

Abstract
Although the Na+/K+ pump is one of the key mechanisms responsible for maintaining cell volume, we have observed experimentally that cell volume remained almost constant during 90 min exposure of guinea pig ventricular myocytes to ouabain. Simulation of this finding using a comprehensive cardiac cell model (Kyoto model incorporating Cl− and water fluxes) predicted roles for the plasma membrane Ca2+-ATPase (PMCA) and Na+/Ca2+ exchanger, in addition to low membrane permeabilities for Na+ and Cl−, in maintaining cell volume. PMCA might help maintain the [Ca2+] gradient across the membrane though compromised, and thereby promote reverse Na+/Ca2+ exchange stimulated by the increased [Na+]i as well as the membrane depolarization. Na+ extrusion via Na+/Ca2+ exchange delayed cell swelling during Na+/K+ pump block. Supporting these model predictions, we observed ventricular cell swelling after blocking Na+/Ca2+ exchange with KB-R7943 or SEA0400 in the presence of ouabain. When Cl− conductance via the cystic fibrosis transmembrane conductance regulator (CFTR) was activated with isoproterenol during the ouabain treatment, cells showed an initial shrinkage to 94.2 ± 0.5%, followed by a marked swelling 52.0 ± 4.9 min after drug application. Concomitantly with the onset of swelling, a rapid jump of membrane potential was observed. These experimental observations could be reproduced well by the model simulations. Namely, the Cl− efflux via CFTR accompanied by a concomitant cation efflux caused the initial volume decrease. Then, the gradual membrane depolarization induced by the Na+/K+ pump block activated the window current of the L-type Ca2+ current, which increased [Ca2+]i. Finally, the activation of Ca2+-dependent cation conductance induced the jump of membrane potential, and the rapid accumulation of intracellular Na+ accompanied by the Cl− influx via CFTR, resulting in the cell swelling. The pivotal role of L-type Ca2+ channels predicted in the simulation was demonstrated in experiments, where blocking Ca2+ channels resulted in a much delayed cell swelling.
INTRODUCTION
The Na+/K+ pump is one of the constitutive proteins present in almost all mammalian cells. It maintains concentration gradients of Na+ and K+ across the cell membrane by exchanging three Na+ for two external K+ ions, using energy from the hydrolysis of one ATP molecule. Thereby, it has an essential role in regulating the cell volume (Balshaw et al., 2001). Recently, Armstrong (2003) proposed a simple mathematical model of cell volume regulation in skeletal muscle, which satisfies the predictions of Donnan equilibrium (Boyle and Conway, 1941). We have also constructed a basic model of Cl− homeostasis and cell volume regulation in cardiac ventricular cells (Terashima et al., 2006), which was composed of background Na+, K+, and Cl− membrane conductances, as well as the Na+/K+ pump and NKCC1 (Na+/K+/2 Cl− cotransporter 1). According to these model analyses, the mechanisms of cell volume regulation are detailed as follows. The [K+] gradient, created by the Na+/K+ pump across the membrane, is the main determinant of a negative Vm. This negative Vm expels Cl− out of the cell through Cl− channels, compensating for the continuous Cl− influx via Cl−-coupled transporters, such as NKCC1. Thereby, the pump maintains cellular osmolarity at the physiological level to keep the cell volume intact. Accordingly, the time course of cell swelling caused by blocking the Na+/K+ pump largely depends on redistribution of Cl− across the membrane and therefore on the membrane Cl− permeability, since the overall total ion flux must obey macroscopic electroneutrality. Membrane Na+ permeability also determines the time course of cell swelling indirectly, through impeding the redistribution of K+ across the membrane during the Na+/K+ pump block (Terashima et al., 2006). This general mechanism was experimentally supported by Dierkes et al. (2006) in leech Retzius neurons. Contrary to the above theoretical expectations, however, it is well known that cardiac cell volume hardly changes during Na+/K+ pump blockade (Pine et al., 1980; Drewnowska and Baumgarten, 1991; Wright and Rees, 1998). However, mechanisms underlying this preservation of cell volume have not yet been elucidated on a quantitative basis.
To clarify mechanisms of cellular responses, such as cell volume regulation, which are accomplished by the complex interactions of many factors, mathematical model analysis is indispensable. To date, several computer models of membrane excitation have been published for ventricular myocytes (see Noble and Rudy, 2001 for review; and the Kyoto model proposed by Matsuoka et al., 2003, 2004). However, neither Cl− homeostasis nor cell volume regulation was included in these models. Previously, we extended the algorithm of calculating the Cl− and water fluxes established in the basic model to the comprehensive cardiac cell model, which included mechanisms underlying membrane excitation, intracellular Ca2+ dynamics, and contraction (Terashima et al., 2006). In the present study, we have fine tuned this model, based on the additional experimental data of cardiac ion and water fluxes, and analyzed the mechanisms of cell volume regulation by conducting computer simulations in parallel with experimental validation, by measuring cell area, as an index of cell volume, and Vm using a voltage-sensitive fluorescent dye, di-8-ANEPPS. We confirmed that the Na+/K+ pump block caused a strong membrane depolarization, accompanied by minimal change in cell volume, as described in the literatures (Pine et al., 1980; Drewnowska and Baumgarten, 1991; Wright and Rees, 1998). In addition, we found that cell volume changed in a biphasic manner when the membrane Cl− conductance was increased by pharmacological intervention. The involvement of multiple factors such as PMCA, Na+/Ca2+ exchange, L-type Ca2+ channels, and Ca2+-activated background cation current Il (Ca) in cell volume regulation were quantitatively examined, in addition to the contributions of membrane Na+, K+, and Cl− conductances.
MATERIALS AND METHODS
Parameter Set in the Kyoto Model
Based on additional experimental data, we have improved our ventricular cell model (Terashima et al., 2006), which had been implemented with algorithms of Cl− and water fluxes. A complete list of equations composing the Kyoto model is available in Tables S1–S13 (available at http://www.jgp.org/cgi/content/full/jgp.200609646/DC1), together with abbreviations. Fig. 1 shows a schematic diagram of the Kyoto model. The channels and transporters focused in the present study are boxed off. The extracellular ion concentrations were set (in mM) for [Na+]o at 141, [K+]o 5.4, [Ca2+]o 1.8, [Cl−]o 140, and [LA]o 10, except for the simulations of drug effects (Fig. 3 B and Figs. 4, 5, and 8) where [Ca2+]o was set at 20 μM. The steady state is well established with 2.5 Hz stimulation, as well as under quiescent conditions. The ion concentrations under these conditions shown in Table I are physiologically relevant, as described in detail below. Additionally, the configurations of the action potential, intracellular Ca2+ transient and major ionic currents under 2.5-Hz stimulation shown in Fig. 2, confirm the validity of membrane excitability, as formulated in the Kyoto model. All calculations were conducted using the Euler method, with the adaptive time step with simBio (Sarai et al., 2006b), and the source code of the Kyoto model is available from http://www.sim-bio.org/. All parameters related to volume regulation, except the background Cl− conductance and NKCC1, are based on experimental findings as described below.
Figure 1.

Schematic diagram of the Kyoto model. The channels and transporters focused on in relation to the volume regulation were boxed off. For abbreviations, see the abbreviations list and Table S1 in the online supplemental material.
Figure 3.

Experimental recordings of the cell area and Vm during the Na+/K+ pump block (A) and simulation of the Na+/K+ pump block using the Kyoto model (B). (A) Top panel shows measurements of Vm in five representative cells (colored lines) with 40 μM ouabain. The control records obtained in the absence of ouabain were averaged and represented as mean ± SEM (n = 4). Measurements of cell area (n = 5) with 40 μM ouabain were shown in the middle panel with different colors. The control records obtained in the absence of ouabain were presented as mean ± SEM in the bottom panel (n = 5). The joining error bars represent contour of the plots. When the error bars are not shown, they are smaller than the symbols. (B) The simulation of the corresponding experimental condition was performed by applying 40 μM ouabain at time 0 to the Kyoto model and the time courses of Vt expressed in percent of original Vt (top) and Vm (bottom) were shown.
Figure 4.

Sensitivity analyses of varying magnitude of membrane Cl− or Na+ conductance, which is involved in the cell volume as well as Vm modulation evoked by the Na+/K+ block. Throughout the recording time, no electrical stimulation was applied. The [Ca2+]o of 20 μM was used (see text for detail). The steady state in model parameters was established before applying ouabain. (A) The relative magnitude of PClb was varied at constant × 1 PbNSC as indicated in the graph at time 0 simultaneously with the start of the Na+/K+ pump block (40 μM ouabain). In parallel to the PClb alteration, MNKCC1 was also scaled to maintain [Cl−]i at 30 mM. Changes in Vt and Vm at various PClb were plotted with the corresponding colors of numerals as indicated in the upper graph. (B) At time 0, the PbNSC was varied at constant × 1 PClb as indicated by numerals simultaneously with the Na+/K+ pump block.
Figure 5.

Simulation of the Na+/K+ pump block with or without PMCA. At time 0, 40 μM ouabain was applied. The amplitude factor of PMCA was reduced to zero at time 0 in the case of simulation without PMCA. Changes in Vm (thin lines) as well as ENaCa (thick lines) were calculated with (top) or without (bottom) PMCA in A. [Ca2+]i in B and Vt in C. Black and blue lines are simulated results with and without PMCA, respectively. Without PMCA, the Vm completely overlapped with the trace of ENaCa after 42 min. The Vt at 120 min was 102.1% and 103.6% with and without PMCA, respectively. Note that the [Ca2+]i (19.50 μM) in the absence of PMCA is nearly equilibrated with the [Ca2+]o 42 min after the onset of ouabain, while [Ca2+]i in the presence of PMCA is 0.18 μM at 120 min.
Figure 8.

Model prediction for the experimental observations in Fig. 7. At time 0, 40 μM ouabain and 1 μM isoproterenol were applied simultaneously. Changes in Vt (A), amounts of intracellular ions expressed as Eq × 10−12 (B), amounts of intracellular cation (blue) or anion (red) (C), Vm (D), ICaL (E), [Ca2+]i (F), and Il (Ca) (G) were demonstrated. Since the present simulation ignores minor changes in anion concentrations accompanying the Ca2+ binding to proteins and Ca2+ flux into SR, there is a slight difference between total amounts of cation and anion.
TABLE I.
Steady-state Variables
| Steady-state values without stimulation |
End diastolic values during 2.5 Hz stimulation |
|
|---|---|---|
| Vm (mV) | −87.80 | −87.34 |
| [Na+]i (mM) | 2.13 | 6.37 |
| [K+]i (mM) | 147.30 | 142.95 |
| [Cl-]i (mM) | 29.94 | 57.71 |
| [Ca2+]i (mM) | 2.14 × 10−5 | 4.67 × 10−5 |
| [LA]i (mM) | 118.83 | 91.16 |
| [ATPtotal]i (mM) | 6.97 | 6.96 |
| Vt (μm3) | 16,000 | 19,886 |
Figure 2.

Action potential, Ca2+ transient and major ionic currents and transporters of the Kyoto model during a steady cycle of 2.5 Hz stimulation. For abbreviations, see Abbreviations list and Table S1 in the online supplemental material. To facilitate comparing relative magnitude of the Ca2+ flux through PMCA (μmol/liter cytosol/s), the Ca2+ flux was expressed in pA/pF, neglecting the antiport with H+.
(1) Water Flux.
The water flux Jwater is driven by the difference in total ion concentrations ([Total ions], sum of Na+, K+, Cl−, Ca2+, and LA across the cell membrane; see Table S4 in the online supplemental material).
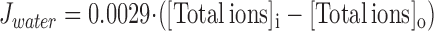 |
A hydraulic conductivity of 0.0029 μm3/mM/ms is determined from experimental recordings of osmotic cell swelling (Wang et al., 1997; Sasaki et al., 1999), which is comparable to the experimental data reported by Suleymanian and Baumgarten (1996) and by Ogura et al. (2002).
(2) Cl− Fluxes.
The total Cl− flux is composed of ICFTR, IVRCC, IClb, and NKCC1. The kinetic schemes of these components are the same as used by Terashima et al. (2006), but their amplitudes have been slightly modified by model fitting to new experimental findings in the present study. There are large variations in the ICFTR current density in accordance with the variable expression levels of CFTR mRNA in guinea pig ventricular myocytes (James et al., 1996). Here, we set PCFTR so that the current density of ICFTR is within the range of the reported values (James et al., 1996) (see Table S8 in the online supplemental material). The Kyoto model can reproduce the current–voltage relationships recorded at various concentrations of isoproterenol in guinea pig ventricular cells obtained by Tareen et al. (1992) (see Fig. 3 in Terashima et al., 2006).
The volume-regulated Cl− current IVRCC is a major factor determining the regulatory volume decrease, albeit that the extent of spontaneous regulatory volume decrease is quite small in guinea pig ventricular myocytes (Yamamoto et al., 2004). We have formulated IVRCC according to the epithelial model of Strieter et al. (1990) and the experimental data reported by Shuba et al. (1996) in guinea pig ventricular myocytes (see Fig. 2 in Terashima et al., 2006 and see Table S8 in the online supplemental material). PVRCC is set so that it can reproduce well the volume–osmolarity relationship described in guinea pig ventricular myocytes by Sasaki et al. (1999) (see Fig. S1 in the online supplemental material).
There are few data estimating the amplitude of the background membrane Cl− conductance, IClb, and flux via Cl−-coupled transporters in guinea-pig ventricular myocytes. We tentatively used the NKCC1 kinetic model developed by Benjamin and Johnson (1997) to represent Cl−-coupled transporters (Terashima et al., 2006). The amplitude factors PClb and MNKCC1 were model adjusted, so that the almost constant cell volume observed during Na+/K+ pump inhibition in this study could be reproduced (Figs. 3 and 4) (see Tables S8 and S10 in the online supplemental material). As a result, [Cl−]i without stimulus in the Kyoto model is 30 mM (Table I), comparable to 20–30 mM [Cl−]i in quiescent sheep Purkinje fibers (Vaughan-Jones, 1982). In addition, higher stimulus frequencies increase [Cl−]i in the Kyoto model (Table I), consistent with the rapid pacing-induced accumulation of Cl− in dog atrial myocytes (Akar et al., 2003).
(3) Background Na+ and K+ Fluxes.
We set the amplitude factors of the background cation conductance IbNSC and Na+/K+ pump based on the experimental reports of Kiyosue et al. (1993) and Nakao and Gadsby, (1989) (Tables S9 and S10 in the online supplemental material). An IbNSC of −0.18 pA/pF at −50 mV in the Kyoto model is the same as that reported in guinea pig ventricular myocytes (Kiyosue et al., 1993). In addition, the current density and current–voltage relationship of the Na+/K+ pump in guinea pig ventricular myocytes (Nakao and Gadsby, 1989) can also be reproduced by the Kyoto model (Fig. S2 in the online supplemental material; the current density at 0 mV is 1.3 pA/pF in guinea pig ventricular myocytes and 1.2 pA/pF in the Kyoto model). The current densities of the Na+/K+ pump and background inward Na+ current determined by Gao et al. (1995) in the guinea pig ventricular myocytes were 0.25 ± 0.09 pA/pF and 0.75 ± 0.26 pA/pF at −60 mV, respectively. The corresponding values of the Kyoto model are 0.44 pA/pF and 0.42 pA/pF, respectively, both of which are in the same range as the experimental values. [Na+]i at rest and with 2.5 Hz stimulus in the Kyoto model are 2.13 and 6.37 mM, respectively (Table I), both of which are within the range of the experimental data of ∼3 and 6.5 mM obtained in guinea pig papillary muscle (Wang et al., 1988).
The equations for Il (Ca) are the same as that used in Matsuoka et al. (2003), where the dependence on [Ca2+]i determined in guinea pig ventricular myocytes (Ehara et al., 1988) was used (see Table S9 in the online supplemental material). The maximum activation of Il (Ca) produces 8.6 nS/cell, which is within the lower range of the experimental estimation from single channel recordings, 7.2 to 72 nS/cell (Ehara et al., 1988).
(4) Ca2+ Fluxes.
Influx through L-type Ca2+ channels is balanced by the efflux via Na+/Ca2+ exchange and PMCA in cardiac myocytes. Recently, model analysis by Sarai et al. (2006a) suggested that involvement of PMCA was required to simulate the rhythmic contractions observed in NCX-knockout mouse. We used the same kinetic model for PMCA as reported by Luo and Rudy (1994) (see Table S10 in the online supplemental material). Most recently, the relative contribution of PMCA to the total Ca2+ extrusion was reported to be 7.1% in guinea pig ventricular myocytes at 37°C (Mackiewicz and Lewartowski, 2006). Based on this report, we tentatively assumed that the contribution of PMCA to total Ca2+ extrusion is 9% (10% of Na+/Ca2+ exchanger).
It is well established that the activity of the Na+/Ca2+ exchanger is regulated by both [Na+]i and [Ca2+]i in the excised membrane patch (Hilgemann et al., 1992a,b), and that these Na+-dependent inactivation and Ca2+-dependent activation mechanisms are relevant to [Ca2+]i dynamics in guinea pig ventricular myocytes (Fujioka et al., 2000; Kuratomi et al., 2003). The kinetic model of these regulatory processes proposed by Fujioka et al. (2000) is used in the present study (see Table S10 in the online supplemental material), but the stoichiometry is tentatively fixed to the standard 3 Na+:1 Ca2+ exchange. Using this model, the INaCa current density of 4.4 pA/pF at 50 mV is comparable to that observed in guinea pig ventricular myocytes, 4–6 pA/pF assuming the experimental condition of high [Ca2+]i (0.8 μM) (Lin et al., 2006). Furthermore, the partial inactivation of Na+/Ca2+ exchange at resting [Ca2+]i prevents [Ca2+]i from decreasing too low during quiescence. In the Kyoto model, [Ca2+]i are 21 and 46 nM at rest and at end-diastole during 2.5-Hz stimulation, respectively (Table I), both of which are only slightly smaller than those measured in quiescent and stimulated guinea pig ventricular myocytes, ∼50 and 100 nM, respectively (Beuckelmann and Wier, 1988; Gannier et al., 1996).
In the present model simulation, spontaneous Ca2+ release is attenuated tentatively by removing the [Ca2+]i-dependent term of k1, the rate constant determining the transition from the closed state to the open state in the RyR channel model (see Table S11 in the online supplemental material). As described in Results, the spontaneous and repetitive cell contractions were largely inhibited during the application of ouabain by omitting CaCl2 from the external saline, while some cells still contracted repetitively at the terminal phase of the simultaneous application of ouabain and isoproterenol. In accordance with the changes in RyR channel model, several amplitude factors regarding the Ca2+ handling have been slightly modified, so as not to affect the configurations of both the action potential and intracellular Ca2+ transient.
Simulation of Drug Effects on Ventricular Myocytes
According to the experimental conditions used in the present study, all simulations of drug effects (Fig. 3 B and Figs. 4, 5, and 8) were performed with no stimulus and a [Ca2+]o of 20 μM, assuming this level of residual Ca2+ in the nominally Ca2+-free Tyrode solution.
The dose-dependent inhibition of the Na+/K+ pump by ouabain is determined based on the report by Baker et al. (1969) (Table S10 in the online supplemental material), a blockade as much as 98.5% of the Na+/K+ pump by 40 μM ouabain is comparable to the experimental data, where the complete inhibition of the Na+/K+ pump was accomplished by >50 μM ouabain in the guinea pig ventricular myocytes (Gao et al., 2002).
In the present study, we used 1 μM isoproterenol in experiments and simulations. Isoproterenol of 1 μM induces nearly full activation of ICFTR in the Kyoto model. Isoproterenol has also been reported to activate ICaL by three- to fourfold (Delpech et al., 1995; Findlay, 2002). For the activation of the L-type Ca2+ channel, we simply assume a threefold increase in the amplitude of ICaL during β-adrenergic stimulation.
It should be noted that changes in [ATP]i in the Kyoto model were negligibly small, in all conditions of simulation performed in the present study, excluding possible modulations of ATP-dependent mechanisms, such as the active ion transporters, the L-type Ca2+ channel, and the CFTR Cl− channel.
Single Cell Preparation
Single ventricular myocytes were obtained by treating guinea pig hearts with collagenase as previously described (Powell et al., 1980; Wang et al., 1997; Sasaki et al., 1999). The experimental protocols were approved by the Animal Research Committee in the Graduate School of Medicine, Kyoto University.
Solutions and Drugs
To avoid spontaneous contractions caused by blocking Na+/K+ pump, a nominally Ca2+-free Tyrode solution was used. The Ca2+-free Tyrode solution contained (in mM) NaCl 140, NaH2PO4 0.33, KCl 5.4, MgCl2 0.45, glucose 5.5, and HEPES 5 (pH 7.4). The modified D-MEM solution was prepared by adding 20 mM NaCl and 25 mM HEPES to D-MEM (without NaHCO3; MP Biomedicals) (pH 7.4).
Drugs used were 40 μM ouabain (Na+/K+ pump inhibitor; Sigma-Aldrich), 1 μM isoproterenol (β-adrenergic agonist for CFTR activation; Sigma-Aldrich), 20 μM KB-R7943 (Na+/Ca2+ exchange blocker; Tocris Cookson Inc.), 1 μM SEA0400 (Na+/Ca2+ exchange blocker; a gift from Taisho Pharmaceutical Company, Ltd.), and 5 μM nifedipine (L-Type Ca2+ channel blocker; Sigma-Aldrich). KB-R7943, SEA0400, and nifedipine were dissolved in DMSO as stock solutions and diluted with Tyrode solution. The final concentration of DMSO was 0.1%.
Measurement of Cell Area as an Index of Cell Volume
The measurement of cell area provides an estimate for determining changes in volume of cardiac myocytes (Yamamoto et al., 2001; Walsh and Zhang, 2005). The myocytes were settled onto the glass bottom of a recording chamber mounted on an inverted microscope (Eclipse TE2000; Nikon) equipped with a 40× oil objective and were superfused with nominally Ca2+-free Tyrode solution at 36–37°C. Images of the cell were obtained every 30 s by a cooled CCD digital camera (ORCA-ER; Hamamatsu Photonics), and the cell area was calculated using Image-Pro Plus version 5.1 software (Media Cybernetics). The cell images during cell contractions were excluded from the data.
Measurement of Vm Using a Voltage-sensitive Dye, di-8-ANEPPS
Changes in Vm were monitored by dual-wavelength ratio imaging of a voltage-sensitive fluorescent dye, di-8-ANEPPS (Invitrogen) (Bedlack, 1992). Although small downward drifts in the fluorescence ratio of di-8-ANEPPS were observed during the long-term measurements (see Fig. 3 A), this method has advantages over the standard method of measuring Vm using glass electrodes. First, we can completely avoid the ion diffusion through the electrode tip between the pipette solution and the cytosol. Also, continuous recording of Vm is feasible by using the Vm-sensitive dye. Myocytes were loaded with di-8-ANEPPS according to the manufacture's protocol. In brief, myocytes were incubated for 20 min in the modified D-MEM solution containing 1 μM di-8-ANEPPS at 16°C and rinsed twice. Fluorescent image pairs of single cells with excitation at 440 ± 10 and 535 ± 25 nm from a 100-W xenon arc lamp were acquired (575 nm dichroic mirror and emission of >590 nm) every 30 s using the cooled CCD digital camera. For each cell, a pair of background images, which were adjacent to the target cell, but contained no other cells, was taken for background subtraction. The fluorescent ratio R is defined as the ratio of the fluorescence intensity at an excitation wavelength of 440 nm to that at 535 nm. The use of the ratiometric method avoids any effects of small variations in dye concentration on the fluorescence results. To convert the fluorescence ratio R to a Vm value in mV, whole-cell voltage clamp was performed using a pipette solution containing (in mM) 145 KCl, 1 MgCl2, 1 EGTA, and 5 HEPES (pH 7.2). In brief, the Vm was clamped to different levels ranging from −120 to +20 mV, and pairs of fluorescence images were taken at each potential and analyzed as described above. The calibration curve is shown in Fig. S3. The R values plotted against the Vm showed a linear relationship (correlation coefficient, r = 0.84, P <0.01), while a relatively large variation in the absolute value of R for each Vm was observed between the individual myocyte (see large error bars in the Fig. S3). The Vm value obtained in each experiment was calculated using the following equation:
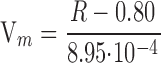 |
Statistics
Results are represented as mean ± SEM. Statistical analyses were performed by one-factor ANOVA using StatView (SAS Institute Inc.). Multiple and two-group comparisons were performed according to Student-Newman-Keul's method and Student's t test, respectively. P < 0.05 was considered significant.
Online Supplemental Material
Lists of abbreviations and all equations used in the Kyoto model are available as Tables S1–S13 in the online supplemental material (http://www.jgp.org/cgi/content/full/jgp.200609646/DC1). In addition, the steady-state variables such as ionic currents, flux, and metabolite concentrations are in Table S14. The volume–osmolarity relationship (Fig. S1) and the current–voltage relationship of Na+/K+ pump (Fig. S2) in the Kyoto model, and the calibration curve for di-8-ANEPPS (Fig. S3) are also presented.
RESULTS
Maintenance of Cell Volume during the Membrane Depolarization Induced by Applying Ouabain to Ventricular Myocytes
Membrane depolarization takes the pivotal role in coupling blockade of the Na+/K+ pump to cell swelling (Armstrong 2003; Terashima et al., 2006). In experiments shown in the top panel of Fig. 3 A, Vm was measured with or without the continuous application of 40 μM ouabain. The control without ouabain was shown as mean ± SEM. Treatment with 40 μM ouabain caused an obvious membrane depolarization, with a large variation in the time course among different cells, as shown by five representative recordings in the top panel of Fig. 3 A. On average, the peak depolarization to −40.0 ± 7.1 mV was attained 66.5 ± 6.8 min (n = 10) after applying ouabain. This average value is in agreement with the reported Vm (∼−33 mV at 1 h or ∼−20 mV at 2 h after applying ouabain) measured by penetrating the intracellular microelectrode into the full-thickness section of ventricular wall, where the variation in Vm or membrane conductance, if any, among intact cells should be automatically averaged through the electrical coupling via gap junction (Pine et al., 1980). In contrast to Vm, no obvious cell swelling was observed as shown in the middle panel of Fig. 3 A, where five representative recordings of the cell area during Na+/K+ pump block are shown. In all experiments, a continuous but small decrease was observed. This decrease was also observed in the control experiments without applying ouabain (Fig. 3 A, bottom). The cell area at 90 min was 98.6 ± 0.6% of the original area in the control group (n = 5) and 96.9 ± 0.7% in the ouabain-treated group (n = 5). These findings clearly indicate that cell swelling should be quite small during the 90-min application of ouabain, which is in agreement with previous reports (Pine et al., 1980; Drewnowska and Baumgarten, 1991). The median time course of depolarization was simulated well by the Kyoto model, as shown in Fig. 3 B. Indeed, the increase in cell volume is much delayed and small, compared with the membrane depolarization. Cell volume is 101.3% of the original value 90 min after applying 40 μM ouabain, indicating quite a small change in cell area.
Role of Membrane Cl− and Na+ Conductances in Cell Volume Regulation As Revealed by Simulation
Mechanisms underlying the almost constant cell volume accompanying the variable time courses of membrane depolarization during the Na+/K+ pump blockade were quantitatively examined by varying the membrane conductances for Cl− and Na+ in the simulation. In Fig. 4 A, membrane Cl− conductance was changed by scaling PClb and MNKCC1 simultaneously to keep control [Cl−]i at the physiological level of 30 mM. In Fig. 4 B, membrane Na+ conductance was changed by varying the magnitude of PbNSC. It is obvious that the cell swells to a larger extent with increased membrane Cl− and Na+ conductances during Na+/K+ pump block, and that the rate of cell swelling is mainly determined by the membrane Cl− conductance (Fig. 4 A, top). Decreasing the Na+ conductance delays the onset of cell swelling, leaving the rate of swelling nearly constant (Fig. 4 B, top). These results support the notion that the rate of Cl− influx is rate limiting in cell swelling, since the overall total flux obeys macroscopic electroneutrality (Armstrong 2003; Terashima et al., 2006). Although there are limited experimental data on the magnitude of membrane background Cl− conductance, the simulation of experimental results (Fig. 3 A) using the comprehensive cell model suggests that the membrane Cl− conductance is small in guinea pig ventricular myocytes.
It is also obvious that the experimental variation in the time course of membrane depolarization can be explained by varying the membrane Na+ conductance, not by varying the Cl− conductance, as shown in the bottom panels of Fig. 4. Here, membrane depolarization is caused by the redistribution of K+ across the membrane, i.e., by changes in the K+ equilibrium potential. To satisfy electroneutrality, this redistribution of K+ should be coupled with the counter movement of Na+ or with the parallel flux of Cl−. The electrochemical driving force for Cl− is much smaller than that for Na+, because ECl deviates only slightly from Vm by the Cl− influx through NKCC1. Therefore, changing the membrane Cl− conductance causes only marginal variations in the rate of K+ redistribution, while that of Na+ greatly affects the K+ redistribution. The accelerated membrane depolarization caused by increasing PbNSC shortens the critical time when Vm becomes more positive than ECl. This results in the overall Cl− influx through channels as well as NKCC1, and the simultaneous cation influx, since the total ionic flux obeys macroscopic electroneutrality. Thereby, intracellular osmolarity increases to evoke cell swelling (Fig. 4 B, top). These simulations suggest that the experimental variation in Vm (Fig. 3 A) might be caused by variation in membrane Na+ conductance between 0.5 and 2 times the control. Indeed, Kiyosue et al. (1993) reported a considerable variation in the current density of the background Na+ conductance among myocytes dissociated by enzymatic treatment. The largest cell swelling in this range of PbNSC is 2.3% at 90 min, which is within the extent of the spontaneous decrease in cell area observed in experiments (Fig. 3 A). It may be possible that relatively small volume changes could not be resolved by measuring the cell area.
Role of PMCA and Na+/Ca2+ Exchanger in Cell Volume Regulation As Predicted by Simulation
The variation in Vm at the quasi-steady state established by the continuous application of ouabain, as seen in the bottom panel of Fig. 4 B, is caused by a trace of Na+/K+ pump activity (1.5% of control) remaining during drug application. [Na+]i at 120 min are 142.3, 140.8, 137.2, 127.0, and 110.3 mM at ×5, 2, 1, 0.5, and 0.2 of PbNSC in the simulation, respectively. Indeed, the variation is much smaller when the Na+/K+ pump activity is completely blocked (unpublished data). This prompted us to examine the role of the other active transporter PMCA in determining Vm as well as cell volume during Na+/K+ pump block. To visualize the role of PMCA, simulations were performed with and without PMCA. As clearly demonstrated in Fig. 5 B, [Ca2+]i is maintained at a low level (0.18 μM) by active Ca2+ extrusion via PMCA. Omitting PMCA from the Kyoto model results in a marked Ca2+ accumulation (19.50 μM, Fig. 5 B) when an ICaL window current is induced by the membrane depolarization after ∼40 min of ouabain treatment. Ca2+ accumulation increases membrane Na+ conductance, through activation of Il (Ca) (13-fold increase), resulting in the jump of Vm at ∼40 min, as shown in the bottom panel of Fig. 5 A. Thus, in the absence of PMCA, Na+, K+, and Ca2+ are passively redistributed across the membrane, and Vm as well as ENaCa become almost zero, resulting in a greater cell swelling (the blue line in Fig. 5 C). In contrast, PMCA maintains ENaCa (∼−115 mV in the top panel of Fig. 5 A) more negative than Vm in combination with the increased [Na+]i and membrane depolarization even after 40 min. Thereby, Na+ is extruded via reversed Na+/Ca2+ exchange throughout the period of ouabain treatment. This reverse mode of Na+/Ca2+ exchange, in addition to less activation of Il (Ca), retards Na+ accumulation within the cell and results in less membrane depolarization during Na+/K+ pump block (Fig. 5 A). The increase in cell volume decreased to about half by PMCA, after 120 min inhibition of the Na+/K+ pump (Fig. 5 C). We conclude that PMCA partially substitutes for the Na+/K+ pump in extruding Na+ out of the cells by using the ATP. Our experimental finding that no contracture was observed during Na+/K+ pump inhibition supports this hypothesis.
The above working hypothesis was tested, experimentally, as shown in Fig. 6. When 20 μM KB-R7943, a blocker of Na+/Ca2+ exchange (Elias et al., 2001; Iwamoto, 2004), was applied 60 min after ouabain treatment, the cell started to swell (Fig. 6 A, top). Essentially the same result was obtained by using a more specific and potent blocker of Na+/Ca2+ exchange, SEA0400 (Matsuda et al., 2001; Iwamoto, 2004) (Fig. 6 A, bottom). To evaluate the effects of these Na+/Ca2+ exchange blockers superimposed on the continuous decrease in cell area, the cell area at the end of 60 min treatment with blockers was normalized with respect to the cell area at the beginning of treatment with these drugs (Fig. 6 B, shaded bars). For comparison, the cell area at 120 min normalized by the 60-min area without the drug treatment (open bar) or only with ouabain (filled bar) are shown. As shown in Fig. 6 B, blocking Na+/Ca2+ exchange significantly increased the cell area, indicating operation of the reversed mode of exchanger during ouabain treatment. These experimental results verify the theoretical predictions of the model simulation.
Figure 6.

Experimental validation of the role of Na+/Ca2+ exchanger in the cell volume regulation. On the top of 40 μM ouabain, 20 μM KB-R7943 or 1 μM SEA0400 was applied at 60 min. (A) A representative time course of cell area with the use of KB-R7943 (top) or SEA0400 (bottom). A dotted line was fitted by eye over the initial half of the experimental time. (B) The cell area at 120 min was expressed as percent of that at 60 min. Each column represents mean ± SEM (n = 5–13). *, P < 0.01, significantly different from cells treated with normal Tyrode solution for 120 min (open bar). ¶, P < 0.01, significantly different from cells treated with ouabain for 120 min (filled bar).
Experimental Demonstration of Cell Swelling Induced by Blocking Na+/K+ Pump in the Presence of an Increased Cl− Conductance
So far it has been demonstrated that the cell swelling induced by Na+/K+ pump block is largely dependent on the magnitude of the membrane Cl− conductance and that this Cl− conductance is extremely small in the guinea pig ventricular myocytes. If so, activation of CFTR Cl− channels by β-adrenergic stimulation should induce cell swelling (Tatsumi et al., 2002). To confirm this assumption, we examined the effects of simultaneous application of 40 μM ouabain and 1 μM isoproterenol on guinea pig ventricular myocytes. As shown in Fig. 7, cell swelling occurred after a delay of 52.0 ± 4.9 min (n = 8). The change of cell area was biphasic. First it gradually decreased to 94.2 ± 0.5% of the original cell area and then increased finally to 104.9 ± 3.1% 30 min after starting to swell (Fig. 7 A). In addition, a sudden jump of Vm after the gradual depolarization to the peak potential of −30.8 ± 4.4 mV (n = 5) was observed (Fig. 7 B). It should be noted that there were no significant differences between the time when the rapid swelling started, 52.0 ± 4.9 min, and the time when the sudden jump of Vm occurred, 58.8 ± 2.2 min (P = 0.80). These findings were contrasting to the data in Fig. 3 A, where no change in cell area and no Vm jump were observed.
Figure 7.

Experimental recordings of the cell area and Vm during the Na+/K+ pump block with increased membrane Cl− conductance. The ventricular cells were incubated with 40 μM ouabain as well as 1 μM isoproterenol, and the cell area (A) and Vm (B) were measured in different cells (n = 5–8). Mean ± SEM of time and cell area measured just before a start of obvious cell swelling were superimposed on the representative recording (A). Mean ± SEM of time just after the Vm jump and that of peak Vm were superimposed on the representative recording (B).
Simulation of the Biphasic Volume Change Induced by Ouabain and Isoproterenol and its Experimental Validation
The Kyoto model could reproduce the experimentally observed volume change, a gradual decrease followed by a rapid increase with a sudden jump in Vm (Fig. 8, A and D). The initial decrease in cell volume is due to enhanced Cl− efflux through CFTR, activated (from 0 to 1.3 nS) by applying 1 μM isoproterenol (Fig. 8 B, see red line). It should be noted that Cl− efflux is accompanied by an equal amount of cation efflux (expressed as a sum of fluxes of Na+, K+, and 2 · Ca2+), satisfying macroscopic electroneutrality (Fig. 8 C). The Cl− efflux, however, is truncated by the continuous membrane depolarization caused by the loss of K+ during the Na+/K+ pump block, and thereby the initial volume decrease is saturated (Fig. 8, A, B, and D). The minimum cell volume is 94.4% of the original volume (Fig. 8 A). During the course of membrane depolarization, the window current of L-Type Ca2+ channel, which is also magnified threefold by β-adrenergic stimulation, is gradually activated over the potential range less negative than −40 mV, as indicated in Fig. 8 E, resulting in a rapid accumulation of [Ca2+]i at 42 min (Fig. 8 F). This increase of [Ca2+]i activates Il (Ca) (Fig. 8 G) and triggers a sudden jump in Vm to a level more positive than ECl (Fig. 8 D). Thereby, the rapid and marked cell swelling is induced through an accumulation of Cl−, as well as Na+ and K+ (Fig. 8, A and B).
To experimentally test the working hypothesis of the involvement of ICaL in initiating the rapid swelling, we applied a specific blocker of L-type Ca2+ channels, nifedipine, simultaneously with ouabain and isoproterenol, to the myocytes (Fig. 9). It was obvious that the swelling phase was significantly depressed in the presence of 5 μM nifedipine (cell area at 30 min after the start of swelling was 98.0 ± 0.7% and 104.9 ± 3.1% of original area with and without nifedipine, respectively, n = 8–9, P = 0.037), leaving the first gradual shrinkage phase intact (average cell area of 95.5 ± 0.7% original; Fig. 9). These results strongly supported the notion that [Ca2+]i accumulation via the window current of L-type Ca2+ channel caused the rapid swelling triggered by membrane depolarization, as predicted by the model simulation.
Figure 9.

Experimental validation of the ICaL involvement in the cell swelling as predicted in Fig. 8. 5 μM nifedipine was applied to block ICaL simultaneously with 40 μM ouabain and 1 μM isoproterenol. A representative record is shown.
DISCUSSION
The Extremely Small Membrane Cl− Permeability Contributes To Stabilizing the Volume of Guinea Pig Ventricular Myocytes
The present study conducted experiments as well as simulations using our comprehensive cardiac cell model, the Kyoto model, to analyze mechanisms underlying cell volume regulation in guinea pig ventricular myocytes. The general mechanisms of cell volume regulation (Armstrong 2003; Terashima et al., 2006) were applicable to the cardiac myocytes, in which cell swelling was minimal during Na+/K+ pump block for 1–2 h. It is the extremely small membrane Cl− conductance that is responsible for the negligible cell swelling of guinea pig ventricular myocytes. The rate of [Cl−]i accumulation calculated in the simulation (0.025 mM/min) is ∼100-fold smaller than the experimental rate (2.7 ± 1.6 mM/min) described in the leech Retzius neurons, where significant swelling was observed during ouabain treatment (Dierkes et al., 2006). The notion that membrane Cl− conductance in the guinea pig ventricular myocytes should be small is consistent with the experimental findings that no obvious effect of varying the extra- or intracellular Cl− concentrations was observed on the membrane current–voltage relationship in the absence of β-adrenergic stimulation (guinea pig ventricular myocytes; Bahinski et al., 1989; Matsuoka et al., 1990). We conclude that cardiac cells have a great advantage in protecting themselves against swelling under certain pathological conditions where the activity of the Na+/K+ pump is impaired.
PMCA in Conjunction with Na+/Ca2+ Exchanger Plays an Important Role in Cell Volume Regulation
It has been assumed that PMCA plays only a complementary role for the Na+/Ca2+ exchanger in extruding Ca2+ out of cardiac cells (Bers et al., 1996; Choi and Eisner, 1999; Mackiewicz and Lewartowski, 2006). However, we recently proposed by means of a quantitative model analysis, that the Ca2+ dynamics could be retained within normal bounds by increasing PMCA activity even in the absence of the Na+/Ca2+ exchanger, as achieved experimentally in the NCX knockout mouse (Sarai et al., 2006a). The present study Na+/K+ pump blockade also reveals a pivotal role for PMCA in impeding membrane depolarization, as well as cell swelling by preventing a massive Na+ accumulation in conjunction with the reverse mode of the Na+/Ca2+ exchanger. This function of PMCA in cell volume regulation was established in the present study by using the Na+/Ca2+ exchange blockers KB-R7943 and SEA0400. To our knowledge, this is the first evidence that highlights the coordinated action of PMCA and the Na+/Ca2+ exchanger in the regulation of cardiac cell volume. Interestingly, an essentially similar mechanism has been described in a particular kind of red blood cell that has no detectable Na+/K+ pump but can maintain cell volume. In these cells, Ca2+ is extruded primarily by a PMCA with a much higher activity than in other cells, such as cardiac cells. Then driven by the energy stored in the electrochemical Ca2+ gradient, the Na+/Ca2+ exchanger serves the unusual role of extruding Na+ in exchange for entering Ca2+, to keep the cell volume constant (Fujise et al., 1991; Milanick and Frame, 1991; Blaustein and Lederer, 1999). It may be concluded that cell volume regulation through PMCA is one of the fundamental mechanisms for all kinds of mammalian cells, although it is not prominent under normal physiological conditions. Considering the high [Ca2+]i caused by the Na+/K+ pump block as well as the low [Ca2+]o of 20 μM in our experimental conditions, compared with the values in resting myocytes with a normal [Ca2+]o of 1.8 mM, it may be possible that the transport via PMCA was enhanced under the present experimental conditions.
The involvement of PMCA in volume regulation may also be variable in magnitude between different experimental conditions, depending on stimulation by Ca2+-calmodulin and PKA (Dixon and Haynes, 1989). In the simulation shown in Fig. 5, the magnitude of Ca2+ efflux via PMCA during ouabain treatment increased to as much as 5 μmol/liter cytosol/s. This value is comparable to the experimental estimation of the maximum rate of Ca2+ extrusion via PMCA with calmodulin, 2.43 μmol/liter cytosol/s (Dixon and Haynes, 1989; Bers, 2003), suggesting that the present estimation of the PMCA contribution is relevant.
It is well known that a large variation exists in the extent of Na+ overload during ischemia in the isolated perfused heart (Hartmann and Decking, 1999; Varadarajan et al., 2001; Bak and Ingwall, 2003). This variation may be explained by assuming incomplete block of the Na+/K+ pump in a variety of experimental ischemia. In fact, our simulations as well as our experiments (Fig. 3) indicated that [Na+]i could be very variable with different Na+ conductances, provided that only a few percent of Na+/K+ pump activity remains during pump inhibition. The variation in the magnitude of membrane background Na+ conductance in isolated myocytes (Fig. 3) may also affect the time course of Na+ overload during ischemia. However, we do not know whether the variation in the background Na+ conductance (Matsuda, 1983; Ehara et al., 1988, Kiyosue et al. 1993) occurs in the intact heart or the variation was artifactually caused during enzymatic cell dissociation.
The Positive Feedback Cycle between ICaL and Il (Ca) in Determining Cell Swelling
The Ca2+-activated background conductances have been frequently discussed in relation to arrhythmic membrane excitation (Carmeliet, 1999), such as the delayed membrane depolarization, but rarely analyzed in cell volume regulation. The present experimental and simulation study using the comprehensive cardiac cell model has disclosed the cascade of events leading to the final cell swelling during Na+/K+ pump block: gradual membrane depolarization due to redistribution of K+ across the membrane, opening of the window ICaL, an increase in [Ca2+]i, activation of Il (Ca), accelerated depolarization, reversion from Cl− efflux to influx through Cl− channels, increase of intracellular osmolarity due to the Cl− and accompanied cation influxes, and final net influx of water. Once swelling commences, volume-dependent activation of IVRCC further accelerates the swelling. Note that the activation of ICaL and Il (Ca) occurs via a positive feedback cycle mediated by the increase in [Ca2+]i and membrane depolarization, causing the rapid jump in membrane depolarization (Fig. 7). The application of isoproterenol in the present study simply accelerated the process and enhanced the cell swelling by magnifying the Ca2+ influx via ICaL as well as the membrane Cl− conductance. Thus, harmful effects of β-adrenergic stimulation are evident during metabolic impairment.
Activation of an additional conductance by increased [Ca2+]i has been described in cardiac myocytes, the Ca2+-activated Cl− channel ICl (Ca). Although this channel has been extensively characterized using dog and rabbit ventricular myocytes (Zygmunt and Gibbons, 1991; Collier et al., 1996), there is a little information about ICl (Ca) in the guinea pig ventricular myocyte, and it is still controversial with regard to the magnitude of guinea pig ICl (Ca) (Sipido et al., 1995; Nakajima et al., 2002). To examine the possible involvement of ICl (Ca) in volume regulation, we tentatively implemented ICl (Ca) into the Kyoto model and performed simulations of blocking the Na+/K+ pump. The extent of cell swelling was simply augmented with increasing magnitude of ICl (Ca) during ouabain application. The rapid inactivation gate, as suggested by the very brief outward current at the beginning of a depolarizing pulse (Sipido et al., 1993), though not included in the above model simulation, should be beneficial in avoiding the unfavorable role of ICl (Ca) in volume regulation.
Limitations in the Model Simulations and Experiments
The feedback cycle of formulating a working hypothesis through simulation and validating the hypothesis by conducting new experiments, as exemplified in this study, does facilitate understanding of complex physiological and pathophysiological functions, involving the interactions of numerous molecular mechanisms. In this respect, the mechanisms of cell swelling suggested in the present study should be further tested by detailed experimental investigations.
Although our basic model specified for volume regulation disclosed the principal mechanisms (Terashima et al., 2006), a deeper insight into the complicated interactions among multiple molecular mechanisms requires a comprehensive cell model. The Kyoto model has already included most of the ion channels and the major ion transporters on the cardiac cell membrane, mechanisms for the Ca2+ dynamics performed by SR, the intracellular Ca2+ buffers, such as calmodulin and troponin, and the model of oxidative phosphorylation in mitochondria (Korzeniewsky and Zoladz, 2001). The parameters have been adjusted to simulate different kinds of experimental findings, such as membrane excitability, frequency-dependent variation of intracellular ion concentrations, excitation–contraction coupling, and volume regulation, as demonstrated in previous publications (Matsuoka et al., 2003, 2004; Terashima et al., 2006). Thereby, we could derive a conclusion that Ca2+ dynamics had quite an important role in cardiac cell volume regulation indirectly, through affecting Na+ homeostasis. However, obvious limitations in the model analysis of cell swelling are imposed by the lack of calculating the intermediate metabolite concentrations, as well as pH homeostasis, which is composed of acid and base transporters (cotransport or antiport with Na+ or Cl−) and buffering reactions. As shown in Fig. 3 B, a more or less cell swelling (1.3% increase at 90 min) is inevitable in the Kyoto model when the Na+/K+ pump is blocked, whereas no obvious volume increase was detected experimentally (Fig. 3 A). This discrepancy also proposes working hypotheses that additional mechanisms, such as changes in pH homeostasis, metabolism, or unknown osmolyte transporters, which are beyond the scope of the present study, might be involved in keeping the cell volume constant.
We failed to observe a contracture at the onset of rapid swelling during the application of ouabain and isoproterenol (Fig. 7). This finding is inconsistent with the prediction of a [Ca2+]i accumulation temporarily as large as 4 μM (Fig. 8), which should have caused a contracture. We might have missed the contracture because of the 30-s duration between capturing cell images. Alternatively, the [Ca2+]i-dependent activation of PMCA via calmodulin kinase II or activation via PKA, which were not implemented in the Kyoto model, might have prevented [Ca2+]i to rise and cause a contracture.
It should finally be commented that limitations in interpreting the experimental findings also arise by the method of measuring the cell volume, as well as Vm. The measurement of cell area as an index of the cell volume might underestimate the real cell volume change, because it is likely that the cell thickness continuously changed with the experimental period (Sasaki et al., 1999). The apparent hyperpolarization detected by the dye (Fig. 7) on the application of isoproterenol is also different from the membrane depolarization measured by the patch clamp method (Sasaki et al., 1999). However, it is obvious in the top panel of Fig. 3 A that the measured Vm tended to decrease continuously, even without any drugs (∼15 mV decreases during the 90-min measurement). If this trend was subtracted as a measurement error, the sudden jump of Vm shown in Fig. 7 B should have started at −40 mV, which was near the threshold Vm for ICaL window current in the Kyoto model.
Supplemental Material
Acknowledgments
We thank Professor T. Powell and Professor D. Hilgemann for their critical reading of the manuscript and fruitful suggestions, and Professor C.H. Leem for determining the membrane hydraulic conductivity.
This study was supported by the Leading Project for Biosimulation and a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: CFTR, cystic fibrosis transmembrane conductance regulator; ECl, equilibrium potential for Cl− (mV); ENaCa, reversal potential for Na+/Ca2+ exchanger, 3ENa-2ECa (mV); IbNSC, background nonselective cation current (pA/pF); ICaL, L-type Ca2+ current (pA/pF); ICFTR, CFTR Cl− channel current (pA/pF); IClb, background Cl− current (pA/pF); ICl (Ca), Ca2+-activated Cl− channel current (pA/pF); Il (Ca), Ca2+-activated background cation current (pA/pF); INaCa, Na+/Ca2+ exchange current (pA/pF); IPMCA, PMCA current (pA/pF); IVRCC, VRCC current (pA/pF); Jwater, water flux across the cell membrane (μm3/ms); LA, impermeable large anion (mM); MNKCC1, an amplitude factor for NKCC1 (amol): amol = 10−18 mol; NKCC1, Na+/K+/2 Cl− cotransporter 1; PMCA, plasma membrane Ca2+-ATPase; Px, convert factor (pA/pF/mM) for CFX; RyR, ryanodine receptor; SERCA, SR Ca2+ pump; Vm, membrane potential (mV); VRCC, volume-regulated Cl− channel; Vt, total cell volume (μm3); [X]i, intracellular concentration of substance X (mM); [X]o, extracellular concentration of substance X (mM).
References
- Akar, J.G., T.H. Everett, R. Ho, J.C. Craft, D.E. Haines, A.P. Somlyo, and A.V. Somlyo. 2003. Intracellular chloride accumulation and subcellular elemental distribution during atrial fibrillation. Circulation. 107:1810–1815. [DOI] [PubMed] [Google Scholar]
- Armstrong, C.M. 2003. The Na/K pump, Cl ion, and osmotic stabilization of cells. Proc. Natl. Acad. Sci. USA. 100:6257–6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahinski, A., A.C. Nairn, P. Greengard, and D.C. Gadsby. 1989. Chloride conductance regulated by cyclic AMP-dependent protein kinase in cardiac myocytes. Nature. 340:718–721. [DOI] [PubMed] [Google Scholar]
- Bak, M.I., and J.S. Ingwall. 2003. Contribution of Na+/H+ exchange to Na+ overload in the ischemic hypertrophied hyperthyroid rat heart. Cardiovasc. Res. 57:1004–1014. [DOI] [PubMed] [Google Scholar]
- Baker, P.F., M.P. Blaustein, R.D. Keynes, J. Manil, T.I. Shaw, and R.A. Steinhardt. 1969. The ouabain-sensitive fluxes of sodium and potassium in squid giant axons. J. Physiol. 200:459–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balshaw, D.M., L.A. Millette, and E.T. Wallick. 2001. Sodium pump function. In Cell Physiology Sourcebook, A Molecular Approach. Third edition. N. Sperelakis, editor. Elsevier Science Publishing Co. Inc., New York. 261–269.
- Bedlack, R.S., M-d. Wei, and L.M. Loew. 1992. Localized membrane depolarizations and localized calcium influx during electric field-guided neurite growth. Neuron. 9:393–403. [DOI] [PubMed] [Google Scholar]
- Benjamin, B.A., and E.A. Johnson. 1997. A quantitative description of the Na-K-2Cl cotransporter and its conformity to experimental data. Am. J. Physiol. 273:F473–F482. [DOI] [PubMed] [Google Scholar]
- Bers, D.M., J.W.M. Bassani, and R.A. Bassani. 1996. Na-Ca exchange and Ca fluxes during contraction and relaxation in mammalian ventricular muscle. Ann. NY Acad. Sci. 779:430–442. [DOI] [PubMed] [Google Scholar]
- Bers, D.M. 2003. Na/Ca exchange and the sarcolemmal Ca-pump. In Excitation-Contraction Coupling and Cardiac Contractile Force. Second edition. Kluwer Academic Publishers, Norwell, MA. 133–160.
- Beuckelmann, D.J., and W.G. Wier. 1988. Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells. J. Physiol. 405:233–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein, M.P., and W.J. Lederer. 1999. Sodium/calcium exchange: its physiological implications. Physiol. Rev. 79:763–854. [DOI] [PubMed] [Google Scholar]
- Boyle, P.J., and E.J. Conway. 1941. Potassium accumulation in muscle and associated changes. J. Physiol. 100:1–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet, E. 1999. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol. Rev. 79:917–1017. [DOI] [PubMed] [Google Scholar]
- Choi, H.S., and D.A. Eisner. 1999. The role of sarcolemmal Ca2+-ATPase in the regulation of resting calcium concentration in rat ventricular myocytes. J. Physiol. 515:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier, M.L., P.C. Levesque, J.L. Kenyon, and J.R. Hume. 1996. Unitary Cl− channels activated by cytoplasmic Ca2+ in canine ventricular myocytes. Circ. Res. 78:936–944. [DOI] [PubMed] [Google Scholar]
- Delpech, N., H. Soustre, and D. Potreau. 1995. Antagonism of β-adrenergic stimulation of L-type Ca2+ current by endothelin in guinea-pig atrial cells. Eur. J. Pharmacol. 285:217–220. [DOI] [PubMed] [Google Scholar]
- Dierkes, P.W., H.J. Wüsten, G. Klees, A. Müller, and P. Hochstrate. 2006. Ionic mechanism of ouabain-induced swelling of leech Retzius neurons. Pflugers Arch. 452:25–35. [DOI] [PubMed] [Google Scholar]
- Dixon, D.A., and D.H. Haynes. 1989. Kinetic characterization of the Ca2+-pumping ATPase of cardiac sarcolemma in four states of activation. J. Biol. Chem. 264:13612–13622. [PubMed] [Google Scholar]
- Drewnowska, K., and C.M. Baumgarten. 1991. Regulation of cellular volume in rabbit ventricular myocytes: bumetanide, chlorothiazide, and ouabain. Am. J. Physiol. 260:C122–C131. [DOI] [PubMed] [Google Scholar]
- Ehara, T., A. Noma, and K. Ono. 1988. Calcium-activated non-selective cation channel in ventricular cells isolated from adult guinea-pig hearts. J. Physiol. 403:117–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias, C.L., A. Lukas, S. Shurraw, J. Scott, A. Omelchenko, G.J. Gross, M. Hnatowich, and L.V. Hryshko. 2001. Inhibition of Na+/Ca2+ exchanger by KB-R7943: transport mode selectivity and antiarrhythmic consequences. Am. J. Physiol. Heart Circ. Physiol. 281:H1334–H1345. [DOI] [PubMed] [Google Scholar]
- Findlay, I. 2002. β-Adrenergic and muscarinic agonists modulate inactivation of L-type Ca2+ channel currents in guinea-pig ventricular myocytes. J. Physiol. 545:375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujise, H., I. Yamada, M. Masuda, Y. Miyazawa, E. Ogawa, and R. Takahashi. 1991. Several cation transporters and volume regulation in high-K dog red blood cells. Am. J. Physiol. 260:C589–C597. [DOI] [PubMed] [Google Scholar]
- Fujioka, Y., K. Hiroe, and S. Matsuoka. 2000. Regulation kinetics of Na+-Ca2+ exchange current in guinea-pig ventricular myocytes. J. Physiol. 529:611–623. [PubMed] [Google Scholar]
- Gannier, F., E. White, D. Garnier, and J.Y. Le Guennec. 1996. A possible mechanism for large stretch-induced increase in [Ca2+]i in isolated guinea-pig ventricular myocytes. Cardiovasc. Res. 32:158–167. [PubMed] [Google Scholar]
- Gao, J., R.T. Mathias, I.S. Cohen, and G.J. Baldo. 1995. Two functionally different Na/K pumps in cardiac ventricular myocytes. J. Gen. Physiol. 106:995–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, J., R.S. Wymore, Y. Wang, G.R. Gaudette, I.B. Krukenkamp, I.S. Cohen, and R.T. Mathias. 2002. Isoform-specific stimulation of cardiac Na/K pumps by nanomolar concentrations of glycosides. J. Gen. Physiol. 119:297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann, M., and U.K.M. Decking. 1999. Blocking Na+-H+ exchange by cariporide reduces Na+-overload in ischemia and is cardioprotective. J. Mol. Cell. Cardiol. 31:1985–1995. [DOI] [PubMed] [Google Scholar]
- Hilgemann, D.W., A. Collins, and S. Matsuoka. 1992. a. Steady-state and dynamic properties of cardiac sodium-calcium exchange. Secondary modulation by cytoplasmic calcium and ATP. J. Gen. Physiol. 100:933–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann, D.W., S. Matsuoka, G.A. Nagel, and A. Collins. 1992. b. Steady-state and dynamic properties of cardiac sodium-calcium exchange. Sodium-dependent inactivation. J. Gen. Physiol. 100:905–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto, T. 2004. Forefront of Na+/Ca2+ exchanger studies: molecular pharmacology of Na+/Ca2+ exchange inhibitors. J. Pharmacol. Sci. 96:27–32. [DOI] [PubMed] [Google Scholar]
- James, A.F., T. Tominaga, Y. Okada, and M. Tominaga. 1996. Distribution of cAMP-activated chloride current and CFTR mRNA in the guinea pig heart. Circ. Res. 79:201–207. [DOI] [PubMed] [Google Scholar]
- Kiyosue, T., A.J. Spindler, S.J. Noble, and D. Noble. 1993. Background inward current in ventricular and atrial cells of the guinea-pig. Proc. Biol. Sci. 252:65–74. [DOI] [PubMed] [Google Scholar]
- Korzeniewski, B., and J.A. Zoladz. 2001. A model of oxidative phosphorylation in mammalian skeletal muscle. Biophys. Chem. 92:17–34. [DOI] [PubMed] [Google Scholar]
- Kuratomi, S., S. Matsuoka, N. Sarai, T. Powell, and A. Noma. 2003. Involvement of Ca2+ buffering and Na+/Ca2+ exchange in the positive staircase of contraction in guinea-pig ventricular myocytes. Pflugers Arch. 446:347–355. [DOI] [PubMed] [Google Scholar]
- Lin, X., H. Jo, Y. Sakakibara, K. Tambara, B. Kim, M. Komeda, and S. Matsuoka. 2006. β-Adrenergic stimulation does not activate Na+/Ca2+ exchange current in guinea pig, mouse, and rat ventricular myocytes. Am. J. Physiol. Cell Physiol. 290:C601–C608. [DOI] [PubMed] [Google Scholar]
- Luo, C.H., and Y. Rudy. 1994. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ. Res. 74:1071–1096. [DOI] [PubMed] [Google Scholar]
- Mackiewicz, U., and B. Lewartowski. 2006. Temperature dependent contribution of Ca2+ transporters to relaxation in cardiac myocytes: important role of sarcolemmal Ca2+-ATPase. J. Physiol. Pharmacol. 57:3–15. [PubMed] [Google Scholar]
- Matsuda, H. 1983. Effects of intracellular calcium injection on steady state membrane currents in isolated single ventricular cells. Pflugers Arch. 397:81–83. [DOI] [PubMed] [Google Scholar]
- Matsuda, T., N. Arakawa, K. Takuma, Y. Kishida, Y. Kawasaki, M. Sakaue, K. Takahashi, T. Takahashi, T. Suzuki, T. Ota, et al. 2001. SEA0400, a novel and selective inhibitor of the Na+-Ca2+ exchanger, attenuates reperfusion injury in the in vitro and in vivo cerebral ischemic models. J. Pharmacol. Exp. Ther. 298:249–256. [PubMed] [Google Scholar]
- Matsuoka, S., T. Ehara, and A. Noma. 1990. Chloride-sensitive nature of the adrenaline-induced current in guinea-pig cardiac myoyctes. J. Physiol. 425:579–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka, S., N. Sarai, S. Kuratomi, K. Ono, and A. Noma. 2003. Role of individual ionic current systems in ventricular cells hypothesized by a model study. Jpn. J. Physiol. 53:105–123. [DOI] [PubMed] [Google Scholar]
- Matsuoka, S., N. Sarai, H. Jo, and A. Noma. 2004. Simulation of ATP metabolism in cardiac excitation-contraction coupling. Prog. Biophys. Mol. Biol. 85:279–299. [DOI] [PubMed] [Google Scholar]
- Milanick, M.A., and M.D. Frame. 1991. Kinetic models of Na-Ca exchange in ferret red blood cells. Interaction of intracellular Na, extracellular Ca, Cd, and Mn. Ann. N. Y. Acad. Sci. 639:604–615. [DOI] [PubMed] [Google Scholar]
- Nakajima, I., H. Watanabe, K. Iino, T. Saito, and M. Miura. 2002. Ca2+ overload evokes a transient outward current in guinea-pig ventricular myocytes. Circ. J. 66:87–92. [DOI] [PubMed] [Google Scholar]
- Nakao, M., and D.C. Gadsby. 1989. [Na] and [K] dependence of the Na/K pump current-voltage relationship in guinea pig ventricular myocytes. J. Gen. Physiol. 94:539–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble, D., and Y. Rudy. 2001. Models of cardiac ventricular action potentials: iterative interaction between experiment and simulation. Philos. Trans. R. Soc. Lond. A. 359:1127–1142. [Google Scholar]
- Ogura, T., H. Matsuda, S. Imanishi, and T. Shibamoto. 2002. Sarcolemmal hydraulic conductivity of guinea-pig and rat ventricular myocytes. Cardiovasc. Res. 54:590–600. [DOI] [PubMed] [Google Scholar]
- Pine, M.B., D. Kahne, B. Jaski, C.S. Apstein, K. Thorp, and W.H. Abelmann. 1980. Sodium permeability and myocardial resistance to cell swelling during metabolic blockade. Am. J. Physiol. 239:H31–H39. [DOI] [PubMed] [Google Scholar]
- Powell, T., D.A. Terrar, and V.W. Twist. 1980. Electrical properties of individual cells isolated from adult rat ventricular myocardium. J. Physiol. 302:131–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarai, N., T. Kobayashi, S. Matsuoka, and A. Noma. 2006. a. A simulation study to rescue the Na+/Ca2+ exchanger knockout mice. J. Physiol. Sci. 56:211–217. [DOI] [PubMed] [Google Scholar]
- Sarai, N., S. Matsuoka, and A. Noma. 2006. b. simBio: a Java package for the development of detailed cell models. Prog. Biophys. Mol. Biol. 90:360–377. [DOI] [PubMed] [Google Scholar]
- Sasaki, N., M. Takano, T. Mitsuiye, and A. Noma. 1999. Changes in cell volume induced by ion channel flux in guinea-pig cardiac myocytes. Clin. Exp. Pharmacol. Physiol. 26:698–706. [DOI] [PubMed] [Google Scholar]
- Shuba, L.M., T. Ogura, and T.F. McDonald. 1996. Kinetic evidence distinguishing volume-sensitive chloride current from other types in guinea-pig ventricular myocytes. J. Physiol. 491:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido, K.R., G. Callewaert, and E. Carmeliet. 1993. [Ca2+]i transients and [Ca2+]i-dependent chloride current in single Purkinje cells from rabbit heart. J. Physiol. 468:641–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido, K.R., G. Callewaert, F. Porciatti, J. Vereecke, and E. Carmeliet. 1995. [Ca2+]i-dependent membrane currents in guinea-pig ventricular cells in the absence of Na/Ca exchange. Pflugers Arch. 430:871–878. [DOI] [PubMed] [Google Scholar]
- Strieter, J., J.L. Stephenson, L.G. Palmer, and A.M. Weinstein. 1990. Volume-activated chloride permeability can mediate cell volume regulation in a mathematical model of a tight epithelium. J. Gen. Physiol. 96:319–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suleymanian, M.A., and C.M. Baumgarten. 1996. Osmotic gradient-induced water permeation across the sarcolemma of rabbit ventricular myocytes. J. Gen. Physiol. 107:503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tareen, F.M., A. Yoshida, and K. Ono. 1992. Modulation of β-adrenergic responses of chloride and calcium currents by external cations in guinea-pig ventricular cells. J. Physiol. 457:211–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumi, S., S. Matsuoka, and A. Noma. 2002. Role of Na+ pump in volume regulation of cardiac myocytes. Jpn. J. Physiol. 52:S71. [Google Scholar]
- Terashima, K., A. Takeuchi, N. Sarai, S. Matsuoka, E.B. Shim, C.H. Leem, and A. Noma. 2006. Modelling Cl− homeostasis and volume regulation of the cardiac cell. Philos. Transact. A Math. Phys. Eng. Sci. 364:1245–1265. [DOI] [PubMed] [Google Scholar]
- Varadarajan, S.G., J. An, E. Novalija, S.C. Smart, and D.F. Stowe. 2001. Changes in [Na+]i, compartmental [Ca2+], and NADH with dysfunction after global ischemia in intact hearts. Am. J. Physiol. Heart Circ. Physiol. 280:H280–H293. [DOI] [PubMed] [Google Scholar]
- Vaughan-Jones, R.D. 1982. Chloride activity and its control in skeletal and cardiac muscle. Philos. Trans. R. Soc. Lond. B Biol. Sci. 299:537–548. [DOI] [PubMed] [Google Scholar]
- Walsh, K.B., and J. Zhang. 2005. Regulation of cardiac volume-sensitive chloride channel by focal adhesion kinase and Src kinase. Am. J. Physiol. Heart Circ. Physiol. 289:H2566–H2574. [DOI] [PubMed] [Google Scholar]
- Wang, D.Y., S.W. Chae, Q.Y. Gong, and C.O. Lee. 1988. Role of αi Na in positive force-frequency staircase in guinea-pig papillary muscle. Am. J. Physiol. 255:C798–C807. [DOI] [PubMed] [Google Scholar]
- Wang, Z., T. Mitsuiye, S.A. Rees, and A. Noma. 1997. Regulatory volume decrease of cardiac myocytes induced by β-adrenergic activation of the Cl− channel in guinea pig. J. Gen. Physiol. 110:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, A.R., and S.A. Rees. 1998. Cardiac cell volume: crystal clear or murky waters? A comparison with other cell types. Pharmacol. Ther. 80:89–121. [DOI] [PubMed] [Google Scholar]
- Yamamoto, S., T. Ehara, and T. Shioya. 2001. Changes in cell volume induced by activation of the cyclic AMP-dependent chloride channel in guinea-pig cardiac myocytes. Jpn. J. Physiol. 51:31–41. [DOI] [PubMed] [Google Scholar]
- Yamamoto, S., K. Ishihara, T. Ehara, and T. Shioya. 2004. Cell-volume regulation by swelling-activated chloride current in guinea-pig ventricular myocytes. Jpn. J. Physiol. 54:31–38. [DOI] [PubMed] [Google Scholar]
- Zygmunt, A.C., and W.R. Gibbons. 1991. Calcium-activated chloride current in rabbit ventricular myocytes. Circ. Res. 68:424–437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}