

Abstract
Open-channel blockers such as tetraethylammonium (TEA) have a long history as probes of the permeation pathway of ion channels. High affinity blockade by extracellular TEA requires the presence of an aromatic amino acid at a position that sits at the external entrance of the permeation pathway (residue 449 in the eukaryotic voltage-gated potassium channel Shaker). We investigated whether a cation–π interaction between TEA and such an aromatic residue contributes to TEA block using the in vivo nonsense suppression method to incorporate a series of increasingly fluorinated Phe side chains at position 449. Fluorination, which is known to decrease the cation–π binding ability of an aromatic ring, progressively increased the inhibitory constant K i for the TEA block of Shaker. A larger increase in K i was observed when the benzene ring of Phe449 was substituted by nonaromatic cyclohexane. These results support a strong cation–π component to the TEA block. The data provide an empirical basis for choosing between Shaker models that are based on two classes of reported crystal structures for the bacterial channel KcsA, showing residue Tyr82 in orientations either compatible or incompatible with a cation–π mechanism. We propose that the aromatic residue at this position in Shaker is favorably oriented for a cation–π interaction with the permeation pathway. This choice is supported by high level ab initio calculations of the predicted effects of Phe modifications on TEA binding energy.
INTRODUCTION
It is less than a decade since the ion channel field was electrified by the first atomic resolution images of a potassium-selective ion channel, the KcsA channel from the bacterium Streptomyces lividans (Doyle et al., 1998). Since then, several important potassium channel structures have appeared, all but one imaging a channel of bacterial origin. The KcsA structure (and subsequent structures of other potassium channels) was consistent with and rationalized a wide range of biochemical and biophysical studies of potassium channels, clearly establishing its relevance to mammalian potassium channels. This includes the location of a residue associated with the blockade of potassium channels by the external application of the cation TEA. For many years, open-channel blockers such as TEA have been exploited to gain insights into the processes of gating and permeation (Armstrong, 1971; Miller, 1982; MacKinnon and Yellen, 1990; Choi et al., 1991; Heginbotham and MacKinnon, 1992; Hidalgo and MacKinnon, 1995; Ikeda and Korn, 1995; Bretschneider et al., 1999; Jiang and MacKinnon, 2000; Thompson and Begenisich, 2003; Andalib et al., 2004; Chatelain et al., 2005). Tyr82 of KcsA is positioned at the mouth of the channel in a location that appears well suited to binding extracellular TEA. This residue in KcsA aligns with Thr449 in the eukaryotic Shaker potassium channel, and early work established its critical role for external TEA blockade. In particular, high affinity blockade by TEA requires an aromatic amino acid (Tyr or Phe) at this site (MacKinnon and Yellen, 1990; Kavanaugh et al., 1991; Heginbotham and MacKinnon, 1992); the Shaker mutants T449Y and T449F have high TEA affinity, as do other potassium channels that naturally have a Tyr or Phe at the aligned site (Frech et al., 1989; Stuhmer et al., 1989; MacKinnon and Yellen, 1990). Aromaticity is critical, as nonaromatic amino acids either more hydrophobic, such as Leu, Ile, and Val, or more hydrophilic, such as Ser, Thr, Glu, or Lys, lead to low affinity block by extracellular TEA (Heginbotham and MacKinnon, 1992; Molina et al., 1997). The unique requirement of an aromatic amino acid at position 449 led Heginbotham and MacKinnon (1992) to propose that a cation–π interaction was critical to the high affinity binding of TEA. An optimal cation–π interaction is supported only if the cation (in this case TEA) interacts with the face, not the edge, of the aromatic ring, an arrangement we will refer to as en face.
Reported crystal structures of KcsA actually show two orientations for residue Tyr82. In support of the cation–π hypothesis, a recent crystal structure of a noninactivating mutant of KcsA shows an en face orientation of Tyr82 (Fig. 1 B, orange side chain; Cordero-Morales et al., 2006), suggesting that this orientation might occur in eukaryotic potassium channels. However, other reported KcsA structures pose a possible problem. Specifically, the side chain of Tyr82 in many published structures of KcsA (e.g., 1BL8 [Doyle et al., 1998], 1K4C [Zhou et al., 2001], 1R3J [Zhou and MacKinnon, 2003], 2A9H [Yu et al., 2005], and 1ZWI [Cordero-Morales et al., 2006]; in the Protein Data Bank [www.rcsb.org/pdb/]) is aligned such that a TEA positioned to block the channel would not interact with the face of the aromatic ring (Fig. 1, blue side chains). Furthermore, several molecular dynamics studies of KcsA complexed with TEA support this nonoptimal geometry at Tyr82 such that the edges of the tyrosine rings point toward the blocker (Crouzy et al., 2001; Luzhkov and Aqvist, 2001; Guidoni and Carloni, 2002; Luzhkov et al., 2003). Indeed, a subsequent crystal structure of KcsA complexed with the TEA analogue tetraethylarsonium (TEAs) also has this edge-on conformation (Lenaeus et al., 2005). Thus, it was concluded that a cation–π interaction was not involved in binding TEA to KcsA but rather that the crucial Tyr influenced the local hydration structure of the binding site.
Figure 1.
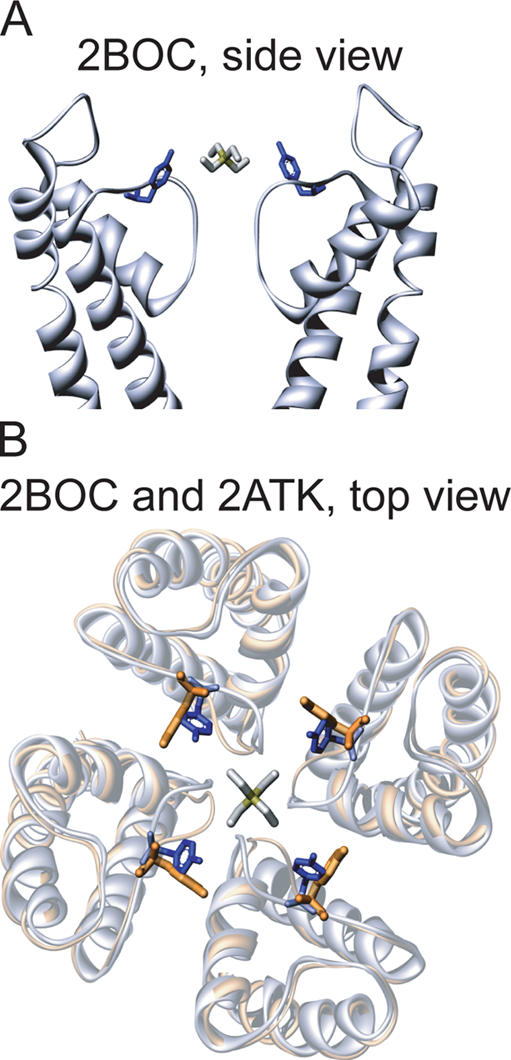
Two orientations of Tyr82 residues of KcsA. Atomic coordinates (2BOC and 2ATK) were obtained from the Protein Data Bank. 2BOC (blue) is wild-type KcsA cocrystallized with TEAs (Lenaeus et al., 2005). 2ATK (orange) is one of two crystal forms of the E71A mutant (Cordero-Morales et al., 2006). (A) Side view of two opposing subunits of 2BOC with TEAs. (B) Top view of 2BOC aligned with 2ATK, showing the approximate en face orientation of Tyr82 in 2ATK.
Therefore, there is support both for and against a cation–π interaction at the aromatic residue at the entrance to potassium channels. Simulation studies of the edge-on KcsA channel argue against a cation–π interaction, but the binding studies of TEA to eukaryotic potassium channels and the recent en face KcsA structure allow for such an interaction. In this study, we aim to test directly for a cation–π mechanism in Shaker potassium channels using a definitive probe developed for functional ion channels and receptors. Modifying the side chain of an aromatic amino acid with fluorine substantially diminishes the cation–π binding ability of the aromatic ring, and multiple substitutions produce additive effects (Dougherty, 1996; Mecozzi et al., 1996a; Ma and Dougherty, 1997). We can achieve such substitutions in Shaker using the nonsense suppression methodology for unnatural amino acid incorporation (Nowak et al., 1995). This methodology has numerous advantages, making it ideal for probing potential cation–π interactions. In comparison with conventional mutagenesis, the steric perturbation introduced by fluorine substitution is universally considered to be minimal. Also, fluorine substitution does not substantially alter the hydrophobicity of the ring. For example, benzene and hexafluorobenzene have nearly identical logP (water/octanol partition) values (Leo et al., 1971). Therefore, induced affinity shifts in cation binding cannot be attributed solely to the effects of hydrophobicity. We have successfully used this fluorination strategy to probe cation-binding sites in the nicotinic acetylcholine receptor, the 5-HT3 (serotonin) receptor, the GABAC receptor, and the NMDA receptor (Zhong et al., 1998; Beene et al., 2002; Mu et al., 2003; Lummis et al., 2005; McMenimen et al., 2006).
In this study, we provide an empirical rationale for the choice between the edge-on and en face orientations by applying the side chain fluorination approach to position 449 of the Shaker channel. We find a compelling correlation between the degree of fluorination of Phe449 and the loss of TEA affinity. The correlation is supported by ab initio quantum mechanical calculations of the two disparate models of the TEA-binding site. We conclude that there is a significant cation–π interaction involved in the binding of TEA to the Shaker channel, suggesting that an aromatic residue at this position adopts an en face orientation.
MATERIALS AND METHODS
Molecular Biology and Unnatural Amino Acids
The channel we used was Shaker H4 with the following three modifications: deletion of residues 6–46 to remove N-type inactivation and deletion of the point mutations C301S and C308S. The in vivo nonsense suppression methodology was performed as described previously (Nowak et al., 1995, 1998). In brief, at site 449 of Shaker, the cDNA was mutated into a TAG nonsense (stop) codon by conventional mutagenesis (Stratagene), and complementary mRNA was transcribed from this cDNA (mMessage mMachine; Ambion). Unnatural amino acids were protected with nitroveratryloxycarbonyl, activated as the cyanomethyl ester, and coupled to the dinucleotide deoxycytosineadenosine (dCA). This aminoacyl dinucleotide was then ligated to a modified tRNA from Tetrahymena thermophila. Deprotection of the aminoacylated tRNA–amino acid was performed by UV irradiation immediately before coinjection with the complementary RNA (cRNA) for the channel into stage V and VI Xenopus laevis oocytes. Typically, 20 ng tRNA–amino acid and 25 ng cRNA were injected in a 50-nl volume.
Electrophysiology
Voltage-clamped potassium currents were recorded with two microelectrodes using either the OpusXpress system (Axon Instruments, Inc.), which includes an automated solution delivery system, or an OC-725C voltage clamp (Warner) in a standard Ringers solution (116 mM NaCl, 2 mM KCl, 1 mM MgCl2, 0.5 mM CaCl2, and 5 mM HEPES, pH 7.5). The chloride salt of TEAs was a gift from A. Gross and M. Lenaeus (Northwestern University Medical School, Chicago, IL).
Computations
For ab initio calculations, we used Gaussian 03 software (Gaussian). To ensure an appropriate level of theory for the examination of cation–π interactions, we optimized the structure of benzene plus a Na+ ion (see Fig. 5 A) with Hartree-Fock (HF) self-consistent field theory using the 6-31G** polarized basis set (Mecozzi et al., 1996a). The same level of theory was used to determine the single point energy of this structure and those containing derivatives of benzene. For binding energy determinations, we used counterpoise correction for basis set superposition errors (Simon et al., 1996), which were always <1 kcal/mol.
Figure 5.
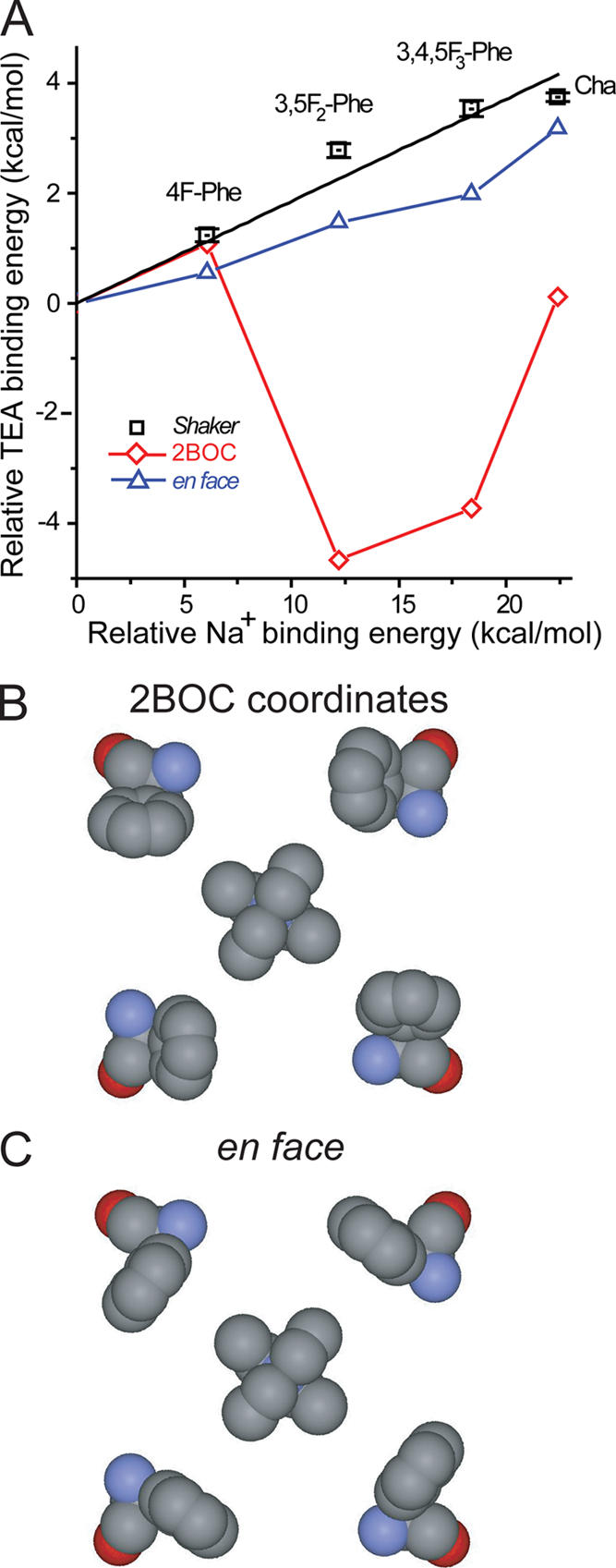
Thermodynamic and ab initio calculations support an en face model of TEA binding to Shaker. (A) Relative binding energy of TEA to Phe derivatives plotted against that calculated for Na+ binding to benzene derivatives. Data obtained experimentally for Shaker (squares) display a linear change (black line; slope = 0.19 ± 0.01, R 2 = 0.88) in binding energy as π electrons are withdrawn by fluorine substitutions. Ab initio calculations for the binding energetics of a reduced system comprised of four aromatics and a single TEA molecule are shown on the same plot for comparison. (B) The reduced system (shown without hydrogen atoms) based on the coordinates of KcsA and TEAs (Lenaeus et al., 2005). These coordinates predict enhanced binding (diamonds in A) as π electrons are removed from the aromatic face, which is a trend inconsistent with experimental data. (C) Conversely, a 60° rotation qualitatively reproduces (triangles in A) the trend of TEA binding energetics obtained experimentally from Shaker.
The 117-atom reduced molecular model of Fig. 5 B is based on the coordinates of TEAs cocrystallized with KcsA (Lenaeus et al., 2005). First, the hydroxyls of Tyr82 residues were removed to convert them into Phe. The arsenic of TEAs was then replaced by a nitrogen, and the structure of the resultant TEA molecule was optimized at the HF/6-31G* level while freezing the position of the central nitrogen and the four Phe82 residues. This optimization preserved both the D 2d conformation of the blocker and the approximate direction of its four alkyl chains. The en face model of Fig. 5 C was generated from the reduced molecular model by a 60° rotation around the CβCγ bond of Phe82. To replace Phe82 with cyclohexylalanine (Cha), we substituted the aromatic ring with cyclohexane and optimized the structure of the ring only (freezing the position of the Cγ carbon) in the context of the reduced model at the HF/STO-3G level. TEA binding energy was then calculated at the HF/6-31G** level as described above for Na+ binding to benzene. Structures are displayed with DS/ViewerPro software (Accelrys).
RESULTS
To test for a cation–π role in the interaction between extracellular TEA and an aromatic residue at position 449 of Shaker, we used Phe derivatives chosen to reduce the negative electrostatic potential on the face of the aromatic ring. Fig. 2 shows examples of inactivation-removed Shaker potassium currents obtained with this method. In all cases, the cRNA encoding for Shaker was identical, containing a UAG codon at position 449. This cRNA was coinjected into oocytes with one of six different constructs of suppressor tRNAs, each containing the anticodon CUA and all but one synthetically acylated with an amino acid. Fig. 2 (A–D) shows families of potassium currents in which the tRNAs were acylated with Phe and three of its fluorinated derivatives. Insets (Fig. 2) show color-coded electrostatic potential surfaces, which were determined by ab initio calculations, of benzene and these fluorinated derivatives. On this color scale, red and blue indicate negative and positive electrostatic potential, respectively. As shown, successive substitution of fluorine atoms for hydrogen atoms on a benzene ring produces a successive decrease in the negative electrostatic potential on the face of the aromatic ring, leading to a monotonic decrease in intrinsic cation binding ability (Mecozzi et al., 1996a,b). Fig. 2 E shows currents from the mutant Cha in which the benzene ring of Phe was replaced by cyclohexane. In general, the currents in all of the mutants we examined had similar kinetics and voltage dependence. Fig. 2 F shows the absence of current from an oocyte coinjected with Shaker 449UAG and a suppressor tRNA that had not been charged with an amino acid. This control ensures that the suppressor tRNA is not reaminoacylated by the cell with a natural amino acid, which could then be incorporated into the channel protein. To control for read-through, namely the possibility that another tRNA species can incorrectly recognize the UAG codon and attach its amino acid at position 449, we injected the cRNA for Shaker 449UAG alone. This control was performed routinely for every experiment and also produced no detectable potassium currents, ruling out read-through.
Figure 2.

Functional expression of unnatural amino acids in Shaker at position 449. (A–E) Representative families of potassium currents elicited by test depolarizations for 10-mV increments between −60 and 50 mV from a holding potential of −80 mV. Leak and capacitance currents were subtracted online with a –P/8 protocol. The label beneath each panel indicates the introduced amino acid. In each case, the inset shows the 6-31G** electrostatic potential surface of benzene derivatives, with red and blue corresponding to −20 and 20 kcal/mol, respectively (Mecozzi et al., 1996a). The numbered positions of the fluorine atoms are shown with respect to the Cγ carbon of Phe. (F) Lack of potassium currents originating from cellular tRNA acylation when T449UAG mRNA was coinjected with an uncharged tRNA.
We measured the block of extracellular TEA in channels containing six different residues at position 449: Phe, Thr (wild type), the three fluorinated derivatives of Phe, and Cha. In all cases, the block was rapid, as typically observed for extracellular TEA (Spruce et al., 1987); that is, the blocker reduced the macroscopic current amplitude with no effect on gating kinetics (Fig. 3, A and B). Fig. 3 C plots the fraction of unblocked channels at 50 mV versus the extracellular TEA concentration, and Table I lists the concentrations producing half block (K i). The pivotal role of the residue at position 449 is evident in this figure and in Table I. The absolute magnitudes and relative K i values for Phe and the native residue (Thr) at this position in Shaker agree with a previous study (Heginbotham and MacKinnon, 1992). The affinities differ by nearly two orders of magnitude between these two natural amino acid residues, supporting the hypothesis that an aromatic residue plays a critical role in TEA binding. For comparison with the study of TEAs cocrystallized with KcsA (Lenaeus et al., 2005), we also measured the potency of this blocker in Shaker. TEAs is a weaker blocker than TEA by a factor of ∼25 in Shaker T449F (unpublished data).
Figure 3.
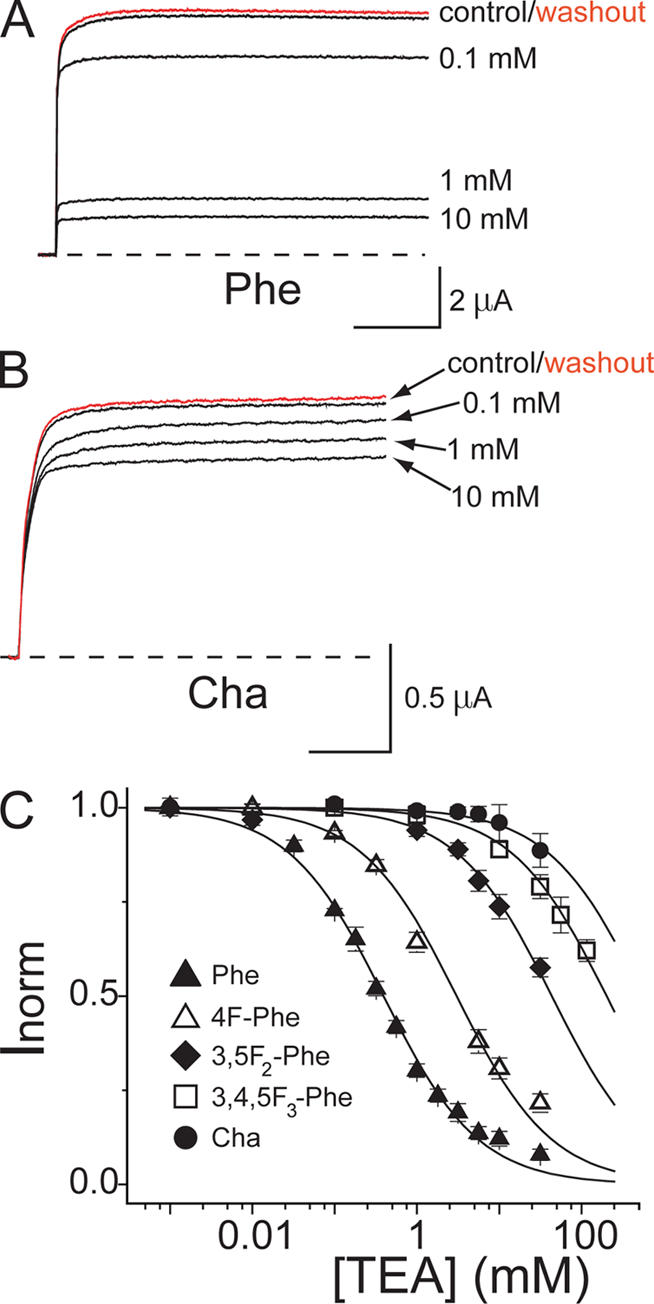
Evidence for cation–π energetics in TEA block. (A and B) Reversible TEA inhibition for Phe and Cha currents at 50 mV. (C) TEA inhibition plots for Shaker channels containing the indicated residue at position 449. Curves are standard binding isotherms fitted to the data. Increased fluorination monotonically increases the dissociation constant K i (Table I). Cha, which is devoid of aromatic character, renders the channel nearly insensitive to TEA.
TABLE I.
Inhibitory Constants for TEA Block Based on Fits of Dose–Response Curves
| T449X, where X= | K i | ΔΔG |
|---|---|---|
| mM | kcal/mol | |
| Phe (n = 10) | 0.39 ± 0.04 | 0 |
| Thr (n = 7) | 33.5 ± 8.9 | 2.58 |
| 4F-Phe (n = 5) | 3.3 ± 0.6 | 1.23 |
| 3,5F-Phe (n = 7) | 47.6 ± 5.9 | 2.78 |
| 3,4,5F-Phe (n = 7) | 175.2 ± 36.2 | 3.54 |
| Cha (n = 3) | 249.2 ± 20.1 | 3.75 |
Based on the curves in Fig. 3 C. K i values are given as the mean ± SEM. Free energy (ΔΔG) is calculated as ΔΔG = RT ln(K i,X/K i,Phe), where RT is 0.58 kcal/mol at room temperature.
One of the distinctions between aromatic and nonaromatic residues at position 449 is the voltage dependence of TEA block. TEA apparently senses 19% of the transmembrane electric field for Thr449 and only 4% for Tyr449 (Heginbotham and MacKinnon, 1992). This suggests that TEA blocks at a more superficial site when an aromatic residue at 449 creates a high affinity binding site. Because the fluorination of Phe reduces TEA affinity, it might also allow TEA to bind at a deeper location in the electric field. However, we found that the voltage dependence of block was shallow (2–8%) for all fluorinated derivatives of Phe449 (Fig. 4), supporting the idea that the blocking site for TEA is the same for all of these derivatives. Note that the measured voltage dependence from the plots in Fig. 4 could be caused by the distribution of K+ ions within the selectivity filter rather than by the position of TEA within the electric field (Spassova and Lu, 1998).
Figure 4.

Voltage dependence of block is similar for all fluorinated derivatives. (A–D) Natural logarithm of the relative fraction of unblocked channels (Fun) versus blocked channels is plotted against membrane potential. [TEA] = 1, 1, 10, and 10 mM for Phe, 4F-Phe, 3,5F2-Phe, and 3,4,5F3-Phe, respectively. Error bars represent SEM.
If an electrostatic component of TEA binding were caused by a cation–π interaction involving the aromatic side chain at residue 449, one would expect increasing the substitution of fluorines to decrease the binding affinity monotonically (Mecozzi et al., 1996a). This is exactly what we observe (Fig. 3 C and Table I). In other systems, such a trend has been interpreted as compelling evidence for a sizable cation–π interaction involving the site undergoing substitution (Zhong et al., 1998; Beene et al., 2002; Mu et al., 2003; Lummis et al., 2005). The first such conclusion, the cation–π interaction between acetylcholine and a Trp residue in the nicotinic acetylcholine receptor (Zhong et al., 1998), was subsequently confirmed by x-ray crystallography (Brejc et al., 2001).
The largest effect on TEA affinity, 640-fold, occurs with the nonaromatic residue Cha. As discussed in the next section, this observation provides further support for a cation–π interaction in TEA binding and contributes valuable information on proposed alternative models underlying TEA affinity.
DISCUSSION
Our data argue that high affinity TEA blockade results, in part, from a significant cation–π interaction between TEA and an aromatic amino acid at position 449 in Shaker. As in other studies of this type (Zhong et al., 1998; Beene et al., 2002; Mu et al., 2003; Lummis et al., 2005; McMenimen et al., 2006), it is not a single mutation that leads to this conclusion but rather a consistent trend across a systematic series of subtle mutations. Fig. 5 A plots the change in free energy of TEA binding (squares) for the Phe derivatives against the effect on the energy of Na+ ion binding to comparable derivatives of benzene. Na+ binding energy in the gas phase was determined by ab initio calculations. The monotonic, nearly linear relationship for Shaker establishes a cation–π contribution to TEA block. Moreover, a previous study shows that fluorination has little effect on nonelectrostatic energetic components of cation binding to a benzene ring (e.g., donor acceptor, charge transfer, and induced dipoles in the aromatic; Ma and Dougherty, 1997).
To estimate the energetic contribution of the cation–π interaction, a comparison of Phe and 3,4,5F3-Phe is useful. Examination of the electrostatic potential surfaces of Fig. 2 shows that three fluorines effectively erase the electrostatic attractiveness of the aromatic ring, and earlier studies showed that trifluorobenzene is a good model for an aromatic that has little or no electrostatic binding ability (Williams, 1993; Mecozzi et al., 1996b). From this perspective, the ΔΔG values of Table I suggest that the cation–π interaction is the dominant factor in distinguishing strong from weak TEA binders in that the energetic consequence of the Phe→F3-Phe mutation (3.5 kcal/mol) is comparable with that of the Phe→Thr mutation (2.6 kcal/mol).
The biggest decrease in TEA affinity is seen for the Phe→Cha mutation (3.8 kcal/mol). This is consistent with both theoretical and experimental studies showing that cyclohexane binds cations more poorly than any of the aromatics considered here (Shepodd et al., 1988; Mecozzi et al., 1996a). Note that the side chains of Phe and Cha, modeled here as benzene and cyclohexane, respectively, are similar in size, shape, and hydrophobicity (Fig. 2). The most noticeable difference, which is in the context of noncovalent binding interactions, is that the negative electrostatic potential that leads to cation binding in benzene disappears in cyclohexane. Thus, the Phe→Cha mutation further confirms that the π character and its associated negative electrostatic potential dominate strong TEA binding when Phe is present at position 449. Also note that cyclohexane is considerably more polarizable than benzene (Craven et al., 1989), ruling out any special role for an induced dipole in the binding of TEA by Phe and derivatives.
Previous computational studies of the KcsA channel are based on structures with Tyr82 in the edge-on orientation and have led others to conclude that the cation–π interaction does not contribute substantially to external TEA blockade (Crouzy et al., 2001; Luzhkov and Aqvist, 2001; Guidoni and Carloni, 2002; Luzhkov et al., 2003). These studies instead emphasize local hydration structure as a crucial component of the energetics of TEA binding, although electrostatic stabilization by the backbone carbonyls of Tyr78 and Gly79 (in KcsA) may also play a role (Guidoni and Carloni, 2002). The distribution of K+ ions within the selectivity filter also appears to affect TEA binding energy (Crouzy et al., 2001; Luzhkov and Aqvist, 2001; Guidoni and Carloni, 2002), another possible source of electrostatic energy. Energetic contributions of local hydration include both electrostatic and hydrophobic forces. However, an essential role of attractive hydrophobic forces between TEA and its binding site is contradicted by earlier data from Shaker, in which relatively hydrophobic residues at site 449 are not generally better at binding TEA (Heginbotham and MacKinnon, 1992). For example, comparison of the essentially isosteric residues Val and Thr shows that polar Thr is associated with a higher TEA affinity (Heginbotham and MacKinnon, 1992). Also, Leu, Ile, and Val are all substantially more hydrophobic than Phe (Radzicka et al., 1988) but produce weak TEA-binding sites. The Phe/Cha pair described here presents another isosteric comparison, and, again, hydrophobics do not explain the results. In addition, the size but not hydrophobicity of the alkylammonium blocker appears to be a critical factor in binding affinity (Hille, 1967; Heginbotham and MacKinnon, 1992). Furthermore, examination of the temperature dependence of TEA block shows that enthalpy rather than entropy largely accounts for the free energy of binding (Heginbotham and MacKinnon, 1992), again arguing against a fundamental role of hydrophobic forces in TEA binding.
Computational Examination of the TEA–Channel Interaction
To further investigate the evidence for a cation–π mechanism, we modeled the TEA–side chain interaction computationally. We were constrained to use KcsA structures for this purpose because it is the only crystallized potassium channel with an aromatic residue (Tyr82) at this position. We specifically started with the structure 2BOC, a cocrystal of KcsA and TEAs (Fig. 1, blue side chain; Lenaeus et al., 2005). KcsA is highly homologous to voltage-gated potassium channels in this region of the protein. In the vicinity of the selectivity filter, KcsA has the sequence TTVGYGDLY, whereas in KV1.2, it is TTVGYGDMV. In addition, the crystallographic distances across the tetramer between the α carbons of these residues agree within 1 Å when comparing KcsA structures with the mammalian KV1.2 (2A79). With respect to a cation–π mechanism, the majority of published structures of KcsA display an unfavorable orientation of the Tyr82 aromatic rings. Moreover, the closest carbon–carbon distance between the blocker and the aromatic side chains (4.1 Å) is larger than typically observed for simple cation–π interactions (e.g., 2.4 Å between Na+ and the center of a benzene ring; Dougherty, 1996; Ma and Dougherty, 1997). To examine the energetic consequences of withdrawing π electrons by fluorinating the aromatic ring of this residue, we constructed a reduced molecular model of TEA and its four coordinating Phe residues based on the 2BOC structure (Lenaeus et al., 2005). In this reduced system (117 atoms; Fig. 5 B), we performed high level ab initio quantum mechanical calculations of the binding energy of TEA. Instead of decreasing the binding, as we observed experimentally (Fig. 5 A, squares), successively fluorinating these Phe residues in silico had a nonmonotonic effect on TEA binding energy (Fig. 5 A, diamonds), the main effect being a substantial increase of TEA affinity in the di- and trifluorinated derivatives.
Our ab initio calculations can be rationalized as follows. The cationic blocker is directed more toward the edge than the face of the aromatic ring (Fig. 5 B). The electrostatic potential along the edge of a simple aromatic ring is positive (Fig. 2 A, inset) and would therefore electrostatically repel TEA. However, fluorination introduces negative electrostatic potential to the edge of the ring via the fluorines (Fig. 2), and this would tend to attract TEA, especially in the three or five position of the aromatic ring (Figs. 1 and 5 B). In fact, the 3,5-difluoro and 3,4,5-trifluoro derivatives of Phe show the most dramatic increases in TEA affinity in the ab initio calculations.
Because these self-consistent theoretical results disagree with our experimental data, we considered the possibility that the aromatic rings were rotated to face the central axis of the pore (Fig. 5 C), as proposed before the crystal structures of KcsA were available (Heginbotham and MacKinnon, 1992; Kumpf and Dougherty, 1993). In this en face orientation, TEA would be attracted to the negative electrostatic potential on the face of the aromatic rings. Fluorination would then decrease the negative electrostatic potential presented to TEA and destabilize its binding, which is a prediction consistent with our experimental data. To quantify this idea, we calculated the energetic consequences of fluorinating the aromatic rings of Phe82 on TEA binding in this conformation. The only modification of the structure of Fig. 5 B was a 60° rotation of the aromatic rings, maintaining all other coordinates of the four Phe residues. With this single change, the results of the ab initio calculations now resemble our experimental results, showing a monotonic destabilization of TEA binding as the aromatic rings are increasingly fluorinated (Fig. 5 A, triangles). Moreover, the rotation itself enhanced TEA affinity by ∼3 kcal/mol. Although we did not examine the optimal dihedral angle in detail, rotations of 40 or 80° were less effective at replicating our experimental data. Given the crudeness of this model and the fact that it is based on a crystal structure of KcsA rather than a eukaryotic voltage-gated potassium channel, the agreement is quite acceptable. Even the larger perturbation associated with the nonaromatic Cha residue is reproduced by the calculations.
Although the 144° χ2 dihedral angle in our en face model is unusual for both Tyr and Phe residues in known protein structures (Dunbrack and Cohen, 1997), an en face orientation of Tyr82 was reported for one of the crystal structures of the functional E71A mutant of KcsA (Cordero-Morales et al., 2006). Fig. 1 B shows this structure (2ATK; orange Tyr82 side chains) aligned with that of KcsA cocrystallized with TEAs (blue Tyr82 side chains). The similarity to our en face model (Fig. 5 C) is apparent. There are other subtle structural changes in this region of the 2ATK structure that enable an en face geometry. We also note that the selectivity filter has conformational variants in wild-type KcsA depending on the concentration of permeant ions (Zhou et al., 2001), supporting the possibility that the functional conformation of Shaker is compatible with an en face orientation of Phe449 residues. It also raises the possibility that the open states of potassium channels in general have conformational flexibility, including the orientation of an aromatic residue at this position.
The slope of the experimental fluorination plot as shown in Fig. 5 A is considered to be a crude indication of the energetic magnitude of the cation–π interaction. The slope of this plot (0.19) is comparable with those seen in studies of agonists such as GABA and serotonin binding to neuroreceptors (Beene et al., 2002; Lummis et al., 2005). However, the receptor experiments measure the binding of an 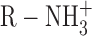 ion to a single aromatic residue. Here, we are simultaneously modifying four aromatic residues. Thus, the slope in the present case suggests that any single TEA–aromatic interaction is not very strong. This is consistent with the low affinity of TEA and the fact that TEA is a very diffuse cation and so is expected to experience a weaker cation–π interaction than more focused cations such as
ion to a single aromatic residue. Here, we are simultaneously modifying four aromatic residues. Thus, the slope in the present case suggests that any single TEA–aromatic interaction is not very strong. This is consistent with the low affinity of TEA and the fact that TEA is a very diffuse cation and so is expected to experience a weaker cation–π interaction than more focused cations such as 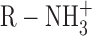 . This may also reflect the fact that TEA cannot make van der Waals contact with all four rings at once in either of the en face conformations we have considered (Fig. 1 B and 5 C), and this should weaken the cation–π interaction. Nevertheless, the magnitude of the cation–π interaction does not fall off steeply with distance (Dougherty, 1996), and, therefore, four nonoptimal interactions can combine to create a moderate effect. It is worth noting that a closer approach of the aromatic side chains to the blocker may be impossible if the open-channel structure is maintained because this residue is only two amino acids downstream from the TVGYGD signature sequence of the selectivity filter. Functional data argue against a large-scale collapse of the aromatics onto the blocker because TEA has no effect on open dwell times of single channels (Spruce et al., 1987).
. This may also reflect the fact that TEA cannot make van der Waals contact with all four rings at once in either of the en face conformations we have considered (Fig. 1 B and 5 C), and this should weaken the cation–π interaction. Nevertheless, the magnitude of the cation–π interaction does not fall off steeply with distance (Dougherty, 1996), and, therefore, four nonoptimal interactions can combine to create a moderate effect. It is worth noting that a closer approach of the aromatic side chains to the blocker may be impossible if the open-channel structure is maintained because this residue is only two amino acids downstream from the TVGYGD signature sequence of the selectivity filter. Functional data argue against a large-scale collapse of the aromatics onto the blocker because TEA has no effect on open dwell times of single channels (Spruce et al., 1987).
Although the calculations we describe do not consider all energetic factors, this is intentional. Our calculations primarily evaluate one component of the binding interaction: the electrostatic attraction between a cation and the side chain (the cation–π interaction). The fact that binding energies correlate so nicely with this term establishes an important role for electrostatic attractions in the binding. We neither rule out nor rule in additional effects such as hydrophobics; we simply note that electrostatics must be important. The functional data on eukaryotic potassium channels show that although there may well be a hydrophobic contribution to binding, it is not the factor that discriminates among residues. Another factor that might contribute to TEA binding in our experiments is the possibility that the electronegative fluorine atoms could influence the local structure of water molecules in the vicinity of the blocker. However, it is not clear how this effect would produce the systematic destabilization of TEA binding that we observe (Figs. 3 C and 5 A). To explore this possibility, we estimated the Gibbs free energy of TEA binding in our reduced edge-on model, accounting both for the gas phase ab initio energies as well as the energies of hydration using a self-consistent reaction field method with the Poisson-Boltzmann solver in the program Jaguar (Schrodinger). As in the gas phase calculations of Fig. 5 A, difluorinating the Phe residue in this conformation enhanced TEA binding (by 2.3 kcal/mol; not depicted), which is again inconsistent with our experimental results and, therefore, tends to rule out the edge-on orientation of Phe449 in Shaker.
How do we resolve our conclusions with earlier works disputing a role for a cation–π mechanism? As noted, previous simulations were based on the analysis of two edge-on crystallographic structures (1BL8 and 2BOC) of the bacterial channel KcsA rather than on a eukaryotic voltage-gated potassium channel or on the en face structure of a KcsA mutant (2ATK). Note that although KcsA contains an aromatic residue at the aligned position (Tyr82), it does not bind TEA strongly. Its K i for TEA block is 3.2 mM (Heginbotham et al., 1999), which is almost 10-fold higher than for Shaker T449F. Also, KcsA Y82T shows a TEA-blocking constant of 143 mM, which is significantly higher than that obtained in Shaker with its wild-type Thr449. Finally, the TEA analogue TEAs used for the 2BOC crystal structure shown in Fig. 1 has a 25-fold larger inhibitory constant than TEA in Shaker T449F, allowing for the possibility that the structure may not be representative of TEA in its binding site. We conclude that inherent but unknown aspects of the KcsA channel, perhaps including a nonoptimal arrangement of Tyr82 residues, underlie a relatively weak TEA-binding site.
In conclusion, we show that in voltage-gated potassium channels, a cation–π interaction makes a substantial contribution to high affinity blockade by extracellular TEA. Evidently, the orientation of an aromatic residue at this position is favorable for a cation–π mechanism. This hypothesis is supported by our experimental and theoretical results, and it provides a simple explanation of the longstanding observation that aromatics at position 449 are necessary and sufficient for high affinity external TEA blockade in voltage-gated potassium channels. We do not yet understand the (presumably subtle) conditions that govern the crystallization of KcsA in one state or the other, but the functional form relevant to Shaker in this region is apparently the en face orientation.
Acknowledgments
We thank Benoit Roux for extensive discussions and advice about Poisson-Boltzmann calculations, Carol Deutsch for comments on the manuscript, Adrian Gross for the gift of a TEAs-Cl sample, and Stephanie Huang, Purnima Deshpande, and Lindsey Ingleby for help with the oocytes.
This research was sponsored by grants from the National Institutes of Health (GM079427, NS11756, and NS34407).
Lawrence G. Palmer served as editor.
Abbreviations used in this paper: Cha, cyclohexylalanine; cRNA, complementary RNA; HF, Hartree-Fock.
References
- Andalib, P., J.F. Consiglio, J.G. Trapani, and S.J. Korn. 2004. The external TEA binding site and C-type inactivation in voltage-gated potassium channels. Biophys. J. 87:3148–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M. 1971. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 58:413–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beene, D.L., G.S. Brandt, W. Zhong, N.M. Zacharias, H.A. Lester, and D.A. Dougherty. 2002. Cation-pi interactions in ligand recognition by serotonergic (5-HT3A) and nicotinic acetylcholine receptors: the anomalous binding properties of nicotine. Biochemistry. 41:10262–10269. [DOI] [PubMed] [Google Scholar]
- Brejc, K., W. J. van Dijk, R.V. Klaassen, M. Schuurmans, J. van Der Oost, A.B. Smit, and T.K. Sixma. 2001. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 411:269–276. [DOI] [PubMed] [Google Scholar]
- Bretschneider, F., A. Wrisch, F. Lehmann-Horn, and S. Grissmer. 1999. External tetraethylammonium as a molecular caliper for sensing the shape of the outer vestibule of potassium channels. Biophys. J. 76:2351–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatelain, F.C., N. Alagem, Q. Xu, R. Pancaroglu, E. Reuveny, and D.L. Minor Jr. 2005. The pore helix dipole has a minor role in inward rectifier channel function. Neuron. 47:833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K.L., R.W. Aldrich, and G. Yellen. 1991. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc. Natl. Acad. Sci. USA. 88:5092–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero-Morales, J.F., L.G. Cuello, Y. Zhao, V. Jogini, D.M. Cortes, B. Roux, and E. Perozo. 2006. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 13:311–318. [DOI] [PubMed] [Google Scholar]
- Craven, I.E., M.R. Hesling, D.R. Laver, P.B. Lukins, G.L.D. Ritchie, and J. Vrbancich. 1989. Polarizability anisotropy, magnetic anisotropy, and quadrupole moment of cyclohexane. J. Phys. Chem. 93:627–631. [Google Scholar]
- Crouzy, S., S. Bernèche, and B. Roux. 2001. Extracellular blockade of K+ channels by TEA: results from molecular dynamics simulations of the KcsA channel. J. Gen. Physiol. 118:207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty, D.A. 1996. Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 271:163–168. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., J.M. Cabral, R.A. Pfuetzner, A.L. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Dunbrack, R.L., Jr., and F.E. Cohen. 1997. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 6:1661–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frech, G.C., A.M. VanDongen, G. Schuster, A.M. Brown, and R.H. Joho. 1989. A novel potassium channel with delayed rectifier properties rat brain by expression cloning. Nature. 340:642–645. [DOI] [PubMed] [Google Scholar]
- Guidoni, L., and P. Carloni. 2002. Tetraethylammonium binding to the outer mouth of the KcsA potassium channel: implications for ion permeation. J. Recept. Signal Transduct. Res. 22:315–331. [DOI] [PubMed] [Google Scholar]
- Heginbotham, L., and R. MacKinnon. 1992. The aromatic binding site for tetraethylammonium ion on potassium channels. Neuron. 8:483–491. [DOI] [PubMed] [Google Scholar]
- Heginbotham, L., M. LeMasurier, L. Kolmakova-Partensky, and C. Miller. 1999. Single Streptomyces lividans K+ channels: functional asymmetries and sidedness of proton activation. J. Gen. Physiol. 114:551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo, P., and R. MacKinnon. 1995. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 268:307–310. [DOI] [PubMed] [Google Scholar]
- Hille, B. 1967. The selective inhibition of delayed potassium currents in nerve by tetraethylammonium ion. J. Gen. Physiol. 50:1287–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, S.R., and S.J. Korn. 1995. Influence of permeating ions on potassium channel block by external tetraethylammonium. J. Physiol. 486:267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., and R. MacKinnon. 2000. The barium site in a potassium channel by x-ray crystallography. J. Gen. Physiol. 115:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh, M.P., M.D. Varnum, P.B. Osborne, M.J. Christie, A.E. Busch, J.P. Adelman, and R.A. North. 1991. Interaction between tetraethylammonium and amino acid residues in the pore of cloned voltage-dependent potassium channels. J. Biol. Chem. 266:7583–7587. [PubMed] [Google Scholar]
- Kumpf, R.A., and D.A. Dougherty. 1993. A mechanism for ion selectivity in potassium channels: computational studies of cation-π interactions. Science. 261:1708–1710. [DOI] [PubMed] [Google Scholar]
- Lenaeus, M.J., M. Vamvouka, P.J. Focia, and A. Gross. 2005. Structural basis of TEA blockade in a model potassium channel. Nat. Struct. Mol. Biol. 12:454–459. [DOI] [PubMed] [Google Scholar]
- Leo, A., C. Hansch, and D. Elkins. 1971. Partition coefficients and their uses. Chem. Rev. 71:525–616. [Google Scholar]
- Lummis, S.C., D.L. Beene, N.L. Harrison, H.A. Lester, and D.A. Dougherty. 2005. A cation:π binding interaction with a tyrosine in the binding site of the GABAc receptor. Chem. Biol. 12:993–997. [DOI] [PubMed] [Google Scholar]
- Luzhkov, V.B., and J. Aqvist. 2001. Mechanisms of tetraethylammonium ion block in the KcsA potassium channel. FEBS Lett. 495:191–196. [DOI] [PubMed] [Google Scholar]
- Luzhkov, V.B., F. Osterberg, and J. Aqvist. 2003. Structure-activity relationship for extracellular block of K+ channels by tetraalkylammonium ions. FEBS Lett. 554:159–164. [DOI] [PubMed] [Google Scholar]
- Ma, J.C., and D.A. Dougherty. 1997. The cation-π interaction. Chem. Rev. 97:1303–1324. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R., and G. Yellen. 1990. Mutations affecting TEA blockade and ion permeation in voltage-activated K+ channels. Science. 250:276–279. [DOI] [PubMed] [Google Scholar]
- McMenimen, K.A., E.J. Petersson, H.A. Lester, and D.A. Dougherty. 2006. Probing the Mg2+ blockade site of an N-methyl-D-aspartate (NMDA) receptor with unnatural amino acid mutagenesis. ACS Chem Biol. 1:227–234. [DOI] [PubMed] [Google Scholar]
- Mecozzi, S., A.P. West Jr., and D.A. Dougherty. 1996. a. Cation-pi interactions in aromatics of biological and medicinal interest: electrostatic potential surfaces as a useful qualitative guide. Proc. Natl. Acad. Sci. USA. 93:10566–10571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecozzi, S., A.P. West Jr., and D.A. Dougherty. 1996. b. Cation-π interactions in simple aromatics: electrostatics provide a predictive tool. J. Am. Chem. Soc. 118:2307–2308. [Google Scholar]
- Miller, C. 1982. Bis-quaternary ammonium blockers as structural probes of the sarcoplasmic reticulum K+ channel. J. Gen. Physiol. 79:869–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina, A., A.G. Castellano, and J. Lopez-Barneo. 1997. Pore mutations in Shaker K+ channels distinguish between the sites of tetraethylammonium blockade and C-type inactivation. J. Physiol. 499:361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu, T.W., H.A. Lester, and D.A. Dougherty. 2003. Different binding orientations for the same agonist at homologous receptors: a lock and key or a simple wedge? J. Am. Chem. Soc. 125:6850–6851. [DOI] [PubMed] [Google Scholar]
- Nowak, M.W., P.C. Kearney, J.R. Sampson, M.E. Saks, C.G. Labarca, S.K. Silverman, W. Zhong, J. Thorson, J.N. Abelson, N. Davidson, et al. 1995. Nicotinic receptor binding site probed with unnatural amino acid incorporation in intact cells. Science. 268:439–442. [DOI] [PubMed] [Google Scholar]
- Nowak, M.W., J.P. Gallivan, S.K. Silverman, C.G. Labarca, D.A. Dougherty, and H.A. Lester. 1998. In vivo incorporation of unnatural amino acids into ion channels in Xenopus oocyte expression system. Methods Enzymol. 293:504–529. [DOI] [PubMed] [Google Scholar]
- Radzicka, A., L. Pedersen, and R. Wolfenden. 1988. Influences of solvent water on protein folding: free energies of solvation of cis and trans peptides are nearly identical. Biochemistry. 27:4538–4541. [DOI] [PubMed] [Google Scholar]
- Shepodd, T.J., M.A. Petti, and D.A. Dougherty. 1988. Molecular recognition in aqueous media: donor-acceptor and ion-dipole interactions produce tight binding for highly soluble guests. J. Amer. Chem. Soc. 110:1983–1985. [Google Scholar]
- Simon, S., M. Duran, and J.J. Dannenberg. 1996. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys. 105:11024–11031. [Google Scholar]
- Spassova, M., and Z. Lu. 1998. Coupled ion movement underlies rectification in an inward-rectifier K+ channel. J. Gen. Physiol. 112:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruce, A.E., N.B. Standen, and P.R. Stanfield. 1987. The action of external tetraethylammonium ions on unitary delayed rectifier potassium channels of frog skeletal muscle. J. Physiol. 393:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhmer, W., J.P. Ruppersberg, K.H. Schroter, B. Sakmann, M. Stocker, K.P. Giese, A. Perschke, A. Baumann, and O. Pongs. 1989. Molecular basis of functional diversity of voltage-gated potassium channels in mammalian brain. EMBO J. 8:3235–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J., and T. Begenisich. 2003. External TEA block of Shaker K+ channels is coupled to the movement of K+ ions within the selectivity filter. J. Gen. Physiol. 122:239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, J.H. 1993. The molecular electric quadrupole moment and solid-state architecture. Accounts Chem. Res. 26:593–598. [Google Scholar]
- Yu, L., C. Sun, D. Song, J. Shen, N. Xu, A. Gunasekera, P.J. Hajduk, and E.T. Olejniczak. 2005. Nuclear magnetic resonance structural studies of a potassium channel-charybdotoxin complex. Biochemistry. 44:15834–15841. [DOI] [PubMed] [Google Scholar]
- Zhong, W., J.P. Gallivan, Y. Zhang, L. Li, H.A. Lester, and D.A. Dougherty. 1998. From ab initio quantum mechanics to molecular neurobiology: a cation-π binding site in the nicotinic receptor. Proc. Natl. Acad. Sci. USA. 95:12088–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y., and R. MacKinnon. 2003. The occupancy of ions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J. Mol. Biol. 333:965–975. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., J.H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]