

Abstract
In the central nervous system, electrogenic sodium- and potassium-coupled glutamate transporters terminate the synaptic actions of this neurotransmitter. In contrast to acidic amino acids, dicarboxylic acids are not recognized by glutamate transporters, but the related bacterial DctA transporters are capable of transporting succinate and other dicarboxylic acids. Transmembrane domain 8 contains several residues that differ between these two types of transporters. One of these, aspartate-444 of the neuronal glutamate transporter EAAC1, is conserved in glutamate transporters, but a serine residue occupies this position in DctA transporters. When aspartate-444 is mutated to serine, cysteine, alanine, or even to glutamate, uptake of d-[3H]-aspartate as well as the inwardly rectifying steady-state currents induced by acidic amino acids is impaired. Even though succinate was not capable of inducing any steady-state transport currents, the dicarboxylic acid inhibited the sodium-dependent transient currents by the mutants with a neutral substitution at position 444. In the neutral substitution mutants inhibition of the transients was also observed with acidic amino acids. In the D444E mutant, acidic amino acids were potent inhibitors of the transient currents, whereas the apparent affinity for succinate was lower by at least three orders of magnitude. Even though L-aspartate could bind to D444E with a high apparent affinity, this binding resulted in inhibition rather than stimulation of the uncoupled anion conductance. Thus, a carboxylic acid–containing side chain at position 444 prevents the interaction of glutamate transporters with succinate, and the presence of aspartate itself at this position is crucial for productive substrate binding compatible with substrate translocation.
INTRODUCTION
Sodium-coupled glutamate transporters serve to keep the synaptic transmitter concentrations below neurotoxic levels. These transporters are key elements in the termination of the synaptic actions of this neurotransmitter. Glutamate uptake is an electrogenic process (Kanner and Sharon, 1978; Brew and Attwell, 1987) in which the transmitter is cotransported with three sodium ions and one proton (Zerangue and Kavanaugh, 1996; Levy et al., 1998), followed by the countertransport of one potassium ion (Kanner and Bendahan, 1982; Pines and Kanner, 1990; Kavanaugh et al., 1997). The mechanism involving two half cycles is supported by the fact that mutants impaired in potassium interaction are locked in an obligatory exchange mode (Kavanaugh et al., 1997; Zhang et al., 1998). Glutamate transporters mediate two distinct types of substrate-induced steady-state current: an inward-rectifying current, reflecting electrogenic sodium-coupled glutamate translocation, and an “uncoupled” sodium-dependent current, which is carried by chloride ions and further activated by substrates of the transporter (Fairman et al., 1995; Wadiche et al., 1995a; Arriza et al., 1997). Moreover, when the membrane voltage is suddenly changed in the absence of substrate, sodium-dependent transient currents are observed (Wadiche et al., 1995b). These transients presumably represent a charge-moving conformational change following binding/debinding of sodium. Addition of a transportable substrate enables electrogenic transport, and thereby converts the transient current into a steady-state current (Wadiche et al., 1995b). On the other hand, nontransportable analogues block the transients, presumably by locking the transporter in one conformation (Wadiche et al., 1995b).
Recently a high-resolution crystal structure of a glutamate transporter homologue, GltPh, from the archeon Pyrococcus horikoshii was published (Yernool et al., 2004). It forms a trimer with a permeation pathway through each of the monomers. The membrane topology of the monomer (Yernool et al., 2004) is quite unusual but is in excellent agreement with the topology inferred from biochemical studies (Grunewald et al., 1998; Slotboom et al., 1999; Grunewald and Kanner, 2000). The monomer contains eight transmembrane domains (TM) and two oppositely oriented reentrant loops, one between domains 6 and 7 and the other between domains 7 and 8. The binding pocket is predominantly formed by TMs 7 and 8 and the two reentrant loops, which in the crystal structure enclose a nonprotein density presumably corresponding to glutamate (Yernool et al., 2004). Importantly, many of the amino acid residues of the transporter, inferred to be important in the interaction with sodium (Zhang and Kanner, 1999; Borre and Kanner, 2001) and potassium (Kavanaugh et al., 1997; Zhang et al., 1998) face the binding pocket and are close to the nonprotein density (Yernool et al., 2004) and this is also true for the side chains of the GltPh equivalents of aspartate-444 and arginine-447, both located in TM 8 of the neuronal transporter EAAC1 (Kanai and Hediger, 1992). Arginine-447 has been demonstrated to be involved in binding the γ-carboxyl group of glutamate (Bendahan et al., 2000). This residue is conserved in eukaryotic and bacterial glutamate transporters as well as in bacterial dicarboxylic acid transporters DctA (Engelke et al., 1989), but not in the two neutral amino acid transporters from the SLC1 family (Fig. 1 A). The substrates of the latter two transporters have in common that they do not possess the extra carboxyl group present in the substrates of the other two classes of transporter. In contrast to amino acids, dicarboxylic acids lack the amino group and this may be correlated with intriguing differences in the TM 8 residues surrounding arginine-447 (Fig. 1 A) between the transporters of dicarboxylic acids and those of amino acids. In this study we have analyzed the impact of mutations in these residues from EAAC1 and document that the presence of a carboxyl group at position 444 prevents the interaction of the transporter with succinate. Moreover the presence of aspartate at this position is crucial for the productive interaction with substrates that leads to transport.
Figure 1.

Amino acid sequence alignment of representative members of the glutamate transporter family and substrate transport by replacement mutants at these positions. (A) Amino acid sequence alignment of the representative members of the glutamate transporter family. EAAC, excitatory amino acid carrier; GLT, glutamate transporter; ASCT, alanine serine cysteine transporter; DctA Ec, dicarboxylate transporter Escherichia coli; DctA Rle, dicarboxylate transporter Rhizobium leguminosarum. (B) Uptake of d-[3H]-aspartate in Xenopus laevis oocytes expressing mutants in the stretch 444DRFRTVV450 (EAAC1 numbering) was done as described under Materials and Methods. Except for arginine-447, each residue of this stretch in EAAC1 was replaced by the equivalent residue of DctA Ec. Aspartate-444 of EAAC1 was changed not only to serine, but also to the other residues indicated. The values are corrected for those obtained with uninjected oocytes and are given as percent of WT uptake. Data shown are mean ± SEM, n = 3. (C) Oocytes expressing WT or the mutants D444S, D444C, and D444E were voltage clamped and gravity perfused with ND96 recording solution (see Materials and Methods) with and without 2 mM L-aspartate. The voltage was stepped from −25 mV to voltages between −100 and +40 mV, in increments of 10 mV. Each potential was held clamped for 250 ms, and the steady-state current from 210 to 240 ms at each potential was averaged. The current in the absence of L-aspartate was subtracted from that in its presence (I) and plotted against the holding potential (Vhold). The values shown are mean ± SEM from three oocytes from different batches that had similar expression levels.
MATERIALS AND METHODS
Site-directed Mutagenesis
The rabbit glutamate transporter EAAC1 with 10 histidines added immediately after the open reading frame followed by the stop codon in the vector pBluescriptSK (Borre and Kanner, 2004) was the construct used for site-directed mutagenesis (Kunkel et al., 1987; Pines et al., 1995). Restriction enzymes NsiI and StuI were used to subclone the fragment carrying the mutation into his-tagged WT EAAC1, which was cloned into the POG1 plasmid. The latter is an oocyte expression vector containing a 5′-untranslated Xenopus β-globin sequence and a 3′-poly (A) signal. The subcloned DNA fragments were sequenced on both strands between the two above-mentioned restriction sites.
cRNA Transcription, Injection, and Oocyte Preparation
cRNA was transcribed using mMESSAGE-mMACHINE (Ambion), injected into Xenopus laevis oocytes, and maintained as described previously (Borre and Kanner, 2001).
Oocyte Electrophysiology and Radioactive Uptake
Xenopus leavis oocytes were placed in the recording chamber, penetrated with two agarose-cushioned micropipettes (1%/2 M KCl, resistance varied between 1 and 3 mΩ), voltage-clamped using GeneClamp 500 (Axon Instruments), and digitized using Digidata 1322 (Axon Instruments), both controlled by the pClamp9.0 suite (Axon Instruments). Voltage jumping was performed using a conventional two-electrode voltage-clamp circuit (Borre and Kanner, 2004). Unless stated otherwise, oocytes were stepped from −100 to +40 mV in +10-mV increments, using −25 mV as holding potential. Each potential was held clamped for 250 ms. For each of the current traces shown, the “on” voltage jumps were at 8 ms, and after 250 ms at the test potential the oocytes were held clamped at the holding potential for another 242 ms before jumping the voltage to the next test potential. The membrane potential was measured relative to an extracellular Ag/AgCl electrode in the recording chamber. Penetrated oocytes were gravity perfused with ND96-recording solution (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH 7.5). In cation substitution experiments, NaCl was replaced by equimolar concentrations of choline chloride. In experiments using the highly permeant thiocyanate anion, all (Fig. 8) or part (Fig. 9) of the NaCl was replaced by NaSCN or by KSCN (Fig. 9 B). In these experiments, an agarose bridge (2%/2 M KCl) connected the extracellular grounding electrode to the recording chamber. Before applying either of the sulfhydryl reagents, (2-trimethylammonium ethyl)methanethiosulfonate (MTSET) or (2-sulfonatoethyl) methanethiosulfonate (MTSES), the flow was stopped and the reagent was added directly into the bath under voltage clamp. In experiments where d,l-TBOA was added, the same procedure was followed. Before applying the reducing agent 1,4 dithiothreitol (DTT) into the bath, the oocyte was unclamped, the grounding electrode was removed, and the flow was stopped. MTSES and/or DTT were allowed to react for 2 min followed by a 5-min washout. The final concentrations in the bath of MTSES and DTT were 2 mM and 50 mM, respectively. For radioactive uptake, five to eight oocytes of each mutant were incubated for 20 min in ND96 containing d-[2,3-3H]-aspartic acid, as described previously (Borre and Kanner, 2001).
Figure 8.

Substrate-induced steady-state currents by WT and D444E transporters in the presence of thiocyanate. Oocytes expressing either WT-EAAC1 (A) or D444E-EAAC1 (B) were voltage clamped and gravity perfused with NaSCN-based buffer as described under Materials and Methods in the absence or presence of 2 mM L-aspartate (triangles), succinate (circles), and GABA (squares) or 10 (A) or 30 μM (B) d,l-TBOA (inverted triangles). The voltage was stepped from −25 mV to voltages between −100 and +40 mV. The currents in the absence of substrate/blocker were subtracted from those in its presence and normalized to the L-aspartate–elicited current at +40 mV (I), and plotted against the holding potential (Vhold). In the three oocytes where currents by L-aspartate and d,l-TBOA were compared directly, the L-aspartate–induced currents ranged from +625 to +837 nA at +40 mV in oocytes expressing WT-EAAC1, whereas in oocytes expressing D444E-EAAC1, the currents suppressed by L-aspartate ranged from +460 to +700 nA. The corresponding values for the currents blocked by d,l-TBOA were +270 to +366 nA for WT-EAAC1 and +600 to +820 nA for D444E-EAAC1. Data shown are mean ± SEM of three oocytes, except for L-aspartate (n = 6).
Figure 9.

Anion selectivity and sodium dependence of D444E transporters. Steady-state currents in oocytes expressing D444E in the presence of 2 mM L-aspartate minus those in its absence were monitored in a chloride-based ND96 external medium (squares) or in media where 9.6 mM NaCl was replaced by an equimolar concentration of the sodium salts of iodide (circles), nitrate (triangles), perchlorate (inverted triangles), or thiocyanate (diamonds). The currents shown are normalized to the value in the thiocyanate medium at +40 mV. The absolute values of the currents in 9.6 mM sodium thiocyanate plus 86.4 mM NaCl ranged from 220 to 320 nA (A). The sodium dependence of the currents suppressed by 2 mM L-aspartate was determined using media containing NaCl at concentrations ranging from 0 to 86 mM (iso-osmotic compensation by choline chloride), 10 mM KSCN, 1.8 mM CaCl2, 1 mM MgCl2, and 5 mM Tris-HEPES, pH 7.5. The magnitude of these currents suppressed by L-aspartate did not change when its concentration was increased to 4 mM, indicating that the concentration of the acidic amino acid was saturating, even at low sodium concentrations. The absolute values of the currents suppressed by L-aspartate in the presence of 86 mM NaCl at +40 mV ranged from 125 to 254 nA (B). Data shown in each of the panels are mean ± SEM of three oocytes.
Data Analysis
All current–voltage relations (Fig. 1 C, Fig. 8, and Fig. 9 A) represent steady-state substrate- elicited net currents ((Ibuffer+substrate) – (Ibuffer)). Except for the data shown in Fig. 2 (A and B), the isolated transient currents were defined as (Ibuffer) − (Ibuffer+substrate/blocker). Transient currents isolated by substrates and current–voltage relations were analyzed by Clampfit version 8.2 or 9.0 (Axon Instruments). The charge movements were quantified by adding the current–time integrals for the voltage of −100 mV and that for +40 mV, with the exception of EAAC1-D444E, where current–time integrals for the voltage of −100 mV and +80 mV were added. Because of the variability in expression level within and between different oocyte batches, in many cases the data have been normalized as indicated in the figure legends. Kinetic parameters were determined by nonlinear fitting to the generalized Hill equation using the build-in functions of Origin 6.1 (Microcal). For determination of apparent affinity for substrate, Imax and K0.5 were allowed to vary and the value of nH was fixed at 1. For the determination of the sodium dependence of the anion currents by the D444E mutant, suppressed by L-aspartate, nH was allowed to vary. The time constants of the substrate/blocker sensitive transient currents were estimated using a nonlinear fitting to a first order decay exponential function by Clampfit.
Figure 2.

Currents in oocytes expressing D444S-EAAC1 transporters. Currents recorded in the absence of substrate during 250-ms voltage pulses from −25 mV to voltages between −100 and +40 mV were subtracted from currents in the same medium containing 2 mM succinate (A and B). In C and D, currents in the presence and absence of 10 mM succinate (C) or 160 μM d,l-TBOA (D) were recorded. The current traces in C and D are from the same cell. For these two panels, the opposite subtraction procedure of that used in A and B was followed. Currents from representative oocytes (n = 3) are shown. The zero current level is indicated by the stippled line.
d-[3H]-aspartate Transport in HeLa Cells
HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FCS, 200 U/ml penicillin, 200 μg/ml streptomycin, and 2 mM glutamine. HeLa cells plated on 24-well plates were infected with the recombinant vaccinia/T7 virus vTF7-3 (Fuerst et al., 1986) and transfected with cDNA, the vector pOG1 with wild-type (WT) or mutant histidine-tagged EAAC1 transporter inserted downstream to the T7 promoter, using the transfection reagent N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate (DOTAP) as previously described (Pines et al., 1995). Uptake of d-[3H]-aspartate into the cells was assayed 18–20 h post transfection. The cells were washed with a solution containing 150 mM choline chloride, 5 mM KPi, pH 7.4, 0.5 mM MgSO4, and 0.3 mM CaCl2 (1 ml/well), and subsequently transport was initiated by addition of 200 μl of an NaCl-based transport solution (150 mM NaCl, with KPi, MgSO4, and CaCl2 as above), containing 0.4 μCi of d-[3H]-aspartate (23.9 Ci/mmol), to each well. Where indicated this radioactive transport solution was supplemented with 5 mM unlabeled succinate. Transport was performed for 10 min at room temperature (22–24°C), and the assay was terminated by washing the cells twice with 1 ml of ice-cold choline-containing solution (see above). Cells were lysed with 1% SDS, and radioactivity was measured by liquid scintillation counting.
RESULTS
Transport in Substitution Mutants
The acidic and neutral amino acid transporters in the SLC1 family have an aspartate residue at position 1 of the seven TM 8 residues depicted in Fig. 1 A, and an arginine residue at position 2. On the other hand, in the dicarboxylic acid transporters DctA these positions are occupied by a serine and a glutamate, respectively. In addition, in the dicarboxylic acid transporters there is an alanine residue at position 5, but a threonine residue in the other transporters (Fig. 1 A). Except for the previously characterized arginine-447 (Bendahan et al., 2000), the other six residues of EAAC1 were mutated to their DctA equivalents and these mutants were expressed in Xenopus oocytes. The uptake of d-[3H]-aspartate by oocytes expressing the V449L and V450T mutants was similar to that observed with oocytes expressing WT, and a modest impact of the R445E and F446A mutations was observed (Fig. 1 B). Uptake by T448A was only ∼20% that of WT, whereas the D444S mutant was virtually devoid of any uptake activity (Fig. 1 B). In addition, when aspartate-444 was mutated to other residues, there was very little uptake, even when the replacement was by similar residues such as glutamate or asparagine (Fig. 1 B). It was difficult to determine, if the residual activity observed with the D444C and D444E mutants (Fig. 1 B) represents uptake rather than noise. However, this issue will be further addressed in the last section of Results.
It is important to note that the radioactive uptake experiments were done using very low substrate concentrations, and therefore uptake rates are way below vmax. For monitoring transport under saturating substrate concentrations, it is much more convenient to measure the substrate-induced steady-state transport currents. This is especially true for mutants with a decreased apparent substrate affinity. In contrast to WT, steady-state currents induced by 2 mM L-aspartate were not observed in the D444S, D444C, and D444E mutants (Fig. 1 C). The same was true when either L-glutamate or D-aspartate was used to induce the transport currents, and transport currents were also not observed with the D444N mutant (unpublished data). Except for the aspartate-444 mutants, L-aspartate induced steady-state transport currents in the replacement mutants at the other positions, including the T448A mutant (unpublished data). The lack of transport activity of the aspartate-444 mutants and the low activity of the T448A mutant are not due to defective expression at the plasma membrane, since surface biotinylation experiments revealed that their surface expression levels were similar or higher than those of WT (done as in Rosental et al., 2006; unpublished data). Therefore, it appears that these mutants have an intrinsic defect in transport.
Substrate-sensitive Transient Currents by the D444S Mutant
No steady-state transport currents in oocytes expressing WT were induced by 2 mM succinate (Fig. 2 A), and the same was true even at concentrations as high as 20 mM (unpublished data). Similar results were also obtained with the R445E, F446A, T448A, V449L, and V450T mutants (unpublished data). Even though succinate did not induce any steady-state currents in the D444S mutant, this mutant exhibited transient currents (Fig. 2 B). These currents represent transient currents blocked by succinate, which were similar to the transient currents suppressed by 160 μM of the nontransportable substrate analogue TBOA (Shimamoto et al., 1998), even though they were somewhat slower (Table I and Fig. 2, C and D; in these panels the subtraction procedure was the opposite of that used in Fig. 2, A and B). The concentration of TBOA used is near saturation in the D444S mutant, which had an IC50 value of ∼30 μM (Table II). GABA, which as succinate, is also not a substrate of the glutamate transporters, was unable to inhibit the transient currents by D444S (Fig. 3, A and B). Glutarate (2 mM) was capable of inhibiting transient currents of similar size as succinate, in contrast to the same concentration of malate or fumarate (unpublished data). The transient currents by D444S were dependent on the presence of sodium and were not observed in the presence of choline (Fig. 3 D). In the presence of lithium, which has been proposed to bind to the same sites as sodium but with a reduced affinity (Larsson et al., 2004), the transient currents were much smaller than in sodium (Fig. 3, A and C). Similar transient currents were not observed with noninjected oocytes (unpublished data).
TABLE I.
Decay Times of Transient Currents Blocked by L-Glutamate, L-Aspartate, D-Aspartate, and Succinate in D444 Mutant Transporters
| L-Glutamate | L-Aspartate | D-Aspartate | Succinate | TBOA | |
|---|---|---|---|---|---|
| D444C | 17.7 ± 1 | 19.3 ± 1 | 18.4 ± 1 | 15.7 ± 1.1 | 4.8 ± 0.3 |
| D444C after MTSES | ND | 10.2 ± 0.4 | ND | 8.7 ± 1.5 | ND |
| D444E | ND | 10.6 ± 1 | ND | 5.1 ± 0.7 | 5.6 ± 1.2 |
| D444S | 11.0 ± 1.2 | 12.2 ± 0.6 | 12.8 ± 0.6 | 9.9 ± 0.7 | 6 ± 0.6 |
Decay times of transients suppressed by 10 μM of d,l-TBOA (160 μM in the case of D444S) or by 10 mM of the other compounds (except for succinate in the case of D444E, where the concentration of succinate was 15 mM) are given in ms and are the mean ± SEM of 3–10 oocytes. In the case of D444S and D444C, the decay time was determined at −100 mV and with D444E at +80 mV.
TABLE II.
IC50 Values for Substrates/Blockers of Charge Movements by Mutant EAAC1 Transporters
| L-Glutamate | L-Aspartate | D-Aspartate | Succinate | TBOA | |
|---|---|---|---|---|---|
| D444A | ND | 720 ± 80 | ND | 870 ± 110 | ND |
| D444C | 29 ± 3 | 12 ± 2 | 8 ± 1 | 840 ± 70 | 1.9 ± 0.2 |
| D444C after MTSES | ND | 112 ± 19 | ND | 716 ± 25 | ND |
| D444E | 24.4 ± 2.2 | 9 ± 1 | ND | >6000 | 1.8 ± 0.4 |
| D444N | 718 ± 217 | 295 ± 45 | ND | NM | ND |
| D444S | 1220 ± 160 | 130 ± 15 | 1370 ± 360 | 1340 ± 110 | 31 ± 8 |
Data shown are mean ± SEM of three to eight oocytes. The IC50 values are given in μM. The charge movements sensitive to a range of concentrations of substrate/blocker (Fig. 4 A, Fig. 5 A, and Fig. 7, A and B) were quantified and the IC50 values were determined as described in Materials and Methods. NM, not measurable, no detectable inhibition was observed.
Figure 3.

Specificity and sodium dependence of the inhibition of transient currents by succinate in D444S-EAAC1. Oocytes expressing D444S-EAAC1 were perfused in the presence and absence of succinate 5 mM (A, C, and D) or GABA 5 mM (B) in media containing either sodium 96 mM (A and B) lithium 96 mM (C), or choline 96 mM (D), and net isolated transient currents were visualized by subtracting the currents in the presence of succinate or GABA from those in their absence. A representative oocyte is shown (n = 3).
In D444S, the magnitude of the charge movements blocked by succinate, was dependent on the concentration of this dicarboxylic acid (Fig. 4 A and Table II) with an IC50 of 1340 ± 110 μM (n = 4). D-aspartate and L-glutamate blocked charge movements of similar magnitude as succinate (Fig. 4 A). The L-glutamate concentration giving a half maximal blockade of the transients was 1220 ± 160 μM (n = 5), similar to the corresponding value for succinate. Remarkably L-aspartate was much more potent than D-aspartate and L-glutamate (Fig. 4 A and Table II). The concentrations, at which a half-maximal effect was obtained, were 1370 ± 360 (n = 5) for D-aspartate and 130 ± 15 μM (n = 6) for L-aspartate (Table II). Despite their ability to inhibit the sodium-dependent transient currents by D444S, the acidic amino acids were unable to induce any inwardly rectifying steady-state currents, even at 10 mM (Fig. 4, B and C).
Figure 4.

Blockade of charge movements in D444S-EAAC1 by succinate and acidic amino acids. The charge moved was calculated from the isolated transient current–time integrals as described under Materials and Methods. The fraction of transient currents blocked at each concentration of the indicated compound was normalized to the transient currents isolated by saturating (10 mM) concentrations of L-glutamate (circles), L-aspartate (triangles), D-aspartate (diamonds), and succinate (squares) and is shown in A. The current–time integrals of the transients ranged from 1.0 to 2.8 nC. Data shown are mean ± SEM of four to six oocytes. The currents blocked by 10 mM D- and L-aspartate in the same oocyte are shown in B and C, respectively.
Transient Currents by Other Aspartate-444 Mutants
Succinate was also able to block the transient currents by oocytes expressing the D444C mutant (Fig. 5 A) with a IC50 value of 840 ± 70 μM (n = 8) (Table II) and the same was true for glutarate (IC50 260 ± 40 μM; n = 4). Also the acidic amino acids were able to inhibit the sodium-dependent transient currents by D444C and their apparent affinity was higher than for D444S, especially in the case of D-aspartate and L-glutamate (Fig. 5 A and Table II) with IC50 values of 8 ± 1 μM (n = 3) and 29 ± 3 μM (n = 4), respectively. In the case of L-aspartate (Fig. 5 B and Table II), the IC50 was 12 ± 2 μM (n = 5). Also in this mutant, succinate was unable to induce steady-state currents (unpublished data) and the same was true for L-aspartate (Fig. 5 B) as well as for D-aspartate and L-glutamate (unpublished data). Also in the D444C mutant, the transient currents inhibited by 10 μM of TBOA (IC50 ∼2 μM, Table II) were faster than those inhibited by the acidic amino acids or by succinate (Fig. 5, B and C, and Table I).
Figure 5.
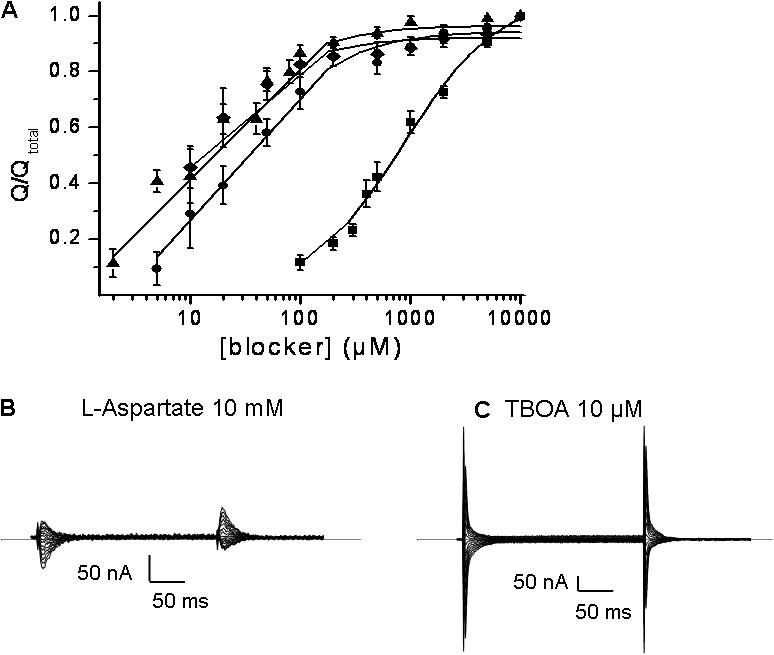
Blockade of charge movements in D444C-EAAC1 by succinate and acidic amino acids. The charge movements were analyzed as described in the legend to Fig. 4. The isolated transient currents were normalized to the transient currents isolated by saturating concentrations (10 mM) of the same compounds (same symbols as in Fig. 4 A) and are shown in A. The current–time integrals of the transients ranged from 1.5 to 7.0 nC. Data shown are mean ± SEM of three to eight oocytes. The currents blocked by 10 mM L-aspartate and 10 μM d,l-TBOA in the same oocyte, typical of three, are shown in B and C, respectively.
The size of the L-aspartate–sensitive transient currents was increased by treatment of the oocytes expressing D444C with 2 mM of the membrane impermeant sulfhydryl reagent MTSES, and this treatment also resulted in an altered voltage dependence of the transients (Fig. 6, A and B). Both of these effects by the sulfhydryl reagent were significantly reversed by treatment with DTT (Fig. 6 C). Similar results were also observed when the effects of MTSES and DTT on the transients suppressed by succinate were tested (unpublished data). These effects were the consequence of modification of the cysteine residue introduced at position 444, because they were not observed with the D444S mutant (Fig. 6, D–F). The IC50 value of D444C for succinate after the modification by MTSES was similar to the value before the sulfhydryl modification (Table II). On the other hand, the IC50 value for L-aspartate was markedly increased after the treatment with MTSES (Table II).
Figure 6.

The effects of MTSES and DTT on the transient currents by D444C. Currents recorded in the presence of 2 mM L-aspartate were subtracted from currents recorded in its absence in oocytes expressing either D444C-EAAC1 (A–C) or D444S-EAAC1 (D–F). The transient currents were measured before (A and D) or after the application of 2 mM MTSES (B and E) and after MTSES and 50 mM DTT (C and F). Typical oocytes are shown (n = 3).
With the D444A mutant, L-aspartate and succinate also inhibited transient currents, with IC50 values of 720 ± 80 and 870 ± 110 μM (n = 4), respectively (Table II). The mutation of aspartate to asparagine is much more conservative than that to serine, cysteine, and alanine, but also with the D444N mutant, no transport currents were observed. The transient currents by D444N could be blocked by the acidic amino acids but not by succinate. Even at 10 mM succinate, no inhibition was observed (Table II).
The transient currents by the D444E mutant were effectively inhibited by L-aspartate with an IC50 of 9 ± 1 μM (n = 3) (Fig. 7 A and Table II). Also L-glutamate inhibited the transients by D444E with an IC50 value of ∼20 μM. On the other hand, succinate was very ineffective in the inhibition of the transient currents (Fig. 7 F) and saturation was not reached, even at 15 mM (Fig. 7 B). Interestingly, the voltage dependence of the transients by this mutant, blocked by L-aspartate, succinate, or TBOA (Fig. 7, C–F), differed from that by the other mutants (Figs. 2–6 ). The observation that the transient currents by D444E are predominantly in the outward direction, indicates that the apparent affinity of the D444E transporters for sodium is increased. Apparently at the holding potential (−25 mV) most of the transporters have already bound sodium, and the transients by this mutant are predominantly the consequence of sodium release from the transporter upon depolarization. Consistent with this idea is the fact that when the external sodium concentration was lowered from 96 to 32 mM, the voltage dependence of the transients became more symmetrical (unpublished data), suggesting that at reduced sodium not all the transporters are in the sodium-bound state at the holding potential. Also in the case of D444E, the transients blocked by 10 μM of TBOA (IC50 ∼2 μM, Table II) decayed somewhat faster than those blocked by 2 mM L-aspartate (Table I).
Figure 7.

Transient currents by D444E-EAAC1 transporters. The concentration dependence of the suppression of the charge movements by L-aspartate (A) or succinate (B) was determined as described under Materials and Methods. Charge moved was normalized to that observed at 1 mM of L-aspartate and 15 mM succinate, respectively. The current–time integrals of the transients ranged from 2.1 to 4.6 nC. Data shown are mean ± SEM of three oocytes. Currents recorded in the presence of L-aspartate at 2 mM (C) or 50 μM (E), of d,l-TBOA at 10 μM (D), or of succinate at 2 mM (F) were subtracted from currents recorded in their absence in oocytes expressing D444E-EAAC1. Representative oocytes are shown (n = 3). The same cell was used in C and D and the same is true for E and F.
Substrate-modulated Anion Conductance by D444E
Even in the absence of coupled transport, the substrates of glutamate transporters can still increase the uncoupled anion conductance (Seal et al., 2001; Borre et al., 2002; Ryan and Vandenberg, 2002). It has been proposed that the substrate, when occupying its binding site on the transporter, may directly gate the anion conductance (Wadiche et al., 1995a). Therefore one might expect that the altered interaction of the aspartate-444 mutants with substrates of the glutamate transporters could lead to altered effects of substrate on the anion conductance. The conductance of thiocyanate is ∼70-fold higher than that of chloride (Wadiche and Kavanaugh, 1998), and in the presence of the former anion, the substrate-induced currents are dominated by the anion conductance, especially at positive potentials. In the presence of thiocyanate, in oocytes expressing WT, 2 mM L-aspartate stimulated the anion conductance. This stimulation of the anion conductance was not seen with GABA or succinate, whereas it was decreased by the blocker TBOA (Fig. 8 A). Remarkably, L-aspartate inhibited the anion conductance of the D444E mutant rather than stimulating it, and as a result, the voltage dependence of the currents, in the presence of L-aspartate minus those in its absence, appears as the mirror image of those by WT (Fig. 8, A and B). Also D-aspartate and L-glutamate inhibited the anion conductance of D444E, in contrast to their stimulatory effect on that of WT (unpublished data). In the presence of thiocyanate, the magnitude and voltage dependence of the steady-state currents blocked by L-aspartate were similar to those blocked by TBOA (Fig. 8 B). Moreover, the selectivity sequence of the substrate-sensitive anion conductance of the D444E mutant (Fig. 9 A) was similar to that of the substrate-activated anion conductance of WT, measured under exactly the same conditions (Rosental et al., 2006). Analysis of the sodium dependence of the L-aspartate–blocked anion currents by D444E yielded a Hill coefficient of 2.7 ± 0.4 (Fig. 9 B), indicating that multiple sodium ions are required for the binding of the substrate/blocker to the D444E mutant. Succinate, at 2 mM only slightly suppressed the transient currents by D444E (Fig. 7 F) and also had only a small effect on the anion conductance of D444E, whereas GABA had no effect at all (Fig. 8 B). Although the D444S and D444C mutants had similar levels of the sodium-dependent anion conductance as WT and D444E, the changes in anion conductance by L-aspartate and succinate in D444S and D444C were very small and difficult to analyze (unpublished data).
d-[3H]-aspartate Transport of D444C and D444E Mutants Expressed in HeLa Cells
As mentioned before, it was difficult to determine if the residual radioactive D-aspartate uptake by the D444C and D444E mutants expressed in oocytes (Fig. 1 B) was real rather than noise. To achieve a better signal to noise ratio, we expressed the mutants in HeLa cells using the vaccinnia/T7 expression system (Fuerst et al., 1986; Pines et al., 1995). The d-[3H]-aspartate uptake by the D444C mutant was ∼5% of that by WT-EAAC1, and the uptake by D444E was even lower, yet still significantly higher than that observed with cells transfected with the vector alone (Fig. 10). No uptake at all was observed in the absence of sodium (choline replacement; unpublished data). In contrast to WT, d-[3H]-aspartate uptake by the D444C mutant was fully inhibited by 5 mM unlabeled succinate, whereas the very low uptake by the D444E mutant was much less sensitive under these conditions (Fig. 10), in harmony with the weak interaction of succinate with the D444E transporters (Table II). In further contrast to WT, no detectable radioactive d-[3H]-aspartate uptake with the mutant was observed in reconstituted proteoliposomes containing internal potassium (unpublished data). In this assay net flux is measured rather than exchange (Kavanaugh et al., 1997).
Figure 10.

Uptake of d-[3H]-aspartate in HeLa cells expressing mutants D444C and D444E. Uptake of d-[3H]-aspartate in HeLa cells transfected with WT-EAAC1, D444C, D444E, and the empty vector were performed in NaCl-containing medium in the absence (open bars) or presence (hatched bars) of 5 mM succinate as described under Materials and Methods. Values were corrected for uptake in cells transfected with the empty vector and given as percent of uptake relative to WT-EAAC1. Values represent the mean ± SEM of three separate experiments, each done in triplicate. The absolute counts of D444C were ∼2.5-fold of those by cells expressing the empty vector, and in the case of D444E, the corresponding value was ∼1.4-fold.
DISCUSSION
Previous attempts to correlate sequence variation in the SLC1 family with the type of substrate used led to the identification of a conserved arginine residue in TM 8, arginine-447 in EAAC1, which is important for the interaction of the transporter with the γ-carboxyl group of glutamate (Conradt and Stoffel, 1995; Bendahan et al., 2000). In this study, a similar rationale was used, and this led to the finding that the presence of aspartate at position 444 is crucial for transport and that the side chain of this residue is a molecular determinant of exclusion of succinate by glutamate transporters (Figs. 2–5 and 7). However, even though the substitution of aspartate-444 to neutral residues led to the ability of succinate to interact with the mutant transporters, these mutants clearly cannot transport either succinate or acidic amino acids (Figs. 2–5 and 7). The lack of transport of acidic amino acids by the mutants can be due to a critical role of aspartate-444 in the correct binding of these substrates and/or in a step subsequent to binding. We refer to correct binding as binding to those determinants enabling the formation of a productive translocation complex resulting in substrate transport. It is possible that when aspartate at position 444 is replaced by a neutral residue, different determinants may ligand the acidic amino acid substrates. In this case, the orientation of the substrate in the binding pocket is likely to be altered so that a productive translocation complex cannot be formed. If we consider the lack of succinate transport by the mutants, the possibility of a role of aspartate-444 in a step subsequent to transport of this dicarboxylic acid seems unlikely, because this residue is not present in the DctA transporters. The lack of succinate transport by the aspartate-444 mutants therefore appears to be due to other determinants that are present in DctA, but not in the acidic amino acid transporting members of the SLC1 family.
It is interesting that in the GltPh structure, the equivalent residues of aspartate-444 and arginine-447 both point toward the binding pocket in the direction of a nonprotein electron density, apparently representing bound substrate (Yernool et al., 2004). In the case of aspartate-444, the importance of this residue in transport is underscored by the finding that even when it is mutated to the very similar glutamate residue, this leads to strongly impaired radioactive substrate uptake (Fig. 1 B and Fig. 10) and defective coupled substrate-induced steady-state transport currents (Fig. 1 C and Fig. 7, C and E).
Nevertheless, the acidic amino acids can still bind to the aspartate-444 mutants. Even though they are not transported, they are able to block the sodium-dependent transient currents in the aspartate-444 mutants (Figs. 4, 5, and 7) and the same is true for succinate in the neutral substitution mutants (Figs. 2–5 ). Such a behavior is a well-known property of nontransportable substrate analogues, which cause the transporter to be fixed in one, blocker-bound conformation. The transient currents are thought to represent a charge-moving conformational change that takes place upon the binding of sodium to the transporter. In the case of the human homologue of the glial glutamate transporter GLT-1 (Pines et al., 1992), the nontransportable substrate analogue kainate is capable to block the sodium-dependent transient currents (Wadiche et al., 1995b), whereas d,l-TBOA does the same in EAAC1 (Shachnai et al., 2005), and the sodium-dependent transients by the GABA transporter GAT-1 are blocked by SKF-89976A and SKF-100330A (Mager et al., 1993; Bismuth et al., 1997). Even though acidic amino acids appear to act on the transients by the aspartate-444 mutants just like the blocker d,l-TBOA, the transients blocked by the latter compound exhibit faster decay rates (Table I). Also the succinate-sensitive sodium-dependent transients of the D444S and D444C mutants are slower than those blocked by d,l-TBOA (Table I). These differences may relate to the number of sodium ions bound to the transporter. TBOA binding to the eukaryotic glutamate transporters requires only one sodium ion (Shimamoto et al., 2007) and the sodium dependence of the TBOA-sensitive leak anion conductance could be fitted by a Hill equation with a Hill coefficient n = 1 (Watzke et al., 2001). On the other hand, binding of the acidic amino acids and succinate may require two or perhaps even three sodium ions. Indeed, using the blockade of the anion conductance (see next paragraph), evidence supporting this idea was obtained with the D444E mutant (Fig. 9 B). The action of succinate as a blocker of the D444C mutant is further emphasized by its ability to block the residual d-[3H]-aspartate uptake by this mutant (Fig. 10).
Binding of acidic amino acids to the D444E mutant is also indicated by its ability to influence the anion conductance (Fig. 8). It has been proposed that the substrate, when occupying its binding site on the transporter, may directly gate the anion conductance (Wadiche et al., 1995a), and this process persists under conditions when transport is blocked (Seal et al., 2001; Borre et al., 2002; Ryan and Vandenberg, 2002). However, substrate suppresses rather than stimulates the uncoupled anion conductance by D444E, and also in this assay its action is similar to that of TBOA (Fig. 8, A and B). This indicates that when aspartate-444 is replaced by the bulkier glutamate, the position of the substrate in the binding pocket is different from that in WT, so that the substrate now acts as a blocker. The orientation of L-aspartate in the binding pocket of D444S and D444C probably differs from that in D444E, because only in the latter mutant does this amino acid efficiently block both the transient and the anion currents. Altogether it appears that aspartate-444 is essential for the correct interaction with acidic amino acids, perhaps by forming a charge pair with their amino group and as a consequence binding leads to transport. When the side chain of this aspartate residue is eliminated by mutation, there are apparently alternative options for the transporter to interact with the amino group, for instance via its main chain carbonyl oxygens. However this altered binding is apparently incompatible with transport and therefore the substrates of WT act as blockers in the aspartate-444 mutants. Succinate does not interact with WT, and in the D444E mutant its apparent affinity is dramatically reduced (Table II). Thus, while the transients suppressed by TBOA are a good tool to compare expression levels in the aspartate-444 mutants, succinate can also be used for this purpose, but only in the neutral substitution mutants. It appears that a carboxyl-containing side chain at position 444 prevents the binding of succinate, but additional, as yet unidentified, determinants are required for the productive binding of succinate to glutamate transporters.
The simplest explanation of our results is that the role of aspartate-444 is at the level of substrate binding. This idea is in beautiful agreement with crystallographic studies of GltPh,, published when this manuscript was in revision (Boudker et al., 2007). In the latter study, cocrystals of GltPh and L-cysteine sulphinic acid, containing an anomalous scattering sulfur atom, were compared with those of the same transporter with L-aspartate bound. Under the assumption that the β-carboxylate of aspartate occupies the same position as the sulfinic group of L-cysteine sulphinic acid, it appears that the GltPh counterpart of aspartate-444 directly interacts with the amino group of the substrate (Boudker et al., 2007). Nevertheless, it cannot be ruled out that aspartate-444 is (also) important in a subsequent step of the transport cycle. It is firmly established that mutant transporters, which have a defective potassium interaction, can still take up radioactive substrate in intact cells via exchange with endogenous substrates (Kavanaugh et al., 1997; Zhang et al., 1998). The aspartate-444 mutants are strongly impaired in d-[3H]-aspartate uptake (Fig. 1 B), and therefore it is possible that aspartate-444 is also important for a step in the sodium-coupled substrate translocation half cycle. It is also possible that aspartate-444 may influence the potassium-dependent reorientation of the empty transporter. D444C exhibits residual uptake of d-[3H]-aspartate (Fig. 10), but experiments with reconstituted proteoliposomes indicate that this uptake is not due to net flux. Even though the very slow radioactive uptake by D444C in whole cells does not permit detection of exchange in the proteoliposomes, it is likely that this uptake in fact reflects exchange, indicating a defective interaction with potassium.
Regarding the external accessibility of the binding pocket, it is important to note that when arginine-447 of EAAC1, which is only three residues away from aspartate-444 and apparently interacts with the γ-carboxyl group of the transported substrate (Bendahan et al., 2000; Yernool et al., 2004), was mutated to cysteine, no impact by MTS reagents on function was observed (Bendahan et al., 2000). On the other hand, position 444 can be reached by impermeant MTS reagents such as MTSES (Fig. 6) and MTSET (not depicted) because D444C, but not D444S, was functionally impacted by these reagents. These observations are consistent with the idea that this position, located in the binding pocket of the GltPh homologue (Yernool et al., 2004), is accessible to the external aqueous medium, at least in the outward-facing form of the transporter. It is interesting to note that the introduction of a negative charge in the D444C mutant alters the voltage dependence of the transients in the same direction as when a glutamate residue replaces a neutral residue at position 444 (Figs. 6 and 7). We have no good explanation for the increased magnitude of the transients observed after MTSES treatment of D444C (Fig. 6, A and B).
It should be noted that in the D444S mutant there is a marked difference in the apparent affinities for L-aspartate on the one hand and L-glutamate and D-aspartate on the other (Fig. 4 and Table II). This difference is much smaller in the D444C mutant (Fig. 5 and Table II). This is probably because the side chains of serine and cysteine, introduced at position 444, interact differently with the transporter substrates tested. When the negative charge at position 444 is taken away, the transporter is better capable of interacting with succinate (Table II), because this charge probably results in an unfavorable interaction of the transporter with the dicarboxylic acids. The D444E mutant, where the negative charge is maintained, shows some interaction with succinate (Table II and Fig. 7 F), perhaps because the position of the negative charge is not precisely the same as in WT. On the other hand, the apparent affinity for the acidic amino acids in the D444E mutant is similar to that of WT. Nevertheless, these substrates are not transported by D444E and instead act as blockers. Thus, the lengthening of the aspartate at position 444 by a methylene group separates the two effects observed in the neutral substitution mutants; the substrate selectivity appears to be unaffected but there is a defect in the translocation step. This is apparently due to strict spatial requirements for the positioning of the carboxyl group at position 444 relative to the substrate. Interestingly, restoration of the negative charge at position 444, via the reaction of the D444C mutant with MTSES, did not result in a decreased sensitivity to succinate and actually increased the IC50 of L-aspartate (Table II). However, in the adduct formed after the reaction with MTSES, the negative charge is at the end of a long linker, and steric constraints probably prevent this charge from occupying a similar position as the β-carboxyl group of aspartate-444 or even of that of the γ-carboxyl of the glutamate side chain in the D444E mutant. The relatively modest resolution of the GltPh structure (Yernool et al., 2004) hampers attempts to rationalize these findings by homology modeling at the present time. This is also the case for the new sodium-bound GltPh structure (Boudker et al., 2007), where two sodium ion binding sites are inferred by using thallium as a sodium analogue. Three sodium ions are translocated with the acidic amino acids by the eukaryotic transporters (Zerangue and Kavanaugh, 1996; Levy et al., 1998), and the absence of the third sodium ion prohibits homology modeling with energy minimization of the aspartate-444 mutants with the various substrates/blockers. It appears therefore that in-depth functional studies of mutants at amino acid residues conserved in the transporter family, in parallel with attempts to obtain additional structures of transporter homologues, will be important to gain further insights into the mechanism of glutamate transport.
Acknowledgments
We thank Annie Bendahan for performing the surface biotinylation experiments and Elia Zomot for his help in the preparation of the figures.
This work was supported by the National Institute of Neurological Disorders and Stroke, National Institutes of Health grant NS 16708 and the European Union Consortium EUGINDAT.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: d,l-TBOA, d,l-threo-β-benzyloxyaspartate; DTT, 1,4 dithiothreitol; EAAC, excitatory amino acid carrier; GLT, glutamate transporter; MTSES, (2-sulfonatoethyl)methanethiosulfonate; MTSET, (2-trimethylammonium ethyl)methanethiosulfonate; TM, transmembrane domain; WT, wild-type.
References
- Arriza, J.L., S. Eliasof, M.P. Kavanaugh, and S.G. Amara. 1997. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc. Natl. Acad. Sci. USA. 94:4155–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendahan, A., A. Armon, N. Madani, M.P. Kavanaugh, and B.I. Kanner. 2000. Arginine 447 plays a pivotal role in substrate interactions in a neuronal glutamate transporter. J. Biol. Chem. 275:37436–37442. [DOI] [PubMed] [Google Scholar]
- Bismuth, Y., M.P. Kavanaugh, and B.I. Kanner. 1997. Tyrosine 140 of the gamma-aminobutyric acid transporter GAT-1 plays a critical role in neurotransmitter recognition. J. Biol. Chem. 272:16096–16102. [DOI] [PubMed] [Google Scholar]
- Borre, L., and B.I. Kanner. 2001. Coupled, but not uncoupled, fluxes in a neuronal glutamate transporter can be activated by lithium ions. J. Biol. Chem. 276:40396–40401. [DOI] [PubMed] [Google Scholar]
- Borre, L., and B.I. Kanner. 2004. Arginine 445 controls the coupling between glutamate and cations in the neuronal transporter EAAC-1. J. Biol. Chem. 279:2513–2519. [DOI] [PubMed] [Google Scholar]
- Borre, L., M.P. Kavanaugh, and B.I. Kanner. 2002. Dynamic equilibrium between coupled and uncoupled modes of a neuronal glutamate transporter. J. Biol. Chem. 277:13501– 13507. [DOI] [PubMed] [Google Scholar]
- Boudker, O., R.M. Ryan, D. Yernool, K. Shimamoto, and E. Gouaux. 2007. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 445:387–393. [DOI] [PubMed] [Google Scholar]
- Brew, H., and D. Attwell. 1987. Electrogenic glutamate uptake is a major current carrier in the membrane of axolotl retinal glial cells. Nature. 327:707–709. [DOI] [PubMed] [Google Scholar]
- Conradt, M., and W. Stoffel. 1995. Functional analysis of the high affinity, Na+-dependent glutamate transporter GLAST-1 by site-directed mutagenesis. J. Biol. Chem. 270:25207–25212. [DOI] [PubMed] [Google Scholar]
- Engelke, T., D. Jording, D. Kapp, and A. Puhler. 1989. Identification and sequence analysis of the Rhizobium meliloti dctA gene encoding the C4-dicarboxylate carrier. J. Bacteriol. 171:5551–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman, W.A., R.J. Vandenberg, J.L. Arriza, M.P. Kavanaugh, and S.G. Amara. 1995. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature. 375:599–603. [DOI] [PubMed] [Google Scholar]
- Fuerst, T.R., E.G. Niles, F.W. Studier, and B. Moss. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. USA. 83:8122–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunewald, M., A. Bendahan, and B.I. Kanner. 1998. Biotinylation of single cysteine mutants of the glutamate transporter GLT-1 from rat brain reveals its unusual topology. Neuron. 21:623–632. [DOI] [PubMed] [Google Scholar]
- Grunewald, M., and B.I. Kanner. 2000. The accessibility of a novel reentrant loop of the glutamate transporter GLT-1 is restricted by its substrate. J. Biol. Chem. 275:9684–9689. [DOI] [PubMed] [Google Scholar]
- Kanai, Y., and M.A. Hediger. 1992. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature. 360:467–471. [DOI] [PubMed] [Google Scholar]
- Kanner, B.I., and A. Bendahan. 1982. Binding order of substrates to the sodium and potassium ion coupled L-glutamic acid transporter from rat brain. Biochemistry. 21:6327–6330. [DOI] [PubMed] [Google Scholar]
- Kanner, B.I., and I. Sharon. 1978. Active transport of L-glutamate by membrane vesicles isolated from rat brain. Biochemistry. 17:3949–3953. [DOI] [PubMed] [Google Scholar]
- Kavanaugh, M.P., A. Bendahan, N. Zerangue, Y. Zhang, and B.I. Kanner. 1997. Mutation of an amino acid residue influencing potassium coupling in the glutamate transporter GLT-1 induces obligate exchange. J. Biol. Chem. 272:1703–1708. [DOI] [PubMed] [Google Scholar]
- Kunkel, T.A., J.D. Roberts, and R.A. Zakour. 1987. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 154:367–382. [DOI] [PubMed] [Google Scholar]
- Larsson, H.P., A.V. Tzingounis, H.P. Koch, and M.P. Kavanaugh. 2004. Fluorometric measurements of conformational changes in glutamate transporters. Proc. Natl. Acad. Sci. USA. 101:3951–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, L.M., O. Warr, and D. Attwell. 1998. Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J. Neurosci. 18:9620–9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager, S., J. Naeve, M. Quick, C. Labarca, N. Davidson, and H.A. Lester. 1993. Steady states, charge movements, and rates for a cloned GABA transporter expressed in Xenopus oocytes. Neuron. 10:177–188. [DOI] [PubMed] [Google Scholar]
- Pines, G., N.C. Danbolt, M. Bjoras, Y. Zhang, A. Bendahan, L. Eide, H. Koepsell, J. Storm-Mathisen, E. Seeberg, and B.I. Kanner. 1992. Cloning and expression of a rat brain L-glutamate transporter. Nature. 360:464–467. [DOI] [PubMed] [Google Scholar]
- Pines, G., and B.I. Kanner. 1990. Counterflow of L-glutamate in plasma membrane vesicles and reconstituted preparations from rat brain. Biochemistry. 29:11209–11214. [DOI] [PubMed] [Google Scholar]
- Pines, G., Y. Zhang, and B.I. Kanner. 1995. Glutamate 404 is involved in the substrate discrimination of GLT-1, a (Na+ + K+)-coupled glutamate transporter from rat brain. J. Biol. Chem. 270:17093–17097. [DOI] [PubMed] [Google Scholar]
- Rosental, N., A. Bendahan, and B.I. Kanner. 2006. Multiple consequences of mutating two conserved β-bridge forming residues in the translocation cycle of a neuronal glutamate transporter. J. Biol. Chem. 281:27905–27915. [DOI] [PubMed] [Google Scholar]
- Ryan, R.M., and R.J. Vandenberg. 2002. Distinct conformational states mediate the transport and anion channel properties of the glutamate transporter EAAT-1. J. Biol. Chem. 277:13494–13500. [DOI] [PubMed] [Google Scholar]
- Seal, R.P., Y. Shigeri, S. Eliasof, B.H. Leighton, and S.G. Amara. 2001. Sulfhydryl modification of V449C in the glutamate transporter EAAT1 abolishes substrate transport but not the substrate-gated anion conductance. Proc. Natl. Acad. Sci. USA. 98:15324–15329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachnai, L., K. Shimamoto, and B.I. Kanner. 2005. Sulfhydryl modification of cysteine mutants of a neuronal glutamate transporter reveals an inverse relationship between sodium-dependent conformational changes and the glutamate-gated anion conductance. Neuropharmacology. 49:862–871. [DOI] [PubMed] [Google Scholar]
- Shimamoto, K., B. Lebrun, Y. Yasuda-Kamatani, M. Sakaitani, Y. Shigeri, N. Yumoto, and T. Nakajima. 1998. DL-threo-β-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol. Pharmacol. 53:195–201. [DOI] [PubMed] [Google Scholar]
- Shimamoto, K., Y. Otsubo, Y. Shigeri, Y. Yasuda-Kamatani, M. Satoh, S. Kaneko, and T. Nakagawa. 2007. Characterization of the tritium-labeled analog of L-threo-β-benzyloxyaspartate binding to glutamate transporters. Mol. Pharmacol. 71:294–302. [DOI] [PubMed] [Google Scholar]
- Slotboom, D.J., I. Sobczak, W.N. Konings, and J.S. Lolkema. 1999. A conserved serine-rich stretch in the glutamate transporter family forms a substrate-sensitive reentrant loop. Proc. Natl. Acad. Sci. USA. 96:14282–14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche, J.I., and M.P. Kavanaugh. 1998. Macroscopic and microscopic properties of a cloned glutamate transporter/chloride channel. J. Neurosci. 18:7650–7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche, J.I., S.G. Amara, and M.P. Kavanaugh. 1995. a. Ion fluxes associated with excitatory amino acid transport. Neuron. 15:721–728. [DOI] [PubMed] [Google Scholar]
- Wadiche, J.I., J.L. Arriza, S.G. Amara, and M.P. Kavanaugh. 1995. b. Kinetics of a human glutamate transporter. Neuron. 14:1019–1027. [DOI] [PubMed] [Google Scholar]
- Watzke, N., E. Bamberg, and C. Grewer. 2001. Early intermediates in the transport cycle of the neuronal excitatory amino acid carrier EAAC1. J. Gen. Physiol. 117:547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yernool, D., O. Boudker, Y. Jin, and E. Gouaux. 2004. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 431:811–818. [DOI] [PubMed] [Google Scholar]
- Zerangue, N., and M.P. Kavanaugh. 1996. Flux coupling in a neuronal glutamate transporter. Nature. 383:634–637. [DOI] [PubMed] [Google Scholar]
- Zhang, Y., and B.I. Kanner. 1999. Two serine residues of the glutamate transporter GLT-1 are crucial for coupling the fluxes of sodium and the neurotransmitter. Proc. Natl. Acad. Sci. USA. 96:1710–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., A. Bendahan, R. Zarbiv, M.P. Kavanaugh, and B.I. Kanner. 1998. Molecular determinant of ion selectivity of a (Na+ + K+)-coupled rat brain glutamate transporter. Proc. Natl. Acad. Sci. USA. 95:751–755. [DOI] [PMC free article] [PubMed] [Google Scholar]