

Abstract
Several naturally occurring polypeptide neurotoxins target specific sites on the voltage-gated sodium channels. Of these, the gating modifier toxins alter the behavior of the sodium channels by stabilizing transient intermediate states in the channel gating pathway. Here we have used an integrated approach that combines electrophysiological and spectroscopic measurements to determine the structural rearrangements modified by the β-scorpion toxin Ts1. Our data indicate that toxin binding to the channel is restricted to a single binding site on domain II voltage sensor. Analysis of Cole-Moore shifts suggests that the number of closed states in the activation sequence prior to channel opening is reduced in the presence of toxin. Measurements of charge–voltage relationships show that a fraction of the gating charge is immobilized in Ts1-modified channels. Interestingly, the charge–voltage relationship also shows an additional component at hyperpolarized potentials. Site-specific fluorescence measurements indicate that in presence of the toxin the voltage sensor of domain II remains trapped in the activated state. Furthermore, the binding of the toxin potentiates the activation of the other three voltage sensors of the sodium channel to more hyperpolarized potentials. These findings reveal how the binding of β-scorpion toxin modifies channel function and provides insight into early gating transitions of sodium channels.
INTRODUCTION
The action potential in excitable cells is characterized by a sharp initial depolarization followed by a slower repolarization of the membrane to resting potentials. Voltage-gated sodium channels generate the rising phase of the action potentials by opening rapidly (Hodgkin and Huxley, 1952). Gating modifier toxins act on the sodium channels to increase ion flux either by favoring channel opening or by slowing their rate of inactivation (Cestèle and Catterall, 2000). β-scorpion toxins, which are also referred to as site 4 toxins, fall in the former category. They increase sodium current by shifting the threshold of activation to hyperpolarized potentials by as much as 20 mV (Cahalan, 1975; Vijverberg et al., 1984; Marcotte et al., 1997; Cestèle et al., 1998).
Much of the recent work on the mode of action of β-scorpion toxins have focused on understanding how these toxins cause their target voltage sensor to activate. Several studies indicate that the β-toxin binding site is formed mainly by residues in the voltage sensor of domain II (Marcotte et al., 1997; Cestèle et al., 1998; Cestèle et al., 2001). Although the binding of the β-scorpion toxin is voltage independent, the full effect of the toxin on the sodium channel is seen only after a brief depolarizing prepulse. According to the “trapping mechanism,” proposed on the basis of effects of the β-toxin CssIV from the scorpion Centruroides suffusus suffusus in neuronal sodium channels, β-toxin binding traps the voltage sensor of DII in the activated state (Cestèle et al., 1998, 2001, 2006). Therefore, a brief depolarizing prepulse is necessary to initially drive the voltage sensors into an activated state. In contrast, Ts1, which has also been classified as a β-scorpion toxin based on its structural homology, competitive binding assay, and its site of action, does not require a depolarizing prepulse to fully affect the sodium channels (Barhanin et al., 1982; Vijverberg et al., 1984; Yatani et al., 1988; Pinheiro et al., 2004). Ts1 is also known as TsVII and Tsγ and is a major toxic component of the venom from the Brazilian scorpion Tityus serrulatus.
Understanding the mechanism of toxin binding to the voltage sensor, however, does not clarify a more fundamental problem. Unlike potassium channel gating modifier toxins, the β-scorpion toxin is believed to bind to only one of the four voltage sensors of the sodium channel (Marcotte et al., 1997; Cestèle et al., 1998, 2001; Cohen et al., 2005). According to the classical models of sodium channel gating, the voltage sensors of the sodium channel activate independently and at least three of them have to be in an activated position for the channel to open (Hodgkin and Huxley, 1952; Armstrong and Bezanilla, 1977; French and Horn, 1983; Patlak, 1991; Keynes, 1994). If only one of them is activated by the β-toxin, the threshold of activation is unlikely to shift significantly since other voltage sensors remain unaffected.
To address this issue, we have used gating charge measurements combined with fluorescence spectroscopy to determine the effect of toxin binding on the voltage-gated sodium channels. Our results demonstrate that the binding of the toxin not only immobilizes domain II voltage sensor, presumably in an activated state, but also causes large hyperpolarizing shifts in the activation of other voltage sensors. These results indicate that cooperative or coupled behavior of voltage sensors is a fundamental property in voltage-gated sodium channels and explains how a site-specific toxin has such profound effects on the voltage-dependent gating process.
MATERIALS AND METHODS
Molecular Biology, Expression, and Labeling
The α-subunit of the rSkM1 was cloned into a pBSTA vector and site-directed mutagenesis was performed as described previously (Chanda and Bezanilla, 2002). The α- and β-subunit cRNA were transcribed in vitro with T7 polymerase (Ambion) and injected in a molar ratio 2:1 into Xenopus oocytes. The oocytes were incubated after injection for 2–5 d at 18°C. Before the fluorescence measurements, the oocytes were incubated overnight in solution containing 100 μM DDT. After removing the DTT, the oocytes were labeled on ice in a depolarizing solution containing 10 μM 5′-tetramethylrhodamine maleimide (TMRM; Molecular Probes) for 20–30 min
Modified Cut Open Oocyte Epifluorescence Setup
The modified cut open setup, described previously (Cha and Bezanilla, 1998), was placed on the stage of an upright microscope (BX50WI; Olympus Optical). The light from a tungsten halogen light source was filtered by a filter 535DS35 and split using a 570DRLP dichroic mirror and was focused using a LUMPlanF1 40X water-immersion objective. The emitted light was filtered using a 565EFLP filter (Chroma Technologies and Omega Optical) and focused onto a PIN-020A photodiode (UDT Technologies) by a microscope condenser lens. The photodiode was connected to the headstage of an integrating Axopatch 1B patch-clamp amplifier (Axon Instruments, Inc.). The current from the photodiode was offset to prevent saturation of the feedback capacitor during acquisition. The excitation light was interrupted with a TTL-triggered VS25 shutter (Vincent Associates) between measurements.
Data Acquisition and Analysis
Fluorescence signals, ionic and gating currents were recorded with a PC44 board (Innovative Integration) and digitized on two 16-bit A/D converters. Fluorescence and gating currents were sampled at 10 μs/point and ionic currents at 5 μs/point. The data acquisition program was developed in house. Linear leak and membrane capacitive currents were subtracted using a P/4 protocol from a subtracting holding potential of −130 mV for ionic currents and 40 mV for gating currents. Fluorescence signals were acquired without subtraction.
The external solution contained 95 mM n-methylglucamine (NMG)-methylsufonate (Mes), 20 mM Na- Mes, 20 mM HEPES, and 2 mM Ca(Mes)2, pH 7.4. The internal solution contained 113 mM NMG-Mes, 2 mM Na-Mes, 20 mM HEPES, and 2 mM EGTA, pH 7.4. For gating current experiments the ionic currents were blocked with 5 μM TTX.
Ts1, purified as described previously (Possani et al., 1981), was diluted in the bath solution containing 1% BSA and applied to the upper and guard chambers. Data were acquired at room temperature, unless otherwise stated.
To ensure that the binding site of the mutants is saturated, we have used previously described electrophysiological criteria. As was previously described by others we find that the α-toxin maximally left shifted the threshold of activation of the wt Na+ channel by 25 mV at 50 nM concentrations. By increasing the concentration to 500 nM, there was no further shift. In three out of four mutants used for fluorescence–voltage measurements in this paper, we find that the 50 nM toxin shifts the activation threshold by 25 mV. In the fourth mutant, we observed that 50 nM toxin shifted the threshold of activation by 10–15 mV. Increasing the toxin concentration to 500 nM, we observed that the activation in this mutant was also left shifted by 25 mV. In our experiments, we have used saturating concentrations of toxin and we have used this criterion of 25-mV shift in the threshold of channel activation as a measure of saturation of the binding site.
Data analysis and acquisition were performed with in-house programs (Gpatch and Analysis). Curve fittings were done on SigmaPlot 8.02 (SPSS, Inc.) and Excel 2000 (Microsoft).
For Fig. 2 C, the following function was fitted to the current
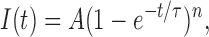 |
(1) |
where I is the current, A is the maximum amplitude, t is the time, τ is the time constant, and n is an empirical constant proportional to the number of states.
Figure 2.

Effect of Ts1 in the Cole-Moore shift. (A) Voltage protocol used to measure the Cole-Moore shift. (B, top) Superimposed sodium currents obtained in the absence of Ts1, following a prepulse to −60 mV (lefthand trace) or −160 (righthand trace). (B, bottom) Superimposed sodium currents obtained in the presence of Ts1 (100 nM), elicited as described above for the control conditions. (C) Superimposed sodium currents (prepulses to −160 mV) recorded in control conditions (blue line) and in the presence of Ts1 (red line). Black lines are the fits to the function in Eq. 1 (see Materials and Methods). Arrows in A and B indicate the beginning of the pulse. (D) Time to the half-maximal sodium current for indicated potentials in control conditions (blue triangles) and in the presence of Ts1 (red circles). The inset shows the time shift necessary to superimpose the currents for the indicated potentials with respect to the currents at −160 mV (control, blue triangles; in presence of Ts1, red circles). Blue and red lines are the fits to the function shown in Eq. 2. The parameters obtained for the curve in the absence of Ts1 were z = 2.3; Vh = 0 mV; the parameters obtained in the presence of Ts1 were z = 4.4; Vh = −17.7 mV.
Based on a two-state model raised to the power of n (1), the time to half maximum of the current t(V) as a function of prepulse voltage was fitted to the data of Fig. 2 D. The expression of t(V) is given by:
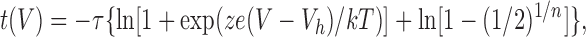 |
(2) |
where τ is the time constant obtained by fitting the traces in Fig. 2 C with Eq. 1, n is the power obtained also with Eq. 1, z is the valence, k and T have their usual meaning, and Vh is an offset.
Charge–voltage relationships in Fig. 3 D were fitted to either a single Boltzmann function
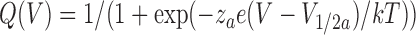 |
(3) |
or a sum of two Boltzmann functions of the form
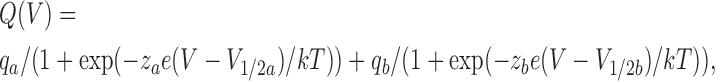 |
(4) |
where qa and qb are the charge amplitudes, za and zb are the valences, e is the electronic charge, V 1/2a and V 1/2b are the half-maximal voltages, and k and T have their usual meaning.
Figure 3.

Effect of Ts1 on gating currents. Superimposed gating current traces elicited by a test pulse to −150 mV (A), −100 mV (B), and 0 mV (C) in control conditions (blue line) and in the presence of 50 nM Ts1 (red line). (A, inset) Voltage protocol used to record the currents. (D) Charge–voltage (Q-V) relationships (normalized to the maximum charge in control conditions) in the presence of Ts1 (red circles) and in control conditions (blue triangles). Solid and dashed traces are the fits to the function in Eqs. 3 and 4, respectively (see Materials and Methods). See Table I for fitted parameters.
Fluorescence–voltage relationships in Fig. 8 were fitted with the following equation:
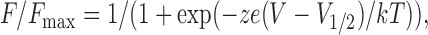 |
(5) |
where z is the valence, e is the electronic charge, and V 1/2 is the half-maximal voltage. Data are shown as mean ± SEM, and Student's t test was used to compare means (*, P < 0.05).
Figure 8.

Effect of Ts1 on S4-DI, S4-DIII, and S4-DIV movements. (A) Voltage protocol used to measure the fluorescence signals. (B–D) F-V relationships for domains I (S216C) (B), III (L1115C) (C), and IV (S1436C) (D) in absence (blue triangles) and presence of Ts1 (50 nM, red circles). Solid lines are the fits to the function in Eq. 5 (see Materials and Methods). See Table II for fitted parameters.
Simulations of the Effect of Gating Modifier Toxin on Eukaryotic Voltage-dependent Sodium Channel
A general model consisting of 16 states is sufficient to illustrate the role of coupling interactions between voltage sensors of the eukaryotic sodium channel. To simplify the model, the inactivated states were ignored and it was assumed that when all four voltage sensors are in an activated position the channel is open. The number of interaction terms necessary to define all the interactions equals to (n − 1)!, where n is the number of interacting units or sensors. When the voltage sensors move completely independent of each other, the interaction terms become equal to 1. If they are greater than 1, then the interaction is positive and if less than 1, the coupling is negative.
For simulation, the midpoint of activation for all the four voltage sensors was set to −50 mV with an apparent valence, z = 1. The intrinsic voltage-dependent rate constants were kept equal. Immobilization of S4-DII voltage sensor was simulated by moving the activation midpoint of S4-DII voltage sensor to −500 mV. To simulate independent movement of the four sensors, interaction terms were kept equal to 1. For the cooperative model, the following values of the interaction terms were used: n 12 = 100; n 13 = 10; n 14 = 50; n 23 = 100; n 24 = 10; n 34 = 75.
RESULTS
Length of the Cole-Moore shift Is Reduced in Toxin-modified Channels
β-Scorpion toxins, including Ts1 toxin used in this study, have been shown to shift the voltage dependence of the sodium currents to more hyperpolarizing direction and reduce the peak amplitude (Marcotte et al., 1997; Cestèle and Catterall, 2000). Fig. 1 compares the traces of sodium currents obtained from oocytes injected with wild-type rat skeletal muscle sodium channels, Nav 1.4. The currents were recorded at −72 (B), −56 (C), and 0 mV (D) before and after the treatment with 50 nM of Ts1. Fig. 1 E compares the conductance–voltage (G x V) curves obtained before and after the treatment with 25 nM of Ts1. As shown previously, the bound toxin was found to induce the appearance of sodium current at more negative potentials compared with control conditions, as well as a slight decrease in the peak of the current amplitude at more depolarized potentials (Marcotte et al., 1997). These experiments were performed at 7°C to improve voltage control and time resolution.
Figure 1.

Effect of Ts1 in the sodium currents activation. (A) Voltage protocol used to record the inward sodium currents. (B) Superimposed sodium currents obtained at −72 mV before (blue trace) and after (red trace) the treatment with 50 nM of Ts1. (C) Superimposed sodium currents obtained at −56 mV before (blue trace) and after (red trace) the treatment with Ts1 (50 nM). (D) Superimposed sodium currents obtained at 0 mV before (blue trace) and after (red trace) the treatment with Ts1 (50 nM). (E) Superimposed G-V curves obtained before (blue symbols) and after (red symbols) the treatment with Ts1 (25 nM).
Voltage-gated sodium channels typically go through a number of closed states before channel opening, which is reflected as the sigmoidal lag typically seen in ionic currents. The length of this lag phase is related to a summation of the inverse rates between closed states. (Cole and Moore, 1960). If Ts1 immobilizes the S4 domain II in an activated position, the number of closed states the channel traverses before opening must be reduced. Alternatively, the toxin binding could shift the voltage dependence of the gating charge movement to a more hyperpolarized potential. In this scenario, the number of states would remain the same but their voltage dependence would be shifted to hyperpolarized potentials. These two possibilities can be distinguished by measuring the voltage dependence of Cole-Moore shifts. In the first case, the length of the Cole-Moore shift will remain shorter over all the potential range whereas, in the second case, the shift would be equal at extreme hyperpolarizations.
We measured the effect of 100 nM Ts1 on the sigmoidal lag phase of ionic currents in response to a conditioning pulse (Cole and Moore, 1960). These experiments were conducted at 7°C to improve time resolution (Taylor and Bezanilla, 1983). The ionic currents were elicited with a 10-ms test pulse to −20 mV following a 2-ms prepulse ranging from −160 to +60 mV. Fig. 2 B shows representative traces comparing ionic currents in response to prepulses of −160 and −60 mV before and after toxin application. The length of the lag phase measured at −20 mV following a −160 mV prepulse was reduced from ∼400 μs in unmodified channels to 350 μs in toxin-modified channels. The reduction of the lag phase in ionic currents suggests that the toxin-modified channels have fewer accessible closed states than the unmodified channels. Furthermore, comparison of ionic currents recorded after a conditioning pulse to −160 mV reveals that in the presence of Ts1, the currents reach the half peak ∼600 μs faster (Fig. 2 C). These ionic current traces were fitted to an exponential function raised to “n” power, where n is an indicator of the number of states. The time constant of the ionic current trace in absence of toxin was 1.7 ms. The value of τ was fixed and the exponent n was varied for fitting the ionic current traces in presence of toxin. In absence of bound Ts1, the n value was equal to 5.3, whereas in the presence of Ts1 this value was reduced to 4.3. The reduction of the exponent agrees well with the prediction that the number of closed states in the toxin-bound channel has decreased. It is also possible that toxin binding reduces the transit time through one or more closed states without eliminating them completely. This possibility cannot be ruled out from the Cole-Moore experiments. Fig. 2 D shows a plot of time to half peak for different conditioning pulses in sodium channels in the presence and absence of toxin. Although Ts1 does not change the voltage dependence, the time to half peak is faster over the whole potential range in the presence of Ts1. These results show that even under conditions when the channels were activated from the farthest closed state (at −160 mV), the lag phase in the ionic current remained shorter in presence of the toxin. The Cole-Moore experiments support the idea that the effect of the toxin is to drive the voltage sensor in domain II to an activated state, either decreasing the number of closed states associated with the movement of DII sensor or speeding the kinetic of the transitions between these states.
Ts1-modified Channels Move Additional Gating Charge at Hyperpolarized Potentials
Trapping the DII sensor in the open state should also reduce the total gating charge associated with voltage-dependent movement of the sensors. We measured gating currents from the sodium channels in the absence and presence of Ts1. Fig. 3 shows the effect of 50 nM Ts1 on gating currents, recorded after a strong hyperpolarizing prepulse at room temperature (20°C) (5 μM TTX were used to block ionic currents). Fig. 3 (A–C) compares the gating currents recorded in absence and in the presence of Ts1, at −150, −100, and 0 mV, respectively. Note that while the toxin increases the size of the gating currents at negative potentials, at potentials more depolarized than 0 mV the situation is reversed. Comparison of the charge–voltage (Q-V) relationships reveals that, in the presence of toxin, the total amount of charge is reduced by 20% after a strong hyperpolarizing prepulse to −180 mV (Fig. 3 D), indicating that within experimental limits, a fraction of gating charge is immobilized. This is evident from the shallow baseline at the foot of the Q-V curve. This reduction in total charge is expected from the trapping model because the immobilized sensor does not contribute to gating charge movement.
Previous measurements of gating currents from β-scorpion toxin–bound sodium channels showed such a reduction in gating charge (Meves et al., 1987). However, those measurements were performed after a positive prepulse, which was required for toxin binding. As the positive prepulse produces charge immobilization (Armstrong and Bezanilla, 1977) the measured charge movement under those conditions did not reveal the full effect of the toxin. In addition to the charge reduction similar to that seen by Meves et al. (1987), we also observed a leftward shift of the charge–voltage curve to hyperpolarized potentials. The Q-V relationship of unmodified channels is well described by a single Boltzmann distribution. The Ts1-modified channels require a double Boltzmann fit to fully describe the Q-V curve over the whole voltage range. The single Boltzmann fit is poor especially at hyperpolarized potentials. The parameters of the fits show that a small component (17%) of gating charge appears dramatically shifted to hyperpolarized potential (Table I). Previous measurements (Meves et al., 1987) of gating currents in presence of toxin did not reveal this feature since the gating currents were recorded from a holding potential of −90 mV. By −90 mV, the toxin-modified voltage-gated sodium channels have already moved ∼25% of its total charge, therefore measurements starting at those potentials will not show the full effect of the toxin. Taken together, the charge–voltage relationship shows that although binding of toxin to the channel reduces 20% of the total gating charge, it also shifts the movement of a component of gating charge far (−114.5 mV) into hyperpolarized potentials. This shift in charge– voltage curve indicates that in addition to trapping the domain II voltage sensor in activated state, the toxin exerts additional effects on the sodium channel gating process.
TABLE I.
Fitting Parameters for the Charge–Voltage (Q-V) Curves
| Before Ts1 (n = 3) | After Ts1 (n = 3) | |
|---|---|---|
| Amp(a) | – | 0.17 ± 0.03 |
| Z(a) | – | 0.77 ± 0.13 |
| V1/2 (a) | – | −114.5 ± 0.48 |
| Amp(b) | 1 | 0.83 ± 0.09 |
| Z(b) | 1.32 ± 0.11 | 1.28 ± 0.02 |
| V1/2 (b) | −38.9 ± 2.9 | −39.6 ± 2.72 |
Amp refers to fractional amplitudes of each component when the curve was fitted to double Boltzmann, Z is the apparent valence, and V1/2 is the midpoint of activation.
Ts1 Binding Is Mainly to Domain II
Before testing the effect of Ts1 binding on each of the sodium channel voltage sensors we wanted to clarify whether Ts1 has a single binding site on the sodium channel. Previous work by Chahine and coworkers (Marcotte et al., 1997) have shown that a chimera consisting of skeletal muscle DII with the other three domains from cardiac sodium channel binds to Ts1 toxin as efficiently as the wild-type skeletal muscle sodium channels. The cardiac sodium channel showed no effect at 100 nM concentration whereas the skeletal muscle showed full effect at 50 nM of Ts1. Mutagenesis studies with a homologous toxin CssIV have identified a number of residues on the extracellular loop regions of the sodium channel that are likely to be involved in interaction with β-scorpion toxins (Cestèle et al., 1998). Mutation of the glycine 845 on S4-DII to asparagine seems to have the largest effect and reduces the binding affinity by 13-fold. Additionally, the mutation of the glutamates 779 (S1-S2DII loop) and 837 (S3-S4DII loop) to cysteine, as well the leucine 840 (S3-S4DII loop), caused a significant reduction of binding affinity for CssIV (Cestèle et al., 2006). These reductions, although significant, do not rule out the possibility that the β-toxin has additional low affinity binding sites on the other voltage-sensing domains. In our initial experiments with labeled channels, we found that the labeling of the mutant R663C on DII with TMRM eliminated the Ts1 binding even at high concentrations. Fig. 4 A shows current recordings of the R663C mutant labeled with TMRM in presence of 1 μM of Ts1 toxin. There is no increase of the currents by the toxin while, in contrast, only 50 nM of toxin induces a large increase of the current in the WT channel (see Fig. 1). This suggests that the Ts1 binding affinity has been considerably reduced (at least 20-fold) by modification of substituted cysteine 663 with rhodamine. These results indicate that binding of Ts1 to a site in the S3–S4 loop of domain II is required for its action.
Figure 4.

Effect of Ts1 on S4-DII movement. Effect of Ts1 in R663C channels. Superimposed traces of the TMRM-labeled R663C channel recorded at −50 mV (first panel) and −40 mV (second panel) before (blue traces) and after the treatment with 1 μM of Ts1 (red traces).
Ts1 Binding Immobilizes Voltage Sensor of Domain II
Although binding of the toxin reduces the gating charge associated with the voltage-dependent conformational change, it remains unclear whether the specific reduction of total charge is associated with a particular domain or involves all domains. Since the residues on S3–S4 loop of domain II in sodium channels bind to β-scorpion toxin, we first measured the effect of this toxin on the voltage sensor of domain II.
We used site-specific fluorescence measurements that, in contrast to gating current measurements, monitor conformational changes associated with voltage-dependent movement in the neighborhood of the fluorophore (Cha et al., 1999; Chanda and Bezanilla, 2002). Thus, by measuring fluorescence changes induced by voltage steps in each of the voltage sensors in presence and in absence of toxin we can track the specific conformational changes induced by the toxin. These measurements were performed at room temperature by labeling with TMRM mutant channels containing a cysteine in the S3−S4 linkers of the sodium channel. We first measured the fluorescence of TMRM attached to a cysteine replacing a serine in position 660. This position is in the S3−S4 linker of domain II where the toxin binding site is located. The presence of the fluorescent label may be expected to either reduce or completely eliminate toxin binding. However, we found that increasing the toxin concentration 10 times, from 50 to 500 nM, restored the original effect of the toxin (see Fig. 5). Voltage-dependent fluorescence changes were measured using the voltage protocol shown in Fig. 6 A. Fig. 6 (B–E) compares the fluorescence signals in control conditions and in the presence of Ts1, obtained at +50, 0, −50, and −100 mV, respectively. Ts1 abolished the fluorescence changes at all potentials analyzed, suggesting that the S4-DII was immobilized as a result of toxin binding (Fig. 7 A). To discard the possibility that the inhibition of the fluorescence changes observed in the presence of Ts1 is due to a quenching effect, we measured the absolute fluorescence values before and after the treatment with the toxin. Fig. 7 B shows that the treatment with 500 nM of Ts1 actually unquenches the fluorescence, increasing the absolute values. An alternative explanation would be that the toxin prevents the change in environment around the fluorophore that is required to produce the voltage-dependent fluorescence change. Although the fluorescence experiments alone do not distinguish between these two possibilities, the gating current measurements (Fig. 3; Meves et al., 1987) and the Cole-Moore experiments (Fig. 2) favor the interpretation that the S4-DII is immobilized in the active state because Ts1 eliminates a fraction of the charge and the channel activates at more negative potentials.
Figure 5.

Effect of Ts1 on S4-DII movement: S660C channel. (A) Superimposed traces of the TMRM-labeled S660C channel in control conditions (blue traces) and in the presence of Ts1 (0.5 μM, red traces), recorded at −80 mV (first panel), −60 mV (second panel), −40 (third panel), and −20 mV (fourth panel). The currents were recorded by applying the voltage protocol described in Fig. 1 A. (B) G-V curves obtained before (blue triangles) and after (red circles) the treatment with Ts1 (0.5 μM).
Figure 6.

Effect of Ts1 on S4-DII movement. (A) Voltage protocol used to measure the fluorescence signals. Superimposed fluorescence traces recorded at +50 mV (C), 0 mV (D), −50 mV (E), and −100 mV (F) in control conditions (blue lines) and in the presence of Ts1 (0.5 μM, red lines). The fluorescence increases in the same direction of the arrow in the scale bar.
Figure 7.

Effect of Ts1 on S4-DII movement. (A) Fluorescence–voltage (F-V) relationships obtained before (blue triangles) and after (red circles) the treatment with Ts1 (mean ± SEM; n = 3). The protocol used to measure the fluorescence signals was described in Fig. 6 A. (B) Absolute fluorescence traces obtained at +60 mV before (blue lines) and after (red lines) the treatment with Ts1. When error bars are not visible they are smaller than the symbols.
Voltage Sensors of Domains I, III, and IV Activate at More Hyperpolarized Potentials in Presence of Ts1
What is the physical origin of additional charge movement induced by the toxin at hyperpolarized potentials? It is possible that the toxin binding, in addition to immobilizing S4-DII, shifts the voltage-dependent movement of other voltage sensors to hyperpolarized potentials, giving rise to an additional charge movement seen at those potentials. In other words, is β-scorpion toxin an allosteric rather than a site-specific modulator of voltage gating? We investigated this possibility by labeling each one of the S3–S4 linkers of the other three domains and measured the fluorescence changes induced by voltage in presence and absence of the toxin. Fig. 8 summarizes the effects of Ts1 (50 nM) on the fluorescence changes from voltage sensors of domains I, III, and IV, obtained at room temperature. Fig. 8 A shows the voltage protocol used to record the traces. Except for the 20-ms test pulses, which in this case varied from −220 to +60 mV in 20-mV steps, the protocol was the same as described in Fig. 6 A. Fig. 8 B shows the effect of Ts1 on the fluorescence–voltage (F-V) relationship of the S4-DI (S216C). In the presence of Ts1, V1/2 was shifted by 58 mV in the hyperpolarizing direction (see Table II). Fig. 8 C shows that in presence of Ts1, the V1/2 of the F-V curve of the S4-DIII (L1115C) was shifted 38 mV to the left (see Table II). The V1/2 of the F-V curve of the S4-DIV (S1436C) was shifted by 17 mV to the left (see Table II) in the presence of toxin (Fig. 8 D). These measurements reveal that the β-scorpion toxin binding to S4-DII allosterically activates the movement of all the other voltage sensors in the sodium channel. It is interesting to note that the maximal effect of this activation shift is seen in the neighboring voltage sensors of domains I and III rather than on the distant S4-DIV. The large hyperpolarizing shifts of the voltage sensor movement of domains I, III, and IV that result from toxin binding in domain II can partly explain the additional gating charge movement seen at hyperpolarized potentials in the presence of β-scorpion toxin. These results support the model that the voltage sensors of the sodium channel are intrinsically tightly coupled. (Chanda et al., 2004).
TABLE II.
Fitting Parameters for the Fluorescence–Voltage (F-V) Curves
| Before Ts1
|
After Ts1
|
|||
|---|---|---|---|---|
| Z | V 1/2 | Z | V 1/2 | |
| S4-DI (S216C) | ||||
| n = 4 before Ts1 | 0.76 ± 0.03 | −67.5 ± 4.4 | 0.81 ± 0.05 | −125.2 ± 1.4a |
| n = 5 after Ts1 | ||||
| S4-DIII (L1115C) | ||||
| n = 3 before Ts1 | 1.30 ± 0.01 | −79.0 ± 3.6 | 0.84 ± 0.04a | −116.9 ± 2.1a |
| n = 3 after Ts1 | ||||
| S4-DIV (S1436C) | ||||
| n = 3 before Ts1 | 1.18 ± 0.08 | −70.2 ± 1.9 | 0.91 ± 0.06 | −87.3 ± 3.1a |
| n = 3 after Ts1 | ||||
Z is the apparent valence and V 1/2 is the midpoint of activation.
Values significantly different from “Before Ts1” group (P < 0.05).
A comparison of shift in Q-V curve and the fluorescence–voltage curves show that in the presence of toxin, the Q-V curve appears to have a small additional component (17%) with a midpoint of 114.5 mV. This shift in the Q-V curve appears to be smaller than expected since our data shows that all the fluorescence–voltage curves of all the three domains that are not immobilized is shifted to the left in the presence of the β-scorpion toxin. We do not yet fully understand the reasons for this discrepancy. A possible explanation is based on the fact that fluorescence–voltage curves measure a local effect while the gating current measures a global effect that depends on the contribution of each of the voltage sensors to the total gating current. It is also possible that the mutation and labeling with fluorophores may to some extent affect the coupling between the domains although at gross level these modifications did not appear to significantly alter the conductance– and charge–voltage relationships (Chanda and Bezanilla, 2002).
Chanda and coworkers have previously shown that when one of the charges in S4-DII was neutralized the F-V curves of the S4-DI, S4-DIII and S4-IV were shifted to more depolarizing potentials, indicating cooperativity between the voltage sensors. However, the maximum shift observed in those experiments was ∼37 mV in the F-V curves of S4-DI. On the other hand, our results show a 58-mV shift in the activation of S4-DI. The larger shift observed in the presence of Ts1 compared with the shift obtained upon a distal charge neutralization could be due to immobilization of S4-DII in activated state when Ts1 is bound. These large shifts in movement of other voltage sensors strongly suggest that the early gating transitions in the sodium channels are coupled.
DISCUSSION
Ts1 Is an Allosteric Modulator of Voltage-dependent Gating of Na Channels
Radiolabeled toxin binding experiments combined with molecular genetic techniques have previously localized the main toxin-binding site to the S3–S4 loop in domain II of the α-subunit of voltage-gated sodium channels (Cestèle et al., 1998, 2001). Based on these studies and electrophysiological measurements, it has been suggested that the β-scorpion toxin activates the voltage-gated sodium channel by binding to the voltage sensor of domain II in activated state and immobilizing it in that position (Cestèle et al., 1998, 2001, 2006; Cohen et al., 2005). However, there are important differences between Centruroides toxin used by Cestèle and coworkers and Ts1 toxin used by us. To see the effect of Centruroides toxin on neuronal Na channel currents Cestele et al. have to use a depolarizing conditioning pulse, suggesting the Css IV toxin traps the voltage sensor of DII only when it reaches activated position. In contrast, the maximal effect of Ts1 can be seen even without a prior depolarizing pulse, suggesting that Ts1 may bind to the channels even at rest. The actual mechanism of Ts1 action on the S4-DII remains to be elucidated. Here, we examine a secondary effect of Ts1 binding, i.e., how Ts1 binding to S4-DII affects the gating transitions in other voltage sensors.
Extensive previous work has shown allosteric interactions among receptor sites for different gating modifier toxins on the sodium channel, including those for scorpion toxins (for review see Catterall et al., 2007). More recent molecular studies indicate that these toxin receptor sites are located in different domains of the sodium channel, indicating there are allosteric interactions among different domains (Catterall et al., 2007). The present results also show interaction between domains but we think that they are due to an intrinsic cooperativity between sodium channel domains. The recent work by Cohen et al. (2006) showed that the binding of site-4 (like β-scorpion toxin, which binds S4-DII) positively increases the binding affinity of site-3 toxin (which binds to S4-DIV). This is opposite of what we would expect from our data. Our data here show that site-4 toxin also stabilizes the S4-DIV in activated position. Since site-3 toxin stabilizes S4-DIV in resting position, we would expect that the binding affinity of site-3 toxin would be reduced in presence of site-4 toxin and not increased. One interpretation that may reconcile our fluorescence data with previous reports of allosteric interactions between different sodium channel toxins is that the S4-DIV voltage sensor moves in two steps (Horn et al., 2000). It has been suggested that the first step is favored for activation whereas the second step is favored for inactivation. The α-toxin may bind to the intermediate that would favor activation of other voltage gates while preventing inactivation. Although this interpretation could explain how α-toxin binding increases the binding affinity of β-toxin while slowing down the inactivation of the channel, it still does not account for one result that was the centerpiece of the Cohen et al. (2006) paper. They describe a mutant β-toxin that binds to the Na+ channel with an affinity comparable to the wild type. This mutant toxin did not have any measurable effect on the gating behavior of the channel but was found to increase the binding affinity of α-toxin. This would suggest that the cooperative binding of α and the β toxins may be unrelated to channel gating or allosteric coupling between different domains. It will be interesting to determine the basis of cooperativity between these two toxins that bind to the sodium channel.
To gain physical insight on our data, we have constructed a 16-state gating scheme (Fig. 9 and Table III) considering two contrasting gating models. In the first model, the voltage sensors were considered to be independent of each other, whereas in the second case, the voltage sensors were coupled to each other. In the independent model, the immobilization of the voltage sensor of domain II by the toxin reduces the total gating charge but does not affect the midpoint of the Q-V curve (Fig. 10 A). The effect on the G-V curve is also very small since the model requires all four voltage sensors to be activated for the channel to open (Fig. 10 B). Note that in the sodium channels at least the first three voltage sensors must be activated for the pore to open (Hanck and Sheets, 1995; Sheets and Hanck, 1995; Chanda and Bezanilla, 2002). As expected, in the independent model, immobilization of only one of the voltage sensors by toxin does not affect the movement of the other voltage sensors. In the cooperative model, when voltage sensor of domain II is immobilized in the activated position by the toxin, not only is the total charge reduced but also the Q-V curve is shifted to the left (Fig. 10 C). The G-V curve of the toxin-bound channel is dramatically shifted to the left in the cooperative model (Fig. 10 D). Binding of the toxin to one of the domains shifts the voltage dependence of not only the DII voltage sensor but also the voltage sensors of the other three domains.
Figure 9.

A general kinetic model for gating in a voltage-gated ion channel. C0 corresponds to the fully deactivated state, with all four S4 segments in the resting position. C1, C2, C3, and C4 correspond to the states where voltage sensors from domains I, II, III, and IV are activated. The numbers in the index refers to voltage sensors that are in activated position. C1234 corresponds to the fully activated state and the open channel form, with all the S4 segments in the activated position.
TABLE III.
Expressions for Forward and Backward Rate Constants for the Proposed Cooperative Model
| Forward rate constants | Backward rate constants | ||||||
|---|---|---|---|---|---|---|---|
| C0-C1 | αf | C12-C123 | n 13 n 23γf | C1-C0 | αb | C123-C12 | γb |
| C0-C2 | βf | C12-C124 | n 14 n 24δf | C2-C0 | βb | C124-C12 | δb |
| C0-C3 | γf | C13-C123 | n 12 n 23βf | C3-C0 | γb | C123-C13 | βb |
| C0-C4 | δf | C13-C134 | n 14 n 34δf | C4-C0 | δb | C134-C13 | δb |
| C1-C12 | n 12βf | C23-C123 | n 12 n 13αf | C12-C1 | βb | C123-C23 | αb |
| C1-C13 | n 13γf | C23-C234 | n 24 n 34δf | C13-C1 | γb | C234-C23 | δb |
| C1-C14 | n 14δf | C14-C124 | n 12 n 24βf | C14-C1 | δb | C124-C14 | βb |
| C2-C12 | n 12αf | C14-C134 | n 13 n 34γf | C12-C2 | αb | C134-C14 | γb |
| C2-C23 | n 23γf | C24-C124 | n 12 n 14αf | C23-C2 | γb | C124-C24 | αb |
| C2-C24 | n 24δf | C24-C234 | n 23 n 34γf | C24-C2 | δb | C234-C24 | γb |
| C3-C13 | n 13αf | C34-C234 | n 23 n 24βf | C13-C3 | αb | C234-C34 | βb |
| C3-C23 | n 23βf | C34-C134 | n 13 n 14αf | C23-C3 | βb | C134-C34 | αb |
| C3-C34 | n 34δf | C123-C1234 | n 14 n 24 n 34δf | C34-C3 | δb | C1234-C123 | δb |
| C4-C14 | n 14αf | C124-C1234 | n 13 n 23 n 34γf | C14-C4 | αb | C1234-C124 | γb |
| C4-C24 | n 24βf | C134-C1234 | n 12 n 23 n 24βf | C24-C4 | βb | C1234-C134 | βb |
| C4-C34 | n 34γf | C234-C1234 | n 12 n 13 n 14αf | C34-C4 | γb | C1234-C234 | αb |
n is the interaction term between different voltage sensors. Interaction terms: S4 domains I and II, n12; S4 domains I and III, n13; S4-domains I and IV, n14; S4 domains II and III, n23; S4 domains II and IV, n24; S4 domains III and IV, n34.
Figure 10.

Modulation of gating in independent and allosteric models. Simulations of the charge–voltage (A) and conductance–voltage curves (B) for the model with independent voltage sensors. Charge–voltage (C) and conductance–voltage (D) curves in the allosteric model, where all four voltage sensor movements are coupled. Toxin-modified data are shown as red lines, whereas the unmodified traces are shown as blue lines.
The finding that β-scorpion toxin is an allosteric rather than a site-specific modulator of voltage sensor movement is a direct consequence of the intrinsic cooperativity between the voltage sensors in the sodium channel. These results highlight fundamental differences between voltage-gated sodium and voltage-gated potassium channels. Cooperativity in potassium channels is limited to transitions associated with the final opening of the channel pore, indicating that the interaction between subunits is via the opening of the conduction pore (Zagotta et al., 1994; Schoppa and Sigworth, 1998; Ledwell and Aldrich, 1999; Horn et al., 2000; Pathak et al., 2005). In contrast, our data show that the shift in the activation of the sodium channel occurs at hyperpolarized potentials where the open probability is near zero. This means that the interaction between domains is occurring at the level of voltage sensor movement at transitions far removed from pore opening. In addition, the immobilization of S4-DII causes ∼60 mV shift in S4-DI activation (Fig. 8 B). This perhaps is the strongest evidence that the early gating transitions corresponding to the movement of S4-DI is modified by toxin binding to S4-DII.
Most classical models of the sodium channel beginning with the Hodgkin-Huxley description have assumed that the voltage sensors move independent of each other (Hodgkin and Huxley, 1952; Armstrong and Bezanilla, 1977; French and Horn, 1983; Patlak, 1991; Keynes, 1994), although cooperativity was included in the squid sodium channel model of Vandenberg and Bezanilla (1991). Recent gating models of voltage-gated potassium channels have included cooperativity of gating transitions in the final pore opening step (Zagotta et al., 1994; Schoppa and Sigworth, 1998). Since the pore segments from neighboring subunits/domains are in direct contact with the each other, these transitions are expected to be cooperative. The initial movement of voltage sensors in the Shaker potassium channels, however, has been shown to occur independent of each other (Horn et al., 2000; Mannuzzu and Isacoff, 2000; Pathak et al., 2005). This idea of independent voltage sensor movements is consistent with the crystal structure of Kv1.2, which shows that the neighboring voltage sensors in the open state of the channel are sufficiently far apart (53 Å) to have any direct interactions (Long et al., 2005). Nonetheless, our findings clearly demonstrate that, unlike voltage-dependent potassium channels, the voltage sensor movements in the eukaryotic voltage-gated sodium channels are tightly coupled.
The molecular interfaces that are involved in the coupling between voltage sensors are not known but it is expected that the structure of the voltage-gated sodium channel will provide some insights into how the sensors interact. Recently, Sack and Aldrich (2006) have shown that a gating modifier toxin induces cooperativity in early transitions of the Shaker potassium channel. Understanding how this gating modifier toxin causes the normally independent gating transitions to become coupled may provide further clues to understanding cooperative phenomenon in voltage-gated ion channels.
It is important to note that the positive cooperativity in sodium channels not only enhances the effect of the toxin but also accelerates the kinetics of sodium activation. Therefore, we speculate that this specialization of the sodium channel was necessary for generation of nerve impulses since sodium channels have to open before potassium channels to initiate an action potential.
Acknowledgments
We acknowledge Dr. L.D. Possani (Institute of Biotechnology, UNAM, University City, Mexico) for providing us with samples of the toxin Ts1. We thank Drs. A.M. Correa, R. Olcese, and the members of Bezanilla and Correa laboratories for their comments. Part of this work was carried out in the Departments of Physiology and Anesthesiology at UCLA.
Supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (research fellowship to F.V. Campos and P.S.L. Beirão), American Heart Association Scientist Development grant (B. Chanda), and National Institutes of Health grant GM30376 (F. Bezanilla).
Olaf S. Andersen served as editor.
Abbreviations used in this paper: Mes, methylsufonate; TMRM, tetramethylrhodamine maleimide.
References
- Armstrong, C.M., and F. Bezanilla. 1977. Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol. 70:567–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin, J., J.R. Giglio, P. Leopold, A. Schmid, S.V. Sampaio, and M. Lazdunski. 1982. Tityus serrulatus venom contains two classes of toxins. Tityus gamma toxin is a new tool with a very high affinity for studying the Na+ channel. J. Biol. Chem. 257:12553–12558. [PubMed] [Google Scholar]
- Cahalan, M.D. 1975. Modification of sodium channel gating in frog myelinated nerve fibres by Centruroides sculpturatus scorpion venom. J. Physiol. 244:511–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall, W.A., S. Cestele, V. Yarov-Yarovoy, F.H. Yu, K. Konoki, and T. Scheuer. 2007. Voltage gated ion channels and gating modifier toxins. Toxicon. 49:124–141. [DOI] [PubMed] [Google Scholar]
- Cestèle, S., Q. Yusheng, J.C. Rogers, H. Rochat, T. Scheuer, and W.A. Catterall. 1998. Voltage sensor-trapping: enhanced activation of sodium channels by β-scorpion toxin bound to the S3–S4 loop in domain II. Neuron. 21:919–931. [DOI] [PubMed] [Google Scholar]
- Cestèle, S., T. Scheuer, M. Mantegazza, H. Rochat, and W.A. Catterall. 2001. Neutralization of gating charges in domain II of the sodium channel α-subunit enhances voltage sensor trapping by a β-scorpion toxin. J. Gen. Physiol. 118:291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestèle, S., and W.A. Catterall. 2000. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie. 82:883–892. [DOI] [PubMed] [Google Scholar]
- Cestèle, S., V. Yarov-Yarovoy, Y. Qu, F. Sampieri, T. Scheuer, and W.A. Catterall. 2006. Structure and function of the voltage sensor of sodium channels probed by a β scorpion toxin. J. Biol. Chem. 281:21332–21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha, A., and F. Bezanilla. 1998. Structural implications of fluorescence quenching in the Shaker K1 channel. J. Gen. Physiol. 112:391–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha, A., P.C. Ruben, A.L. George, E. Fujimoto, and F. Bezanilla. 1999. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na channel fast inactivation. Neuron. 22:73–87. [DOI] [PubMed] [Google Scholar]
- Chanda, B., and F. Bezanilla. 2002. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J. Gen. Physiol. 120:629–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda, B., O.K. Asamoah, and F. Bezanilla. 2004. Coupling interactions between voltage sensors of the sodium channel as revealed by site-specific measurements. J. Gen. Physiol. 123:217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, L., I. Karbat, N. Gilles, N. Ilan, M. Benveniste, D. Gordon, and M. Gurevitz. 2005. Common features in the functional surface of scorpion β toxins and elements that confer specificity for insect and mammalian voltage-gated sodium channels. J. Biol. Chem. 280:5045–5053. [DOI] [PubMed] [Google Scholar]
- Cohen, L., N. Lipstein, and D. Gordon. 2006. Allosteric interactions between scorpion toxin receptor sites on voltage-gated Na channels imply a novel role for weakly active components in arthropod venom. FASEB J. 20:1933–1935. [DOI] [PubMed] [Google Scholar]
- Cole, K.S., and J.W. Moore. 1960. Potassium ion current in the squid giant axon: dynamic characteristic. Biophys. J. 1:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French, R.J., and R. Horn. 1983. Sodium channel gating: models, mimics, and modifiers. Annu. Rev. Biophys. Bioeng. 12:319–356. [DOI] [PubMed] [Google Scholar]
- Hanck, D.A., and M.F. Sheets. 1995. Modification of inactivation in cardiac sodium channels: ionic current studies with Anthopleurin-A toxin. J. Gen. Physiol. 106:601–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin, A.L., and A.F. Huxley. 1952. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117:500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn, R., S. Ding, and H.J. Gruber. 2000. Immobilizing the moving parts of voltage-gated ion channels. J. Gen. Physiol. 116:461–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keynes, R.D. 1994. The kinetics of voltage-gated ion channels. Q. Rev. Biophys. 27:339–434. [DOI] [PubMed] [Google Scholar]
- Ledwell, J.L., and R.W. Aldrich. 1999. Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol. 113:389–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, S.B., E.B. Campbell, and R. MacKinnon. 2005. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 309:897–903. [DOI] [PubMed] [Google Scholar]
- Mannuzzu, L.M., and E.Y. Isacoff. 2000. Independence and cooperativity in rearrangements of a potassium channel voltage sensor revealed by single subunit fluorescence. J. Gen. Physiol. 115:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte, P., L.Q. Chen, R.G. Kallen, and M. Chahine. 1997. Effects of Tityus serrulatus scorpion toxin gamma on voltage-gated Na+ channels. Circ. Res. 80:363–369. [DOI] [PubMed] [Google Scholar]
- Meves, H., N. Rubly, and D.D. Watt. 1987. Gating current experiments on frog nodes of Ranvier treated with Centruroides sculpturatus toxins or aconitine. Pflugers Arch. 409:381–393. [DOI] [PubMed] [Google Scholar]
- Pathak, M., L. Kurtz, F. Tombola, and E. Isacoff. 2005. The cooperative voltage sensor motion that gates a potassium channel. J. Gen. Physiol. 125:57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patlak, J. 1991. Molecular kinetics of voltage-dependent Na+ channels. Physiol. Rev. 71:1047–1080. [DOI] [PubMed] [Google Scholar]
- Pinheiro, C.B., S. Marangoni, M.H. Toyama, and I. Polikarpov. 2004. Structural analysis of Tityus serrulatus Ts1 neurotoxin at atomic resolution: insights into interactions with Na+ channels. Acta Crystallogr. D Biol. Crystallogr. 59:405–415. [DOI] [PubMed] [Google Scholar]
- Possani, L., W.E. Steinmetz, M.A. Dent, A.C. Alagon, and K. Wuthrich. 1981. Preliminary spectroscopic characterization of six toxins from Latin American scorpions. Biochim. Biophys. Acta. 669:183–192. [DOI] [PubMed] [Google Scholar]
- Sack, J.T., and R.W. Aldrich. 2006. Binding of a gating modifier toxin induces intersubunit cooperativity early in the Shaker K channel's activation pathway. J. Gen. Physiol. 128:119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppa, N.E., and F.J. Sigworth. 1998. Activation of Shaker potassium channels. III. An activation gating model for wild-type and V2 mutant channels. J. Gen. Physiol. 111:313–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets, M.F., and D.A. Hanck. 1995. Voltage-dependent open-state inactivation of cardiac sodium channels: gating current studies with Anthopleurin-A toxin. J. Gen. Physiol. 106:617–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, R.E., and F. Bezanilla. 1983. Sodium and gating current time shifts resulting from changes in initial conditions. J. Gen. Physiol. 81:773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg, C.A., and F. Bezanilla. 1991. A sodium channel gating model based on single channel, macroscopic ionic, and gating currents in the squid giant axon. Biophys. J. 60:1511–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijverberg, H.P., D. Pauron, and M. Lazdunski. 1984. The effect of Tityus serrulatus scorpion toxin gamma on Na channels in neuroblastoma cells. Pflugers Arch. 401:297–303. [DOI] [PubMed] [Google Scholar]
- Yatani, A., G.E. Kirsch, L.D. Possani, and A.M. Brown. 1988. Effects of New World scorpion toxins on single-channel and whole cell cardiac sodium currents. Am. J. Physiol. 254:H443–H451. [DOI] [PubMed] [Google Scholar]
- Zagotta, W.N., T. Hoshi, and R.W. Aldrich. 1994. Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J. Gen. Physiol. 103:321–362. [DOI] [PMC free article] [PubMed] [Google Scholar]