

Abstract
Lanthanide gadolinium (Gd3+) blocks CaV1.2 channels at the selectivity filter. Here we investigated whether Gd3+ block interferes with Ca2+-dependent inactivation, which requires Ca2+ entry through the same site. Using brief pulses to 200 mV that relieve Gd3+ block but not inactivation, we monitored how the proportions of open and open-blocked channels change during inactivation. We found that blocked channels inactivate much less. This is expected for Gd3+ block of the Ca2+ influx that enhances inactivation. However, we also found that the extent of Gd3+ block did not change when inactivation was reduced by abolition of Ca2+/calmodulin interaction, showing that Gd3+ does not block the inactivated channel. Thus, Gd3+ block and inactivation are mutually exclusive, suggesting action at a common site. These observations suggest that inactivation causes a change at the selectivity filter that either hides the Gd3+ site or reduces its affinity, or that Ca2+ occupies the binding site at the selectivity filter in inactivated channels. The latter possibility is supported by previous findings that the EEQE mutation of the selectivity EEEE locus is void of Ca2+-dependent inactivation (Zong Z.Q., J.Y. Zhou, and T. Tanabe. 1994. Biochem. Biophys. Res. Commun. 201:1117–11123), and that Ca2+-inactivated channels conduct Na+ when Ca2+ is removed from the extracellular medium (Babich O., D. Isaev, and R. Shirokov. 2005. J. Physiol. 565:709–717). Based on these results, we propose that inactivation increases affinity of the selectivity filter for Ca2+ so that Ca2+ ion blocks the pore. A minimal model, in which the inactivation “gate” is an increase in affinity of the selectivity filter for permeating ions, successfully simulates the characteristic U-shaped voltage dependence of inactivation in Ca2+.
INTRODUCTION
In CaV1.2 channels, Ca2+ selectivity and block by various polyvalent metal ions are mediated by carboxyl side chains of the four glutamates (EEEE locus) that form ion-binding site(s) (for review see Sather and McCleskey, 2003). A somewhat overlooked observation that a glutamate to glutamine substitution in the S5-S6 loop of the third repeat (EEEE to EEQE modification) eliminates Ca2+-dependent inactivation (Zong et al., 1994) strongly indicated that the selectivity locus plays an important role in Ca2+-dependent inactivation. Previously, we showed that Ca2+-dependent inactivation of CaV1.2 channels specifically prevents permeation of Ca2+, but not alkali metal ions (Babich et al., 2005). We concluded that Ca2+-dependent inactivation controls Ca2+ conductance by affecting the selectivity mechanism rather than by occluding the pore at a cytoplasmic inactivation gate. This leads to the idea that perhaps the selectivity filter is the gate of Ca2+-dependent inactivation and that Ca2+-dependent inactivation specifically prevents Ca2+ permeation by stabilizing a high Ca2+ affinity state of the selectivity filter.
To test this hypothesis, we analyzed how blockage of the channel by lanthanide gadolinium (Gd3+) depends on inactivation and vice versa. This approach eliminates problems that can occur when Ca2+ affinity of inactivated channels is assessed by simple manipulations of extracellular Ca2+, as this by itself might change the affinity of the selectivity filter.
Like many other trivalent metal ions, Gd3+ is a potent blocker of Ca2+ channels. At concentrations of 10–100 nM, it reduces the peak and accelerates decay of ionic current during depolarization. This accelerated decay has been proposed to be due to an increase of the potency of block of open rather than closed channels (Biagi and Enyeart, 1990; Obejero-Paz et al., 2004), or because trivalent metal ions accelerate inactivation by acting at a site that is different from the blocking site (Beedle et al., 2002).
The results of the accompanying paper (Babich et al., 2007) explain the voltage-dependent enhancement of Gd3+ block by linking it to activation directly, rather than via inactivation. Similarly, inactivation also increases with voltage because it is also linked to activation. In addition, Gd3+ block is relieved at high positive voltages, generating a characteristic U-shaped dependence on voltage. The complex voltage dependence of Gd3+ block is very similar to that of Ca2+-dependent inactivation, which parallels Ca2+ influx rather than voltage (Brehm and Eckert, 1978). This makes it tempting to suggest that Gd3+ binding stabilizes channels in an inactivated state, similar to the action of other Ca2+ channel blockers (e.g., dihydropyridines). However, we (Babich et al., 2007) showed that the U-shaped voltage dependence of Gd3+ block is not affected by tampering with regulation of inactivation by calmodulin (Lee et al., 1999; Peterson et al., 1999; Qin et al., 1999; Zuhlke et al., 1999). The results presented below demonstrate that Gd3+ block actually reduces Ca2+-dependent inactivation. Moreover, the reverse is also true; Ca2+ inactivation reduces Gd3+ block. Thus, although inactivation is not a prerequisite of the U-shaped voltage dependence of Gd3+ block, both inactivation and Gd3+ block are linked to activation and depend on electrodiffusion into the pore in a similar fashion.
Since Gd3+ block is strongly influenced by permeant ions, we suggest that Ca2+-dependent inactivation reduces Gd3+ binding by increasing the occupancy of the binding site by permeant ion(s). Based on our findings, we developed a model that successfully describes the U-shaped voltage dependence of Ca2+-dependent inactivation as a result of an increase of the affinity of the selectivity filter to Ca2+. This view does not contradict the effects of Ca2+/calmodulin on inactivation of these channels, but rather places the mechanistic focus of the permeability changes at the selectivity filter.
MATERIALS AND METHODS
Channel expression and patch-clamp technique were as described in the accompanying paper (Babich et al., 2007).
Online Supplemental Material
The additional material (available at http://www.jgp.org/cgi/content/full/jgp.200709734/DC1) contains script text files that were used to run the simulations described in the text. The file cain.par contains the equations, parameters of the model, and extensive comments. The file cain2.par is a modification of cain.par to model the effect of Ca2+ accumulation. The calculation program CalC for the scripts is available at http://web.njit.edu/∼matveev/. Fig. S1 illustrates simulation of the dependence of inactivation kinetics on series resistance. Fig. S2 illustrates simulation of the dependence of inactivation kinetics on single-channel current. Fig. S3 illustrates simulation of inactivation in Ba2+.
RESULTS
Gd3+ Block Prevents Ca2+-dependent Inactivation
Although both inactivation and blockage reduce ionic currents, it is possible to evaluate their specific contributions using pulse protocols that exploit the removal of Gd3+ block at large positive voltages. Thus, we used a 20-ms pulse to 200 mV to unblock Gd3+ during development of both inactivation and Gd3+ block. The pulse to 200 mV was applied at different times during a much longer voltage pulse to 20 mV that activated maximal Ca2+ currents (Fig. 1 A). In the absence of Gd3+ (Fig. 1 B), the peaks of tail currents after the pulses to 200 mV reflected the onset of inactivation that occurred during the step from the holding potential to 20 mV before the pulses to 200 mV were applied. The peaks were somewhat larger than the current just before the pulses to 200 mV were applied, reflecting a small additional increase in the degree of activation between 20 and 200 mV. At 50 nM Gd3+ (Fig. 1 C), the tail currents increased in comparison with those in the absence of the blocker. Because the submicromolar amounts of Gd3+ do not affect the voltage dependence of activation (Babich et al., 2007), the increase was unlikely to be due an additional activation at 200 mV. Since the pulse to 200 mV removes Gd3+ from open-blocked channels, the magnitude of tail currents in the presence of Gd3+ reflects the number of channels that were opened and opened-blocked just before the unblocking pulse was applied. The current activated by stepping from −90 to 20 mV further decreased and decayed much more quickly when 1 μM Gd3+ was added to the bathing solution (Fig. 1 D), but the tail currents caused by stepping from 200 to 20 mV increased even more in comparison with those in Fig. 1, B and C. Similar observations were seen in six cells. Without exception, addition of Gd3+ reduced currents activated by the test depolarization but increased the tail currents after the pulse to 200 mV.
Figure 1.

Tail currents reveal that Gd3+ reduces inactivation in Ca2+. (A) Voltage-pulse protocol used. 20-ms step to 200 mV was applied to relieve Gd3+ block at different times of the pulse to 20 mV. (B) Currents from a cell bathed in solution with 10 mM Ca2+ and 0 Gd3+. The peaks of tail currents in response to stepping from 200 to 20 mV followed the time course of inactivation. The dashed line through the peaks is the best fit by an exponential: I = −I0 + ΔI (1 − e−kt), where I0 = 979 pA, ΔI = 669 pA, and k = 0.010 ms−1. The ratio between the numbers of inactivated and noninactivated channels after 500 ms at 20 mV can be estimated by ΔI/(I0 − ΔI). On average, it was 2.1 ± 0.17 (n = 6). (C) Currents from the same cell bathed in solution with 10 mM Ca2+ and 50 nM Gd3+. The dashed line through the peaks is the best fit with I0 = 986 pA, ΔI = 552 pA, and k = 0.011 ms−1. The averaged ΔI/(I0 − ΔI) ratio was 1.24 ± 0.14 (n = 6). (D) Currents from the same cell bathed in solution with 10 mM Ca2+ and 1 μM Gd3+. The dashed line through the peaks is the best fit with I0 = 948 pA, ΔI = 328 pA, and k = 0.007 ms−1. The averaged ΔI/(I0 − ΔI) ratio was 0.51 ± 0.13 (n = 6).
The increase of the number of channels that were opened and opened-blocked (O+OB) reflected a reduction of inactivation due to the presence of Gd3+. In principle, Gd3+ could bind to inactivated channels as well. Thus, the extent of inactivation calculated as 1 − (O+OB) reflects the number of inactivated and potentially inactivated-blocked channels. A minimal Scheme 1 to describe the interaction between inactivation and Gd3+ is that of state-dependent binding of Gd3+.
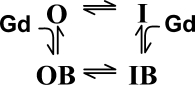 |
(SCHEME 1) |
The observation that O+OB increases with [Gd3+] indicates that inactivation is reduced in blocked states, i.e., the rate of the OB→IB transition is small and the reverse transition is not changed. This is consistent with the idea that block reduces Ca2+ influx needed for the Ca2+/calmodulin regulation.
Gd3+ Block Does Not Increase Inactivation in Ba2+
Experiments illustrated in Fig. 2 analyze how Gd3+ block affects inactivation of Ba2+ currents. The test pulse from −90 to 0 mV activated maximal Ba2+ current. The steps from 0 to 200 mV and back to 0 mV caused tail currents, whose peaks indicate the degree of inactivation (I + IB). To avoid current rundown due to intracellular accumulation of Ba2+ during prolonged pulses, only two sets of pulses with 5-ms and 500-ms-long initial steps to 0 mV were taken in each cell. The magnitude of the tail currents (i.e., degree of inactivation) did not decrease in the presence of Gd3+ even though the blocker reduced and accelerated decay of currents during the pulse from −90 to 0 mV. In three cells tested by the same experimental protocol as on Fig. 2, the tail currents elicited by stepping from 200 to 0 mV did not change in the presence of Gd3+. In two cells, addition of Gd3+ caused a small (∼10%) increase of the tails consistent with inactivation in Ba2+ also depending to small degree on ion influx (Ferreira et al., 1997). These results show that the effects of Gd3+ block on inactivation are more pronounced for Ca2+, rather than Ba2+, conductance.
Figure 2.

Inactivation of Ba2+ currents in the presence of Gd3+. Currents were elicited similar to that in Fig 1. The 20-ms step to 200 mV was applied after 5 ms at 0 mV (traces a and c), or after 500 ms at 0 mV (traces b and d). Without the blocker (traces a and b), the peaks of the tails differ because of inactivation. With 25 nM Gd3+ (traces c and d), currents elicited by the step from −90 to 0 mV were smaller and decayed more rapidly. However, the 200-mV pulse after 5ms at 0 mV relieved the tonic Gd3+ block to reveal the magnitude of the unblocked current (compare traces a and c). Gd3+ did not change the magnitude of tail currents after 500 ms at 0 mV (compare traces b and d). The ratio between the numbers of inactivated and noninactivated channels after 500 ms at 0 mV was estimated directly from the traces as indicated. On average, it was 0.43 ± 0.11 (n = 5) without Gd3+ and 0.35 ± 0.09 after addition of 25 nM of Gd3+.
Ca2+-dependent Inactivation Prevents Gd3+ Block
The data in Fig. 1 indicate that blocked channels do not inactivate and the distribution in the OB↔IB step of Scheme 1 is shifted toward the OB state. Therefore, Gd3+ binding to the inactivated state could significantly increase the portion of open-blocked channels (I→IB→OB) in addition to the direct block (O→OB). In other words, binding of Gd3+ to inactivated channels would increase the apparent efficiency of Gd3+ for block of open channels as defined by the O/OB ratio. In the experiment in Fig. 3, we assessed whether the block could occur through binding of Gd3+ to inactivated channels by using conditions in which inactivation is greatly reduced. For this purpose, we included a mutant calmodulin CAM1234 with low affinity to Ca2+ in our expression system. The presence of CAM1234 dramatically reduces Ca2+-dependent inactivation (Peterson et al., 1999). In Fig. 3, the pulse protocol was the same as in Fig. 1, but only traces for the 500-ms first pulse to 20 mV are shown. Despite the dramatic difference in inactivation between the wild-type and the CAM1234 mutant in the absence of Gd3+ (compare black traces), both tonic block (reduction of peak currents) and use-dependent block (acceleration of current decay) were nearly the same (compare gray traces). This is expected since Gd3+ prevents inactivation. Importantly, the O/OB ratio determined from the peak currents following the pulse to 200 mV was not significantly different in cells with normal or mutated calmodulin. Therefore, inactivation did not change the reequilibriation between opened and opened-blocked channels. We conclude that there is no significant occupancy of an IB state, i.e., Gd3+ does not bind to inactivated channels.
Figure 3.
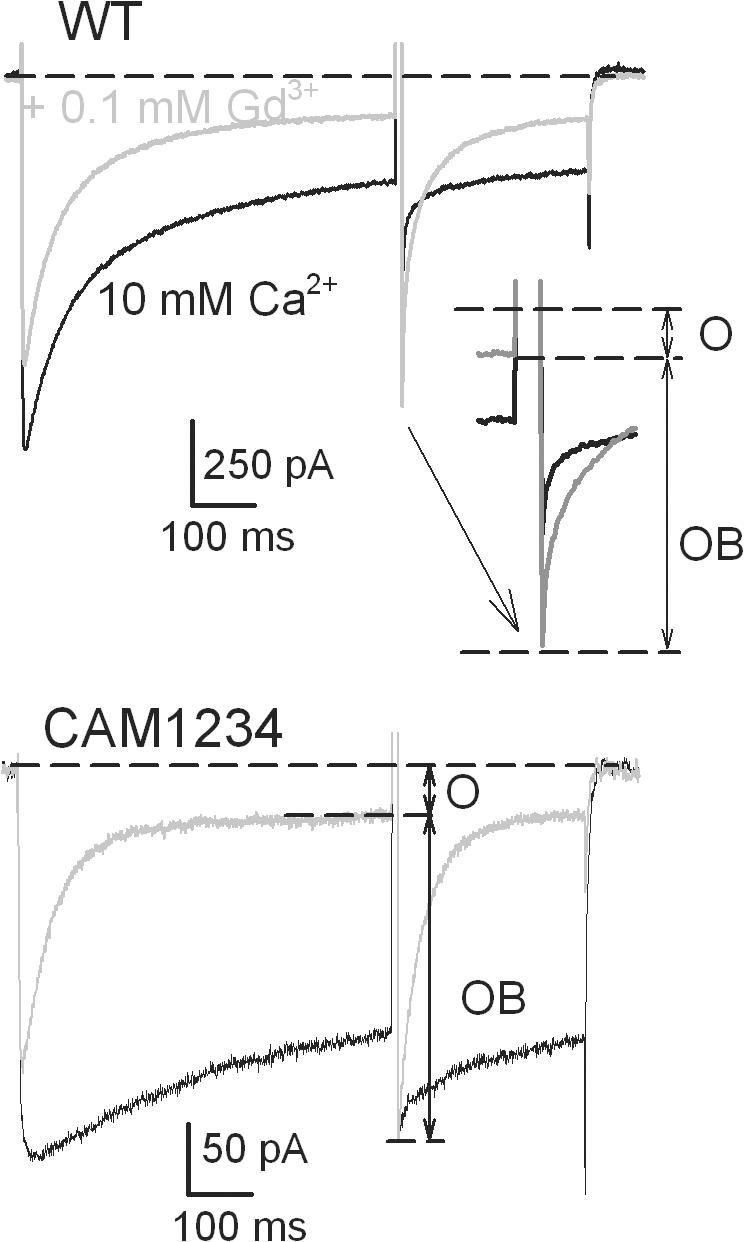
The CAM1234 mutation of calmodulin reduced inactivation of Ca2+ currents, but it did not alter open-channel block by Gd3+. Currents were elicited by the same pulse protocol as in Fig. 1 A. Only traces with 500-ms pulse from −90 to 20 mV are shown. Although in the absence of Gd3+ (black traces) inactivation of currents at 20 mV was much less in the cell with CAM1234, application of 0.1 μM Gd3+ (gray traces) reduced the peak and accelerated the decay of currents similarly in the wild-type cell and in the cell with the mutated calmodulin. Since the step to 200 mV did not relieve inactivation (Fig. 1 B), but relieved Gd3+ block (see accompanying paper Babich et al., 2007), the ratio between the numbers of open and open-blocked channels after 500 ms at 20 mV can be simply estimated from the current before the step to 200 mV and from the peak of the tail current on the return from 200 to 20 mV, as indicated. On average, the ratio was 0.161 ± 0.012 in the wild-type cells (n = 3) and 0.156 ± 0.014 (n = 4) in cells with CAM1234.
DISCUSSION
Hypothesis: Ca2+ Ions Occlude the Selectivity Locus during Ca2+-dependent Inactivation
Our observations indicate that Ca2+-dependent inactivation and Gd3+ block of CaV1.2 channels are mutually exclusive, suggesting direct or allosteric action at a common site. A possible mechanism is that Ca2+-dependent inactivation decreases Gd3+ binding, and that Gd3+ occupancy of the site blocks inactivation. The simplest form of such a mechanism is one in which inactivation increases occupancy of the Gd3+ site by Ca2+.
Several lines of data suggest that the selectivity filter itself is the site of interaction between Gd3+ and Ca2+-dependent inactivation. Gd3+ block is thought to occur at the selectivity filter and its potency is strongly influenced by competition with permeant ions. Inactivation has been shown to specifically reduce Ca2+ permeability, but not permeability of monovalent ions in the absence of Ca2+ (Babich et al., 2005). Importantly, the EEQE mutation at the selectivity locus eliminates Ca2+-dependent inactivation while allowing Ca2+ to permeate (Zong et al., 1994).
Thus, our studies of Gd3+ block suggest a determining role for the selectivity filter in Ca2+-dependent inactivation of these channels, in addition to the well-established role of calmodulin in enhancing or stabilizing the inactivated state. These ideas prompted us to determine whether a minimal model of this mechanism could account for key features of inactivation.
The Minimal Model of Ion-dependent Inactivation
Starting from the idea that Ca2+-dependent inactivation stabilizes a state of the selectivity filter with higher affinity to Ca2+, we built a minimal model of inactivation that successfully describes the U-shaped dependence of inactivation on voltage, the signature of the current-dependent mechanism. The model incorporates only very basic assumptions about gating, ion flux, and Ca2+ binding in the pore.
The model (Fig. 4) assumes that when Ca2+ is not in the pore, voltage-dependent inactivation obeys the four-step Charge1-Charge2 schema (Brum and Rios, 1987; Shirokov et al., 1992). The left–right transition from R (resting) to A (activated) states is the opening of the voltage-dependent gate; the down–up transition from P (primed) to I (inactivated) states is the inactivating step. Ca2+ binding to the pore (front-back transition) interferes with any of the four states, making it a three-particle allosteric mechanism.
Figure 4.

A model of ion-dependent inactivation.
The pore of noninactivated channels is assumed to have an apparent affinity for Ca2+ in the 10 mM range that is sufficient to provide the 106 s−1 throughput/flux rate corresponding to the observed 0.1–1 pA single- channel currents. According to our interpretation of experimental results, the affinity increases in inactivated channels. Even in inactivated states, the microscopic kinetic steps describing Ca2+ binding are rapid in comparison with other transitions that correspond to the channel's conformational changes. Therefore, the rapid equilibration approximation is used to describe Ca2+ binding. This simplification allows the U-shaped voltage dependence of inactivation to be accounted for based on the fundamentals of the link between activation/ inactivation and ionic flux without making specific structural assumptions about location of the interaction.
The rapid equilibration approximation of the intrapore ion binding has been used to describe H+ blockage of Na+ channels (Woodhull, 1973) and Ca2+ blockage of the EEEE locus in the pore of cyclic nucleotide-gated channels (Seifert et al., 1999).
Because Ca2+-dependent inactivation does not change the voltage dependence of the intramembrane charge movements in noninactivated and inactivated channels (Isaev et al., 2004), Ca2+ binding is proposed to affect inactivation steps (P↔I), but not transitions R↔A, that correspond to movement of the voltage sensor. When Ca2+ ion is in the pore, the inactivation onset rate (P→I) is increased by a factor γ (20–100 in calculations). Because of microscopic reversibility, this is equivalent to increasing the affinity of inactivated channels to Ca2+ by the same factor (i.e., effective KD.eff. = KD/γ in I states).
In addition to being coupled to the states of the channel, Ca2+ binding in channels with open voltage-dependent gate (A states) is affected by voltage directly because the site is in the permeation pathway. If the on-off rates of Ca2+ binding to the pore are independent of the direction from/to which Ca2+ ions move to/from the channel at 0 mV, then the effect of voltage might be accounted for by using the effective dissociation constant written as:
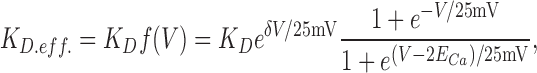 |
where δ is the portion of the electric field at the Ca2+ binding site, ECa is the equilibrium potential for Ca2+, and the voltage steepness factor 25 mV approximates RT/F at room temperature. The formula for f(V) is an extension of the Woodhull theory of voltage-dependent block (Woodhull, 1973) for permeating ion.
Opening of the gate and low affinity of the pore correspond to the open noninactivated channel. In the AR states Ca2+ can move through the channel. When the gate is open, but the channel is inactivated (the AI state), Ca2+ flux through the channel is small because the pore has higher affinity to Ca2+. The amplitude of single-channel current through the single site was calculated as:
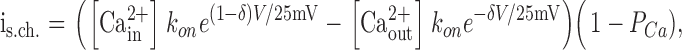 |
where PCa is the probability that the site is occupied by Ca2+. It is equal to
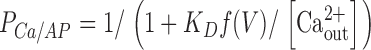 |
in the state AP, and to
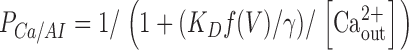 |
in the state AI.
Other constants and details of the model are described in the script file (text file cain.par in the supplemental material, available at http://www.jgp.org/cgi/content/full/jgp.200709734/DC1) that was used to run the calculation with the CalC modeling program (Matveev et al., 2004).
Currents simulated for a set of voltage pulses applied from −100 mV are shown in Fig. 5 A. They are the sums of currents through channels in noninactivated and inactivated states. Simulated currents through inactivated states (Fig. 5 B) are very small. The rate of inactivation (determined by fitting a sum of an exponential and a constant) was maximal (Fig. 5 D) at voltages where peak current (Fig. 5 C) was maximal. Therefore, the simulation demonstrates that the U-shaped voltage dependence of inactivation can arise simply from Ca2+ binding more potently to the pore of inactivated channels and thus preventing the influx. Inactivation increases at voltages where the channels activate because the state at which Ca2+ blocks the pore is more likely when the voltage sensor is at the cis/active position. Inactivation decreases at more positive voltages because diffusion of Ca2+ that stabilizes the high affinity inactivated state of the site within the pore is reduced.
Figure 5.

Simulated Ca2+ currents using the single-site approximation to calculate single-channel currents. The “inactivation-binding” coupling factor was γ = 50. (A) Total currents. Voltage steps were from the holding potential −100 mV to −60–90 mV, increment 10 mV. (B) Currents through inactivated channels. (C) Peak current–voltage relationship. (D) Rates of the best fits by the sum of an exponential and a constant to the decay phase of currents in A.
In several studies, it has been shown that the maximum rate of inactivation does not seem to correspond to the peak of the ionic current (e.g., Noceti et al., 1998). Instead, the maximal rate of inactivation occurs 10–20 mV more negative than the voltage of maximal currents. Since the driving force for Ca2+ influx increases with voltage becoming more negative, this could be taken as evidence that the accumulation of Ca2+ on the intracellular side of the channel, rather than its diffusion into the pore and an immediate effect on gating, is critical for the U-shaped voltage dependence of inactivation. However, the kinetics of inactivation at voltages of the negative resistance slope of the current–voltage relationship are extremely sensitive to the error in voltage clamp due to finite series resistance. The model readily reproduces the negative shift of the voltage dependence of inactivation rate by accounting for the realistic values of series resistance, membrane capacitance, and magnitudes of transmembrane currents (see Fig. S1).
Although it is not required to explain the U-shaped voltage dependence of inactivation, possible contribution of the “local” accumulation of Ca2+ can be added on to the model (Fig. S2). In this case, the “inactivation-block coupling” factor γ is increased proportionally to the magnitude of the single-channel current when the channel is in conductive states. This modification also describes the observation that the rate of inactivation is maximal at voltages more negative than those causing peak currents.
To test whether or not the model critically depends on how the amplitude of single-channel current was calculated, we also used other formulations that represent two extreme cases: ohmic (long-channel approximation) and constant field (GHK, or short-channel approximation). While these two approaches require experimentally determined scaling factors, the single-site formulation described above calculates ionic current/flux using the same Ca2+ on–off rates that account for the interaction with inactivation. All three formulations give similar result; inactivation of simulated ionic currents has a pronounced U-shaped voltage dependence.
Conclusion
Involvement of the pore structure in the C-type/slow inactivation of voltage-gated K+ and Na+ channels has been well established (Lopez-Barneo et al., 1993; Baukrowitz and Yellen, 1995; Balser et al., 1996; Starkus et al., 1997; Kiss et al., 1999; Starkus et al., 2000; Loots and Isacoff, 2000; Kuo et al., 2004, see comment by Kass, 2004; Berneche and Roux, 2005). Although participation of the selectivity filter in inactivation gating was originally proposed for Ca2+ channels (Brum et al., 1988; Pizarro et al., 1989), understanding of its mechanisms had been delayed apparently because it was thought to be a property of voltage-dependent inactivation, which is much slower and therefore of a lesser physiological significance than Ca2+-dependent inactivation. Nevertheless, some studies that analyzed mutations in the selectivity locus of Ca2+ channels clearly showed its involvement in rapid inactivation of L-type (Yatani et al., 1994; Zong et al., 1994) and T-type (Talavera et al., 2003) channels.
The idea that during inactivation Ca2+ blocks the channel at the selectivity filter does not contradict the view that the calmodulin tethered to the intracellular side of the channel is an important regulator of inactivation. To explain a more rapid inactivation and a more pronounced U-shape of voltage dependence in Ca2+, rather than in Ba2+, the “inactivation-block coupling” factor γ in our minimal model of ion-dependent inactivation should be greater in Ca2+ than in Ba2+ (Fig. S3). This is in agreement with our previous suggestion (Isaev et al., 2004) that Ca2+/calmodulin controls inactivation by stabilizing the inactivated state(s) with Ca2+ bound to the pore. However, according to our view, the U-shaped voltage dependence of inactivation does not occur from the Ca2+/calmodulin interaction. It results from Ca2+ blockage of the selectivity filter, or, more specifically, the effect of voltage on Ca2+ accessibility to it.
In the last few years, the study of Ca2+-dependent inactivation has been driven mostly by the analysis of the role of calmodulin. Our work demonstrates that other Ca2+ site(s) are involved as well. The mechanism that actually stops Ca2+ influx through inactivated channels has not been identified, but our data and modeling suggest that intrapore Ca2+ itself is key. Our observations strongly indicate that changes in the selectivity filter play a fundamental role in the block of Ca2+ influx during Ca2+-dependent inactivation.
Although further studies are needed to determine specifics of the molecular mechanism that alters Ca2+ binding to the selectivity filter during activation/inactivation gating, the theory described here provides a relatively simple framework for explaining how auxiliary subunits and calmodulin fine tune ion- and voltage- dependent inactivation by directly influencing movements of the S5-pore-S6 bundles of the α1 subunit.
Supplemental Material
Acknowledgments
The authors are grateful to John Reeves for valuable suggestions and discussions.
The work was supported by National Institutes of Health grants MH62838 to R. Shirokov and National Science Foundation grant DMS-0417416 to V. Matveev.
Olaf S. Andersen served as editor.
O. Babich's present address is Molecular Pharmacology Department, AstraZeneca R&D, Södertälje, Sweden.
References
- Babich O., D. Isaev, and R. Shirokov. 2005. Role of extracellular Ca2+ in gating of CaV1.2 channels. J. Physiol. 565:709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babich, O., J. Reeves, and R. Shirokov. 2007. Block of CaV1.2 channels by Gd3+ reveals pre-opening transitions in the selectivity filter. J. Gen. Physiol. 129:461–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balser, J.R., H.B. Nuss, N. Chiamvimonvat, M.T. Perez-Garcia, E. Marban, and G.F. Tomaselli. 1996. External pore residue mediates slow inactivation in μ1 rat skeletal muscle sodium channels. J. Physiol. 494:431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berneche, S., and B. Roux. 2005. A gate in the selectivity filter of potassium channels. Structure. 13(4):591–600. [DOI] [PubMed] [Google Scholar]
- Baukrowitz, T., and G. Yellen. 1995. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 15:951–960. [DOI] [PubMed] [Google Scholar]
- Beedle, A.M., J. Hamid, and G.W. Zamponi. 2002. Inhibition of transiently expressed low- and high-voltage-activated calcium channels by trivalent metal cations. J. Membr. Biol. 187:225–238. [DOI] [PubMed] [Google Scholar]
- Biagi, B.A., and J.J. Enyeart. 1990. Gadolinium blocks low- and high-threshold calcium currents in pituitary cells. Am. J. Physiol. 259:C515–C520. [DOI] [PubMed] [Google Scholar]
- Brehm, P., and R. Eckert. 1978. Calcium entry leads to inactivation of calcium channel in Paramecium. Science. 202:1203–1206. [DOI] [PubMed] [Google Scholar]
- Brum, G., and E. Rios. 1987. Intramembrane charge movement in frog skeletal muscle fibres. Properties of charge 2. J. Physiol. 387:489–517. (published erratum appears in J. Physiol. 1988. 396:581) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum, G., R. Fitts, G. Pizarro, and E. Rios. 1988. Voltage sensors of the frog skeletal muscle membrane require calcium to function in excitation-contraction coupling. J. Physiol. 398:475–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, G., J. Yi, E. Rios, and R. Shirokov. 1997. Ion-dependent inactivation of barium current through L-type calcium channels. J. Gen. Physiol. 109:449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaev, D., K. Solt, O. Gurtovaya, J.P. Reeves, and R. Shirokov. 2004. Modulation of the voltage sensor of L-type Ca2+ channels by intracellular Ca2+. J. Gen. Physiol. 123:555–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass, R.S. 2004. Sodium channel inactivation goes with the flow. J. Gen. Physiol. 124:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss, L., J. LoTurco, and S.J. Korn. 1999. Contribution of the selectivity filter to inactivation in potassium channels. Biophys. J. 76:253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, C.C., W.Y. Chen, and Y.C. Yang. 2004. Block of tetrodotoxin-resistant Na+ channel pore by multivalent cations: gating modification and Na+ flow dependence. J. Gen. Physiol. 124:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, A., S.T. Wong, D. Gallagher, B. Li, D.R. Storm, T. Scheuer, and W.A. Catterall. 1999. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 399:155–159. [DOI] [PubMed] [Google Scholar]
- Loots, E., and E.Y. Isacoff. 2000. Molecular coupling of S4 to a K+ channel's slow inactivation gate. J. Gen. Physiol. 116:623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo, J., T. Hoshi, S.H. Heinemann, and R.W. Aldrich. 1993. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1:61–71. [PubMed] [Google Scholar]
- Matveev, V., R.S. Zucker, and A. Sherman. 2004. Facilitation through buffer saturation: constraints on endogenous buffering properties. Biophys. J. 86:2691–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noceti, F., R. Olcese, N. Qin, J. Zhou, and E. Stefani. 1998. Effect of Bay K 8644(−) and the β2a subunit on Ca2+-dependent inactivation in α1C Ca2+ channels. J. Gen. Physiol. 111:463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obejero-Paz, C.A., I.P. Gray, and S.W. Jones. 2004. Y3+ block demonstrates an intracellular activation gate for the α1G T-type Ca2+ channel. J. Gen. Physiol. 124:631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, B.Z., C.D. DeMaria, J.P. Adelman, and D.T. Yue. 1999. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 22:549–558. [DOI] [PubMed] [Google Scholar]
- Pizarro, G., R. Fitts, I. Uribe, and E. Rios. 1989. The voltage sensor of excitation-contraction coupling in skeletal muscle. Ion dependence and selectivity. J. Gen. Physiol. 94:405–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, N., R. Olcese, M. Bransby, T. Lin, and L. Birnbaumer. 1999. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc. Natl. Acad. Sci. USA. 96:2435–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather, W.A., and E.W. McCleskey. 2003. Permeation and selectivity of calcium channels. Annu. Rev. Physiol. 65:133–159. [DOI] [PubMed] [Google Scholar]
- Seifert, R., E. Eismann, J. Ludwig, A. Baumann, and U.B. Kaupp. 1999. Molecular determinants of a Ca2+-binding site in the pore of cyclic nucleotide-gated channels: S5/S6 segments control affinity of intrapore glutamates. EMBO J. 18:119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokov, R., R. Levis, N. Shirokova, and E. Rios. 1992. Two classes of gating current from L-type Ca channels in guinea pig ventricular myocytes. J. Gen. Physiol. 99:863–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus, J.G., L. Kuschel, M.D. Rayner, and S.H. Heinemann. 1997. Ion conduction through C-type inactivated Shaker channels. J. Gen. Physiol. 110:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus, J.G., S.H. Heinemann, and M.D. Rayner. 2000. Voltage dependence of slow inactivation in Shaker potassium channels results from changes in relative K+ and Na+ permeabilities. J. Gen. Physiol. 115:107–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera, K., A. Janssens, N. Klugbauer, G. Droogmans, and B. Nilius. 2003. Pore structure influences gating properties of the T-type Ca2+ channel α1G. J. Gen. Physiol. 121:529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhull, A.M. 1973. Ionic blockage of sodium channels in nerve. J. Gen. Physiol. 61:687–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatani, A., A. Bahinski, M. Wakamori, S. Tang, Y. Mori, T. Kobayashi, and A. Schwartz. 1994. Alteration of channel characteristics by exchange of pore-forming regions between two structurally related Ca2+ channels. Mol. Cell. Biochem. 140:93–102. [DOI] [PubMed] [Google Scholar]
- Zong, S., J. Zhou, and T. Tanabe. 1994. Molecular determinants of calcium-dependent inactivation in cardiac L-type calcium channels. Biochem. Biophys. Res. Commun. 201:1117–1123. [DOI] [PubMed] [Google Scholar]
- Zuhlke, R.D., G.S. Pitt, K. Deisseroth, R.W. Tsien, and H. Reuter. 1999. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 399:159–162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}