

Abstract
The closed KcsA channel structure revealed a crossing of the cytosolic ends of the transmembrane helices blocking the permeation pathway. It is generally agreed that during channel opening this helical bundle crossing has to widen in order to enable access to the inner cavity. Here, we address the question of whether the opening of the inner gate is sufficient for ion conduction, or if a second gate, located elsewhere, may interrupt the ion flow. We used fluorescence lifetime measurements on KcsA channels labeled with tetramethylrhodamine at residues in the C-terminal end of TM2 to report on the opening of the lower pore region.
We found two populations of channels with different fluorescence lifetimes, whose relative distribution agrees with the open probability of the channel. The absolute fraction of channels found with an open bundle crossing is too high to explain the low open probability of the KcsA-WT channel. We found the same distribution as in the WT channel between open and closed bundle crossing for two KcsA mutants, A73E and E71A, which significantly increase open probability at low pH. These two results strongly suggest that a second gate in the ion permeation pathway exists. The location of the mutations A73E and E71A suggests that the second gate may be the selectivity filter, which resides in an inactivated state under steady-state conditions. Since the long closed times observed in KcsA-WT are not present in KcsA-A73E or -E71A, we propose that KcsA-WT remains predominantly in a state with an open bundle crossing but closed (inactivated) second gate, while the mutations A73E and E71A sharply decrease the tendency to enter in the inactivated state, and as a consequence, the second gate is predominantly open at steady state. The ability to monitor the opening of the bundle crossing optically enables the direct recording of the movement of the pore helices while the channel is functioning.
INTRODUCTION
In the crystal structure of KcsA (Doyle et al., 1998), the TM2 helices cross over and form a tight bundle toward the intracellular face of the channel. Proton-dependent gating (Cuello et al., 1998; Heginbotham et al., 1999; Meuser et al., 1999) takes place through a significant rearrangement of the TM2 moving away from the fourfold axis of symmetry, as demonstrated from site-directed spin labeling in combination with electron paramagnetic resonance (EPR) spectroscopy (Perozo et al., 1998, 1999; Liu et al., 2001; Sompornpisut et al., 2001) and site-directed mass tagging (Kelly and Gross, 2003). This movement seems to be consistent with the open state structure of the calcium-dependent potassium channel from Methanobacterium thermoautrophicum, MthK, showing a wide open intracellular pore (16 Å) (Jiang et al., 2002b) and the archaea voltage-dependent K+ channel KvAP (Jiang et al., 2003a,b), although distance determinations suggest that helix bundle opening in KcsA might be significantly smaller (Liu et al. 2001). These structures clearly show that, due to a kink in TM2, the C-terminal end of the helices is pointing outward away from the symmetry axis of the channel. By extension, it is generally agreed that in Kv channels the equivalent TM2 helix bundle, formed by the S6 segment, also moves through an equivalent mechanism (Liu et al., 1997; Del Camino et al., 2000; Del Camino and Yellen, 2001).
The opening of the bundle crossing, as a prerequisite for ion conduction, is supported by the interaction of domains modulating gating with the lower pore region. It is suggested, for instance, that the RCK domains of MthK channels, which form a ring below the intracellular pore entrance, pull the pore helices apart upon Ca2+ binding (Jiang et al., 2002a). Similarly, the S4–S5 linker, which is the covalent connection between pore region and voltage sensor of voltage-dependent channels, has been shown to be essential for voltage sensing (McCormack et al., 1991; Schoppa and Sigworth, 1998; Sanguinetti and Xu, 1999; Chen et al., 2001; Decher et al., 2004). The S4–S5 linker is bound to the N terminus of S5 (TM1 in KcsA) and interacts directly with the C terminus of S6, as shown in chimeric channels (Lu et al., 2002) and as suggested based on the crystal structure (Long et al., 2005). In hyperpolarization-activated cyclic nucleotide-gated channels, Decher et al. (2004) have shown that the cyclic nucleotide binding domain (C terminal to S6, TM2 in KcsA) interacts with the S4–S5 linker, again supporting a physical interaction of the ligand binding sites and voltage sensors with the intracellular endings of the pore helices.
The above results demonstrate the movement of the helix bundle and corroborate that this opening is the actual modulated entity. Nevertheless, in spite of demonstrated conformational rearrangements in TM2, the KcsA channel shows an extremely low open probability, even at low pH (Cuello et al., 1998; Heginbotham et al., 1999), which is not reflected in spectroscopic ensemble measurements looking at the bundle crossing opening (Perozo et al., 1999; Liu et al., 2001). Other previous experimental results cannot be explained by a single gate located near the bundle crossing, indicating that the ion conduction pathway may be interrupted by a second gate located in or close below the selectivity filter (below meaning closer to the cytosol; this convention will be used throughout the paper). For instance, accessibility studies in different potassium channels have shown that the nonconducting state of the channel did not fully coincide with the closing of the bundle crossing, but that some compounds could still enter the inner cavity dependent on their size (Flynn and Zagotta, 2001; Bruening-Wright et al., 2002; Phillips and Nichols, 2003; Proks et al., 2003). Interestingly, the same compound, MTSEA, showed state-dependent modulation in KIR 6.2 (Phillips and Nichols, 2003), but not in KIR 2.1 (Xiao et al., 2003), suggesting that the opening of the bundle crossing varies between channels. This is in accordance with the findings of Webster et al. (2004), in which the lower pore radius in the closed state in Shaker was smaller than in its prokaryotic relatives KcsA and MthK.
The location of the putative second gate is suggested to be at the selectivity filter (Sun et al., 1996; Liu et al., 2001; Zhou et al., 2001; Bruening-Wright et al., 2002; Claydon et al., 2003). One of the first indications that the selectivity filter participates in gating came from the findings that the type of permeant ion (Swenson and Armstrong, 1981; Spruce et al., 1989; Demo and Yellen, 1992; LeMasurier et al., 2001) and mutations in the filter (Lu et al., 2001) and the p-loop (Sun et al., 1996; Proks et al., 2001) influence the gating behavior of the channels. The strongest indications for a gating mechanism of the selectivity filter are the occurrence of subconductance levels in the early openings and during deactivation in drk1 (Chapman et al., 1997) and in the T442S mutant Shaker channels (Zheng and Sigworth, 1998). Finally, C-type inactivation, which is thought to be a nonconductive state of the selectivity filter, is strongly dependent on the occupancy of the selectivity filter and is thought to involve small structural changes (Lopez-Barneo et al., 1993; Starkus et al., 1997). Direct measurements of the flexibility of the selectivity filter were done using EPR measurements (Perozo et al., 1999) and x-ray crystallography (Zhou et al., 2001). Zhou et al. (2001) even found a closed selectivity filter in the crystal structure at low potassium concentrations and suggested that this conformation explains channel flickering or the permeant ion effect on gating. Recent results by Cordero-Morales et al. (2006) showed that a pH decrease in KcsA causes a transient current, and they suggested, based on EPR spectroscopy, that is caused by a second gate, explaining the low open probability in the steady state.
In this work, we set out to directly measure the opening of the bundle crossing using fluorescence spectroscopy and correlate it with the conducting state of the channel. We constructed KcsA mutants with cysteines in the bundle crossing and reacted them with fluorescent probes, which allowed us to determine the state of the lower pore region as a function of the open probability using fluorescence lifetime measurements. Fluorescence lifetime measurements have the advantage over previous spectroscopic methods that it allows us to distinguish between different populations of the investigated sample, since each population gives origin to different time constants. In this way, it is possible to estimate what fraction of the total population dwells in a specific state.
We have shown previously that the fluorescence intensity of the dye tetramethyl rhodamine (TMR) is modulated by the environment (Cha and Bezanilla, 1997; Blunck et al., 2004). With TMR attached to specific sites of ion channels expressed in cells or Xenopus oocytes, the dynamic changes of the fluorescence intensity may be followed directly. As KcsA is difficult to express in cells or oocytes, the fluorescence must be observed in a reconstituted system and we chose to measure KcsA channels in a fluorescence spectrometer. Here, changing the pH is accompanied with errors in the sample concentration either due to dilution of small samples or due to independent samples of small volumes. To circumvent these problems, one may determine the lifetime of the fluorophores linked to the protein that is directly dependent on their quantum yield. If a fluorophore is excited, it transits into the excited state (Fig. 1 A). Each fluorophore has a characteristic dwell time in the excited state before it relaxes back into the ground state. This so-called lifetime τ is the reciprocal of the sum of all transition rates back to the ground state according to a standard kinetic model (Lakowicz, 1999). The quantum yield (QY), and therefore also the fluorescence intensity, is the fraction of the fluorescent (f) out of all transitions back to the ground state and, hence, directly proportional to τ (see Appendix). If the fluorophore comes into proximity of a quenching molecule, the lifetime and the QY (fluorescence intensity) decrease. But since the lifetime τ is a property of the fluorophore, it is not dependent on the actual concentration in the solution. In this paper, we use frequency domain techniques to determine the lifetimes of fluorophores attached at the bundle crossing to the KcsA channel under conditions that render the TM2 bundle open or closed.
Figure 1.

Principle of lifetime measurements in the frequency domain. (A) If a fluorophore absorbs a photon (hν) it is excited to the S1 state. Each fluorophore has a characteristic dwell-time τ in the S1 state before it relaxes back to the ground state S0. This can occur by emitting a photon (red) by nonradiative deexcitation (knr) or by quenching (q). (B) If the amplitude of the excitation light (blue) is sinusoidally modulated, the emission will follow this time course with a time delay (due to the dwell time in S1), resulting in the phase shift ΔΦ. Due to a low-pass filtering effect, the amplitude of the emission is reduced (ΔM). (C) Labeling efficiency: comparison of labeling of single-cysteine mutant and WT channel. The labeling ratio of mutant to WT is ∼13.
MATERIALS AND METHODS
Chemicals and Solutions
All chemicals were purchased from VWR or Fisher Scientific if not stated otherwise. Tetramethylrhodamine-5-maleimide (TMRM; Molecular Probes) was dissolved in DMSO to a final concentration of 50 mM directly before addition to the protein. Synthetic lipids were purchased from Avanti Polar Lipids, Inc. and stored, dissolved in chloroform at a concentration of 25 mg/ml, at −80°C. Prior to use, the chloroform was evaporated under a steady N2 flow and lipids are redissolved in the respective buffer or decane for bilayer experiments. Lipids used were POPE, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine; POPG, 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]; POPS, 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-l-serine]. n-Dodecyl-β-d-maltopyranoside (DDM) was purchased by Anatrace and stored in a 20% stock solution in H2O at −20°C.
Protein Purification, Labeling, and Reconstitution
Cysteine mutants of KcsA channels were engineered and the proteins were isolated and purified as described previously (Cortes and Perozo, 1997; Perozo et al., 1998). All subsequent steps in the protein preparation were performed on ice under light not exciting the fluorophore (λ ≥ 610 nm). The protein in detergent was incubated with a 10-fold excess of TMRM under an N2 atmosphere on ice for 12–16 h. Excess dye was removed by gel filtration using a Sephadex G50 column (PD10; Amersham Biosciences). 0.5-ml fractions were collected and protein concentration determined using Coomassie Plus protein assay (Pierce Biotechnology).
Unilammelar lipid vesicles (POPE:POPG = 3:1) were formed by sonication (80 kHz) in PBS (for Biobead saturation) or PBS + 0.05% DDM. The proteins were mixed with the lipid and detergent emulsion in a 20:1 mass-of-lipids:mass-of-proteins ratio and sonicated shortly. Biobeads SM-2 (Bio-Rad Laboratories) were washed and saturated with lipids without detergent for 20 min before use. The protein/lipid/detergent mixture was incubated three successive rounds (2 h at RT, 2 h at RT, 12 h at 4°C) with biobeads in a ratio 1:100 mass-of-detergent:mass-of-biobeads to remove all detergent. The samples were either used directly or aliquoted and stored under N2 atmosphere at −80°C.
Frequency Domain Lifetime Measurements
The lifetimes of fluorophores may be measured either in the time or in the frequency domain. In the time domain, one determines the fluorescence decay after an exciting pulse and determines the time constant of the exponential decay of emission. In the frequency domain, one uses amplitude-modulated light of different frequencies ω. Due to the time delay between absorbing and emitting a photon (the lifetime τ) there is a time delay between the excitation and emission light. In amplitude-modulated light, this delay appears as a phase shift and a decrease in modulation amplitude (Fig. 1 B, and see below).
With increasing modulation frequency ω, the phase shift increases, while the modulation amplitude of the emission decreases (Fig. 1 B, and see Eqs. 1 and 2). The reason for the phase shift is relatively plausible: while the lifetime τ remains constant for a given environment, the time it takes for one wave to complete (2π/ω) decreases with increasing ω. Therefore, τ increases in relation to (2π/ω). Since the fluorophore acts as a lowpass filter with the lifetimes as time constants, the modulation amplitude decreases with increasing modulation frequency (see also Appendix).
Fluorescence was recorded with a Fluorolog 3 spectrofluorimeter (Spex Industries), equipped with a double-grating monochromator, Glan Thompson polarizers, and a cuvette holder thermostated at 25°C. 20 μl of sample was diluted in 450 μl of the appropriate buffer and sonicated to equilibrate internal and external solution. Fluorescence was excited at 513 nm and emission filtered using a 565 EFXF101 long pass filter (Omega Optical Inc.). Excitation was modulated at 10 different frequencies (10–200 MHz, in logarithmic intervals) and phase shift and modulation amplitude at each frequency determined. As a standard, we used Ludox (0.0 ns; Sigma-Aldrich). The data were fitted to one- or, if necessary, two-component models using Datamax32 (Horiba Jobin Yvon Inc.) or MicroCal Origin (Originlab Corp.) analysis software. Modulation amplitude (M) and phase shift (Φ) were simultaneously fitted to identical parameters using (Lakowicz, 1999)
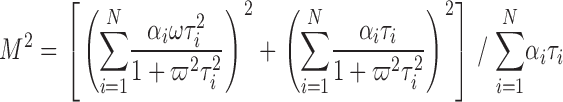 |
(1) |
and
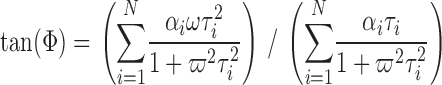 |
(2) |
with
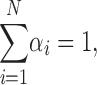 |
αi = relative amplitudes of lifetimes, τi = lifetimes, N = number of lifetimes, and ω = modulation frequency.
Solutions were 10 mM K3-citrate + 3 mM K2HPO4 and were adjusted to the correct pH with NaOH or H3PO4, respectively. All bilayer and lifetime measurements were done in the same solutions.
Rb+ Flux Measurements
Purified KcsA was reconstituted into asolectin liposomes and 86Rb+ fluxes were performed using the protocol previously described for KcsA (Cuello et al., 1998). Vesicles containing 86Rb+ were measured using a scintillation counter.
Bilayer Experiments
Bilayers were formed in a 150-μm hole in a horizontal bilayer configuration, similar to that described in Pantoja et al. (2001). The holes were pretreated with lipids to facilitate bilayer formation. A mass:mass = 1:3 mixture of POPS:POPE was dissolved in n-decane to a final concentration of 25 mg/ml. To form the bilayer, a clean glass rod is dipped into the lipid–decane mixture and brushed over the hole in the chip.
Voltage was applied and current measured using an Axon 200B patch-clamp amplifier (Axon Instruments). Bilayer formation was monitored optically and by capacitance measurement. The samples containing the reconstituted channel proteins were sonicated for ∼10 s before application to the bilayer. They were applied by dipping a glass rod into the vesicle preparation and brushing over a formed bilayer, or by pipetting small amounts directly onto the bilayer. Fusion was assisted by establishing an osmotic gradient. Current recording and analysis was done using in house written software.
Liposome Patch-Clamp
Patch-clamp measurements on liposomes were done as described previously (Delcour et al., 1989; Cortes et al., 2001; Cordero-Morales et al., 2006). Protein was reconstituted in a 1:10,000 (mass of protein:mass of lipids) ratio. The suspension was centrifuged for 1 h at 100,000 g. Subsequently, the pellet (∼10 mg lipid) was resuspended in rehydration buffer (60 μl). Before patch-clamp experiments, drops of the suspension were dried 24 h in a desiccation chamber and rehydrated for 5 h in 20 μl of rehydration buffer.
RESULTS
Dependence of Tetramethylrhodamine Fluorescence on the Dielectric Constant and pH
The lifetime is characteristic for each fluorophore and may be influenced by several factors. If the fluorophore gets exposed to a dynamic quencher (O2, specific amino acids) the transition from the excited state (S1) to the ground state (S0) can occur using this pathway. Hence, the rate of transition is increased and the fluorescence lifetime decreased (Marme et al., 2003) (see Appendix).
From previous measurements on Shaker potassium channels (Mannuzzu et al., 1996; Cha and Bezanilla, 1997), we know that TMRM is sensitive to changes in the hydrophobicity of the surrounding medium or the presence of quenching amino acid residues such as Glu, Asp (Blunck et al., 2004), Trp, Tyr, Phe, or His (Marme et al., 2003). To characterize the effect of the dielectric constant on the lifetime of TMRM, we determined fluorescence lifetimes of TMRM in different solvents. For all conditions, the phase shift increases significantly in the observed frequency range, while the modulation amplitude decreases. The H2O curves are shifted to higher frequencies, indicating that the fluorescence lifetime in water is shorter than in the other solvents. Shorter lifetimes act only at higher frequencies as a low pass filter. As described in the Materials and methods, phase shift and modulation amplitude can directly be fitted to models containing a varying number of lifetimes. As a result, lifetimes, τi, and the relative amplitudes, αi, can be obtained. The data here could be interpreted using a single lifetime for TMRM (Fig. 2 A). The lifetime was dependent on the solvent and shifted from 1.62 ns in H2O to 3.18 ns in octanol (methanol, 2.65 ns; ethanol, 2.97 ns). Evidently, we would detect a transition of TMRM from an exposed hydrophilic to a protected hydrophobic environment, e.g., due to a transition from solution to membrane. The presence of a quenching amino acid would also reduce the lifetime of the fluorophore (Lakowicz, 1999). Since we want to use the pH dependence of KcsA to manipulate the open probability of the channel, we measured the pH dependence of the fluorescence lifetime of TMRM (0.2 μM) dissolved in a suspension of unilammelar vesicles (10 mg lipids/ml solution) as a control (Fig. 2 B). The lifetime remained constant (1.47 ± 0.02 ns) between pH 2.0 and pH 6.0. Again, the data could be fitted with a single component model that was used to directly determine the lifetimes τ (Fig. 2 C). At pH 7.0, a small increase to 1.68 ns was observed. At pH 8.0 and pH 9.0, the lifetime only increased very slightly.
Figure 2.

(A and B) Lifetime measurements of TMR-maleimide (0.2 μM) in different solvents (A) and at different pH in a 0.5 mg/ml lipid vesicles solution (B). Shown are the data (symbols) as well as the fits (lines). The fit results of B are shown in C. For each given pH or solvent, the modulation amplitude (decreasing) and phase shift (increasing) are shown. (C) Lifetimes obtained from measurements in B. Modulation amplitude and phase shift could be fitted with a single lifetime model. (D) Positions of the attachment sites for the fluorophores. Q119C and G116C are in the bundle crossing, L90C is on top of TM2, where only small movements are expected.
Gating Behavior of KcsA-Single Cysteine Mutants
To attach the fluorescent probe at a specific position in the KcsA channel, we took advantage of an extensive cysteine mutant library developed earlier (Perozo et al., 1998) and used maleimide chemistry to couple the fluorescent probe to its selected position. Cysteines were introduced at position L90C, on top of TM2, and at positions G116C and Q119C, at the C-terminal end of TM2, right below the bundle crossing pointing inwards (Fig. 2 D). According to EPR measurements of KcsA, the largest conformational changes are expected near the bundle crossing (position 112–119) (Perozo et al., 1999; Liu et al., 2001). Only very small movements were expected for the upper TM2, consistent with the MthK open channel structure. All single cysteine mutations did not seem to influence the gating behavior significantly. We tested the function of the labeled channels in planar lipid bilayer and found normal conductance and a very low open probability (<0.1) (unpublished data). This is the upper limit (N p0) for the open probability, since we could not determine the exact number of channels in the bilayer. The openings showed typical flickering of the channel, and short openings were followed by prolonged closed states of the channel. The pH dependence, determined in Rb+ flux measurements, did not shift for the mutants with respect to KcsA-WT.
Fluorescence Lifetimes of KcsA-WT and -L90C TMRM Are Not pH Dependent
As a consequence of conformational changes, a fluorophore attached to a protein may be moved from its original position to a new environment. During this translocation, the fluorophore may become quenched either because it gets into close proximity of a quenching amino acid or because it gets exposed to water. In both cases, a decrease in TMR fluorescence lifetime would be observed. To ensure that no intrinsic lifetime changes of the fluorophore in the fluorescence signals from KcsA channels are observed, we used KcsA-WT and -L90C as controls. While the WT channel would indicate any unspecific background labeling, KcsA-L90C is labeled at a position in the upper TM2, where only very little movement is expected. Although the total fluorescence intensity of KcsA-WT was much lower, we could not establish reliable intensity relations in the small volumes used for the spectrometric measurements. In bulk measurements after removal of excess dye, however, we found that the fluorescence intensity of the cysteine mutants is ∼13 times higher than the WT channel (Fig. 1 C) before reconstitution using biobeads. The biobeads remove additional excess label, so that the ratio is further increased. The remaining background fluorescence was likely caused by unspecific bound fluorophores in the vesicles or proteins. The modulation amplitude and phase shift (Fig. 3 A) could be fitted to a one-component model with τ = 2.9 ns. The lifetime did not significantly change at different pH. KcsA-L90C showed much higher fluorescence compared with KcsA-WT, consistent with the specific binding of the maleimide group to the cysteine. Still, the modulation amplitude and phase shift could be fitted to a single component model (Fig. 3 B). The lifetime found is τ = 2.73 ns at pH 9.0 and changes slightly to 2.47 ns at pH 2.0 (Fig. 4 A). The values are comparable with TMRM dissolved in methanol, indicating that the fluorophore is at least partly protected from the aqueous solution surrounding it.
Figure 3.

Lifetime measurements of KcsA mutants at different pH (N = 3). (A) KcsA-WT (background fluorescence), (B) KcsA-L90C, (C) KcsA-Q119C, and (D) KcsA-G116C. KcsA-WT and –L90C could be fitted with a single lifetime model, KcsA-Q119C and -G116C had to be fitted to a double lifetime model (see Materials and methods for details). Shown are the data (symbols) as well as the fits (lines). For each given pH, the modulation amplitude (decreasing) and phase shift (increasing) are shown.
Figure 4.

(A) pH dependence of lifetimes obtained from measurements in Fig. 2 using a single lifetime (L90C) or double lifetime (Q119C, G116C) model. (B and C) Correlation of fraction of fast lifetime to the open probability of KcsA obtained from Rb+ flux measurements as a function of pH for KcsA-G116C (B) and -Q119C (C). The data were fitted to a single Boltzmann function assuming a two-state model.
We had to consider the possibility of aggregated protein, since we do not know how these would behave as a function of pH. Before reconstitution, all aggregated proteins have been removed by centrifugation. In the native bilayer environment, it is not very likely that the channels aggregate. Nevertheless, since we do not find a significant effect of pH on the fluorescence of L90C, we can deduce that denaturated or aggregated protein, which may still be present, merely adds to the background fluorescence and does not disturb our pH- dependent measurements.
Another point to consider is that the channels reconstitute in both directions. KcsA has a preferred inside-out direction, but some proteins are also oriented the other way. Since we equilibrate the pH before the measurements, the orientation does not influence the results.
Fluorescence Lifetimes of KcsA-Q119C and -G116C TMRM Show pH-dependent Distribution
The lifetime measurements of the two mutants KcsA-Q119C and KcsA-G116C show a different shape (Fig. 3, C and D), when compared with the signal from KcsA-L90C. Accordingly, the modulation amplitudes and phase shifts had to be fitted to a two-component model, with time constants τ1 = 3.6 ns and τ2 = 0.7 ns (G116C) and τ1 = 3.0 ns and τ2 = 1.2 ns (Q119C) at pH 7. Fitting with a third component did not further improve the quality of the fit or resulted in two equal time constants. The fast time constant is even faster than TMRM in aqueous solution, indicating that an additional quencher is close to the fluorophore in this state.
While the time constants do not change significantly at different pH (Fig. 4 A), the observed shift in the mean lifetime is caused by a change in the relative proportion of the two lifetimes. The presence of two lifetimes indicates that the fluorophores are divided into two populations, each with a different lifetime. The pH dependence of the distribution between the two populations suggests that they might represent the closed and open state of the bundle crossing. To compare the fluorescence with the open probability, we plotted the pH dependence of the fraction of the fast time constant along with the relative open probability obtained from Rb+ flux experiments (Fig. 4, B and C). As the pH is decreased, the fraction of the fast component of KcsA-G116C increases and correlates very well with the open probability of the channel (Fig. 4 B; pH1/2 = 5.75 [fast comp.], pH1/2 = 6.0 [open prob.]). This suggests that fluorophores with a short decay time correspond to channels with an open bundle crossing while fluorophores with a slow decay correspond to a closed bundle crossing. The pH-dependent shift from the slow to the fast population of KcsA-Q119C is shifted toward lower pH (pH1/2 = 4.74; Fig. 4 C) and does not fully saturate even at pH 2, suggesting that the lifetime in Q119C might additionally be influenced by another process. This may be a further opening of the channel, or a secondary effect on the fluorophore itself. The signal of KcsA-G116C, on the other hand, seems to be purely governed by the opening of the channel. We therefore concentrated the following studies on this position.
Since KcsA-WT and KcsA-L90C do not show any pH-dependent fluorescence change, the fluorescence change of KcsA-G116C has to be caused by the fluorophore covalently linked to the cysteine in the protein. Nevertheless, we have to validate whether the two states of the fluorophores correspond to the open and closed channel. A strong indication is that the pK of the fluorescence change corresponds to the pK of the H+ sensor of KcsA (Fig. 4 B). But this could have two implications. One possibility is that the H+ sensor is causing the fluorescence change directly. In this case, the state (protonated or deprotonated) determines the fluorescence of the fluorophore. The other possibility, however, is that the H+ sensor is opening the bundle crossing and that the conformational change of the bundle crossing is causing the fluorescence change. Neither the mutation G116C nor Q119C had an influence on the pH dependence of the channel so that we may conclude that these positions are not directly involved in pH sensing. Since the fluorophore in the closed position only comes into contact with residues of the lower TM2 pointing inward, the fluorescence has to be caused by the conformation of the lower bundle crossing, not the protonation state of the H+ sensor.
Nevertheless, since we also have background fluorescence (Fig. 1 C and Fig. 3 A), there remains the possibility that the fluorophore is statically quenched in either the closed or the open state. Statically quenched fluorophores do not get excited and would therefore not be visible in the fluorescence measurements (Lakowicz, 1999). If the fluorophores would be statically quenched in either the closed or the open state, the fraction of the fast component would indicate not the relative population of the open channel, but the development of the open or closed state of the channel with respect to the background. Therefore in this case, we still would monitor the conformational state of the bundle crossing.
Since the fluorophores come to very close proximity in the closed position, there is the possibility of self-quenching (Zhuang et al., 2000). However, self-quenching leads to a shorter fluorescence lifetime and, therefore, would lead to an increase in the fast component at higher pH (closing of the channel), which we did not observe. (The case that the fluorophores are so close together that the quenched lifetime cannot be detected by our acquisition system anymore would be equivalent to static quenching; see above.)
The values of the two time constants indicate that the fluorophore is protected from a quenching environment in the closed position and becomes more exposed in the open state. Since in the open state the fast time constant is faster than TMRM in aqueous solution, an additional quencher must be in close proximity to the fluorophore when the channel is open.
The Fluorescence Anisotropy of KcsA-G116C and -Q119C
If the fluorophore transits from a protected to an unprotected position during the opening of the bundle crossing, there is the possibility that the mobility of the fluorophore is altered during the transition. We determined the anisotropy of the fluorophore at different pH for KcsA-G116C and -Q119C. The anisotropy was around 0.24 for both mutants, which is typical for fluorophores bound to proteins (Eftink et al., 1990), but we could not find a significant pH dependence. The values for G116C varied between 0.22 and 0.24. The scattering of the anisotropy values of Q119C was larger (<0.06), but also showed no clear trend with pH changes.
KcsA-A73E Shows an Increased Open Probability
Cordero-Morales et al. (2006) have recently described the mutation E71A, which increases the open probability drastically from ∼0.1–0.15 (WT) to 0.95 by dramatically slowing down KcsA entry into a C-type inactivated state in the selectivity filter (Fig. 5 D). Here, we further describe the mutation A73E at the bottom of the selectivity filter that shows a higher open probability (0.8) in the bilayer (Fig. 5 A). This mutation had been initially identified from 86Rb+ flux analyses and patch clamp measurement as one that increases the mean open time of KcsA (Cuello, L.G., and E. Perozo. 2002. Biophys. J. 82:174a; Cordero-Morales et al., 2006). But in contrast to the E71A mutant, A73E still enters a prolonged closed state (Fig. 5 A, arrows), suggesting at least a partial entry into the inactivated state. The overall dwell time is reduced in the closed state and increased in the open state. In contrast, the probability of E71A to enter the prolonged closed state is extremely low (Cordero-Morales et al., 2006).
Figure 5.

(A) Bilayer measurements of KcsA-G116C (top) and KcsA-G116C-A73E (bottom) at pH 2. Bilayers were formed from POPE:POPS = 1:1 mixture in decane. The arrows indicate some of the occasions where A73E transits into the prolonged closed state. The dotted line named C indicates the closed level. (B) pH-dependent shift of the fast component for KcsA-G116C (black), -G116C-A73E (red), and -G116C-E71A (blue). The fraction of the fast component is shown in the inset. They have been baselined to pH 7 in order to compare the shift. At pH 7, WT and mutant channels are in the closed position. The data were fitted to a single Boltzmann function assuming a two-state model. (C) Lifetime measurements of KcsA-G116C-A73E (top) and -G116C-E71A (bottom) (N=3). Shown are the data (symbols) as well as the fits (lines). For each given pH, the modulation amplitude (decreasing) and phase shift (increasing) are shown. (D) For comparison, single channel recording of E71A in liposomes is shown. E71A does not enter a prolonged closed state.
pH Shift of Fluorescence Lifetimes Is Not Dependent on the Open Probability of the Mutants
Fig. 5 (B and C) shows the lifetime measurements and the relative distribution of the two double mutants KcsA-E71A-G116C and KcsA-A73E-G116C, compared with the single mutant. The lifetimes of the two double mutants revealed very similar values to those of the single mutant KcsA-G116C, characterized by the presence of two different components (around 1 and 3 ns). E71A-G116C has identical values of the fraction of the fast component to G116C. A73E-G116C shows a higher fraction of the slow component (Fig. 5 B, inset), but the shift from high to low pH remains the same (Fig. 5 B). The pH dependence of the fraction of the fast component of all three mutants (G116C, E71A-G116C, and A73E-G116C) superimposes with the open probability of the channels measured with Rb+ flux. While it may be possible that the higher fraction of the slow component originates from additional background labeling in the lipids, it cannot originate from the higher open probability of this mutant as the slow component is favored by the closed state.
DISCUSSION
In this paper, we set out to use fluorescence lifetime spectroscopy to monitor the state of the bundle crossing in KcsA. For the channels labeled in the bundle crossing, we found two different lifetimes, one at τ ≈ 3 ns and another at τ ≈ 1 ns. Upon pH changes, these lifetimes remain constant, but their relative amplitude shifts with the same pK value as the open probability of the channels measured with Rb+ flux. We therefore assigned the fast time constant to the open state and the slower time constant to the closed state of the bundle crossing. The distribution of the lifetimes suggests that during steady-state gating, the movement of the TM2 helix bundle is not the main determinant of the open probability. The pore helix mutants E71A and A73E have been shown to gate with a much higher open probability than that observed in WT-KcsA, yet we found no change in the relative amplitudes of the lifetimes of fluorophores attached to the end of the TM2 helix bundle. Likewise, the fraction of the fast component in KcsA-WT is too high to account for its low open probability. These data can only be explained if the open probability is controlled by the actions of a second gate located in series with the bundle crossing and suggests that the opening of the bundle crossing does not automatically lead to ion conduction (Fig. 6 A).
Figure 6.

(A) Model for the gating of KcsA. The opening of the bundle crossing is the pH-dependent step, but the ion flow can be obstructed by a second gate in the pathway. This second gate is modulated by the mutation A73E and E71A. (B) Structure of the selectivity filter of KcsA (1J95). E71A and A73E are positioned right behind the selectivity filter, which can undergo a conformational change to a nonconductive state. The ribbon shows TM2; M96 and G99 are highlighted in green. (C) Schematic of the mechanism as to how KcsA-A73E may increase the open probability. The larger glutamate (blue sphere) prevents the selectivity filter (orange) from relaxing and thus inactivating.
In KcsA-WT, the equilibrium between closed and open state of this second gate is predominantly shifted toward a nonconductive conformation, which is the cause for the low open probability. This nonconductive conformation shows all the hallmarks of C-type inactivation as recently determined by patch clamp methods (Cordero-Morales et al., 2006). The mutations E71A and A73E shift the equilibrium between a closed and open second gate toward the open state, relieving steady-state inactivation and allowing a much higher open probability. Nevertheless, since all channels have the same pH dependence for the open probability, we can conclude that the opening of the bundle crossing is the pH-dependent step. The pH-dependent opening of the bundle crossing determines how much time the channel dwells on the “right” side in Fig. 6 A, while the transition of the second gate reduces this probability by a factor β/(α+β) so that the resulting open probability lies close to 0 for WT, but is much higher for the mutants.
The position of the mutations E71A and A73E in the pore helix and in close proximity to the selectivity filter signature sequence (Fig. 6 B) suggest that this region of the channel may be the actual gate as it is suggested for C-type inactivation. Cordero-Morales et al. (2006) have proposed that the carboxyl–carboxylate interaction between E71 and D80 in the upper region of the selectivity filter destabilizes the open state and promotes inactivation. So that when this interaction is disrupted, filter reorientations and changes in local dynamics would prevent the conformational changes that lead to C-type inactivation. The mutation A73E, on the other hand, cannot straightforwardly be interpreted with the same mechanism. The mutation A73E adds another charge to the lower selectivity filter and increases the size of the residue (Fig. 6 B). Since A73C also increases the open probability (Cuello, L.G., and E. Perozo. 2002. Biophys. J. 82:174a), the volume change associated with the mutation might be the decisive parameter for the effects on inactivation (and hence, the open probability). The larger size of glutamate as opposed to alanine may stabilize the open conformation of the selectivity filter.
In the model (Fig. 6 A), we assume that the two gates are independent of each other. Nevertheless, since the selectivity filter seems to inactivate after opening of the bundle crossing (Cordero-Morales et al., 2006), there may be a coupling between both gates, where the closed position of the bundle crossing stabilizes the conductive state of the selectivity filter. The position of A73 could be an indicator that the coupling occurs at this position. If the TM2 moves to close the lower pore (Perozo et al., 1999; Jiang et al., 2002b) the residues adjacent to A73, M96, and G99 (Fig. 6 B) may push against A73, thereby stabilizing the selectivity filter in its conductive configuration and inhibiting entry into the inactivated state, as discussed above. When the TM2 is in the open position, A73 could relax back so that the selectivity filter may transit into the inactivated state. By exchanging the alanine to the more voluminous glutamate, this relaxation is hindered when TM2 is in the open position due to the larger size of the glutamate side chai n so that the open state of the selectivity filter is favored (Fig. 6 C, bottom). This mechanism is consistent with findings in the bacterial NaChBac channel. The inactivation of NaChBac has been shown to occur independently of the position of the voltage sensor and could be considered to be C-type inactivation (Blunck et al., 2004; Pavlov et al., 2005). Zhao et al. (2004) could prevent inactivation by the mutation G219P. The G219 in NaChBac corresponds to G99 of KcsA, suggesting that the interaction between G99 and A73 mediates coupling between the two gates.
There has been increasing evidence that the selectivity filter is not as rigid as previously supposed and here we provide evidence that the selectivity filter indeed gates. In addition, it seems that the state of the selectivity filter is not fully independent of the lower gate, although coupling of the gate in the selectivity filter with the opening of the lower bundle crossing is not fully understood. The recording of the state of the bundle crossing by fluorescence as described in this paper simultaneously with the ionic current through the pore is expected to provide more mechanistic details of channel gating and the relation between activation and slow inactivation (Blunck, R., J.L. Vazquez-Ibar, Y.S. Liu, E. Perozo, and F. Bezanilla. 2003. Biophys. J. 84:124).
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant GM30376 (F. Bezanilla), CRC 202965 (R. Blunck), and NIH grant GM057846 (E. Perozo).
Olaf S. Andersen served as editor.
APPENDIX
Fluorescence Lifetimes
If the fluorophore is excited to the S1 state, it may transit back to the ground state via different pathways: by emitting a photon (fluorescence), by transferring the energy to the surrounding without radiation (nonradiative deexcitation), or by transferring the energy to a different molecule (quenching, energy transfer). Each of these processes has different probabilities so that the fluorescence decay after an excitation pulse is given by
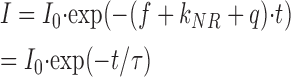 |
(3) |
with
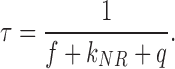 |
(3a) |
The quantum yield QY, to which the fluorescence intensity is proportional, is defined as the number of emitted photons per number of absorbed photons. Therefore, it may be calculated as the fraction of the fluorescent transitions f out of all transitions back to the ground state. Thus,
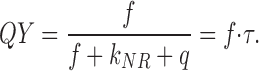 |
(4) |
Eqs. 3a and 4 demonstrate why the lifetime is a measure of the quenching of the fluorophore. If a quencher comes into close proximity of a fluorophore, the probability for a quenching q increases, while f and kNR remain constant. Therefore, τ and QY decrease.
Phase Shift and Amplitude Modulation in the Frequency Domain
As mentioned above, the lifetime of the fluorophore is a property of the fluorophore and is therefore independent of the modulation frequency. Nevertheless, the phase shift gives the ratio between the lifetime τ of the fluorophore and the time tf one modulation wave requires to complete (2π/ω; Fig. 1). If one considers a population with a single lifetime τ, Eq. 2 becomes relatively simple:
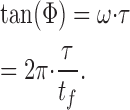 |
(5) |
Thus, with increasing ω, decreasing tf, Φ increases.
To understand the decrease of the modulation, one has to consider that the fluorescence at each time point is the sum of the responses of all previous excitation times. The fluorophore acts as a lowpass filter with the lifetimes as time constants. As
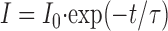 |
(3′) |
is the response to a single excitation pulse, the response at any time point is the sum of many decays, so that
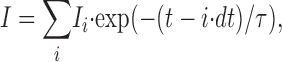 |
(6) |
where dt is a time increment and Ii changes sinusoidal proportional to the excitation intensity. If the modulation amplitude is high, Ii changes rapidly so that the maximal value for I includes terms with low amplitude. If Ii, on the other hand, varies slowly (low frequency ω), the maximal value for I will contain mainly large amplitudes Ii. Fig. 7 illustrates this for the case where one considers only three terms of Eq. 6. As a consequence, the modulation amplitude of the emission decreases with increasing modulation frequency.
Figure 7.

The fluorescence intensity at a time t, I(t), contains contributions from previous time points. Therefore, the contributions Ii at low frequencies (gray) have a higher amplitude than at high frequencies (red).
Abbreviations used in this paper: EPR, electron paramagnetic resonance; POPE, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine; POPS, 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-l-serine]; QY, quantum yield; TMR, tetramethyl rhodamine; TMRM, tetramethylrhodamine-5-maleimide.
References
- Blunck, R., D.M. Starace, A.M. Correa, and F. Bezanilla. 2004. Detecting rearrangements of shaker and NaChBac in real-time with fluorescence spectroscopy in patch-clamped mammalian cells. Biophys. J. 86:3966–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruening-Wright, A., M.A. Schumacher, J.P. Adelman, and J. Maylie. 2002. Localization of the activation gate for small conductance Ca2+-activated K+ channels. J. Neurosci. 22:6499–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha, A., and F. Bezanilla. 1997. Characterizing voltage-dependent conformational changes in the Shaker K+ channel with fluorescence. Neuron. 19:1127–1140. [DOI] [PubMed] [Google Scholar]
- Chapman, M.L., H.M. VanDongen, and A.M. VanDongen. 1997. Activation-dependent subconductance levels in the drk1 K channel suggest a subunit basis for ion permeation and gating. Biophys. J. 72:708–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J., J.S. Mitcheson, M. Tristani-Firouzi, M. Lin, and M.C. Sanguinetti. 2001. The S4-S5 linker couples voltage sensing and activation of pacemaker channels. Proc. Natl. Acad. Sci. USA. 98:11277–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claydon, T.W., S.Y. Makary, K.M. Dibb, and M.R. Boyett. 2003. The selectivity filter may act as the agonist-activated gate in the G protein-activated Kir3.1/Kir3.4 K+ channel. J. Biol. Chem. 278:50654–50663. [DOI] [PubMed] [Google Scholar]
- Cordero-Morales, J.F., L.G. Cuello, Y. Zhao, V. Jogini, D.M. Cortes, B. Roux, and E. Perozo. 2006. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 13:311–318. [DOI] [PubMed] [Google Scholar]
- Cortes, D.M., and E. Perozo. 1997. Structural dynamics of the Streptomyces lividans K+ channel (SKC1): oligomeric stoichiometry and stability. Biochemistry. 36:10343–10352. [DOI] [PubMed] [Google Scholar]
- Cortes, D.M., L.G. Cuello, and E. Perozo. 2001. Molecular architecture of full-length KcsA: role of cytoplasmic domains in ion permeation and activation gating. J. Gen. Physiol. 117:165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello, L.G., J.G. Romero, D.M. Cortes, and E. Perozo. 1998. pH-dependent gating in the Streptomyces lividans K+ channel. Biochemistry. 37:3229–3236. [DOI] [PubMed] [Google Scholar]
- Decher, N., J. Chen, and M.C. Sanguinetti. 2004. Voltage-dependent gating of hyperpolarization-activated, cyclic nucleotide-gated pacemaker channels: molecular coupling between the S4-S5 and C-linkers. J. Biol. Chem. 279:13859–13865. [DOI] [PubMed] [Google Scholar]
- Del Camino, D., and G. Yellen. 2001. Tight steric closure at the intracellular activation gate of a voltage-gated K(+) channel. Neuron. 32:649–656. [DOI] [PubMed] [Google Scholar]
- Del Camino, D., M. Holmgren, Y. Liu, and G. Yellen. 2000. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 403:321–325. [DOI] [PubMed] [Google Scholar]
- Delcour, A.H., B. Martinac, J. Adler, and C. Kung. 1989. Modified reconstitution method used in patch-clamp studies of Escherichia coli ion channels. Biophys. J. 56:631–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demo, S.D., and G. Yellen. 1992. Ion effects on gating of the Ca(2+)-activated K+ channel correlate with occupancy of the pore. Biophys. J. 61:639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, D.A., C.J. Morais, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Eftink, M.R., L.A. Selvidge, P.R. Callis, and A.A. Rehms. 1990. Photophysics of indole-derivatives - experimental resolution of la and Lb transitions and comparison with theory. J. Phys. Chem. 94:3469–3479. [Google Scholar]
- Flynn, G.E., and W.N. Zagotta. 2001. Conformational changes in S6 coupled to the opening of cyclic nucleotide-gated channels. Neuron. 30:689–698. [DOI] [PubMed] [Google Scholar]
- Heginbotham, L., M. LeMasurier, L. Kolmakova-Partensky, and C. Miller. 1999. Single Streptomyces lividans K(+) channels: functional asymmetries and sidedness of proton activation. J. Gen. Physiol. 114:551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. a. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. b. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, V. Ruta, M. Cadene, B.T. Chait, and R. MacKinnon. 2003. a. X-ray structure of a voltage-dependent K(+) channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., V. Ruta, J. Chen, A. Lee, and R. MacKinnon. 2003. b. The principle of gating charge movement in a voltage-dependent K(+) channel. Nature. 423:42–48. [DOI] [PubMed] [Google Scholar]
- Kelly, B.L., and A. Gross. 2003. Potassium channel gating observed with site-directed mass tagging. Nat. Struct. Biol. 10:280–284. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J.R. 1999. Principles of Fluorescence Spectroscopy. Second edition. Kluwer Academic/Plenum Publishers, New York. 698 pp.
- LeMasurier, M., L. Heginbotham, and C. Miller. 2001. KcsA: it's a potassium channel. J. Gen. Physiol. 118:303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., M. Holmgren, M.E. Jurman, and G. Yellen. 1997. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 19:175–184. [DOI] [PubMed] [Google Scholar]
- Liu, Y.S., P. Sompornpisut, and E. Perozo. 2001. Structure of the KcsA channel intracellular gate in the open state. Nat. Struct. Biol. 8:883–887. [DOI] [PubMed] [Google Scholar]
- Long, S.B., E.B. Campbell, and R. MacKinnon. 2005. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 309:897–903. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo, J., T. Hoshi, S.H. Heinemann, and R.W. Aldrich. 1993. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1:61–71. [PubMed] [Google Scholar]
- Lu, T., A.Y. Ting, J. Mainland, L.Y. Jan, P.G. Schultz, and J. Yang. 2001. Probing ion permeation and gating in a K+ channel with backbone mutations in the selectivity filter. Nat. Neurosci. 4:239–246. [DOI] [PubMed] [Google Scholar]
- Lu, Z., A.M. Klem, and Y. Ramu. 2002. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J. Gen. Physiol. 120:663–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannuzzu, L.M., M.M. Moronne, and E.Y. Isacoff. 1996. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 271:213–216. [DOI] [PubMed] [Google Scholar]
- Marme, N., J.P. Knemeyer, M. Sauer, and J. Wolfrum. 2003. Inter- and intramolecular fluorescence quenching of organic dyes by tryptophan. Bioconjug. Chem. 14:1133–1139. [DOI] [PubMed] [Google Scholar]
- McCormack, K., M.A. Tanouye, L.E. Iverson, J.W. Lin, M. Ramaswami, T. McCormack, J.T. Campanelli, M.K. Mathew, and B. Rudy. 1991. A role for hydrophobic residues in the voltage-dependent gating of Shaker K+ channels. Proc. Natl. Acad. Sci. USA. 88:2931–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuser, D., H. Splitt, R. Wagner, and H. Schrempf. 1999. Exploring the open pore of the potassium channel from Streptomyces lividans. FEBS Lett. 462:447–452. [DOI] [PubMed] [Google Scholar]
- Pantoja, R., D. Sigg, R. Blunck, F. Bezanilla, and J.R. Heath. 2001. Bilayer reconstitution of voltage-dependent ion channels using a microfabricated silicon chip. Biophys. J. 81:2389–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov, E., C. Bladen, R. Winkfein, C. Diao, P. Dhaliwal, and R. French. 2005. The pore, not cytoplasmic domains, underlies inactivation in a prokaryotic sodium channel. Biophys. J. 89:232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo, E., D.M. Cortes, and L.G. Cuello. 1998. Three-dimensional architecture and gating mechanism of a K+ channel studied by EPR spectroscopy. Nat. Struct. Biol. 5:459–469. [DOI] [PubMed] [Google Scholar]
- Perozo, E., D.M. Cortes, and L.G. Cuello. 1999. Structural rearrangements underlying K+-channel activation gating. Science. 285:73–78. [DOI] [PubMed] [Google Scholar]
- Phillips, L.R., and C.G. Nichols. 2003. Ligand-induced closure of inward rectifier Kir6.2 channels traps spermine in the pore. J. Gen. Physiol. 122:795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proks, P., C.E. Capener, P. Jones, and F.M. Ashcroft. 2001. Mutations within the P-loop of Kir6.2 modulate the intraburst kinetics of the ATP-sensitive potassium channel. J. Gen. Physiol. 118:341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proks, P., J.F. Antcliff, and F.M. Ashcroft. 2003. The ligand-sensitive gate of a potassium channel lies close to the selectivity filter. EMBO Rep. 4:70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti, M.C., and Q.P. Xu. 1999. Mutations of the S4-S5 linker alter activation properties of HERG potassium channels expressed in Xenopus oocytes. J. Physiol. 514(Pt 3):667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppa, N.E., and F.J. Sigworth. 1998. Activation of Shaker potassium channels. II. Kinetics of the V2 mutant channel. J. Gen. Physiol. 111:295–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sompornpisut, P., Y.S. Liu, and E. Perozo. 2001. Calculation of rigid-body conformational changes using restraint-driven Cartesian transformations. Biophys. J. 81:2530–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruce, A.E., N.B. Standen, and P.R. Stanfield. 1989. Rubidium ions and the gating of delayed rectifier potassium channels of frog skeletal muscle. J. Physiol. 411:597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus, J.G., L. Kuschel, M.D. Rayner, and S.H. Heinemann. 1997. Ion conduction through C-type inactivated Shaker channels. J. Gen. Physiol. 110:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Z.P., M.H. Akabas, E.H. Goulding, A. Karlin, and S.A. Siegelbaum. 1996. Exposure of residues in the cyclic nucleotide-gated channel pore: P region structure and function in gating. Neuron. 16:141–149. [DOI] [PubMed] [Google Scholar]
- Swenson, R.P., Jr., and C.M. Armstrong. 1981. K+ channels close more slowly in the presence of external K+ and Rb+. Nature. 291:427–429. [DOI] [PubMed] [Google Scholar]
- Webster, S.M., D. Del Camino, J.P. Dekker, and G. Yellen. 2004. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature. 428:864–868. [DOI] [PubMed] [Google Scholar]
- Xiao, J., X.G. Zhen, and J. Yang. 2003. Localization of PIP2 activation gate in inward rectifier K+ channels. Nat. Neurosci. 6:811–818. [DOI] [PubMed] [Google Scholar]
- Zhao, Y., V. Yarov-Yarovoy, T. Scheuer, and W.A. Catterall. 2004. A gating hinge in Na+ channels; a molecular switch for electrical signaling. Neuron. 41:859–865. [DOI] [PubMed] [Google Scholar]
- Zheng, J., and F.J. Sigworth. 1998. Intermediate conductances during deactivation of heteromultimeric Shaker potassium channels. J. Gen. Physiol. 112:457–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y., J.H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]
- Zhuang, X., T. Ha, H.D. Kim, T. Centner, S. Labeit, and S. Chu. 2000. Fluorescence quenching: a tool for single-molecule protein-folding study. Proc. Natl. Acad. Sci. USA. 97:14241–14244. [DOI] [PMC free article] [PubMed] [Google Scholar]