schultz, M. Varadi, and A. Schwartz. 1991. Acceleration of activation and inactivation by the β subunit of the skeletal muscle calcium channel. Nature. 352:159–162. [DOI] [PubMed] [Google Scholar]
- Varadi, G., Y. Mori, G. Mikala, and A. Schwartz. 1995. Molecular determinants of Ca2+ channel function and drug action. Trends Pharmacol. Sci. 16:43–49. [DOI] [PubMed] [Google Scholar]
- Wakamori, M., G. Mikala, A. Schwartz, and A. Yatani. 1993. Single-channel analysis of a cloned human heart L-type Ca2+ channel α1 subunit and the effects of a cardiac β subunit. Biochem. Biophys. Res. Commun. 196:1170–1176. [DOI] [PubMed] [Google Scholar]
- Walker, D., D. Bichet, S. Geib, E. Mori, V. Cornet, T.P. Snutch, Y. Mori, and M. De Waard. 1999. A new β subtype-specific interaction in α1A subunit controls P/Q-type Ca2+ channel activation. J. Biol. Chem. 274:12383–12390. [DOI] [PubMed] [Google Scholar]
- Walker, D., and M. De Waard. 1998. Subunit interaction sites in voltage-dependent Ca2+ channels: role in channel function. Trends Neurosci. 21:148–154. [DOI] [PubMed] [Google Scholar]
- Wei, S.-k., H.M. Colecraft, C.D. DeMaria, B.Z. Peterson, R. Zhang, T.A. Kohout, T.B. Rogers, and D.T. Yue. 2000. Ca2+ channel modulation by recombinant auxiliary β subunits expressed in young adult heart cells. Circ. Res. 86:175–184. [DOI] [PubMed] [Google Scholar]
- Wei, X., A. Neely, A.E. Lacerda, R. Olcese, E. Stefani, E. Perez Reyes, and L. Birnbaumer. 1994. Modification of Ca2+ channel activity by deletions at the carboxyl terminus of the cardiac α1 subunit. J. Biol. Chem. 269:1635–1640. [PubMed] [Google Scholar]
- Wei, X., A. Neely, R. Olcese, W. Lang, E. Stefani, and L. Birnbaumer. 1996. Increase in Ca2+ channel expression by deletions at the amino terminus of the cardiac α1C subunit. Receptors Channels. 4:205–215. [PubMed] [Google Scholar]
- Wei, X.Y., E. Perez Reyes, A.E. Lacerda, G. Schuster, A.M. Brown, and L. Birnbaumer. 1991. Heterologous regulation of the cardiac Ca2+ channel α1 subunit by skeletal muscle β and γ subunits. Implications for the structure of cardiac L-type Ca2+ channels. J. Biol. Chem. 266:21943–21947. [PubMed] [Google Scholar]
- West, A.E., W.G. Chen, M.B. Dalva, R.E. Dolmetsch, J.M. Kornhauser, A.J. Shaywitz, M.A. Takasu, X. Tao, and M.E. Greenberg. 2001. Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. USA. 98:11024–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, M.E., D.H. Feldman, A.F. McCue, R. Brenner, G. Velicelebi, S.B. Ellis, and M.M. Harpold. 1992. Structure and functional expression of α1, α2, and β subunits of a novel human neuronal calcium channel subtype. Neuron. 8:71–84. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, H., M. Hara, M. Strobeck, K. Fukasawa, A. Schwartz, and G. Varadi. 1998. Multiple modulation pathways of calcium channel activity by a β subunit. Direct evidence of β subunit participation in membrane trafficking of the α1C subunit. J. Biol. Chem. 273:19348–19356. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, H., M. Okuda, G. Mikala, K. Fukasawa, and G. Varadi. 2000. Cloning of the β2a subunit of the voltage-dependent calcium channel from human heart: cooperative effect of α2/δ and β2a on the membrane expression of the α1C subunit. Biochem. Biophys. Res. Commun. 267:156–163. [DOI] [PubMed] [Google Scholar]
- Yasuda, T., L. Chen, W. Barr, J.E. McRory, R.J. Lewis, D.J. Adams, and G.W. Zamponi. 2004. Auxiliary subunit regulation of high-voltage activated calcium channels expressed in mammalian cells. Eur. J. Neurosci. 20:1–13. [DOI] [PubMed] [Google Scholar]
- Zerangue, N., B. Schwappach, Y.N. Jan, and L.Y. Jan. 1999. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron. 22:537–548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.