

Abstract
Muscle degeneration and myotonia are clinical hallmarks of myotonic dystrophy type 1 (DM1), a multisystemic disorder caused by a CTG repeat expansion in the 3′ untranslated region of the myotonic dystrophy protein kinase (DMPK) gene. Transgenic mice engineered to express mRNA with expanded (CUG)250 repeats (HSA LR mice) exhibit prominent myotonia and altered splicing of muscle chloride channel gene (Clcn1) transcripts. We used whole-cell patch clamp recordings and nonstationary noise analysis to compare and biophysically characterize the magnitude, kinetics, voltage dependence, and single channel properties of the skeletal muscle chloride channel (ClC-1) in individual flexor digitorum brevis (FDB) muscle fibers isolated from 1–3-wk-old wild-type and HSA LR mice. The results indicate that peak ClC-1 current density at −140 mV is reduced >70% (−48.5 ± 3.6 and −14.0 ± 1.6 pA/pF, respectively) and the kinetics of channel deactivation increased in FDB fibers obtained from 18–20- d-old HSA LR mice. Nonstationary noise analysis revealed that the reduction in ClC-1 current density in HSA LR FDB fibers results from a large reduction in ClC-1 channel density (170 ± 21 and 58 ± 11 channels/pF in control and HSA LR fibers, respectively) and a modest decrease in maximal channel open probability(0.91 ± 0.01 and 0.75 ± 0.03, respectively). Qualitatively similar results were observed for ClC-1 channel activity in knockout mice for muscleblind-like 1 (Mbnl1 ΔE3/ΔE3), a second murine model of DM1 that exhibits prominent myotonia and altered Clcn1 splicing (Kanadia et al., 2003). These results support a molecular mechanism for myotonia in DM1 in which a reduction in both the number of functional sarcolemmal ClC-1 and maximal channel open probability, as well as an acceleration in the kinetics of channel deactivation, results from CUG repeat–containing mRNA molecules sequestering Mbnl1 proteins required for proper CLCN1 pre-mRNA splicing and chloride channel function.
INTRODUCTION
Myotonic dystrophy type 1 (DM1) is the most common adult-onset form of muscular dystrophy with an estimated frequency of 1 in 7,400 (Harper, 1989). DM1 is characterized by dominantly inherited ocular cataracts, cardiac conduction defects, hypogonadism, muscle weakness, and an increase in skeletal muscle excitability resulting in prominent myotonia. Interestingly, DM1 is not caused by mutations in the genes that encode either the skeletal muscle sodium (SCN4A) or chloride (CLCN1) channel. Rather, DM1 is linked to a trinucleotide (CTG) repeat expansion in the 3′ untranslated region of the myotonic dystrophy protein kinase gene (DMPK) (Brook et al., 1992). The trinucleotide repeat expansion leads to the production of DMPK transcripts that are not exported from the nucleus (Taneja et al., 1995; Davis et al., 1997), and thus result in a decrease in DMPK translation and expression (Furling et al., 2001). Mounsey et al. (2000) found that sodium channels in skeletal muscle fibers obtained from both Dmpk +/− and Dmpk −/− mice exhibit incomplete inactivation, late channel reopenings, and a persistent sodium current. These results suggest that altered sodium channel inactivation contributes to increased muscle excitability and susceptibility to myotonia in DM1 (Mounsey et al., 2000). However, Dmpk-dependent changes in skeletal muscle sodium channel inactivation are apparently not sufficient for increased susceptibility to myotonia since Dmpk −/− mice are not myotonic. Moreover, a second form of DM (DM2) results from a similar repeat expansion (CCTG) in intron 1 of a completely different gene (zinc finger protein-9 or ZNF9 gene) (Liquori et al., 2001). Changes in the activity of a small conductance, calcium-activated potassium channel have also been suggested to contribute to the increased incidence of myotonia in DM (Behrens et al., 1994; Kimura et al., 2003).
An attractive alternative hypothesis for DM pathogenesis in humans involves an RNA transdominant mechanism in which CUG repeat–containing mRNA transcripts from the mutant DMPK allele accumulate in the nucleus and sequester critical regulators of RNA processing for a specific subset of genes (e.g., CLCN1) (Mankodi et al., 2002). To determine the effects of CUG repeat expansion on skeletal muscle, Mankodi et al. (2000) developed a transgenic mouse model of DM1 (HSA LR) that exhibits high level muscle-specific expression of mRNA for human skeletal actin (HSA) harboring ∼250 CUG repeats in the 3′ untranslated region of the gene. HSA LR mice exhibit prominent myotonia and aberrant splicing of Clcn1 pre-mRNA, including a high incidence of inclusion of a novel exon (exon 7a) that results in a frame shift and premature termination in the ClC-1 protein. Additionally, 76% of Clcn1 from HSA LR mice was aberrantly spliced with 69% resulting in various premature terminations. Similar transdominant alterations in CLCN1 mRNA splicing were also observed in muscle obtained from human DM1 and DM2 patients (Mankodi et al., 2002). Although the truncated ClC-1 products do not form functional heterodimeric chloride channels, coexpression with wild-type ClC-1 results in dominant-negative effects on chloride channel activity (i.e., reduced current density and accelerated channel deactivation) (Berg et al., 2004).
Muscleblind-like 1 (MBNL1) proteins are regulators of mRNA splicing (Ho et al., 2004) that bind with high affinity to expanded CUG or CCUG repeat RNA. MBNL1 proteins are sequestered into nuclear RNA foci in skeletal muscle of HSA LR mice (CUG), as well as DM1 (CUG) and DM2 (CCUG) patients (Mankodi et al., 2001). In support of a pathogenic role for loss of MBNL1 function in DM pathogenesis, skeletal muscle of Mbnl1 knockout mice (Mbnl1 ΔE3/ΔE3) also exhibits prominent myotonia, aberrant Clcn1 mRNA splicing, and reduced ClC-1 protein expression (Kanadia et al., 2003). While evidence supports an overall reduction in total membrane chloride conductance (G Cl) in skeletal muscle of HSA LR mice (Mankodi et al., 2000), the effects of CUG repeat expansion and Mbnl1 deficiency in mice on the functional expression, kinetics, voltage dependence, and single channel properties of muscle ClC-1 channels have not been determined.
The aim of this study was twofold. First, we set out to characterize the functional expression and biophysical properties (kinetics, voltage dependence, single channel properties) of ClC-1 channels in single skeletal muscle fibers in whole-cell patch clamp experiments. Chloride channel conductance in skeletal muscle has been reported previously using double sucrose (Duval and Leoty, 1980), vaseline gap (Fahlke and Rudel, 1995), and two-electrode (Chen and Jockusch, 1999) voltage clamp techniques (for review see Bretag, 1987). However, no studies have characterized macroscopic (or unitary) ClC-1 properties in whole-cell patch clamp experiments of native skeletal muscle preparations. This is presumably because ClC-1 current density is thought to be too high and adult muscle fibers too large to provide an adequate voltage clamp in most preparations. Here we circumvented this problem by using very low resistance pipettes (∼0.5 MΟ) to patch clamp relatively small, freshly dissociated flexor digitorum brevis (FDB) muscle fibers from young (10–21 d old) mice. This approach enabled biophysical characterization of ClC-1 channel activity in native mammalian skeletal muscle fibers. In addition, we provide a comprehensive comparison of endogenous ClC-1 biophysical properties with that observed following heterologous (in HEK293 cells) and homologous (in skeletal myotubes) expression of the cloned murine ClC-1 channel (mClC-1). A second aim of this study was to compare the expression and biophysical properties of ClC-1 channel activity in FDB fibers obtained from WT mice with those observed in two different mouse models of human myotonic dystrophy (HSA LR and Mbnl1 ΔE3/ΔE3 mice).
MATERIALS AND METHODS
Construction and Expression of mClC-1 in HEK293 Cells and Skeletal Myotubes
A cDNA encoding the murine skeletal muscle chloride channel (mClC-1; NM_013491) was generated using reverse transcription of mRNA isolated from skeletal muscle of WT mice as previously described (Mankodi et al., 2002). Murine ClC-1 exhibits 87% and 96% amino acid identity with human and rat ClC-1, respectively. The cDNA was ligated into the Nhe1/Xho1 sites of pShuttle-IRES-hrGFP-1 (Stratagene). HEK293 cells (Stratagene) were transfected at 50% confluence using Lipofectamine 2000 (Invitrogen) according to manufacturer recommendations. Primary cultures of myotubes were prepared from myoblasts obtained from skeletal muscle of newborn type 1 ryanodine receptor null (dyspedic) mice as previously described (Nakai et al., 1996). Dyspedic myotubes were used as a homologous mClC-1 expression system because they lack endogenous chloride currents (see Fig. 2 B, upright triangles) and remain quiescent during nuclear cDNA microinjection. Expression of mClC-1 in dyspedic myotubes was achieved by nuclear microinjection of 0.02 μg/μl mClC-1-IRES-hrGFP-1 6–8 d after the initial plating of myoblasts. Expressing HEK293 cells and skeletal myotubes were identified 24–72 h after transfection or nuclear microinjection, respectively, by monitoring for GFP fluorescence. GFP-positive cells/myotubes were then used in whole-cell patch clamp experiments as described below.
Figure 2.

Macroscopic properties of expressed and native ClC-1 currents. Voltage dependence of average instantaneous (open symbols) and steady-state (closed symbols) ClC-1 currents recorded from mClC-1–expressing HEK293 cells (A; n = 12), mClC-1–expressing skeletal myotubes (B; n = 4), and native FDB fibers obtained from 18–20-d-old WT mice (C; n = 8). Instantaneous currents measured in control noninjected myotubes did not display appreciable ClC-1 currents (upright triangles). (D) Superimposed and normalized instantaneous current–voltage relationships obtained from mClC-1– expressing HEK293 cells (squares), myotubes (inverted triangles), and native WT FDB fibers (circles). (E) Average relative Po-V curves for mClC-1–expressing HEK293 cells (squares), myotubes (inverted triangles), and native WT FDB fibers (circles) obtained from tail currents elicited at −100 mV. Smooth curves through each PO-V dataset were generated using a modified Boltzmann equation (Eq. 3).
Preparation of FDB Muscle Fibers
Skeletal muscle fibers were isolated from FDB muscle obtained from 10–20-d-old WT, HSA LR, and Mbnl1 ΔE3/ΔE3 mice, as previously described (Beam and Knudson, 1988). All animals were housed in a pathogen-free area at the University of Rochester School of Medicine and Dentistry (URSMD). Animals were anesthetized and killed by procedures that were reviewed and approved by the University Committee on Animal Resources at URSMD. In brief, FDB muscles were dissected and mechanically cleaned of connective tissue in a Ringer solution containing (in mM): 146 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, pH 7.4 with NaOH (Beam and Knudson, 1988). Muscles were then shaken for 40 min at 37°C in 1 mg/ml collagenase A (Roche) dissolved in Ringer's solution. Individual FDB fibers were then dissociated by trituration using Pasteur pipettes of decreasing bore size. Only fibers exhibiting clear striations and clean surfaces were chosen for electrophysiological recordings. All experiments were conducted at room temperature on fibers obtained within 8 h of isolation.
Macroscopic Recordings of ClC-1 Currents
ClC-1 currents were measured using the whole-cell variant of the patch clamp technique (Hamill et al., 1981) and recorded in an external solution consisting of (in mM) 145 TEA-Cl, 10 CaCl2, 10 HEPES, and 3 μM nifedipine (pH 7.4). Low resistance patch pipettes (0.5 MΩ for FDB fibers/myotubes and 1–2 MΩ for HEK293 cells) were filled with an internal recording solution consisting of (in mM) 110 Cs-aspartate, 30 CsCl, 5 MgCl2, 10 Cs2-EGTA, and 10 HEPES (pH 7.4). These conditions provide adequate block of sodium (0 Na), calcium (nifedipine), and potassium (0 K/TEA/Cs) currents. In the cell-attached configuration, seal resistances of ≥5 GΩ were achieved in all experiments. After establishment of the whole-cell configuration in FDB fibers and expressing myotubes, a dialysis period of 8–10 min was used to ensure complete exchange with the internal solution. Ionic currents were compensated for series resistance (>90%), filtered at 2 kHz using an Axopatch 200A amplifier (Molecular Devices), digitized at 10 kHz using a 16-bit converter (Digidata 1322A; Axon Instruments), and acquired/analyzed using the pCLAMP 9 software suite (Molecular Devices).
Chloride currents were elicited using a voltage protocol (see Fig. 1, top) similar to that used widely by other investigators studying ClC-1 channels expressed in heterologous systems. From a holding potential of −40 mV (near the calculated chloride equilibrium potential of −35.8 mV), an initial 200-ms depolarization to +60 mV was used to fully activate ClC-1 channels, followed by a second 250-ms (350 ms for Mbnl1 ΔE3/ΔE3 fibers) voltage step of variable amplitude (between −140 mV and +60 mV in 10-mV increments), and then a final 200-ms voltage step to −100 mV (−110 mV for Mbnl1 ΔE3/ΔE3 fibers). This voltage protocol was first delivered in the absence and then following the addition of 1 mM 9-anthracene carboxylic acid (9AC), a blocker of ClC-1 channels. Only experiments with an input resistance of ≥100 MΩ after the addition of 9AC were included in this study. Offline subtraction of currents recorded in the presence of 9AC from those recorded in its absence (9AC-sensitive currents) was used to eliminate residual leak and capacitative currents. Thus, 9AC-sensitive currents were used to define ClC-1 channel activity in this study. After block of ClC-1 currents, total cell capacitance (Cm), uncompensated series resistance (Rs), and the time constant for membrane charging (τm) were determined by integration of the capacity transient resulting from the average of five 10-mV depolarizing pulses applied from a holding potential of −80 mV (Table I). Current density (pA/pF) was then calculated for each record in order to compare data across cells of different sizes. This approach enabled quantification of several fundamental features of ClC-1 activity, including (a) peak current density, (b) reversal potential, (c) kinetics of channel deactivation (i.e., two exponential fitting of current decay during the variable voltage step), and (d) the voltage dependence of relative channel open probability (from peak currents during the final voltage step). Instantaneous ClC-1 current density was plotted versus membrane voltage (Vm) and fitted according to
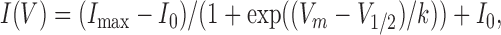 |
(1) |
where I max is the maximum current at the test potential (Vm), I 0 is a constant offset, V 1/2 is the half-maximal activation voltage, and k is a slope factor. ClC-1 currents during the variable voltage step used to quantify the kinetics of current deactivation were fitted according to the following two-exponential function:
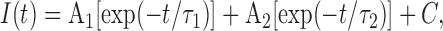 |
(2) |
where I(t) represents current amplitude at any time t during the pulse, A1 and A2 represent the steady-state current amplitudes of each component with their respective time constants (τ1 and τ2), and C represents a time-independent current amplitude. For each test potential, the relative contribution of each current amplitude (A1, A2, and C) was calculated by dividing the absolute value by the sum of all three components (e.g., A1/Atotal = A1/[A1 + A2 + C]). To determine the effect of current magnitude on channel deactivation, we analyzed the deactivation kinetics at −100 mV after 200-ms prepulses to potentials ranging from +60 mV to −80 mV (in 10-mV increments) in 18–20-d-old WT FDB fibers. The current magnitude decreased as the prepulse became more negative due to a decrease in steady-state channel open probability during the prepulse. The dependence of the kinetics of channel deactivation across different current magnitudes within the same cell was then quantified using Eq. 2, as described above.
Figure 1.

Chloride channel currents from mClC-1–expressing HEK293 cells, mClC-1–expressing mouse skeletal myotubes, and native FDB fibers obtained from 18–20-d-old WT mice. Representative whole-cell currents recorded first in the absence (Raw) and then in the presence of 1 mM 9AC (9AC) using an identical voltage protocol (top). ClC-1 currents were then quantified after offline subtraction of 9AC-insensitive currents from raw currents (9AC sensitive). (Inset) Capacitative current recorded from the WT FDB fiber resulting from the average of five 10-mV depolarizations delivered from a −80-mV holding potential. The time constant the capacitative current relaxation was fitted (thick line) to a first-order exponential function (τm = 339 μs). Cell capacitance and access resistance deduced from the fit were 671 pF and 509 kΩ, respectively. The scale bars for the inset are 6 nA (vertical) and 2 ms (horizontal). The dashed lines represent the zero current level.
TABLE I.
Parameters of Linear Properties and Relative Open Probabilities
| Linear properties | Porel | |||||
|---|---|---|---|---|---|---|
| n | Cm | Rs | Tau | V 1/2 | k | |
| pF | kΩ | μs | mV | mV | ||
| mClC-1 HEK cells | 12 | 13 ± 1 | 495 ± 32 | 176 ± 5 | −61.5 ± 4.2 | 25.0 ± 0.7 |
| mClC-1 myotubes | 4 | 184 ± 41 | 1,039 ± 93 | 219 ± 9 | −74.8 ± 1.7 | 25.1 ± 3.1 |
| WT FDB fibers (18–20 d) | 8 | 620 ± 68 | 391 ± 77 | 487 ± 63 | −66.0 ± 5.6 | 29.0 ± 3.2 |
| HSA LR FDB fibers (18–20 d) | 15 | 704 ± 25 | 393 ± 47 | 488 ± 27 | −60.8 ± 4.4 | 27.7 ± 2.9 |
| Mbnl1 +/+ FDB fibers (9–14 d) | 22 | 512 ± 37 | 811 ± 50 | 377 ± 25 | −72.3 ± 2.6 | 27.0 ± 0.9 |
| Mbnl1 ΔE3/ΔE3 FDB fibers (9–14 d) | 16 | 511 ± 41 | 787 ± 60 | 358 ± 60 | −75.8 ± 2.1 | 23.4 ± 1.1 |
The voltage dependence of relative channel open probability (Porel) was calculated from the normalized tail current amplitude elicited in the final voltage step (at −100 mV) and fitted according to
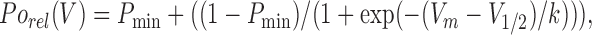 |
(3) |
where P min is an offset, V m is the membrane potential, V 1/2 is the potential at which Po(V) = (Po max − P min)/2, and k is a slope factor.
Nonstationary Noise Analysis
Determination of ClC-1 single channel current (i), maximal channel open probability (Po max), and the number of functional ClC-1 channels (N) was calculated using nonstationary noise analysis (Sigworth, 1980). Mean current (I) and variance (σ2) were calculated from 70–120 current traces (delivered at 0.2 Hz) during channel deactivation resulting from a 200-ms test pulse to −140 mV after a 100-ms prepulse to +60 mV. Baseline variance was measured from a 200-ms pulse to the channel reversal potential (−32 mV) delivered immediately after the test pulse. Mean background and capacitative currents calculated by averaging >20 test pulses recorded in the presence of 1 mM 9AC were used for offline subtraction. Variance was obtained by averaging the squared difference of consecutive 9AC-subtracted traces (Heinemann and Conti, 1992). To eliminate variance due to linear capacitative currents, variance measurements were initiated after >94% of the capacitance transient had decayed. The mean current (I)–variance (σ) relationships were fitted to the following parabolic equation:
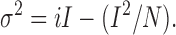 |
(4) |
The number of ion channels (N) and the unitary current (i) were used to calculate maximal channel open probability from the peak current (Imax) according to
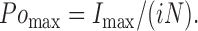 |
(5) |
Conductance values were calculated from i assuming a linear current–voltage curve at negative membrane potentials (Pusch et al., 1994). Data were analyzed using the Ana (written and provided by M. Pusch, Institute of Biophysics, Genova, Italy, http://www.ge.cnr.it/ICB/conti_moran_pusch/programs-pusch/software-mik.htm), Excel, pCLAMP, and Sigmaplot 8.0 software packages. All results are given as means ± SEM with statistical significance (P < 0.05) determined using a two-tailed, unpaired Student's t test.
RESULTS
Comparison of Macroscopic ClC-1 Current Properties in Wild-Type FDB Fibers with mClC-1–expressing HEK293 Cells and Skeletal Myotubes
A major goal of this study was to quantify the biophysical properties of ClC-1 channel activity in native skeletal muscle fibers using the whole-cell patch clamp technique. To minimize problems related to voltage clamp of large ClC-1 currents, we used low resistance (∼0.5 MΩ) patch pipettes to voltage clamp FDB fibers from young (10–21 d old) wild-type mice. This preparation has been used previously to monitor the function and biophysical properties of native L-type Ca2+ channels (Beam and Knudson, 1988; Wang et al., 1999). The voltage dependence and kinetics of ClC-1 channel activation and deactivation was assessed using a voltage protocol (Fig. 1, top; see also Materials and methods) that fully activates ClC-1 channels with a strong depolarization, followed by a second voltage step of variable amplitude, and then a final voltage step to −100 mV. This protocol was delivered in the absence and then presence of 9AC (1 mM). 9AC-sensitive currents were used throughout this study as an indication of ClC-1 channel activity. Using this approach, the voltage dependence and kinetics of macroscopic 9AC-sensitive ClC-1 currents were similar in wild-type FDB fibers and in mClC-1–expressing HEK293 cells or skeletal myotubes (Figs. 1–3 ). Fig. 1 shows representative macroscopic ClC-1 currents recorded from an mClC-1–expressing HEK293 cell (left), an mClC-1–expressing skeletal myotube (middle), and a native FDB fiber obtained from a 19-d-old wild-type mouse (right). Currents are shown under control conditions (top), in the presence of 1 mM 9AC (middle), and following subtraction of 9AC-insensitive currents (bottom). Peak currents at −140 mV in control FDB fibers were reduced ∼98% by 1 mM 9AC. Current density for all experiments was calculated by dividing currents by the cell capacitance determined from the average of five 10-mV depolarizing pulses from a −80 mV holding potential (inset to 9AC data in Fig. 1, right). The average cell capacitance of HEK293 cells, myotubes, and 18–20-d-old wild-type FDB fibers was 13 ± 1 pF (n = 12), 184 ± 41 pF (n = 4), and 620 ± 68 pF (n = 8), respectively (Table I).
Figure 3.

Deactivation kinetics of expressed and native ClC-1 channels. Voltage dependence for the relative contributions of the slow (A), fast (B), and nondeactivating (C) gating components of ClC-1 deactivation in mClC-1–expressing HEK293 cells (squares; n = 12), mClC-1–expressing myotubes (triangles; n = 4), and native FDB fibers obtained from 18–20-d-old WT mice (circles; n = 8). (D) Voltage dependence of the time constants for the fast (lower symbols) and slow (upper symbols) components of ClC-1 deactivation. *, P ≤ 0.05 HEK293 cells compared with WT FDB fibers.
The voltage dependence of the average instantaneous (open symbols) and steady-state (closed symbols) components of 9AC-sensitive currents were similar in mClC-1–expressing HEK293 cells (Fig. 2 A), mClC-1–expressing myotubes (Fig. 2 B), and native wild-type FDB fibers (Fig. 2 C). This similarity is emphasized upon superimposing normalized instantaneous current–voltage relationships (Fig. 2 D). Similarly, the voltage dependence of ClC-1 relative channel open probability (Porel) assessed during the final voltage step was comparable in wild-type FDB fibers (circles), mClC-1–expressing HEK293 cells (squares), and skeletal myotubes (inverted triangles). Porel–V relationships were well described by a modified Boltzmann function (Eq. 3) with similar values for V 1/2 and k for FDB fibers (n = 8), HEK293 cells (n = 12), and myotubes (n = 4) (Table I). Similar values for the voltage dependence of Porel have also been reported by others after heterologous expression of human ClC-1 (Pusch et al., 1995).
Initial inspection of the 9AC-sensitive currents shown in Fig. 1 suggested that the rate of ClC-1 deactivation was somewhat faster after expression of mClC-1 in HEK293 cells compared with that observed in either mClC-1–expressing myotubes or native FDB fibers. Therefore, as a fourth measure of ClC-1 function (in addition to instantaneous, steady-state, and Porel–voltage relationships), we also compared the voltage dependence of the kinetics of ClC-1 channel deactivation in native FDB fibers and after expression of mClC-1 in HEK293 cells and skeletal myotubes. An adequate quantitative description of ClC-1 deactivation kinetics requires fitting the deactivation time course with the sum of two exponentials (Warnstedt et al., 2002). Thus, we used a biexponential fitting paradigm to extract the steady-state amplitudes (Afast, Aslow, and C) and corresponding deactivation time constants (τfast and τslow) that comprise the macroscopic current decay. Fig. 3 depicts the voltage dependence of the relative proportion of slow (A), fast (B), and nondeactivating (C) amplitudes of the total current and the two time constants of deactivation (D) for mClC-1–expressing HEK293 cells (squares), mClC-1–expressing myotubes (triangles), and native wild-type FDB fibers (circles). The fast, slow, and nondeactivating components of mClC-1 exhibit similar voltage dependence in HEK293 cells, myotubes, and FDB fibers and are also similar to that observed for human ClC-1 (Warnstedt et al., 2002). However, across most voltages, the relative amplitude of the slow component in mClC-1–expressing HEK293 cells is significantly (P < 0.05) reduced and the fast component increased compared with mClC-1–expressing myotubes and native FDB fibers. In addition, the fast time constant of channel deactivation was also faster in mClC-1–expressing HEK293 cells over the majority of test potentials compared with that observed in native FDB fibers. Thus, an increase in the relative proportion of fast gating and a decrease in the fast time constant of channel deactivation account for the increase in the rate of macroscopic mClC-1 deactivation observed in mClC-1–expressing HEK293 cells. Together, results presented in Figs. 1–3 provide strong evidence regarding the feasibility of obtaining reliable measures of macroscopic ClC-1 function in FDB fibers obtained from young mice.
Comparison of ClC-1 Properties in FDB Fibers Obtained from Wild-Type and HSALR Mice
A second aim of this study was to determine if increased muscle hyperexcitabilty and myotonia observed in HSALR (Mankodi et al., 2000) and Mbnl1 ΔE3/ΔE3 (Kanadia et al., 2003) mice arises from defects in endogenous ClC-1 channel activity. Fig. 4 depicts representative 9AC-sensitive ClC-1 currents recorded from FDB fibers obtained 18-d-old wild-type (Fig. 4 A) and HSA LR (Fig. 4 B) mice. Average ClC-1 current density measured from 18–20-d-old HSA LR FDB fibers exhibited all of the hallmarks of classic ClC-1 currents, including prominent inward rectification of the instantaneous current with a reversal potential near ECl (Fig. 4 C), a weak voltage dependence to Porel (Fig. 4 D), and rapid and voltage-dependent channel deactivation similar to that observed in wild-type FDB fibers (Fig. 5). The most obvious difference observed in HSA LR FDB fibers was a dramatic reduction in ClC-1 current magnitude compared with age-matched wild-type FDB fibers. The reduction in ClC-1 current magnitude was not due to differences in cell size since total membrane capacitance was not significantly different in FDB fibers obtained from 18–20-d wild-type and HSA LR mice (Table I). Peak ClC-1 current density (measured at −140 mV) was reduced ∼70% in FDB fibers obtained from 18–20-d-old HSA LR mice (−49.8 ± 3.7 pA/pF, n = 8, and −14.0 ± 1.6 pA/pF, n = 15, for wild-type and HSA LR fibers, respectively). Maximal inward steady-state current density was also decreased to a similar degree (unpublished data). Additionally, the reduction in peak ClC-1 current density could not be attributed to a hyperpolarizing shift in the voltage dependence of channel activation (as occurs for certain ClC-1 mutations in dominant myotonia congenita) (Warnstedt et al., 2002), since the voltage dependence of Porel was not significantly different for FDB fibers obtained from 18–20-d-old wild-type (filled circles) and HSA LR (open squares) mice (Fig. 4 D and Table I).
Figure 4.

Macroscopic ClC-1 current density is dramatically reduced in FDB fibers of HSA LR mice. Representative family of ClC-1 currents recorded from FDB fibers of 19-d-old WT (A) and HSA LR (B) mice elicited using an identical voltage protocol (see Fig. 1, top). The dashed lines represent the zero current level. (C) Voltage dependence of average instantaneous current–voltage relationship in FDB fibers obtained from 18–20-d-old WT (closed circles; n = 8) and HSA LR (open squares; n = 16) mice. The apparent depolarizing shift in the reversal potential of the instantaneous ClC-1 current in FDB fibers from HSALR mice most likely arises from difficulty in resolving the true reversal potential in face of small, nearly undetectable, outward currents in these fibers. (D) Average relative Po-V curves for FDB fibers obtained from 18–20-d-old WT (closed circles; n = 8) and HSA LR (open squares; n = 16) mice. Smooth curves through each PO-V dataset were generated using a modified Boltzmann equation (Eq. 3).
Figure 5.

Deactivation kinetics of ClC-1 currents in FDB fibers obtained from WT and HSA LR mice. (A) Superimposed traces of normalized ClC-1 current deactivation (solid lines) elicited at −100 mV in FDB fibers obtained from 19-d-old WT and HSA LR mice and fit with a second order exponential (left; white dashed lines). ClC-1 currents from representative WT (middle) and HSA LR (right) FDB fibers fit with a biexponential function and replotted on log-linear plots. Fractional amplitudes of the extracted slow, fast, and nondeactivating components of the currents are shown for clarity. (B–D) Voltage dependence of the average relative contribution of the slow (B), fast (C), and steady-state (D) components of ClC-1 deactivation in FDB fibers obtained from 18–20-d-old WT (open circles, n = 8) and HSA LR (open squares, n = 16) mice. (E) Voltage dependence of the fast and slow time constants of ClC-1 deactivation in FDB fibers obtained from 18–20-d-old WT (open circles, n = 8) and HSA LR (open squares, n = 16) mice. *, P ≤ 0.05.
Kinetic Analysis of ClC-1 Deactivation in FDB Fibers from Wild-Type and HSALR Mice
Berg et al. (2004) found that two of the ClC-1 splice variants that commonly occur in DM1 produce both a dominant-negative reduction in wild-type ClC-1 currents and an acceleration in channel deactivation. Therefore, we tested if a similar alteration in ClC-1 deactivation kinetics is also observed in the HSA LR mouse model of DM1 by comparing the kinetic parameters of ClC-1 channel deactivation in FDB fibers obtained from 18–20-d-old wild-type and HSA LR mice. Normalization of channel deactivation in wild-type and HSA LR fibers reveals that the deactivation time course at −100 mV is indeed faster in HSA LR fibers (Fig. 5 A left, dashed lines). Nevertheless, a quantitative description of the time course of ClC-1 deactivation still requires the sum of two exponential functions in both wild-type and HSA LR FDB fibers. The biexponential nature of channel deactivation is best appreciated by replotting the currents from wild-type (Fig. 5 A, middle) and HSA LR (Fig. 5 A, right) fibers on a semilogarithmic scale. In this analysis, the steady-state amplitudes of the slow (Aslow), fast (Afast), and nondeactivating (C) components are represented by their respective Y intercepts. The voltage dependence of the relative contributions of Aslow (Aslow/Atotal; where Atotal = Aslow + Afast + C), Afast (Afast/Atotal), and C (C/Atotal) to channel deactivation and the magnitude of the two deactivation time constants (τslow and τfast) are summarized in Fig. 5 (B–E, respectively). The results show that a significant acceleration in channel deactivation observed at intermediate voltages (−80 to −120 mV) in HSA LR FDB fibers arises from an increase in the fractional contribution of the fast component of channel deactivation coupled with a parallel decrease in the relative contribution of the slow component of channel deactivation. Specifically, ClC-1 channels in HSA LR FDB fibers exhibit a significant increase in fast gating by up to ∼15% at −100 mV without a significant effect on the relative contribution of the nondeactivating component. On the other hand, the time constants of channel deactivation (τslow and τfast) were not different across these intermediate voltages, although τfast was somewhat reduced at very negative potentials in HSA LR fibers. These data indicate that faster channel deactivation in HSA LR fibers at intermediate voltages arises from an increase in the relative contribution of the fast gating component of channel deactivation.
Kinetic Analysis after Prepulse-dependent Reduction in ClC-1 Cl− Current Density in Wild-Type FDB Fibers
The increase in the contribution of the fast component of channel deactivation could arise either from dominant-negative effects on channel gating of novel splice isoforms present in HSA LR fibers (Berg et al., 2004) or as a result of the dramatically different ClC-1 current magnitudes found in wild-type and HSA LR fibers. To test the latter, we quantified ClC-1 deactivation at −100 mV in 18–20-d-old wild-type FDB fibers after 200-ms prepulses to potentials ranging from +60 mV to −80 mV (10-mV increments). Using this approach, we were able to compare the kinetics of ClC-1 deactivation during a single test pulse (−100 mV) across a broad range of different ClC-1 current densities within the same cell. Fig. 6 A shows a family of ClC-1 test pulse currents obtained from a 19-d-old wild-type FDB fiber. The prepulse dependence of the time-dependent ClC-1 current (i.e., the Afast and Aslow components) during the test pulse is shown in Fig. 6 B. Superimposing the largest ClC-1 current (using a +60 mV prepulse) with the normalized current after a 50% reduction in open probability (using a −70-mV prepulse) reveals that the deactivation time course is essentially identical for the two markedly different ClC-1 currents (Fig. 6 B, inset). In fact, prepulse- dependent changes in ClC-1 current magnitude did not significantly affect the relative contribution of the fast and slow gating component of deactivation or their respective time constants (Fig. 6 C). The relative contributions of the fast and slow components of channel deactivation (at −100 mV) and the values of their respective time constants in WT (+60 mV prepulse), 50% reduced WT (−70 mV prepulse), HSA LR (+60 mV prepulse), and Mbnl1 ΔE3/ΔE3 (+60 mV prepulse) are compared in Fig. 6 D. These results indicate that faster kinetics of deactivation observed in HSA LR and Mbnl1 ΔE3/ΔE3 does not result from a current-dependent effect on channel gating.
Figure 6.

Kinetics of ClC-1 deactivation after prepulse-dependent current reduction. (A) Representative family of ClC-1 currents (at−100 mV) recorded from an FDB fiber obtained from a 19-d-old WT mouse after 200-ms prepulses to potentials ranging from +60 to −140 mV (in 10-mV increments). (B) Voltage dependence of average time-dependent currents (Afast+Aslow) after prepulse-dependent current reduction. (Inset) Normalized ClC-1 currents (solid lines) fit with a second order exponential in control (60 mV prepulse; circles) and after ∼50% (average = 48.8 ± 0.1%, n = 8) reduction in current magnitude (−70 mV prepulse; squares). Black dashed line indicates the zero current level. (C, bottom) Voltage dependence of the average relative contribution of the fast (closed circles) and slow (open circles) components of ClC-1 deactivation (C, lower) and their respective time constants (C, top). (D) Average relative contribution of the fast and slow components of the time-dependent current (Afast+Aslow) and their respective time constants to ClC-1 deactivation (at −100 mV) in FDB fibers obtained from WT mice (+60 mV prepulse; black, n = 8), WT mice after ∼50% reduction in ClC-1 current magnitude (−70 mV prepulse; red, n = 8), HSA LR mice (+60 mV prepulse; green, n = 14), Mbnl1 ΔE3/ΔE3 mice (+60 mV prepulse; white; n = 11). *, P ≤ 0.05 compared with WT +60 mV prepulse.
Nonstationary Noise Analysis in FDB Fibers from Wild-Type, HSA LR, and Mbnl1 ΔE3/ΔE3 Mice
The absence of a significant difference in the voltage dependence of Porel between FDB fibers obtained from wild-type and HSA LR mice indicates that the observed reduction in ClC-1 current density in HSA LR fibers must result from a reduction in the number of functional channels (N), single channel conductance (γ), and/or maximal channel open probability (Pomax). To determine which of these parameters account for the ∼70% reduction in ClC-1 current density in HSA LR fibers, nonstationary noise analysis was performed from whole-cell patch clamp measurements of ClC-1 channel activity in FDB fibers of 12-d-old wild-type and 18–21-d-old HSA LR mice (Fig. 7). Fig. 7 A shows representative mean 9AC-sensitive ClC-1 currents recorded from 92 and 79 consecutive 200-ms voltage steps to −140 mV after a 200-ms prepulse at +60 mV from FDB fibers obtained from a 12-d-old wild-type (left) and a 19-d-old HSA LR (right) mouse, respectively. The corresponding time course of the current variance for each experiment is shown in Fig. 7 B and the mean– variance relationships are plotted in Fig. 7 C. The mean–variance plots were fitted according to Eq. 4 in order to extract the values of i, N, and Pomax (the latter from Eq. 5). Average values for i, γ, and Pomax obtained from native FDB fibers using this approach (Table II) are similar to those reported previously after heterologous expression of hClC-1 in tSA201 (Hebeisen et al., 2004), Xenopus oocytes (Rychkov et al., 1998), and HEK 293 cells (Pusch et al., 1994; Rosenbohm et al., 1999). We found that single channel ClCl-1 current amplitude (0.25 ± 0.03 pA and 0.30 ± 0.03 pA, respectively) and conductance (2.3 ± 0.02 pS and 2.8 ± 0.03 pS, respectively) were not significantly different between FDB fibers obtained from wild-type and HSA LR mice. A small, but significant (P < 0.01), reduction in maximum channel open probability was observed in HSA LR FDB fibers (0.91 ± 0.01 and 0.75 ± 0.03 for wild-type and HSA LR fibers, respectively). However, the major effect observed was a dramatic reduction (65%) in ClC-1 channel density HSA LR fibers (170 ± 21 and 58 ± 11 channels/pF for wild-type and HSA LR fibers, respectively), which was primarily responsible for the ∼70% reduction in ClC-1 current density (−37.8 ± 2.6 and −12.0 ± 1.4 pA/pF for wild-type and HSA LR fibers, respectively) observed in these experiments. Since there is an approximately twofold increase in macroscopic ClC-1 current density between postnatal day 12 and day 18 in HSALR mice (unpublished data), the observed reduction in peak ICl reported in Table II likely reflects a lower estimate of the decrease in ClC-1 activity. Qualitatively similar effects on ClC-1 channel magnitude (Fig. 8, A and B), voltage dependence of Porel (Fig. 8 C), deactivation kinetics (Fig. 6 D, white bars), and single channel properties assessed from nonstationary noise analysis (Fig. 8, D and E; Table II) were observed in experiments conducted in 12-d-old wild-type control and Mbnl1 knockout (Mbnl1 ΔE3/ΔE3) mice, a second mouse model for DM that exhibits aberrant Clcn1 splicing and prominent myotonia (Kanadia et al., 2003).
Figure 7.

Nonstationary noise analysis during ClC-1 deactivation in FDB fibers obtained from wild-type and HSA LR mice. (A) Representative mean currents in FDB fibers obtained from 12-d-old WT (left) and 18-d-old HSA LR (right) mice elicited using a 200-ms voltage step to −140 mV after a 100-ms prepulse to +60 mV. The initial 2 ms of each voltage step is blanked and the total duration of the traces truncated to 400 ms for clarity. (B) Variance time course for the currents shown in A. (C) Mean–variance relationships for the experiments shown in A and fit by Eq. 4 (see Table II for average values of fitted parameters).
TABLE II.
Parameters of Nonstationary Noise Analysis
| WT (n = 8) | HSA LR (n = 17) | Mbnl1 +/+ (n = 5) | Mbnl1 ΔE3/ΔE3 (n = 4) | |
|---|---|---|---|---|
| 12 d | 18–21 d | 12 d | 12 d | |
| Capacitance (pF) | 404 ± 26 | 722 ± 29b | 596 ± 46 | 439 ± 33a |
| i (pA) | 0.25 ± 0.03 | 0.30 ± 0.03 | 0.23 ± 0.04 | 0.26 ± 0.04 |
| g (pS) | 2.3 ± 0.02 | 2.8 ± 0.03 | 2.2 ± 0.04 | 2.5 ± 0.04 |
| Pomax | 0.91 ± 0.01 | 0.75 ± 0.03b | 0.86 ± 0.01 | 0.76 ± 0.03b |
| N | 69,165 ± 9,131 | 44,868 ± 9,892 | 173,908 ± 32,520 | 52,387 ± 9,589b |
| N/pF | 170 ± 21.0 | 58 ± 11.0b | 298 ± 65.0 | 121 ± 22.0a |
| Peak I Cl (pA/pF) | −37.8 ± 2.6 | −12.0 ± 1.4b | −60.8 ± 4.4 | −23.6 ± 3.2b |
Separate WT controls were conducted for HSA LR and Mbnl1 ΔE3/ΔE3 because the two mouse lines are on different genetic backgrounds (FVB and C57Bl6, respectively).
P < 0.05.
P < 0.01.
Figure 8.

Macroscopic ClC-1 current density is reduced in FDB fibers from Mbnl1 ΔE3/ΔE3 mice. (A) Representative family of ClC-1 currents recorded from FDB fibers obtained from 14-d-old WT (left) and Mbn1l ΔE3/ΔE3 (right) mice. (B) Average instantaneous ClC-1 current–voltage curves for fibers obtained from 9–14-d-old WT (closed circles, n = 22) and Mbnl1 ΔE3/ΔE3 (open triangles, n = 16) mice. (C) Average relative Po-V curves for FDB fibers obtained from 9–14-d-old WT (closed circles) and Mbnl1 ΔE3/ΔE3 (open triangles) mice. Smooth curves through each PO-V dataset were generated using a modified Boltzmann equation (Eq. 3). (D and E) Representative mean–variance relationships for FDB fibers obtained from 12-d-old WT (D) and Mbnl1 ΔE3/ΔE3 (E) mice elicited using a 200-ms voltage step to −140 mV after a 100-ms prepulse to +60 mV (see Table II for average values of fitted parameters).
DISCUSSION
The pathogenic mechanism for myotonia in DM has long been debated. Incomplete sodium channel inactivation (Mounsey et al., 2000) and an increase in the activity of small conductance, calcium-activated potassium channels have been suggested to contribute to increased muscle excitability and susceptibility to myotonia in DM (Renaud et al., 1986; Behrens et al., 1994; Kimura et al., 2003). Alternatively, myotonia in DM has also been suggested to arise from a dramatic reduction in transmembrane chloride conductance that results from expanded CUG repeat-containing mRNA molecules sequestering/inactivating MBNL1 proteins required for proper splicing of CLCN1 pre-mRNA (Mankodi et al., 2002; Kanadia et al., 2003). Our results obtained from two murine models for myotonia in DM1 are consistent with the later mechanism leading to a marked pathogenic reduction in the number of functional ClC-1 channels in the sarcolemma of skeletal muscle. Important novel contributions of this study include (a) the first complete biophysical characterization of ClC-1 function in native skeletal muscle fibers using macroscopic whole-cell and nonstationary noise analysis approaches; (b) functional characterization of a newly cloned murine ClC-1 channel (mClC-1) and demonstration that heterologous expression of mClC-1 in HEK293 cells and homologous expression in skeletal myotubes faithfully reproduces the biophysical properties of ClC-1 measured from native FDB fibers; (c) demonstration that increased muscle excitability in two mouse models of DM (HSA LR and Mbnl1 ΔE3/ΔE3 mice) is associated with a severe reduction in sarcolemmal ClC-1 activity stemming primarily from a decrease in the number of functional ClC-1 channels; and (d) evidence for reduced maximum ClC-1 channel open probability and accelerated kinetics of channel deactivation in FDB fibers of HSA LR and Mbnl1 ΔE3/ΔE3 mice. Together, these results provide strong support for the toxic RNA model for DM pathogenesis in which myotonia results from a transdominant effect of CUG repeat–containing mRNA on MBNL1-mediated control of proper splicing of CLCN1 pre-mRNA.
Biophysical Characterization of ClC-1 Channel Function in Native FDB Fibers
Macroscopic ClC-1 channel activity in skeletal muscle has previously been measured using double sucrose (Duval and Leoty, 1980), vaseline gap (Fahlke and Rudel, 1995), and two-electrode (Chen and Jockusch, 1999) voltage clamp techniques. However, under certain conditions, whole-cell patch clamp experiments can afford superior control over the intracellular environment, blockade of overlapping ionic currents, and provide enhanced voltage clamp speed and uniformity. Unfortunately, whole-cell voltage clamp measurements of ClC-1 channel activity have only been previously reported after heterologous expression of cloned ClC-1 channels (Steinmeyer et al., 1991; Pusch et al., 1994; Warnstedt et al., 2002). Here we report the first complete biophysical characterization of the voltage dependence and kinetics of ClC-1 channel activity using whole-cell voltage clamp and nonstationary noise analysis approaches in native skeletal muscle fibers. In our experiments, ClC-1 currents were carefully isolated by eliminating sodium (0 extracellular sodium), potassium (0 extracellular potassium, extracellular TEA, and intracellular cesium), and calcium (3 μM nifedipine) currents. In addition, for all experiments, leak and capacitative currents were removed by offline subtraction of 9AC-insensitive currents. The resulting 9AC-sensitive currents exhibited all of the hallmarks of ClC-1 currents observed for heterologously (e.g., Xenopus oocytes, Sf9 cells, COS-7 cells, HEK293 cells) expressed cloned ClC-1 channels (Figs. 1–3 ). Specifically, similar to heterologously expressed ClC-1 channels, ClC-1 currents in native FDB fibers exhibited chloride selectivity (Erev being close to ECl) and instantaneous and steady-state current–voltage relationships with characteristic inward rectification and “S”-shaped voltage dependence, respectively (Fig. 2, A–D). Moreover, relative Po–voltage relationships based on normalized isochronal measurements of tail current amplitudes exhibited sigmoidal voltage dependence with similar slope and V1/2 values (Fig. 2 E and Table I). Finally, ClC-1 current deactivation in native FDB fibers and mClC-1–expressing myotubes and HEK293 cells were all well described by a biexponential function (Fig. 3). These results demonstrate the reliability of our biophysical characterization of ClC-1 activity in native FDB fibers.
Coonan and Lamb (1998) suggested that a second chloride channel (insensitive to extracellular 9AC) may contribute to the total chloride conductance in rat skeletal muscle. Our results in which 1 mM 9AC rapidly blocked 98% of all chloride conductance under our recording conditions (Fig. 1, right), argues against a significant contribution of 9AC-insensitive chloride channels in mouse FDB fibers. However, we cannot rule out a potential role of calcium-activated chloride channels that would likely be inhibited/blocked under our recording conditions (i.e., in the presence of 10 mM intracellular EGTA).
Influence of Cellular Context on ClC-1 Activity
We carefully compared the biophysical properties (voltage dependence of instantaneous, steady-state, Porel, and kinetics of channel deactivation) of ClC-1 channel activity in native murine FDB fibers with that of mClC1 expressed in both HEK293 cells and primary cultures of skeletal myotubes. To our knowledge, this study is the first to provide a comprehensive, side-by-side comparison of the functional properties of exogenously expressed mClC-1 channels to that of native ClC-1 currents. We compared murine ClC-1 channel properties under three separate cellular contexts (native FDB fibers and mClC-1 expressed in HEK293 cells and primary mouse myotubes). Not surprisingly the macroscopic properties of mClC-1 expressed in HEK293 cells reported here are nearly identical to those previously reported for heterologous expression of cloned human (87% identical) and rat (96% identical) ClC-1 recorded under similar conditions (Rychkov et al., 1998). We also characterized mClC-1 properties after homologous expression in primary skeletal myotubes since myotubes represent an early developmental state of muscle that lacks endogenous ClC-1 channel expression (Fig. 2 B, upright triangles) (Conte Camerino et al., 1989; Wischmeyer et al., 1993; Bardouille et al., 1996). Thus, comparing mClC-1 properties after heterologous expression in HEK293 cells with that observed in native FDB fibers and after homologous expression in myotubes could reveal potential “muscle-specific” modulatory effects on ClC-1 function that may not be faithfully reproduced using standard heterologous expression systems (e.g., HEK293 cells). Overall, ClC-1 functional properties (e.g., reversal potential, inward rectification of the instantaneous I-V relationship, voltage dependence of steady-state activation and Porel, deactivation kinetics) were remarkably similar in HEK293 cells, myotubes, and native FDB fibers (Figs. 1–3 ). However, we found that the relative rate of ClC-1 channel deactivation across most voltages was significantly slower in FDB fibers and mClC-1–expressing myotubes (Fig. 3). Thus, it is possible that muscle-specific regulatory mechanisms absent in HEK293 cells may influence ClC-1 gating. Nevertheless, the overall similarity of ClC-1 biophysical properties in native murine FDB fibers and mClC-1–expressing myotubes and HEK293 cells indicates that basal ClC-1 function is not markedly influenced by cellular context and provides strong justification for the evaluation of mClC-1 function using heterologous expression systems.
Previous work indicates that ClC-1 transcription in native tissue is strongly dependent on muscle electrical activity. During muscle denervation, there is a rapid decrease in the levels of ClC-1 transcript in parallel with a reduction in ClC-1 channel activity (Conte Camerino et al., 1989; Klocke et al., 1994; Chen and Jockusch, 1999). Papponen et al. (2005) used antibodies raised against the C terminus of ClC-1 to suggest that ClC-1 protein is not detectable in the sarcolemma of freshly dissociated FDB fibers. This study also found that overnight incubation with staurosporin was required for reinsertion of ClC-1 channels into the sarcolemma. Additionally, a recombinant Semliki Forest virus was used to express myc-tagged ClC-1 proteins in myoblasts and myotubes derived from rat L6 myogenic cells. Surprisingly, ClC-1 proteins were found to be localized to unidentified intracellular vesicles and not in the sarcolemma after expression in L6 myoblasts and myotubes (Papponen et al., 2005). However, in these studies, sarcolemmal ClC-1 expression was only assessed using imaging approaches and was not corroborated using electrophysiology recordings of transmembrane ClC-1 currents. Under our experimental conditions, we were able to measure robust ClC-1 channel activity in both freshly dissociated FDB fibers and after nuclear injection of mClC-1 cDNA in primary mouse skeletal myotubes. Since our experiments provide no measure of ClC-1 channel activity before fiber isolation, significant ClC-1 internalization during dissociation is possible. However, we observed no significant time-dependent decrease in ClC-1 current density up to 8 h after fiber isolation and only a 16% decrease >24 h after isolation (unpublished data).
Role of ClC-1 Channel Dysfunction in Two Mouse Models of Myotonic Dystrophy
Transmembrane chloride conductance (GCl) comprises 70–80% of the total resting membrane conductance in mammalian skeletal muscle, the remainder of which results from background potassium channels (Rudel and Lehmann-Horn, 1985). This large GCl acts to stabilize the resting membrane potential. During activation of skeletal muscle, action potentials rapidly propagate deep into the muscle fiber along the transverse tubule (t-tubule) membrane system. The extensive t-tubule invaginations create a diffusion-limited space that can result in significant potassium accumulation during trains of muscle excitation (Neelands et al., 2001). In the absence of a large GCl, this potassium accumulation depolarizes the membrane potential and promotes muscle excitability. Thus, ClC-1 channels in skeletal muscle provide a critical short-circuit current that acts to counteract the excitatory influence of excessive potassium accumulation within the t-tubule system. Reduction of GCl by >50% is sufficient to promote myotonia in both humans and experimental animals (Bryant and Morales-Aguilera, 1971; Rudel and Lehmann-Horn, 1985), as demonstrated through partial ClC-1 current blockade using aromatic monocarboxylic acid channel blockers (Furman and Barchi, 1978). Moreover, mutations in CLCN1 result in both dominant and recessive myotonia congenita (Pusch, 2002). Dominant mutations in ClC-1 typically produce a depolarizing shift in the voltage dependence of ClC-1 activation, which results in a decrease in ClC-1 open probability at the resting membrane potential, and consequently an increased susceptibility to myotonia in goats (Beck et al., 1996) and humans (Rudel et al., 1988). In general, recessive mutations result in nonfunctional channels and an elimination of sarcolemmal ClC-1 conductance. However, DM has not been linked to mutations in CLCN1 or any other ion channel gene.
Our results indicate that macroscopic ClC-1 current density (measured at −140 mV) was reduced 70% and 61% in freshly dissociated FDB fibers isolated from 18–20-d-old HSA LR and Mbnl1 ΔE3/ΔE3 (Figs. 4 and 8) mice, respectively. This reduction in ClC-1 function may actually be an underestimate of the effect observed at physiologic potentials since a greater degree of inward rectification was observed for ClC-1 currents recorded from FDB fibers of HSA LR and Mbnl1 ΔE3/ΔE3 mice (Fig. 4 C and Fig. 8 B). Nonstationary noise analysis revealed that this reduction in macroscopic ClC-1 current density arises from a marked decrease in number of functional channels and a minor decrease (16%) in Pomax (Table II). However, since the small reduction in Pomax occurred in the absence of a significant shift in the voltage dependence of the Porel (Fig. 4 D and Fig. 8 C), a decrease in Pomax can only contribute minimally to the observed reduction in macroscopic ClC-1 current. In addition, no change in single channel conductance (γ) was observed in FDB fiber obtained from HSA LR or Mbnl1 ΔE3/ΔE3 mice. Rather, the observed reduction in macroscopic ClC-1 current density in HSA LR and Mbnl1 ΔE3/ΔE3 fibers resulted primarily from a reduction in ClC-1 channel density in both animal models (Table II). This reduction in the number of functional ClC-1 channels could arise from a decrease in the amount of full-length ClC-1 protein, dominant-negative effects of truncated isoforms on the full-length protein (Berg et al., 2004), or a combination of both. Distinguishing between the relative impact of these potential mechanisms will require determining the expression levels of full-length and truncated ClC-1 proteins and also assessing the effects of truncated isoforms on full-length protein expression, stability, and function.
Although ClC-1 currents in FDB fibers obtained from HSA LR and Mbnl1 ΔE3/ΔE3 mice exhibited very similar properties to those observed for wild-type fibers, ClC-1 currents from the animal models deactivated significantly faster than wild-type fibers. This difference in the rate of channel deactivation resulted from parallel shifts in the relative proportion of the fast and slow components of channel deactivation with minimal effects on the time constants of each component. Aberrant splicing of Clcn1 pre-mRNA leads to the production of truncated transcripts (Mankodi et al., 2002), that have been shown to both lower ClC-1 current density and modify the rate of channel deactivation (Berg et al., 2004). The most prominent aberrant splice product (44 ± 3%) identified in HSA LR muscle results from the inclusion of an additional exon cassette between exons 6 and 7 (named exon 7a), which leads to the introduction of a premature stop codon (Mankodi et al., 2002). A similar splice product, which includes two additional exon cassettes 6b and 7a, is expressed in human DM1 muscle. The DM1 (exon 6b/7a+) and HSA LR (exon7a+) variants are truncated at similar positions (codon 283 in DM1, codon 290 in HSA LR). Recently, Berg et al. (2004) found that coinjection of Xenopus oocytes with the 283x (exon 6b/7a+) splice variant results in a significant reduction in wild-type hClC-1 current density and an acceleration in channel deactivation. Thus, our findings of a small, but significant, reduction in ClC-1 Pomax observed in nonstationary noise analysis experiments (Table II) and accelerated channel deactivation are consistent with the results of Berg et al. (2004) and support the conclusion that truncated ClC-1 protein products exert a dominant-negative effect on full-length ClC-1 channels in DM1 muscle. In addition, the observed reduction in the number of functional ClC-1 channels might also result from dominant-negative effects of truncated fragments on ClC-1 trafficking, degradation, or function. Together our observation of decreased Pomax and accelerated deactivation kinetics in FDB fibers from HSA LR and Mbnl1 ΔE3/ΔE3 mice coupled with the heterologous expression studies of Berg et al. (2004) suggest that truncated ClC-1 fragments exert dominant-negative effects on ClC-1 channel activity.
Molecular Mechanism for a Pathogenic Reduction in Functional ClC-1 Channel Density in DM1
The role of altered ClC-1 function in mediating the myotonic phenotype of DM1 has long been debated. No mutations in the CLCN1 gene have been identified in DM1 patients, indicating that any effect on ClC-1 function is likely to be indirect. In this study, we found that the number of functional ClC-1 channels in FDB fibers of HSA LR and Mbnl1 ΔE3/ΔE3 mice is reduced to levels sufficient to account for an increased susceptibility to myotonia. Although effects of truncated fragments on functional ClC-1 channel trafficking/degradation cannot be ruled out (see above), the observed reduction in functional ClC-1 channel density (66%) in HSA LR fibers correlates well with the incidence of aberrant Clcn1 pre-mRNA splicing (76%). This supports the idea that the reduction in the number of functional ClC-1 channels primarily reflects a reduction in the level of Clcn1 mRNA that encodes full-length channels (Mankodi et al., 2002). Thus, a major fraction of the reduction in the number of functional ClC-1 channels likely results from a posttranscriptional effect. In any event, the observed reduction in number of functional ClC-1 channels in FDB fibers of HSA LR and Mbnl1 ΔE3/ΔE3 mice is sufficient to account for the prominent myotonia observed in these two murine models of DM.
A central question is how do transcripts containing expanded CUG repeats and MBNL1 deficiency both lead to a similar reduction in Clcn1 mRNA, and subsequently, a parallel decrease in the number of functional ClC-1 channels? One potential mechanism is outlined by the molecular model shown in Fig. 9. This model, which is derived from both prior published reports and the functional results presented in this study, illustrates how proper pre-mRNA splicing of Clcn1 transcripts and subsequent production of sufficient ClC-1 channel activity leads to protection from myotonia in wild-type skeletal muscle (Fig. 9, left) and how this process might be disrupted in DM1 (Fig. 9, right). In normal muscle, MBNL1 proteins translocate to the nucleus and direct proper splicing of a subset of genes that includes CLCN1. Proper excision of introns from CLCN1 pre-mRNA results in the generation of full-length transcripts that are exported from the nucleus and used to produce functional ClC-1 homodimeric channels. In DM1, a CTG repeat expansion in the 3′ untranslated region of the DMPK gene results in the production of mRNA species that contain several hundred to several thousand CUG repeats. The expanded CUG repeats form a secondary hairpin structure that is recognized by MBNL1, a regulator of alternative splicing. The CUG-MBNL1 complex is retained in the nucleus in discrete foci because the amount of expanded CUG repeat RNA is sufficient to sequester MBNL1 protein. The resulting MBNL1 deficiency leads to aberrant splicing of Clcn1 pre-mRNA and inclusion of additional exons that introduce premature stop codons; the predominant splice defect involves inclusion of a portion of intron 7 (termed exon7a). Some of the resulting aberrantly spliced transcripts are degraded through the nonsense-mediated decay pathway while others are exported and direct the translation of nonfunctional truncated ClC-1 protein products. This results not only in a dramatic reduction in the number of functional full-length ClC-1 channels, but also in the accumulation of truncated fragments that potentially exert dominant-negative effects on full-length ClC-1 channel current magnitude (Berg et al., 2004), deactivation kinetics (Berg et al., 2004; Fig. 5 and Fig. 6 D), and maximum channel open probability (Table II). Together, these effects result in a large reduction in transmembrane ClC-1 channel activity, and therefore increased susceptibility to myotonia due to the loss of an essential short-circuit current required to counteract the excitatory influence of potassium accumulation in the t-tubule system during muscle excitation.
Figure 9.

Proposed molecular model for increased muscle excitability in DM1. (Left) Proper CLCN1 pre-mRNA splicing in normal skeletal muscle is regulated by muscleblind-like 1 (MBNL1) proteins, which are shown to bind to hypothetical intronic splice repressor elements in intron 6. (Right) Altered CLCN1 pre-mRNA splicing in DM1 arises from DMPK transcripts with expanded repeats (CUG)n accumulating in nuclear foci and sequestering double-stranded CUG binding proteins, including MBNL1. Depletion of MBNL1 proteins results in inclusion of additional exons (e.g., exon 7a) containing premature termination codons. Additionally, aberrantly spliced CLCN1 transcripts are subsequently exported from the nucleus, degraded through the nonsense-mediated decay pathway, and/or produce truncated proteins that potentially exert additional dominant-negative effects on ClC-1 function (Berg et al., 2004). These effects result in a dramatic reduction in the number of functional ClC-1 channels and a subsequent increase in muscle excitability.
The model depicted in Fig. 9 explains results of this study obtained using two murine models for myotonia in DM1. First, the decrease in the number of functional ClC-1 channels in HSA LR mice likely results from aberrant splicing of Clcn1 pre-mRNA caused by muscle- specific expression of expanded CUG repeat–containing mRNA species that bind/sequester Mbnl1 proteins. Loss of Mbnl1-mediated regulation of proper Clcn1 splicing leads to a severe reduction in the level of full-length Clcn1 mRNA and the production of aberrantly spliced Clcn1 transcripts that encode truncated protein products that may exert additional dominant-negative effects on ClC-1 function; effects that together lead to a subsequent pathogenic reduction in ClC-1 current density. A similar cascade of events would also be expected to occur during Mbnl1 deficiency in skeletal muscle of Mbnl1 ΔE3/ΔE3 mice. Important implications of this pathogenic mechanism are that exogenous delivery of either wild-type ClC-1 channels or Mbnl1 proteins might prove effective in rescuing myotonia in HSA LR and Mbnl1 ΔE3/ΔE3 mice (Kanadia et al., 2006) and, if so, would provide a “proof of concept” for ClC-1 and/or Mbnl1 gene therapeutic approaches for the treatment of myotonia in DM.
Acknowledgments
We would like to thank Dr. Michael Pusch for providing access to and assistance with the use of his Ana analysis software package and to Dr. Ted Begenisich for helpful comments and suggestions for improving the manuscript.
This work was supported by a research grant from the Muscular Dystrophy Association (to R.T. Dirksen), grants from the National Institutes of Health (AR44657 to R.T. Dirksen and AR46806 to C.A. Thornton), a University of Rochester Paul D. Wellstone Muscular Dystrophy Cooperative Research Center grant (AR050762), and a National Institute of Dental and Craniofacial Research training grant (T32DE07202 to J.D. Lueck).
Olaf S. Andersen served as editor.
Abbreviations used in this paper: 9AC, 9-anthracene carboxylic acid; ClC-1, chloride channel type 1; DMPK, myotonic dystrophy protein kinase; DM1, myotonic dystrophy type 1; FDB, flexor digitorum brevis; HSA LR, human skeletal actin long repeat mouse; MBNL1, muscleblind-like 1; SCN4A, skeletal muscle voltage-dependent sodium channel.
References
- Bardouille, C., D. Vullhorst, and H. Jockusch. 1996. Expression of chloride channel mRNA in cultured myogenic cells: a marker of myotube maturation. FEBS Lett. 396:177–180. [DOI] [PubMed] [Google Scholar]
- Beam, K.G., and C.M. Knudson. 1988. Calcium currents in embryonic and neonatal mammalian skeletal muscle. J. Gen. Physiol. 91:781–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, C.L., C. Fahlke, and A.L. George. 1996. Molecular basis for decreased muscle chloride conductance in the myotonic goat. Proc. Natl. Acad. Sci. USA. 93:11248–11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens, M.I., P. Jalil, A. Serani, F. Vergara, and O. Alverez. 1994. Possible role of apamin-sensitive K+ channels in myotonic dystrophy. Muscle Nerve. 17:1264–1270. [DOI] [PubMed] [Google Scholar]
- Berg, J., H. Jiang, C.A. Thornton, and S.C. Cannon. 2004. Truncated ClC-1 mRNA in myotonic dystrophy exerts a dominant-negative effect on the Cl− current. Neurology. 63:2371–2375. [DOI] [PubMed] [Google Scholar]
- Bretag, A.H. 1987. Muscle chloride channels. Physiol. Rev. 67:618–724. [DOI] [PubMed] [Google Scholar]
- Brook, J.D., M.E. McCurrach, H.G. Harley, A.J. Buckler, D. Church, H. Aburatani, K. Hunter, V.P. Stanton, J.P. Thirion, T. Hudson, et al. 1992. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat located at the 3′ end of a transcript encoding a protein kinase family member. Cell. 68:799–808. [DOI] [PubMed] [Google Scholar]
- Bryant, S.H., and A. Morales-Aguilera. 1971. Chloride conductance in normal and myotonic muscle fibres and the action of monocarboxylic aromatic acids. J. Physiol. 219:367–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M.F., and H. Jockusch. 1999. Role of phosphorylation and physiological state in the regulation of the muscular chloride channel ClC-1: a voltage-clamp study on isolated M. interosseus fibers. Biochem. Biophys. Res. Commun. 261:528–533. [DOI] [PubMed] [Google Scholar]
- Conte Camerino, D., A. De Luca, M. Mambrini, and G. Vrbova. 1989. Membrane ionic conductances in normal and denervated skeletal muscle of the rat during development. Pflugers Arch. 413:568–570. [DOI] [PubMed] [Google Scholar]
- Coonan, J.R., and G.D. Lamb. 1998. Effect of transverse-tubular chloride conductance on excitability in skinned skeletal muscle fibres of rat and toad. J. Physiol. 509:551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, B.M., M.E. McCurrach, K.L. Taneja, R.H. Singer, and D.E. Housman. 1997. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl. Acad. Sci. USA. 94:7388–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval, A., and C. Leoty. 1980. Ionic currents in slow twitch skeletal muscle in the rat. J. Physiol. 307:23–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlke, C., and R. Rudel. 1995. Chloride currents across the membrane of mammalian skeletal muscle fibres. J. Physiol. 484:355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furling, D., D. Lemieux, K. Taneja, and J. Puymirat. 2001. Decreased levels of myotonic dystrophy protein kinase (DMPK) and delayed differentiation in human myotonic dystrophy myoblasts. Neuromuscul. Disord. 11:728–735. [DOI] [PubMed] [Google Scholar]
- Furman, R.E., and R.L. Barchi. 1978. The pathophysiology of myotonia produced by aromatic carboxylic acids. Ann. Neurol. 4:357–365. [DOI] [PubMed] [Google Scholar]
- Hamill, O.P., A. Marty, E. Neher, B. Sakmann, and F.J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Harper, P.S. 1989. Myotonic Dystrophy. Second edition. Saunders, London. 316–320.
- Hebeisen, S., A. Biela, B. Giese, G. Muller-Newen, P. Hidalgo, and C. Fahlke. 2004. The role of the carboxy-terminus in ClC chloride channel function. J. Biol. Chem. 279:13140–13147. [DOI] [PubMed] [Google Scholar]
- Heinemann, S.H., and F. Conti. 1992. Nonstationary noise analysis and application to patch clamp recordings. Methods Enzymol. 207:131–148. [DOI] [PubMed] [Google Scholar]
- Ho, T.H., N. Charlet-B, M.G. Poulos, S. Singh, M.S. Swanson, and T.A. Cooper. 2004. Muscleblind proteins regulate alternative splicing. EMBO J. 23:3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanadia, R.N., K.A. Johnstone, A. Mankodi, C. Lungu, C.A. Thornton, D. Esson, A.M. Timmers, W.W. Hauswirth, and M.S. Swanson. 2003. A muscleblind knockout model for myotonic dystrophy. Science. 302:1978–1980. [DOI] [PubMed] [Google Scholar]
- Kanadia, R.N., J. Shin, Y. Yuan, S.G. Beattie, T. Wheeler, C.A. Thornton, and M.S. Swanson. 2006. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. USA. 103:11748–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, T., M.P. Takahashi, H. Fujimura, and S. Sakoda. 2003. Expression and distribution of a small-conductance calcium-activated potassium channel (SK3) protein in skeletal muscles from myotonic muscular dystrophy patients and congenital myotonic mice. Neurosci. Lett. 347:191–195. [DOI] [PubMed] [Google Scholar]
- Klocke, R., K. Steinmeyer, T.J. Jentsch, and H. Jockusch. 1994. Role of innervation, excitability, and myogenic factors in the expression of the muscular chloride channel ClC-1. A study on normal and myotonic muscle. J. Biol. Chem. 269:27635–27639. [PubMed] [Google Scholar]
- Liquori, C.L., K. Ricker, M.L. Moseley, J.F. Jacobsen, and W. Kress. 2001. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 293:864–867. [DOI] [PubMed] [Google Scholar]
- Mankodi, A., E. Logigian, L. Callahan, C. McClain, R. White, D. Henderson, M. Krym, and C.A. Thornton. 2000. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 289:1769–1772. [DOI] [PubMed] [Google Scholar]
- Mankodi, A., C.R. Urbinati, Q.P. Yuan, R.T. Moxley, V. Sansone, M. Krym, D. Henderson, M. Schalling, M.S. Swanson, and C.A. Thornton. 2001. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 10:2165–2170. [DOI] [PubMed] [Google Scholar]
- Mankodi, A., M.P. Takahashi, H. Jiang, C.L. Beck, W.J. Bowers, R.T. Moxley, S.C. Cannon, and C.A. Thornton. 2002. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell. 10:35–44. [DOI] [PubMed] [Google Scholar]
- Mounsey, J.P., D.J. Mistry, C.W. Ai, S. Reddy, and J.R. Moorman. 2000. Skeletal muscle sodium channel gating in mice deficient in myotonic dystrophy protein kinase. Hum. Mol. Genet. 9:2313–2320. [DOI] [PubMed] [Google Scholar]
- Nakai, J., R.T. Dirksen, H.T. Nguyen, I.N. Pessah, K.G. Beam, and P.D. Allen. 1996. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 380:72–75. [DOI] [PubMed] [Google Scholar]
- Neelands, T.R., P.S. Herson, D. Jacobson, J.P. Adelman, and J. Maylie. 2001. Small-conductance calcium-activated potassium currents in mouse hyperexcitable denervated skeletal muscle. J. Physiol. 536:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papponen, H., T. Kaisto, V.V. Myllyla, R. Myllyla, and K. Metsikko. 2005. Regulated sarcolemmal localization of the muscle-specific ClC-1 chloride channel. Exp. Neurol. 191:163–173. [DOI] [PubMed] [Google Scholar]
- Pusch, M. 2002. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum. Mutat. 19:423–434. [DOI] [PubMed] [Google Scholar]
- Pusch, M., K. Steinmeyer, and T.J. Jentsch. 1994. Low single channel conductance of the major skeletal muscle chloride channel, ClC-1. Biophys. J. 66:149–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch, M., K. Steinmeyer, M.C. Koch, and T.J. Jentsch. 1995. Mutations in dominant human myotonia congenita drastically alter the voltage dependence of the ClC-1 chloride channel. Neuron. 15:1455–1463. [DOI] [PubMed] [Google Scholar]
- Renaud, J.F., C. Desnuelle, H. Schmid-Antomarchi, M. Hugues, G. Serratrice, and M. Lazdunski. 1986. Expression of apamin receptor in muscles of patients with myotonic muscular dystrophy. Nature. 319:678–680. [DOI] [PubMed] [Google Scholar]
- Rosenbohm, A., R. Rudel, and C. Fahlke. 1999. Regulation of the human skeletal muscle chloride channel hClC-1 by protein kinase C. J. Physiol. 514:677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel, R., and F. Lehmann-Horn. 1985. Membrane changes in cells from myotonia patients. Physiol. Rev. 65:310–356. [DOI] [PubMed] [Google Scholar]
- Rudel, R., K. Ricker, and F. Lehmann-Horn. 1988. Transient weakness and altered membrane characteristic in recessive generalized myotonia (Becker). Muscle Nerve. 11:202–211. [DOI] [PubMed] [Google Scholar]
- Rychkov, G.Y., M. Pusch, M.L. Roberts, T.J. Jentsch, and A.H. Bretag. 1998. Permeation and block of the skeletal muscle chloride channel, ClC-1, by foreign anions. J. Gen. Physiol. 111:653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth, F.J. 1980. The variance of sodium current fluctuations at the node of Ranvier. J. Physiol. 307:97–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmeyer, K., C. Ortland, and T.J. Jentsch. 1991. Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature. 354:301–308. [DOI] [PubMed] [Google Scholar]
- Taneja, K.L., M. McCurrach, M. Schalling, D. Housman, and R.H. Singer. 1995. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 128:995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z.M., M.L. Messi, and O. Delbono. 1999. Patch-clamp recording of charge movement, Ca2+ current, and Ca2+ transients in adult skeletal muscle fibers. Biophys. J. 77:2709–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnstedt, M., C. Sun, B. Poser, M.J. Escriva, L. Tranebjarg, T. Torbergsen, M. van Ghelue, and C. Fahlke. 2002. The myotonia congenita mutation A331T confers a novel hyperpolarization-activated gate to the muscle chloride channel ClC-1. J. Neurosci. 22:7462–7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischmeyer, E., E. Nolte, R. Klocke, H. Jockusch, and H. Brinkmeier. 1993. Development of electrical myotonia in the ADR mouse: role of chloride conductance in myotubes and neonatal animals. Neuromuscul. Disord. 3:267–274. [DOI] [PubMed] [Google Scholar]