

Abstract
We report the X-ray crystal structures of two different iron(III) porphyrinates: [Fe(OEP)(NO3)] and [Fe(TPP)(NO3)]. The first complex has the nitrate ion coordinated by a single oxygen atom while the second derivative has the nitrate coordinated in a symmetric bidentate fashion. This latter structure is a redetermination that shows some differences from an earlier structure; the difference appears to be the result of an unrecognized nitrate ion disorder in the earlier structure determination. Changes in physical properties of three species ([Fe(TPivP)(NO3)], [Fe(OEP)(NO3)], and [Fe(TPP)(NO3)] as a function of coordination mode were examined by Mössbauer and EPR spectroscopies; EPR spectra appear to be most sensitive to the change in coordination mode.
Keywords: Iron(III) porphyrinates, nitrate binding mode, EPR spectroscopy, Mössbauer spectroscopy, crystal structure, electronic structure
Introduction
Interactions between iron porphyrinates and the nitrate ligand (NO3−) occur in a number of areas of bioinorganic chemistry. Nitrate is a key component of the nitrogen cycle in the fixation of atmospheric nitrogen to a more biologically accessible forms.1 Effects of nitrate on mammalian physiology are also closely studied, particularly in light of its occurrence in drinking waters. Explications of the debate as to the harmful or beneficial effects of nitrate can be found in two recent publications for popular audiences.2,3 Nitrate can also be produced in vitro from either oxidation of coordinated NO4 or NO interaction with coordinated dioxygen to yield nitrate that may initially be coordinated to heme.5,6
In addition to its physiological importance, the nitrate ligand possesses a varied coordination chemistry. A number of coordination modes have been observed upon interaction with a metal ion7 with three of the more common nonbridging modes illustrated in Scheme 1. These coordination geometries range from the monodentate mode (η1) (Scheme 1(a)) through the anisobidentate or asymmetric bidentate (η2) mode (Scheme 1(b)) to the symmetric (η2) bidentate mode Scheme 1(c)). An initial attempt to classify these systems based upon geometry was proposed by Addison et al.8 and later refined by Lever et al.9 who showed such coordination modes could be distinguished on the basis of spectroscopic measurements.
Scheme 1.

Variable coordination modes of the nitrate ligand have been observed in several series of complexes. Driessen et al.10 investigated a series of complexes based upon the tripodal ligand amtd11 and containing a range of metal ions. Depending on the identity of the central metal ion in [M(amtd)(NO3)2], variable coordination modes of the nitrate are observed. A similar result is observed by Parkin et al.12 in the study of model complexes of the enzyme carbonic anhydrase. Once again, the identity of the metal ion in a tripodal ligand frameworks determines the coordination geometry of the nitrate ligand.
Yet, in these examples, comparing the electronic properties of the coordinated ligand across such a series of complexes is complicated by the change in metal identity. In order to accurately investigate the effects on the electronic structure of the complexes upon changes in coordination geometry would require a series of related ligand frameworks (excluding the nitrate), all containing the same central metal, but which still allows variation in nitrate geometry. To our knowledge, the ferric porphyrinate complexes [Fe(por)(NO3)] provide the only example of such a series. Hence we undertook a study of the electronic structure for such a series of complexes aiming to correlate changes in both the Mössbauer and EPR spectra with changes in ligand geometry.
A number of porphyrinate complex structures containing a coordinated nitrate ligand have been reported13 with a range of coordination modes reported therein. Characterization of a five-coordinate monodentate ferrous complex [Fe(TpivPP)(NO3)]− has been published.14 The structures of the ferric porphyrinate complexes [Fe(por)(NO3)] based upon the porphyrinates OEP,15 TPP,16 and TpivPP17 demonstrate the geometries illustrated in Scheme 1. Initial attempts at forming a nitrite-coordinated heme complex had resulted in formation of free nitrate as determined by IR spectroscopy following an oxygen-atom transfer reaction.18 A related reaction utilizing picket fence porphyrin (TpivPP) with a protected ligand binding site resulted in nitrate formation with nitrate coordinated to the heme.17 The crystal structure of [Fe(TpivPP)(NO3)]17 displays a symmetrically coordinated bidentate nitrate ligand with the projected plane of the nitrate eclipsing a trans pair of porphyrin nitrogen atoms. Asymmetric bidentate coordination has been reported for [Fe(TPP)(NO3)]16 with the projected plane of the nitrate bisecting adjacent pairs of porphyrin nitrogen atoms. The triclinic [Fe(OEP)(NO3)]15 exhibits a monodentate nitrate ligand. We have subsequently determined the crystal structure of a monoclinic form of [Fe(OEP)(NO3)](reported herein) which also possess a monodentate nitrate ligand.
Initially we undertook to investigate the effects of nitrate coordination mode upon the electronic structure of the ferric porphyrinates. Based upon the reported variations in coordination geometry, the complexes [Fe(por)(NO3)] provide a means of studying systematic changes in the electronic environment of the iron as the coordination geometry changes. However, seemingly contradictory EPR results were obtained, and led us to reexamine the structure of the asymmetric bidentate-coordinated species [Fe(TPP)(NO3)].16 The structure was redetermined at 100 K with crystals having comparable cell constants to those reported previously. Our new structure demonstrates that, rather than a single asymmetric bidentate coordination mode, the nitrate ligand is in fact disordered over two positions. This new structure is discussed particularly in relation to the previous room temperature example.16 We also present a second crystalline form of [Fe(OEP)(NO3)], which like the previously reported structure,15 contains a monodentate nitrate ligand. In addition to the two new crystal structures, we provide electronic characterization of all three [Fe(por)(NO3)] complexes both in solution and solid state. These reveal marked differences in nitrate coordination geometry between the solid and solution coordination modes for both of the bidentate systems.
Experimental Section
General Information
All reactions were carried out using standard Schlenkware techniques unless otherwise noted. Tetrahydrofuran and hexanes were distilled over sodium and benzophenone prior to use. Methylene chloride was distilled over calcium hydride. H2OEP was purchased from Midcentury Chemicals. H2TPP was synthesized according to the method of Adler et al.19 Metallation reactions to yield [Fe(OEP)(Cl)] and [Fe(TPP)(Cl)] were carried out according to Adler et al.20 H2TpivPP was prepared according to a local modification of the reported synthesis.21 [Fe(TpivPP)(Cl)] was obtained by stirring 150 mg of the free-base overnight with 100 mg of FeCl2 in 25 mL of THF and 0.2 mL of 2,6-lutidine.
Solution UV/Vis spectra were measured on a Perkin-Elmer Lambda 19 UV/Vis/near-IR spectrometer. IR spectra were collected on single crystals ground between two sodium chloride plates with a small amount of Nujol. Spectra were collected on a Nicolet Nexus 870 FT-IR spectrometer. EPR measurements on ground single crystals of [Fe(TpivPP)(NO3)] were made at 77 K on a Bruker ER 100E spectrometer calibrated with a dpph standard. All other EPR measurements were made on a Bruker EMX-EPR spectrometer. Measurements were made on either frozen methylene chloride solutions or finely ground crystalline solids. Samples were prepared for Mössbauer spectroscopy by grinding approximately 40 mg of crystals, immobilizing in Apiezon M grease to form a mull which was subsequently sealed in an airtight Mössbauer cup.
Synthesis of [Fe(OEP)(NO3)]
[Fe(OEP)(Cl)] (109.5 mg, 0.176 mmol) was placed in a Schlenk flask with finely ground AgNO3 (35.8 mg, 0.211 mmol). 25 mL of THF was added and the solution stirred overnight. The solvent was removed in vacuo and the reddish-brown solid redissolved in methylene chloride, filtered to remove the inorganic salts and layered with hexanes. X-ray quality crystals were obtained after 7 days.
Synthesis of [Fe(TPP)(NO3)]
Large single crystals of [Fe(TPP)(NO3)] were obtained using a similar metathesis pathway. 150 mg (0.24 mmol) of [Fe(TPP)(Cl)] was stirred overnight in 15 mL of THF with 48 mg (0.29 mmol) of AgNO3. Subsequent solvent removal, dissolution in methylene chloride and filtration was carried out prior to layering with hexanes. Large single crystals of up to 2.5 mm length were obtained after 5 days. Determination of the cell constants of the crystals revealed comparable values to those previously reported.16
Synthesis of [Fe(TpivPP)(NO3)]
150 mg (0.14 mmol)of [Fe(TpivPP)(Cl)] was placed in a Schlenk flask with 27 mg (0.16 mmol) of AgNO3 and 15 mL of THF. The mixture was stirred overnight and the solvent evaporated prior to redissolving in CH2Cl2 and filtration. UV/Vis spectroscopy of both the THF and CH2Cl2 solutions confirmed the formation of [Fe(TpivPP)(NO3)] yet attempts to obtain a crystalline product were unsuccessful in spite of multiple attempts using variations in crystallization conditions.
X-ray Structure Determination
Structures of [Fe(OEP)(NO3)] and [Fe(TPP)(NO3)] were solved using the direct methods program SHELXS.22 All heavy atoms were located using subsequent difference Fourier syntheses. The structures were refined against F2 with the program SHELXL,23 in which all data collected were used including negative intensities. All nonhydrogen atoms were refined anisotropically. All hydrogen atoms were idealized using the standard SHELXL idealization method. Complete crystallographic details, atomic coordinates, anisotropic thermal parameters and fixed hydrogen coordinates for both are included in the Supporting Information. Table 1 gives brief crystallographic details.
Table 1.
Crystallographic Details for [Fe(OEP)(NO3)] and [Fe(TPP)(NO3)]
| [Fe(OEP)(NO3)] | [Fe(TPP)(NO3)] | |
|---|---|---|
| formula | C36H44FeN5O3 | C44H28FeN5O3 |
| FW, amu | 650.61 | 730.56 |
| a, Å | 13.209(3) | 10.0271(3) |
| b, Å | 13.774(3) | 16.1341(5) |
| c, Å | 19.258(4) | 16.1341(5) |
| β, deg | 105.50(3) | 90.330(2) |
| γ, deg | 90 | 90.330(2) |
| V, Å3 | 3376.2(12) | 3399.24(17) |
| space group | P21/c | P21/n |
| Z | 4 | 4 |
| μ, mm−1 | 0.489 | 0.495 |
| radiation | MoKα, λ̄ = 0.71073 Å | MoKα, λ̄ = 0.71073 Å |
| temperature, K | 293 | 100(2) |
| final R indices [I > 2σ (I)] | R1 = 0.0502, wR2 = 0.1140 | R1 = 0.0447, wR2 = 0.1167 |
| final R indices (all data) | R1 = 0.0802, wR2 = 0.1311 | R1 = 0.0704, wR2 = 0.1405 |
A single crystal of [Fe(OEP)(NO3)] was mounted on a glass fiber with the long axis approximately collinear with the axis of the fiber. All measurements were performed with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) on an Enraf-Nonius FAST area detector diffractometer at 127 K as previously described.24 Intensities of all reflections were reduced using Lorentz and polarization correction factors; the data were also corrected for absorption by an empirical (ψ scans) absorption correction (μ = 0.489 mm−1). Two positions of the methyl carbons of ethyl groups 6 and 7 were observed with this disorder arising from rotation about the Cm–C(6) and Cm–C(7) bonds. This was resolved by allowing the two possible methyl group positions to refine giving relative occupancies of 65 and 35%. Relevant bond lengths and angles for [Fe(OEP)(NO3)] are summarized in Table 2.
Table 2.
Details of FeNO3 Geometry for All [Fe(por)(NO3)] Complexes. Coordination Geometries are Illustrated in Figure 4.
| Complex | Fe–O1a | Fe–O2a | δa,b | N5–O1a | N5–O2a | N5–O3a | ref. |
|---|---|---|---|---|---|---|---|
| [Fe(OEP)(NO3)]c | 1.996(2) | 3.042(2) | 1.076 | 1.301(3) | 1.212(3) | 1.199(3) | 15 |
| [Fe(OEP)(NO3)]d | 2.016(3) | 2.644(3) | 0.628 | 1.206(5) | 1.208(6) | 1.198(4) | tw |
| [Fe(TPP)(NO3)]e | 2.019(4) | 2.323(8) | 0.304 | 1.203(6) | 1.176(6) | 1.188(5) | 16 |
| [Fe(TPP)(NO3)]f | 2.125(3) | 2.268(3) | 0.143 | 1.199(4) | 1.300(3) | 1.217(3) | tw |
| [Fe(TPP)(NO3)]g | 2.117(4) | 2.121(4) | 0.004 | 1.334(4) | 1.271(4) | 1.217(3) | tw |
| [Fe(TpivP)(NO3)] | 2.123(3) | 2.226(3) | 0.103 | 1.271(4) | 1.252(4) | 1.214(3) | 17 |
Value in Å.
Difference between Fe–O1 and Fe–O2.
Monoclinic form.
Triclinic form.
Room temperature structure.
100 K structure, major orientation.
100 K structure, minor orientation.
A single crystal of [Fe(TPP)(NO3)] was placed in inert oil, mounted on a glass fiber attached to a brass mounting pin and transferred to the cold stream of the diffractometer. Crystal data were collected and integrated using a Bruker Apex system with graphite-monochromated MoKα (λ = 0.71073 Å) radiation. Data collection was carried out at 100 K. [Fe(TPP)(NO3)] was found to possess a disordered nitrate ligand with two orientations. The two nitrate orientations were refined and determined to have relative occupancies of 60 and 40%. Oxygen atoms of the major orientation are labelled O1A and O2A whilst the minor orientation is labelled O1B and O2B. The central nitrogen atom and the uncoordinated oxygen of the nitrate ligand did not display any disorder. Relevant bond lengths and angles for [Fe(TPP)(NO3)] are summarized in Table 2.
Results
The crystal structures of two five-coordinate nitrate-ligated iron(III) porphyrinate complexes have been obtained. The first of these, monoclinic [Fe(OEP)(NO3)], represents a second crystalline example of a monodentate nitrate coordinated system in an iron(III) porphyrinate.15 The second structure, [Fe(TPP)(NO3)], is a redetermination of a previously reported room temperature structure.16 Here, we report a revised structure from data collected at 100 K.
Figure 1 shows the ORTEP diagram and a partial labelling scheme for the monoclinic form of [Fe(OEP)(NO3)] clearly illustrating the monodentate coordination geometry of the nitrate ligand. Figure 2a is a schematic representation of the porphyrin core showing the perpendicular displacements from the 24-atom mean plane. The iron is significantly shifted out of the plane in the direction of the nitrate ligand while the porphyrin core shows a modest combined doming/tenting distortion. An ORTEP diagram for [Fe(TPP)(NO3)] illustrating both orientations of the nitrate ligand is shown as Figure 3. The bidentate coordination mode of the nitrate is clearly evident. Figure 2b shows the porphyrin core of [Fe(TPP)(NO3)] which also shows a large displacement of the iron in the direction of the nitrate ligand. Projections of both orientations of the nitrate ligand are illustrated with the coordinating oxygens of the major orientation being labeled O1A, O2A. The minor orientation is rotated approximately 70° from the major with both lying close to a pair of opposite Fe–Np bonds. The dihedral angles(φ) which represent the relative orientation of the nitrate ligand with respect to the closest Fe–Np vector are 13 and 8° for the major and minor orientations respectively. In contrast, for monoclinic [Fe(OEP)(NO3)] structure, φ is 41° with the nitrate ligand lying almost midway between a pair of Fe–Np bonds. For both the OEP and TPP systems, the nitrate plane lies perpendicular to the plane of the porphyrin. Selected bond lengths for these complexes and related systems are given in Table 2.
Figure 1.

ORTEP diagram of the monoclinic form of [Fe(OEP)(NO3)] with 50% probability ellipsoids. The atom labeling scheme is also shown.
Figure 2.
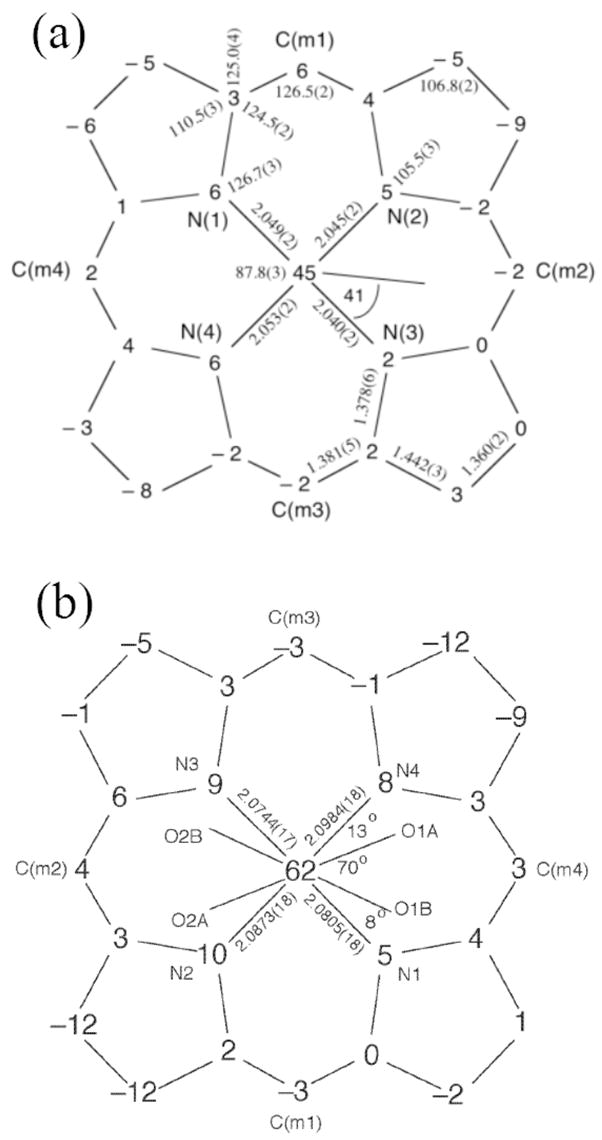
Formal diagram of the porphyrinato core of a) [Fe(OEP)(NO3)] and b) [Fe(TPP)(NO3)] displaying perpendicular displacement (in units of 0.01 Å) of the core atoms from the 24-atom mean porphyrin plane. Positive displacements are towards the nitrate coordinated face of the porphyrin. The projections of the nitrate ligand upon the porphyrin plane are also shown. For [Fe(TPP)(NO3)], the two orientations of the nitrate ligand are shown. (O1A and O2A represent the oxygen atom positions of the major orientation.).
Figure 3.

ORTEP diagram of [Fe(TPP)(NO3)] with 50% probability ellipsoids. Both orientations of the nitrate ligand is shown with the major orientation labelled O1A, O1B.
Table 3 reports UV/vis and IR spectroscopic data for the [Fe(por)(NO3)] complexes. EPR measurements for both ground crystalline solids and solutions were made on all three systems. These data are summarized in Table 4. Solution measurements on all three porphyrin systems made at 4 K are clearly axial with the major feature g⊥ = 5.7 – 6.0 and the minor feature, g||~2. Powder measurements on the monodentate [Fe(OEP)(NO3)] also reveal an axial spectrum with g = 5.8, 2. By contrast, rhombic spectra are observed for the crystalline bidentate TPP and TpivPP complexes with g values as reported in Table 4. Mössbauer spectra were collected at 4.2 K in zero field for ground crystalline samples of all three porphyrinates. These are fit to a S = 5/2 model and data for this fit are given in Table 5. Spectra are illustrated in Figure 6.
Table 3.
UV-Vis and IR Spectroscopic Data for a Series of [Fe(por)(NO3)] Complexes.
| Complex | UV-visa (nm) | ν (NO3)b (cm−1) |
|---|---|---|
| [Fe(OEP)(NO3)] | 385, 502, 534, 583, 636 | 1515, 1385, 1276. |
| [Fe(TPP)(NO3)] | 412, 513, 574, 658 | 1531, 1261, 1070 |
| [Fe(TpivP)(NO3)] | 415, 509, 574, 639 | 1515, 1276, 1071 |
Measured on CH2Cl2 solution.
Measured as KBr disk.
Table 4.
Summary of EPR Data for [Fe(por)(NO3)] Complexes at 4 and 77 K.a
| Complex | Sample | Temp (K) | Spectrum Appearance | g-values |
|---|---|---|---|---|
| [Fe(OEP)(NO3)] | Cryst. powder | 77 | Axial | g⊥ = 5.85 |
| [Fe(OEP)(NO3)] | Cryst. powder | 4 | Axial | g⊥ = 5.81, g|| = 2.018 |
| [Fe(OEP)(NO3)] | Solution | 77 | Axial | g⊥ = 5.99, g|| = 2.00 |
| [Fe(OEP)(NO3)] | Solution | 4 | Axial | g⊥ = 5.86, g|| =1.998 |
|
| ||||
| [Fe(TPP)(NO3)] | Cryst. powder | 77 | Rhombic | g = 7.71, 4.04, 1.78 |
| [Fe(TPP)(NO3)] | Cryst. powder | 4 | Rhombic | g = 8.5, 4.0, 1.8 |
| [Fe(TPP)(NO3)] | Solution | 77 | Axial | g⊥ = 5.65, g|| 2.001 |
| [Fe(TPP)(NO3)] | Solution | 4 | Axial | g⊥ = 5.82, g|| 1.99 |
|
| ||||
| [Fe(TpivPP)(NO3)] | Powderb | 77 | Axial | g⊥ = 5.45, g|| = 1.98 |
| [Fe(TpivPP)(NO3)] | Powderb | 4 | Axial | g⊥ = 5.54, g|| = 1.99 |
| [Fe(TpivPP)(NO3)] | Solution | 77 | Axial | g⊥ = 5.65, g|| = 2.00 |
| [Fe(TpivPP)(NO3)] | Solution | 4 | Axial | g⊥ = 5.71, g|| = 2.00 |
| [Fe(TpivPP)(NO3)] | Cryst. powder | 77 | Rhombic | g⊥ = 6.44, 3.97, 1.9 |
All powder samples were prepared from ground single crystals unless otherwise noted. All solution measurements were carried out in frozen CH2Cl2.
Powder sample of unknown phase.
Table 5.
Mössbauer data for [Fe(por)(NO3)] complexes.
| Complex | Temp (K) | ΔEQ (mm/sec) | δ (mm/sec) |
|---|---|---|---|
| [Fe(OEP)(NO3)] | 4.2 | 1.44 | 0.53 |
| 15 | 1.39 | 0.41 | |
| 50 | 1.39 | 0.42 | |
| 100 | 1.38 | 0.41 | |
| 150 | 1.36 | 0.42 | |
| 200 | 1.38 | 0.38 | |
| RT | 1.37 | 0.36 | |
|
| |||
| [Fe(TPP)(NO3)] | 4.2 | 0.97 | 0.61 |
| 200 | 0.98 | 0.47 | |
| RT | 0.97 | 0.38 | |
|
| |||
| [Fe(TpivPP)(NO3)] | 4.2 | 1.0 | 0.56 |
| 200 | 1.10 | 0.49 | |
| RT | 1.07 | 0.37 | |
Figure 6.
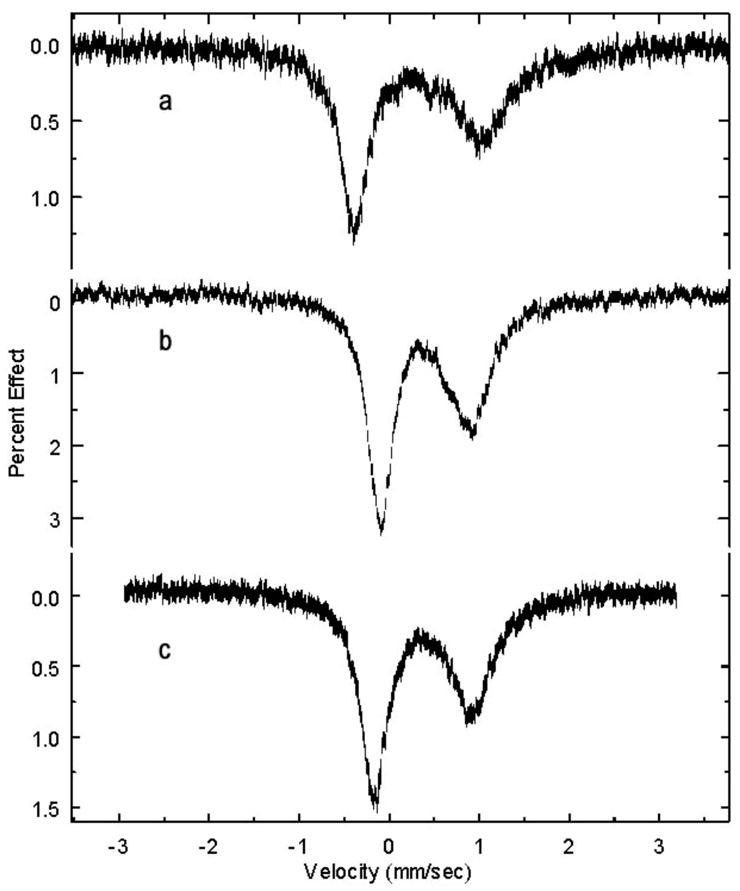
Mössbauer spectra of a) [Fe(OEP)(NO3)], b) [Fe(TPP)(NO3)], and c) [Fe(TpivPP)(NO3)]. Spectra taken at room temperature.
Discussion
Synthetic Aspects
We report a straightforward synthetic route for obtaining ferric iron porphyrinate complexes containing a coordinated nitrate ligand. A simple metathesis reaction yields the desired [Fe(por)(NO3)] complex with the undesirable side-products being removed by a simple filtration. The ease of this synthetic pathway is emphasized when compared with the previous reported methods which either proceeded via an oxygen-atom transfer,17 NO attack of a bridged μ-oxo-dimer15 or in the case of the TPP derivative, reaction of the μ-oxo-dimer with aqueous nitric acid followed by a three-week crystallization.16 In the latter case, we are able to successfully obtain large crystals in under one week. The only problem encountered with the new reaction system lay in the inability to obtain crystals of the picket-fence porphyrin complex [Fe(TpivPP)(NO3)]. We were able to show spectroscopically that the species could be obtained in solution yet were unable to prepare a solid crystalline sample. UV-Vis and IR spectroscopic data for all three systems are given in Table 3. In all cases, IR bands associated with the free nitrate ligand were not found, confirming the coordinated nature of the nitrate in all three cases.
Molecular Structures
We report here a new crystalline form and a redetermined structure for two high-spin ferric (nitrato)-porphyrinates. Schematic representations of the porphyrin core for these systems and all other [Fe(por)(NO3)] complexes are shown in Figure 4. At the onset of the project, it was thought, based on the reported structures15–17 that the series of complexes [Fe(por)(NO3)] exhibited distinct examples of three coordination geometries (Scheme 1). However after evaluation of the EPR data, we decided to redetermine the structure of the asymmetric bidentate coordinated system [Fe(TPP)(NO3)].16 The reported structure16 showed a nitrate ligand lying midway between a pair of Fe–Np bonds with two very different Fe–O bond lengths of 2.019(4) and 2.323(8) Å and δ, the difference between the Fe–O bond lengths, of 0.304 Å. The porphyrin core displays a characteristic saddling distortion and the iron is significantly displaced from the mean porphyrin plane in the direction of the nitrate ligand. A similar saddling distortion and iron out-of-plane displacement is observed in the porphyrin core of our redetermined structure at 100 K Figure 2(b). In fact, if we overlay the porphyrin cores of the RT structure and our newly redetermined structure so as to align the saddling patterns, we observe that the originally reported nitrate ligand orientation lies midway between the two orientations found in the 100 K structure. This is consistent with the very large thermal ellipsoids observed for the two coordinated oxygen atoms of the nitrate. Based upon this observation, we conclude the previously reported nitrate ligand orientation is in fact an averaging of the two orientations we observe in our 100 K structure. These two orientations present a more eclipsed nitrate position both lying approximately 10° from a Fe–Np vector. A similar orientation is observed in the other bidentate system [Fe(TpivPP)(NO3)]17 where the bidentate nitrate lies 10° from the closest Fe–Np vector. However, neither of the two new nitrate orientations in the 100 K structure of [Fe(TPP)(NO3)] display anywhere near the same extent of asymmetric coordination as reported in the RT structure. The major orientation shows the greatest disparity in the two Fe–O bond length (δ = 0.1433 Å) whereas the bond lengths in the minor orientation are practically equivalent (δ = 0.0047 Å). Based upon these results, we reclassify the [Fe(TPP)(NO3)] system as containing a symmetrically coordinated bidentate nitrate ligand.
Figure 4.
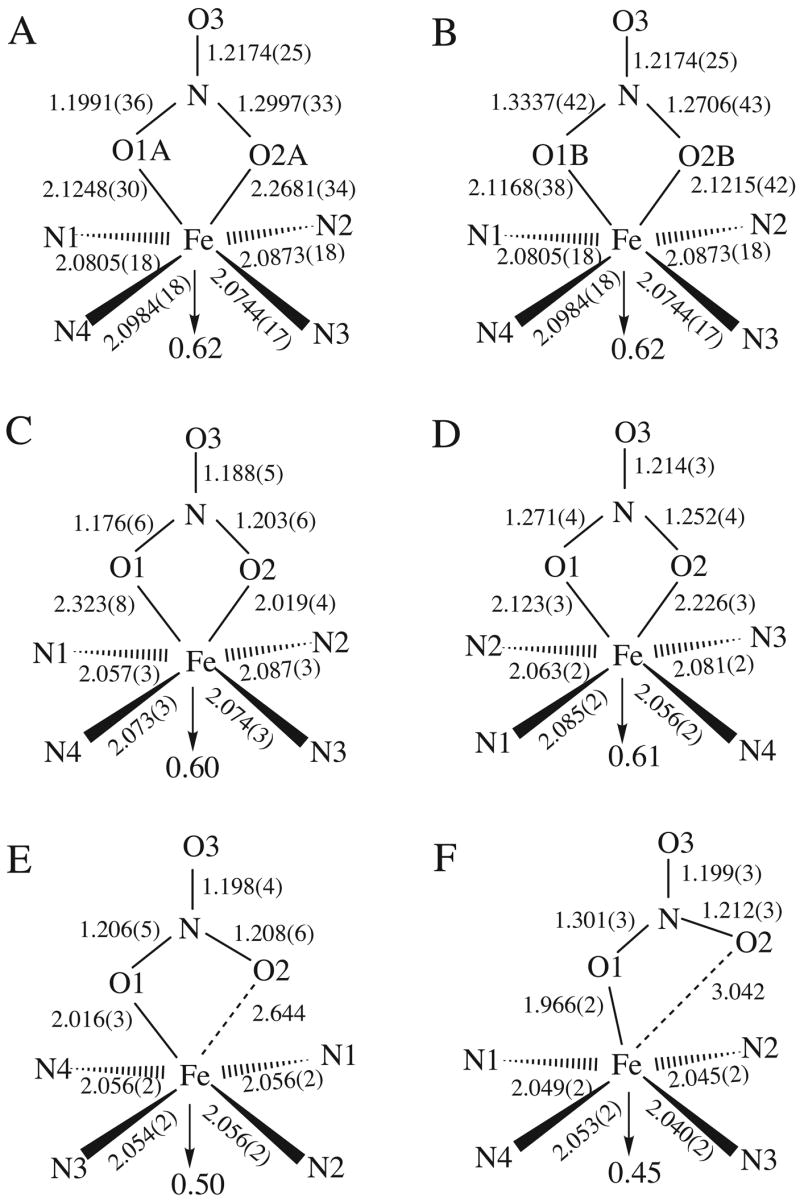
Schematic representations of the N4FeNO3 geometry for all [Fe(por)(NO3)] complexes. Structures represented are (A) [Fe(TPP)(NO3)] 100 K, major orientation; (B) [Fe(TPP)(NO3)] 100 K, minor orientation; (C) [Fe(TPP)(NO3)] Room temperature structure (reference 16); (D) [Fe(TpivPP)(NO3)] (reference 17); (E) [Fe(OEP)(NO3)] triclinic form (reference 15); (F) [Fe(OEP)(NO3)] monoclinic form.
We also observe some disparity in the Fe–O distances in the two structures of [Fe(OEP)(NO3)]. This is most evident in the nonbonded distance between the iron center and the non-coordinating oxygen, O2, in Figure 4(e) and (f). Although the Fe–O bond length for both of the monodentate nitrates are very similar, the nitrate position with respect to the porphyrin core is significantly different. In the previously reported triclinic form,15 the nitrate adopts an orientation similar to the bidentate systems with the Fe–O2 vector perpendicular to the porphyrin plane and passing through the core center where the iron sits. In the monoclinic form, the Fe–O bond lies more perpendicular to porphyrin plane with the N–O(2) bond shifted significantly from the centered normal to the porphyrin plane. This is reflected in a marked increase in the distance between the iron center and the noncoordinated O(3) from 2.644 Å in the triclinic system to 3.042 Å for the monoclinic system. In both cases, the nitrate ligand still lies close to midway between a pair of Fe–Np vectors and equivalent iron out-of-plane displacements are observed.
Figure 4 illustrates the core geometry for all ferric [Fe(por)(NO3)] systems reported to date. There are a number of features common to all the systems. Each system displays a significant out-of-plane displacement of the iron in the direction of the nitrate ligand. large out-of-plane displacements are common in five-coordinate ferric porphyrinates.25 No significant differences are observed in the Fe–Np distances for each system although the distances for the two monodentate OEP derivatives are slightly shorter than the meso-substituted porphyrinates.
The variation in the projection of the nitrate plane onto the plane of the porphyrin has already been discussed briefly. Comparing the data from all systems reveals when the nitrate coordinates in a bidentate fashion, the projection of the ligand plane is close to a Fe–Np vector with a dihedral angle (φ) of ~10°. When the nitrate coordinates in a monodentate fashion, the orientation changes such that it lies midway between the two Fe–Np vectors, possibly to minimize steric interactions between the pyrrolic nitrogens and the uncoordinated oxygen of the nitrate ligand.
EPR Spectroscopy
EPR spectra on both the powder and solutions of all three nitrate complexes were collected at 77 and 4 K. These data are summarized in Table 4 and displayed in Figure 5. For the OEP complexes, the EPR spectra for both solid and solution at both temperatures are axial. At 77 K however, the minor g|| feature was too weak to be detected. When the temperature is reduced to 4 K, both g⊥ and g|| features are evident. The observation of axial spectra for both the solid and the solution is consistent with a monodentate coordination mode in both states. Similar axial spectra are observed in other five-coordinate high-spin systems such as [Fe(por)(Cl)].26 We have already discussed the differences in coordination mode of the nitrate ligand in the TPP and picket-fence derivatives and we would expect the EPR spectra for these to differ from the simple axial spectra observed for the OEP derivative. Had the previously reported coordination mode of the nitrate in [Fe(TPP)(NO3)], the highly asymmetric bidentate geometry, been representative of the nitrate geometry at lower temperatures, we would perhaps have expected to see EPR spectra for this system that differ from both that of the OEP and TpivPP systems. Instead, following our new crystallographic determination, we now expect the EPR spectra of the TPP and TpivPP derivatives to be somewhat similar. Indeed, this is true with the EPR of the ground crystalline material displaying rhombic spectra for both of the bidentate coordinated systems. [Fe(TPP)(NO3)] at 77 K exhibits a complex spectrum from which we identify a major rhombic component has g = 7.71, 4.04 and 1.78 with the largest g-tensor shifting to 8.5 at 4 K. Correspondingly, the largest g-tensor in the EPR spectrum of the ground crystalline [Fe(TpivPP)(NO3)] is smaller than that of the TPP derivative (g = 6.44, 3.97, 1.9). EPR data from a powder sample of [Fe(TpivPP)(NO3)] of unknown phase which was prepared using a crude precipitation technique exhibits a characteristic axial spectrum with g = 5.5, 2 pattern. Based upon our observations from the OEP system, this likely contains a monodentate nitrate ligand. Similarly, axial EPR spectra are observed for solutions of the TPP and picket fence derivatives. This suggests that the coordination mode of the nitrate ligand in both these systems changes to monodentate upon solvation.
Figure 5.
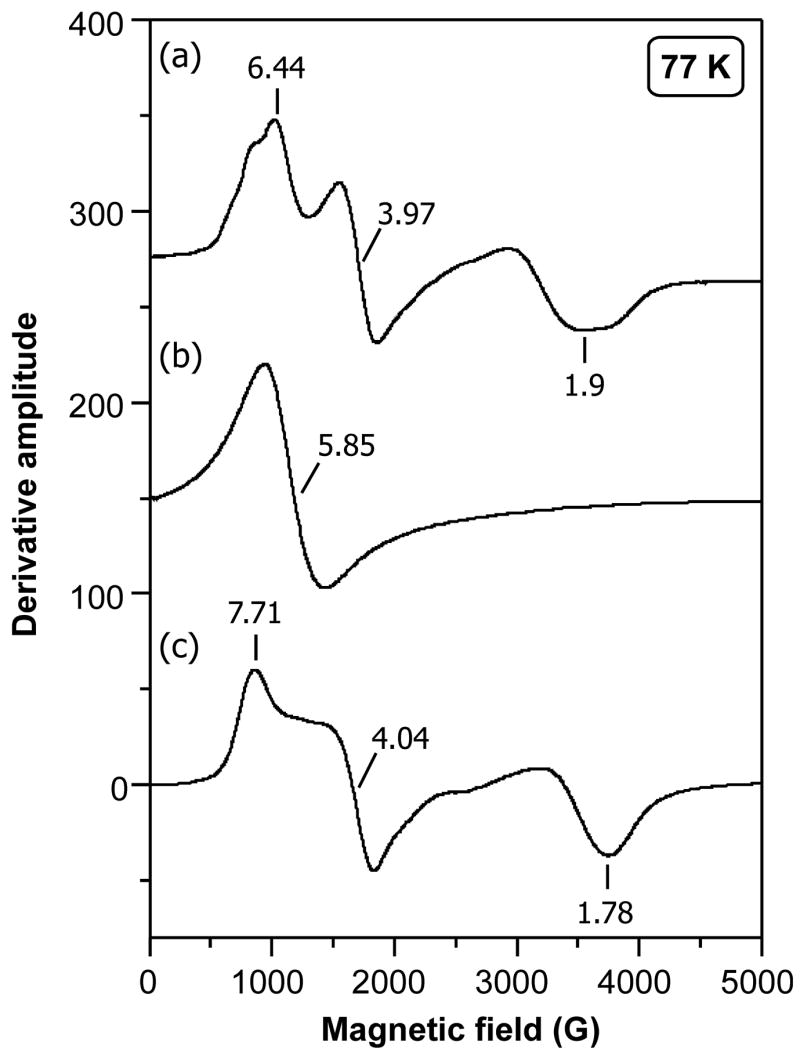
EPR spectra at 77 K of ground single crystals of a) [Fe(TpivPP)(NO3)], b) [Fe(OEP)(NO3)], and c) [Fe(TPP)(NO3)].
Mössbauer Spectroscopy
Table 5 summarizes the Mössbauer measurements carried out on each of the nitrate complexes collected over a range of temperatures. All three systems were studied at 4 K in an applied magnetic field and the spectra could be satisfactorily fit to a S = 5/2 model (Table 6). All three systems displayed asymmetric quadrupole doublets at room temperature. Linewidths broadened such that quadrupole splitting fits were inaccessible in the temperature range from 150 K to below 18 K for the TPP and TpivPP derivatives; the OEP derivative did not show the same broadening behavior. The values for the quadrupole splitting are somewhat larger than other high-spin ferric systems such as [Fe(por)(Cl)] (ΔEQ ~0.9 mm/sec). Since the quadrupole splitting is dependent upon the symmetry of the metal center and we have already observed significant differences in the nature of the g tensors in the ground crystalline samples, we expect here to observe substantial differences in the values of ΔEQ for all 3 systems. Although differences are observed, the fact that the OEP derivative has the largest value of ΔEQ is unexpected. Moreover, there remains some uncertainty about the exact phase for the picket fence porphyrin measurement.
Table 6.
Data from S = 5/2 Fit to 4.2 K Mössbauer spectra of [Fe(por)(NO3)] derivatives.
| Complex | ΔEQ (mm/s) | δ (mm/s) | η | Γ (mm/s) | D (cm−1) | E (cm−1) | Aii/gN βN (T) |
|---|---|---|---|---|---|---|---|
| [Fe(OEP)(NO3)] | 1.44 | 0.53 | 0.06 | 0.52 | 5.7 | 0.37 | −14.9, −17.3, −2.7 |
| [Fe(TPP)(NO3)] | 0.97 | 0.61 | 0.0 | 0.42 | 6.9 | 0.6 | −21.8, −20.0, −10.4 |
| [Fe(TpivPP)(NO3)] | 1.0 | 0.56 | 0.4 | 0.57 | 10 | 0.2 | −23.1, −20.9, −2.0 |
Summary
Nitrate can bind to the iron center in the iron(III) porphyrinates in either a symmetric bidentate or monodentate mode. An earlier report of an asymmetric binding mode is probably not correct. EPR spectra display sensitivity to the coordination mode. An axial high-spin signal is seen for monodentate coordination, whereas a rhombic high-spin spectrum is observed for the bidentate species. Mössbauer spectra do not appear to be as sensitive to the coordination mode.
Supplementary Material
Appendix A. Supplementary data
CCDC Reference Nos. 633779 and 633780 contain the supplementary crystallographic data for this paper. These data are available free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/datarequest/cif or CCDC, 12 Union Road, Cambridge CB2 1EZ, UK on request, quoting the deposition numbers above.
Figure 7.
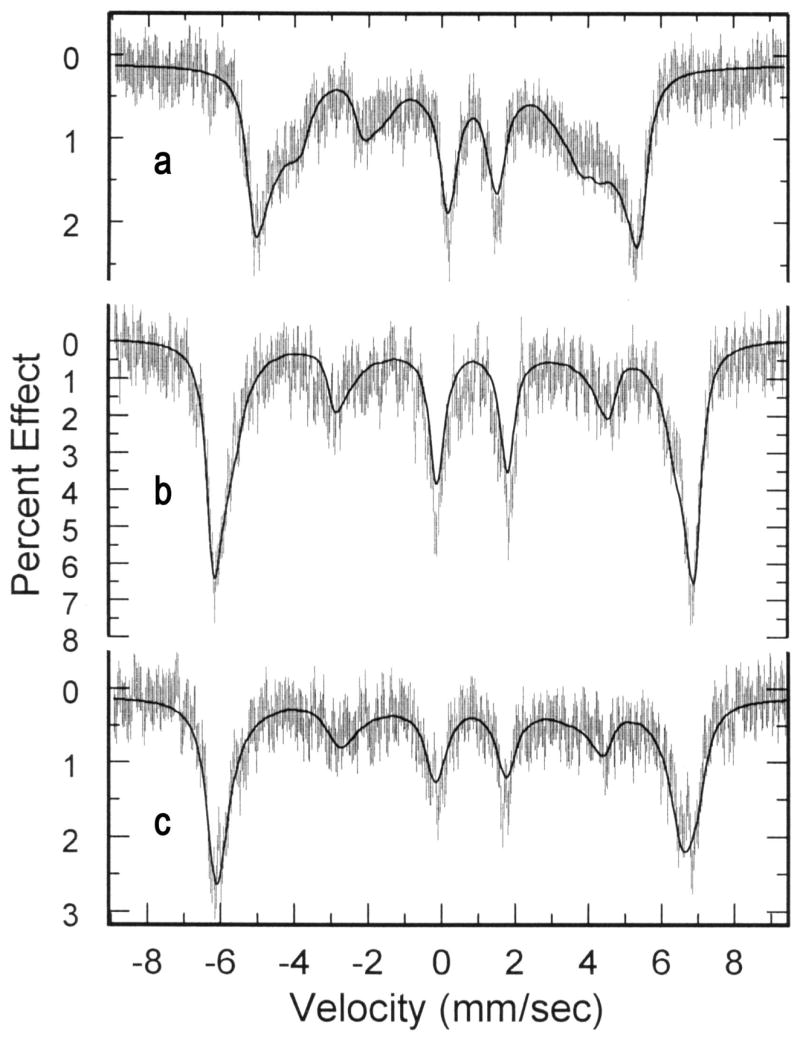
Mössbauer spectra and calculated fits of the spectra of a) [Fe(OEP)(NO3)], b) [Fe(TPP)(NO3)], and c) [Fe(TpivPP)(NO3)]. Spectra taken at 4.2 K and an applied 4.5 T field.
Acknowledgments
We thank the National Institutes of Health for support of this research 11 under Grant GM-38401. Funds for the purchase of the FAST area detector were provided through NIH Grant RR-06709. We thank the NSF for EPR support through instrumentation grant NSF-98-70990.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Averill B. Chem Rev. 1996;96:2951. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]
- 2.Dudley N. Nitrates. The Threat to Food and Water. Green Print; London: 1990. [Google Scholar]
- 3.L’hirondel J, L’hirondel J-L. Nitrate and Man: Toxic, Harmless or Beneficial? CABI Publishing; New York, NY: 2002. [Google Scholar]
- 4.Inoue M, Minamiyama Y, Takemura S. Methods Enzymol. 1996;269:474. doi: 10.1016/s0076-6879(96)69048-x. [DOI] [PubMed] [Google Scholar]
- 5.Doyle MP, Hoekstra JW. J Inorg Biochem. 1981;14:351. doi: 10.1016/s0162-0134(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 6.Eich RF, Li T, Lemon DD, Doherty DH, Curry SR, Aitken JF, Mathews AJ, Johnson KA, Smith RD, Phillips GN, Jr, Olson JS. Biochemistry. 1996;35:6976. doi: 10.1021/bi960442g. [DOI] [PubMed] [Google Scholar]
- 7.Cotton FA, Wilkinson G, Murillo CA, Bochmann M. Advanced Inorganic Chemistry. 6. Wiley-Interscience; New York, NY: 1999. p. 489. [Google Scholar]
- 8.Addison CC, Logan N, Wallwork SC, Garner CD. Quart Rev. 1971;25:289. [Google Scholar]
- 9.(a) Gatehouse BM, Livingstone SE, Nyholm RS. J Chem Soc. 1957:4222. [Google Scholar]; (b) Lever ABP, Mantovani E, Ramaswamy BS. Can J Chem. 1971;49:1957. [Google Scholar]
- 10.(a) Van Driel GJ, Driessen WL, Reedjik J. Inorg Chem. 1985;24:2919. [Google Scholar]; (b) Kleywegt GJ, Wiesmeijer WGR, Van Driel GJ, Driessen WL, Reedjik J. J Chem Soc Dalton Trans. 1985:2717. [Google Scholar]
- 11.Abbreviations used in this paper Por - a generalized porphyrin dianion, OEP dianion of 2,3,7,8,12,13,17,18-octaethylporphyrin, TPP dianion of 5,10,15,20-tetraphenylporphyrin, TpivPP dianion of meso-tetrakis(α-α-α-α-pivalamidophenyl)porphyrin, Np - pophyrinato nitrogen atom, amtd tris-((3,5-dimethylpyrazoyl-1-yl)methyl)amine, dpph diphenylpicrylhydrazl.
- 12.(a) Han R, Parkin G. J Am Chem Soc. 1991;113:9707. [Google Scholar]; (b) Kimblin C, Murphy VJ, Hascall T, Bridgewater BM, Bonanno JB, Parkin G. Inorg Chem. 2000;39:967. doi: 10.1021/ic990682v. [DOI] [PubMed] [Google Scholar]
- 13.Wyllie GRA, Scheidt WR. Chem Rev. 2002;102:1067. doi: 10.1021/cr000080p. [DOI] [PubMed] [Google Scholar]
- 14.Nasri H, Ellison MK, Shaevitz B, Gupta GP, Lang G, Scheidt WR. Inorg Chem. 2006;45:5284. doi: 10.1021/ic052059i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellison MK, Shang M, Kim J, Scheidt WR. Acta Crystallogr Sect C. 1996;C52:3040. doi: 10.1107/s0108270196010062. [DOI] [PubMed] [Google Scholar]
- 16.Phillippi MA, Baenziger N, Goff HM. Inorg Chem. 1981;20:3904. [Google Scholar]
- 17.Munro OQ, Scheidt WR. Inorg Chem. 1998;37:2308. doi: 10.1021/ic970855l. [DOI] [PubMed] [Google Scholar]
- 18.Finnegan MG, Lappin AG, Scheidt WR. Inorg Chem. 1990;29:181. [Google Scholar]
- 19.Adler AD, Longo FR, Finarelli JD, Goldmacher J, Assour J, Korsakoff L. J Org Chem. 1967;32:476. [Google Scholar]
- 20.Adler AD, Longo FR, Kampus F, Kim J. J Inorg Nucl Chem. 1970;32:2443. [Google Scholar]
- 21.Collman JP, Gagne RR, Halbert TR, Lang G, Robinson WT. J Am Chem Soc. 1975;97:1427. doi: 10.1021/ja00839a026. [DOI] [PubMed] [Google Scholar]
- 22.Sheldrick GM. Acta Crystallogr. 1990;A46:467. [Google Scholar]
- 23.Sheldrick GM. SHELXL-97: FORTRAN program for crystal structure refinement. University of Göttingen; Göttingen, Germany: 1997. [Google Scholar]
- 24.Scheidt WR, Turowska-Tyrk I. Inorg Chem. 1994;33:1314. [Google Scholar]
- 25.Scheidt WR. Systematics of the Stereochemistry of Porphyrins and Metalloporphyrins. In: Kadish KM, Smith K, Guilard R, editors. The Porphyrin Handbook. Chapter 16 Academic Press; San Diego, CA and Burlington, MA: 2000. p. 3. [Google Scholar]
- 26.Palmer G. In: Iron Porphyrins Part 2. Lever ABP, Gray HB, editors. Chapter 2 VCH Publishers Inc.; New York: 1983. [Google Scholar]
- 27.Debrunner PG. In: Iron Porphyrins Part 3. Lever ABP, Gray HB, editors. Chapter 2 VCH Publishers Inc.; New York: 1983. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix A. Supplementary data
CCDC Reference Nos. 633779 and 633780 contain the supplementary crystallographic data for this paper. These data are available free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/datarequest/cif or CCDC, 12 Union Road, Cambridge CB2 1EZ, UK on request, quoting the deposition numbers above.