

Abstract
Apolipoprotein E4 (apoE4) encoded by ε4 allele is a strong genetic risk factor for Alzheimer’s disease (AD). ApoE4 carriers have accelerated amyloid β-protein (Aβ) deposition in their brains, which may account for their unusual susceptibility to AD. We hypothesized that the accelerated Aβ deposition in the brain of apoE4 carriers is mediated through cholesterol-enriched low-density membrane (LDM) domains. Thus, the concentrations of Aβ and various lipids in LDM domains were quantified in the brains of homozygous apoE3 and apoE4 knock-in (KI) mice, and in the brains of those mice bred with β-amyloid precursor protein (APP) transgenic mice (Tg2576). The Aβ40 and Aβ42 concentrations and the Aβ42 proportions in LDM domains did not differ between apoE3 and apoE4 KI mice up to 18 months of age. The Aβ40 concentration in the LDM domains was slightly, but significantly higher in apoE3/APP mice than in apoE4/APP mice. The lipid composition of LDM domains was modulated in an apoE isoform-specific manner, but its significance for Aβ deposition remains unknown. These data show that the apoE isoform-specific effects on the Aβ concentration in LDM domains do not occur in KI mouse models.
Keywords: Alzheimer’s disease, amyloid β-protein, apolipoprotein E, knock-in mouse, lipids, low-density membrane domains
Human apolipoprotein E (apoE) alleles encode three isoforms, apoE2, apoE3, and apoE4, which differ from each other by one or two amino acids (either cysteine or arginine at positions 112 and 158) (Weisgraber and Mahley 1996). The presence of apoE4 allele (ε4) markedly increases the risk of Alzheimer’s disease (AD) in a gene dosage-dependent manner (Corder et al. 1993; Strittmatter et al. 1993), and is associated with accelerated deposition of amyloid β-protein (Aβ) in the brain (Schmechel et al. 1993; Morishima-Kawashima et al. 2000; Walker et al. 2000), but presumably not with the extent of deposition (Morishima-Kawashima et al. 2000). This may explain why apoE4 carriers are predisposed to AD, because an earlier onset of Aβ deposition increases the time span for development of AD.
Apolipoprotein E is involved in transporting lipids, especially cholesterol, to the various tissues (Weisgraber 1994). In the CNS, apoE is thought to play an important role in modulating lipid turnover, although few details are known (Weisgraber 1994; Holtzman and Fagan 1998; LaDu et al. 2000; Han 2004). The relationship between the cholesterol concentration and Aβ production or deposition has been claimed repeatedly. Overall, decreased concentrations of cholesterol are associated with decreased Aβ secretion or production, or both (Sparks et al. 1994; Simons et al. 1998; Frears et al. 1999; Refolo et al. 2000; Fassbender et al. 2001). On the contrary, epidemiological studies suggest a potential link between cardiovascular disease and AD (Skoog et al. 1993; Ravona-Springer et al. 2003). Medication with statins that suppress 3-hydroxy-3-methylglutaryl CoA reductase activity is thought to decrease the incidence of AD (Jick et al. 2000; Wolozin et al. 2000), although recent results are conflicting (Li et al. 2004; Rea et al. 2005). It is possible that the effects of apoE4 on Aβ deposition are mediated through cholesterol metabolism in the CNS. This is supported by the observation that the temporal and regional profiles of Aβ deposition are distinctly altered in apoE-null mice (Bales et al. 1997; Irizarry et al. 2000), indicating that apoE has marked effects on the distribution and extent of Aβ deposition in the brain.
A substantial amount of Aβ in the cultured cell and in the brain is localized to low-density membrane (LDM) domains (Lee et al. 1998; Morishima-Kawashima and Ihara 1998; Oshima et al. 2001), which are rich in cholesterol and sphingolipids, and likely represent lipid rafts (Simons and Ikonen 1997). Under pre-symptomatic conditions, one-fourth of the total Aβ in the mouse brain (Kawarabayashi et al. 2004) and one-half of the insoluble Aβ in the human brain (Oshima et al. 2001) are localized to LDM domains. The Aβ42 concentration in LDM domains is proportional to the extent of Aβ42 accumulation in the parenchyma, which in turn correlates with the amyloid burden in a logarithmic manner (Oshima et al. 2001). On the contrary, familial AD-associated mutation of presenilin 2 (N141I) increases the concentration of Aβ42 in LDM domains and modulates their lipid composition (Sawamura et al. 2000). Thus, apoE4 may affect the Aβ42 concentration in LDM domains by altering their cholesterol or other lipid concentrations, and may facilitate Aβ deposition in the brain parenchyma. Here, we report our investigations of the effects of apoE isoforms on the Aβ concentration in LDM domains and their lipid composition, using human apoE3 and apoE4 knock-in (KI) mice.
Materials and methods
Animals
Male homozygous apoE3 and apoE4 KI mice (Hamanaka et al. 2000; Hayashi et al. 2002) were studied at 2 months (n = 3 for each allele), 12 months (n = 3), 18 months (n = 5), and 24 months (n = 3) of age. In these mice, human apoE3 or apoE4 cDNA is targeted to the endogenous mouse apoE gene, and human apoE is expressed under the control of endogenous mouse regulatory elements. The expression levels of each apoE isoform in the brain are similar in the two lines (Mori et al. 2003). The pattern of human apoE staining was similar to the distribution of mouse apoE in the wild-type mouse brain (Hamanaka et al. 2000).
F1 offsprings of each apoE KI line were backcrossed six generations onto the C57BL/6N background following a marker-assisted selection protocol (Wong 2002) to generate the congenic strain. The resultant heterozygous apoE KI mice (C57BL/6N congenic) were bred with SJL inbred mice to produce the mice on mixed C57BL/6N × SJL backgrounds. The heterozygous apoE KI mice (C57BL/6N × SJL backgrounds) were then crossed with Tg2576 mice, which overexpress Swedish mutant β-amyloid precursor protein (APP) under the control of the hamster prion promoter on mixed C57BL/6 × SJL backgrounds (Hsiao et al. 1996). Among offsprings from this mating, the apoE+/−, APP+/− mice were intercrossed with the apoE+/−, APP−/− mice to produce the apoE+/+, APP+/− mice (referred to as apoE3/APP or apoE4/APP mice). The apoE+/+, APP+/− mice were studied at 12 months (n = 5 for each human apoE isoform) and 15 months (n = 6) of age. All procedures were approved by the animal study committee at each institute.
Tissue extraction
Each brain (minus the cerebellum) was homogenized with a motor-driven Teflon-glass homogenizer in Tris-saline (TS: 50 mmol/L Tris-HCl, pH 7.6, and 150 mmol/L NaCl) containing a cocktail of protease inhibitors. The homogenate was centrifuged at 540 000 g for 20 min in a TLX centrifuge (Beckman, Palo Alto, CA, USA). The resulting pellet was washed once with TS, and extracted with 6 mol/L guanidine-HCl in 50 mmol/L Tris-HCl, pH 7.6. The homogenate was centrifuged at 265 000 g for 20 min. The supernatant was diluted to 0.5 mol/L guanidine-HCl, and the concentrations of TS-insoluble Aβ40 and Aβ42 were measured using an enzyme-linked immunosorbent assay (ELISA), as described previously (Morishima-Kawashima et al. 2000).
Detergent-insoluble low-density membrane domains
Low-density membrane fractions were obtained as described previously (Sawamura et al. 2000). The mouse cerebral tissue minus the cerebellum (120 mg) was homogenized in 2 mL of 2-morpholinoethanesulfonic acid (MES)-buffered saline [MBS: 25 mmol/L MES, pH 6.5, and 150 mmol/L NaCl] containing 1% Triton X-100, 1 mmol/L phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin, 10 μg/mL pepstatin, and 10 μg/mL aprotinin. The homogenate was adjusted to 40% sucrose by adding an equal volume of 80% sucrose in MBS, placed at the bottom of an ultracentrifuge tube, and overlaid with 4 mL of 35% sucrose and 4 mL of 5% sucrose in MBS without Triton X-100. The discontinuous gradient was centrifuged at gav 190 000 g; gmax 270 000 g for 20 h in an SW 41 rotor (Beckman) at 4°C. The interface at 5%/35% sucrose (fraction 2) and each of the layers composed of 5%, 35%, and 40% sucrose (fractions 1, 3, and 4, respectively) were collected, diluted threefold with MBS, and centrifuged. The resultant pellets and the pellet (P) obtained from the original sucrose gradient centrifugation were extracted with 6 mol/L guanidine-HCl, and the concentrations of Aβ40 and Aβ42 contained in the extracts were measured by ELISA.
Antibodies and ELISA
The monoclonal antibodies, 4G8 (specific for Aβ17–24) and 6E10 (raised against Aβ1–17), were obtained from Signet Laboratories (Dedham, MA, USA). 82E1 was from IBL (Gunma, Japan). Anti-flotillin antibody was from Transduction Laboratories (Lexington, KY, USA).
The two-site ELISA for Aβ40 and Aβ42 was performed as described (Morishima-Kawashima et al. 2000). The capture antibody used was BNT77 (raised against Aβ11–28, the epitope located in Aβ11–16). The detection antibody was horseradish peroxidase-conjugated BA27 (raised against Aβ1–40 and specific for Aβ40) or BC05 (raised against Aβ35–43 and specific for Aβ42). BC05 weakly cross-reacts with APP (the affinity being 1/300–1/500 times that with Aβ42) (Morishima-Kawashima and Ihara 1998), and the TS-insoluble Aβ42 levels were corrected taking into account the concentrations of APP (Morishima-Kawashima et al. 2000). On the contrary, only a fraction of APP was localized to the LDM domains (Sawamura et al. 2000), and the APP concentrations were 100 times less than those of Aβ42 in LDM domains.
Western blotting
The pellet from fraction 2 prepared from apoE3/APP or apoE4/APP mice was delipidated with chloroform–methanol, and the residue was extracted with formic acid as described previously (Morishima-Kawashima et al. 2000). Following centrifugation, the supernatant was dried up in a Speed Vac (Savant Instruments, Farmingdale, NY, USA) and solubilized with Laemmli’s sample buffer containing 4 mol/L urea. These samples were applied to a 16.5% Tris-tricine gel. Following transfer, the nitrocellulose membrane was dipped in boiled phosphate-buffered saline to enhance Aβ immunoreactivity. The bound antibodies were detected using enhanced chemiluminescence system (Amersham Pharmacia, Buckingham, UK), and the signal was captured using an LAS-1000plus luminescent image analyzer (Fuji Film, Tokyo, Japan). Quantification of Aβ was performed using authentic standard Aβ and 4G8, as previously described (Qi et al. 2003).
To determine the total brain Aβ levels, the brain homogenates from apoE3/APP or apoE4/APP mice were subjected to semi-quantitative western blotting using 82E1.
Immnocytochemistry
Formalin-fixed, paraffin-embedded tissue sections of Tg2576, apoE3/APP, and apoE4/APP mice brains were deparaffinized and immunostained with 4G8 by the avidin–biotin method (Vectastain Elite; Vector Laboratories, Burlingame, CA, USA), with 3,3′-diaminobenzidine as a substrate. The immunostained sections were counterstained briefly with hematoxylin.
Lipid analysis
The pellets obtained from LDM fractions prepared from apoE3/APP or apoE4/APP mice were subjected to lipid analysis. The lipids were quantified using electrospray ionization–mass spectrometry (ESI–MS) as described (Han and Gross 2005a,b). LDM fractions prepared from prefrontal cortexes of non-demented human subjects with apoE3/apoE3 genotype (Morishima-Kawashima et al. 2000; Oshima et al. 2001) were also subjected to lipid analysis using ESI–MS.
Statistical analysis
Data are presented as means ± SD. Variables were compared between apoE3 and apoE4 mice using Student’s t-test. The age dependence of the Aβ42 proportion was evaluated by a Spearman rank correlation test. Statistical analyses were performed using Microsoft Excel 2001 for Student’s t-test and StatView J-5.0 for the Spearman rank correlation test. p < 0.05 was considered significant for the Aβ analyses and p < 0.01 for lipid analysis.
Results
Aβ concentrations in LDM domains from apoE3 and apoE4 KI mice brains
We first assessed the Aβ concentrations in the insoluble fraction of apoE3 or apoE4 KI mice brains. At all the ages examined, the concentration of Aβ40 in the insoluble fraction appeared to be higher in apoE4 mice than in apoE3 mice, but the difference was not significant (data not shown). The concentration of Aβ42 also did not differ significantly between apoE3 and apoE4 KI mice. The concentration of Aβ40 increased markedly up to 18 months of age, whereas that of Aβ42 remained relatively constant (data not shown).
Low-density membrane domains were prepared from mice brains using sucrose density gradient centrifugation in the presence of Triton X-100. Fraction 2 was characterized by the presence of flotillin, a raft marker for neurons, and other subcellular organelle markers distributed among the fractions, as shown previously (data not shown; Oshima et al. 2001). At 2 months of age, a significant amount of Aβ was localized to LDM domains (Fig. 1a). About 40% of the total Triton-insoluble Aβ40 (sum of the insoluble Aβ40 in each fraction) and ~30% of the total Triton-insoluble Aβ42 fractionated into the LDM fraction. The proportion of Aβ partitioned to LDM domains appeared to increase in an age-dependent manner up to 18 months of age. At 12 and 18 months of age, ~50% of the total Triton-insoluble Aβ40 was located in LDM domains, and the concentration of Aβ40 was more than two times higher than at 2 months of age (Figs 1b and c). At 12 months, the Aβ42 concentration was about twice that at 2 months, and increased further to more than three times higher at 18 months of age. The proportion of Aβ42 (Aβ42/sum of Aβ40 and Aβ42) in LDM domains increased in an age-dependent manner (p = 0.0337 for apoE3, and p = 0.0228 for apoE4; Spearman rank correlation test) and amounted to more than 30% at 18 months of age (Fig. 1d). The Aβ42 proportion increased further and reached ~40% at 24 months of age, but without any noticeable isoform-specific effects (Fig. 1d).
Fig. 1.
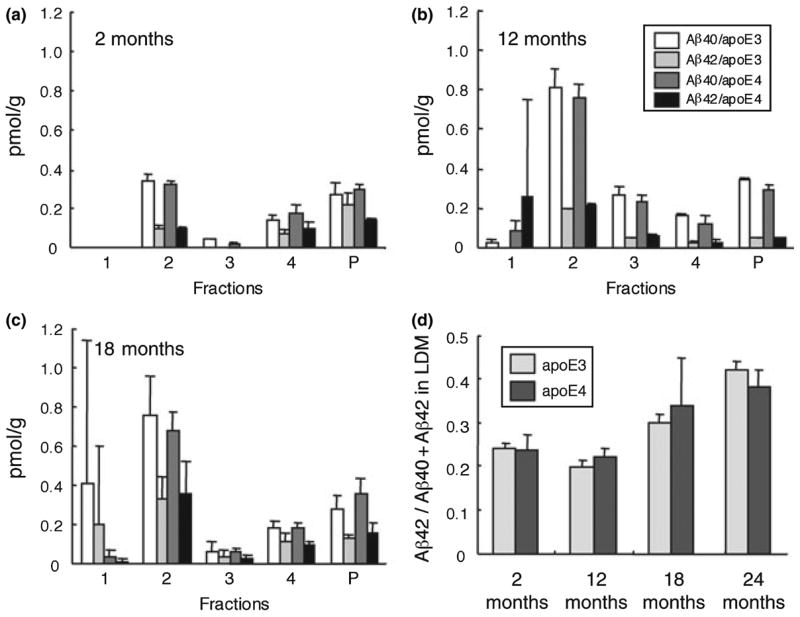
Quantification of Aβ40 and Aβ42 in the fractions obtained by sucrose density gradient centrifugation of the homogenates from apoE3 and apoE4 KI mice at two (a), 12 (b), and 18 months of age (c). Each fraction was diluted and centrifuged (see ‘Materials and methods’ section). The resulting pellet was extracted with guanidine-HCl, and the concentrations of Aβ40 and Aβ42 in the extract were measured by ELISA. Fraction 2 represents low-density membrane (LDM) domains. Data are presented as means ± SD (n = 3 for 2- and 12-month-old mice, n = 5 for 18-month-old mice). Open bars, Aβ40 concentrations in apoE3 mice; light gray bars, Aβ42 concentrations in apoE3 mice; dark gray bars, Aβ40 concentrations in apoE4 mice; black bars, Aβ42 concentrations in apoE4 mice. Panel (d) shows the proportion of Aβ42 to the sum of Aβ40 and Aβ42 in LDM fraction (fraction 2) at each age, including 24 months of age (n = 3). Light gray bars, apoE3 mice; dark gray bars, apoE4 mice.
The Aβ40 concentration in LDM domains tended to be slightly higher in apoE3 mice than in apoE4 mice at all ages examined, but these differences were not significant (Figs 1a–c). Neither the Aβ42 concentration nor its proportion differed significantly between apoE3 and apoE4 mice at any age (Figs 1a–d). Fraction 1 (the top layer) usually contained the least amounts of Aβ and a trace amount of proteins (Oshima et al. 2001). However, the fractions prepared from the brains of one 12-month-old apoE4 mouse and one 18-month-old apoE3 mouse contained markedly high concentrations of Aβ42, and of Aβ40 and Aβ42, respectively, for unknown reasons (hence the large S.D. in Figs 1b and c).
Aβ concentrations in LDM domains from apoE3/APP and apoE4/APP mice
It is possible that a residual heterogeneous genetic background affected the distribution or the concentration of Aβ in LDM domains, because the apoE KI mice we used were not entirely congenic (Lehman et al. 2003). In addition, because the endogenous Aβ concentration in the mouse brain is very low, small differences cannot be quantified accurately. In other words, small differences, if any, must be amplified to become readily detectable. Thus, we established congenic lines for apoE3 and apoE4, and crossed each line with mice overexpressing human Swedish mutant APP (Tg2576).
As expected, the apoE3/APP and apoE4/APP mice had higher Aβ concentrations in the brain. Total Aβ levels were 61.7 ± 8.0, 62.8 ± 3.0, 62.0 ± 15.1, and 62.4 ± 9.0 pmol/g of brain tissue for apoE3/APP and aopE4/APP mice at 12 months of age and apoE3/APP and aopE4/APP mice at 15 months of age, respectively. Thus, the levels of total brain Aβ did not differ significantly between apoE3/APP and aopE4/APP mice at both ages. In contrast, the Aβ40 concentration in LDM domains was slightly, but significantly higher in apoE3/APP mice than in apoE4/APP mice at 12 and 15 months of ages (p = 0.0015 at 12 months, and p = 0.0041 at 15 months; Student’s t-test)(Fig. 2a), which confirmed the trend observed in apoE KI mice as described above. The Aβ42 concentration in LDM domains did not differ significantly between apoE3/APP and apoE4/APP mice at both ages. The Aβ42 proportion (Aβ42/sum of Aβ40 and Aβ42) in LDM domains also did not differ significantly between apoE3/APP and apoE4/APP mice at both ages, and was similar to the proportion in apoE3 and apoE4 mice (Figs 1d and 2b).
Fig. 2.
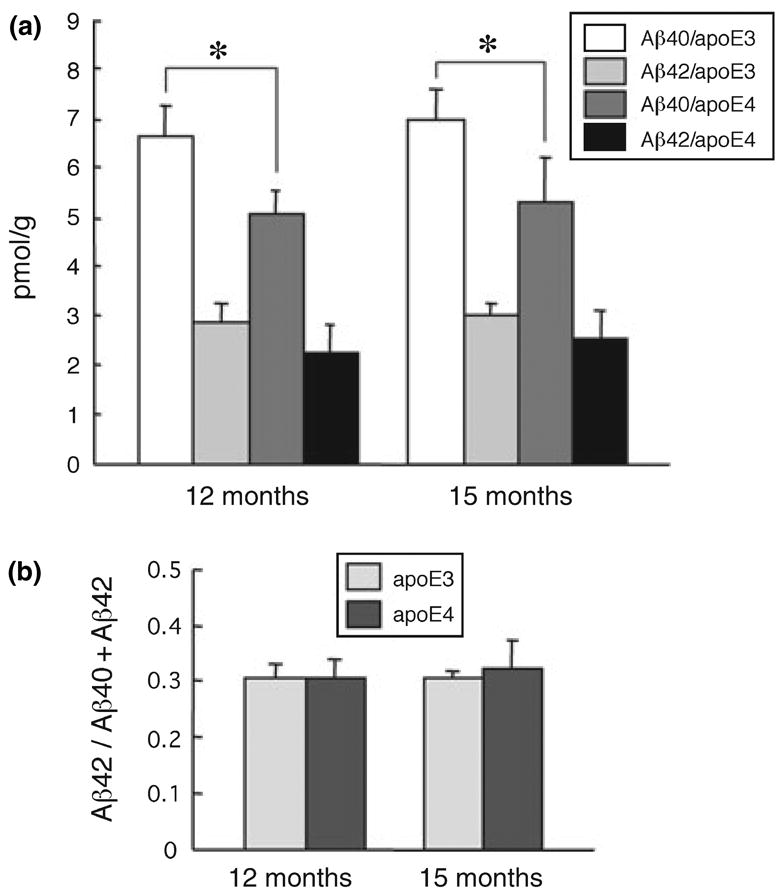
Aβ40 and Aβ42 in the LDM fractions from apoE3/APP and apoE4/APP mice (a). The low-density membrane (LDM) fractions were obtained from the mice at 12 and 15 months of age and the concentrations of Aβ40 and Aβ42 were measured by ELISA. Data are presented as means ± SD (n = 5 for 12-month-old mice, n = 6 for 15-month-old mice). *p < 0.01 (Student’s t-test). Open bars represent Aβ40 concentrations in apoE3/APP mice; light gray bars, Aβ42 concentrations in apoE3/APP mice; dark gray bars, Aβ40 concentrations in apoE4/APP mice; black bars, Aβ42 concentrations in apoE4/APP mice. Panel (b) shows the proportion of Aβ42 to the sum of Aβ40 and Aβ42 in the LDM fraction. Light gray bars represent apoE3/APP mice; dark gray bars represent apoE4/APP mice.
Immunocytochemistry showed scarcely Aβ deposits in low-power fields of the brain at these ages, which contrasts with the brain of Tg2576 mice at the same age that exhibited many Aβ deposits (Fig. 3). This is consistent with the observation that the expression of human apoE significantly delays Aβ deposition in the APP transgenic mouse brain (Holtzman et al. 1999; Fagan et al. 2002). Thus, taken together with the total Aβ levels, these mice are at the very early stage of Aβ deposition even at 12 and 15 months of age.
Fig. 3.
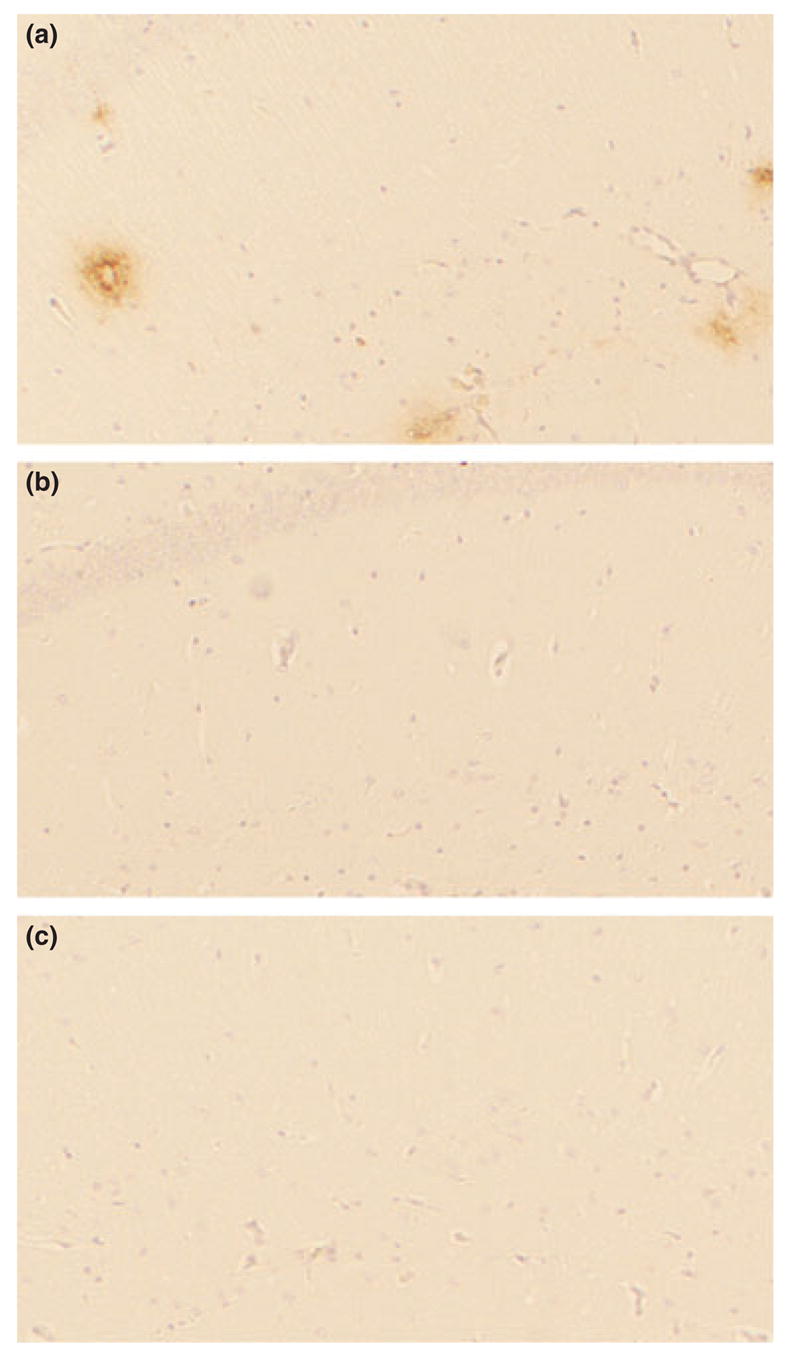
Immunostaining with 4G8 of the sections from Tg2576 and apoE/APP mice brains. The coronal tissue sections including the hippocampus CA1 region from Tg2576 (a), apoE3/APP (b), and apoE4/APP mice (c) aged 15–16 months were immunostained with 4G8. Whereas many Aβ deposits were observed in the CA1 of Tg2576 mice brains, they were scarcely observed in the corresponding regions of apoE3/APP and apoE4/APP mice brains.
Low-density membrane domains prepared from the brain of Tg2576 mice contain a large amount of sodium dodecyl sulfate (SDS)-stable Aβ dimers, which may be involved in the accelerated Aβ deposition (Kawarabayashi et al. 2004). SDS-stable Aβ dimers are observed often in the cortex of aged human brains by western blotting (Enya et al. 1999; Morishima-Kawashima et al. 2000) and may have toxic effects on neurons (Walsh et al. 2002). Because the ELISA quantifies only SDS-dissociable Aβ monomers, and hardly SDS-stable Aβ dimers (Enya et al. 1999), the latter were assessed by western blotting. SDS-stable Aβ dimers were undetectable in any LDM fraction, except for a fraction from one apoE3/APP mouse at 15 months of age, in which 4G8 revealed a large accumulation of Aβ dimers (Fig. 4). The amount of Aβ monomers detected by western blotting corresponded to the sum of Aβ40 and Aβ42 quantified by ELISA, confirming the accuracy of both methods to quantify the Aβ in LDM domains.
Fig. 4.
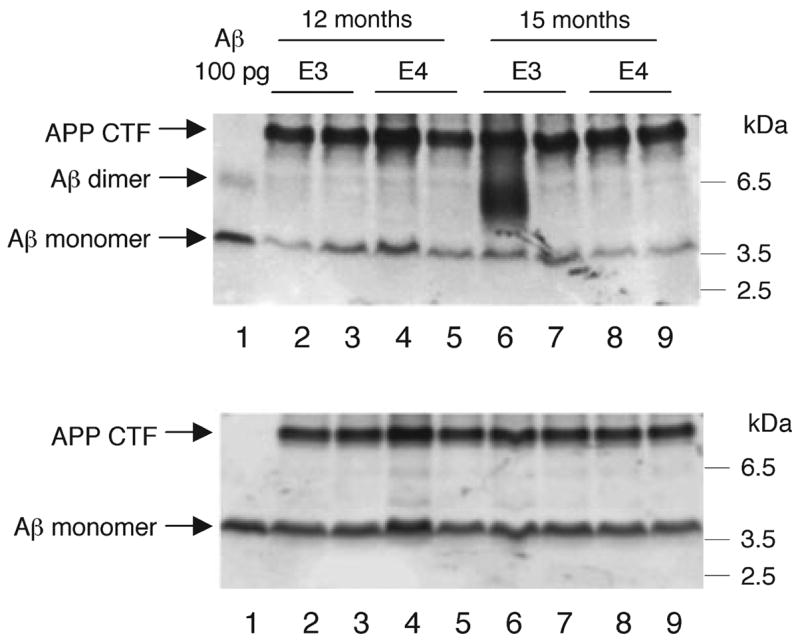
Western blotting for Aβ of the low-density membrane (LDM) fraction from apoE/APP mice. The LDM fractions were prepared from apoE3/APP and apoE4/APP mice brains at 12 and 15 months of age, and equal aliquots from each mouse brain (corresponding to 3 mg of brain tissue) were subjected to western blotting with 4G8 (upper panel) and 6E10 (lower panel). Representative examples (two out of five or six mice for each line at each age) are shown. Synthetic Aβ40 (100 pg) was loaded in lane 1 on each panel. The Aβ monomer was observed at 4 kDa in all mice with both antibodies. By using 4G8, only one apoE3/APP mouse aged 15 months gave a broad band at ~6 kDa, corresponding to SDS-stable Aβ dimers (lane 6 on the upper panel). 6E10 was unable to detect these Aβ dimers, and thus they are presumably truncated at the N-terminus to a substantial extent.
Altered lipid composition of LDM domains from apoE4/APP mice
We next examined the lipid composition of LDM domains from apoE/APP mice brains using ESI–MS (Table 1). The cholesterol concentration in LDM domains did not differ significantly between apoE3/APP and apoE4/APP mice, suggesting that cholesterol metabolism in the CNS differs from the peripheral metabolism (Han 2004). In contrast, the concentrations of some lipids in LDM domains were altered in apoE4/APP mice. Phosphatidylinositol concentration was significantly lower in apoE4/APP mice than in apoE3/APP mice at 12 months of age. Phosphatidylglycerol was lower and sphingomyelin was higher in apoE4/APP mice at 15 months of age. Phosphatidylcholine was also higher in apoE4/APP mice at both 12 and 15 months of age. Although some alterations were significant at only one age, such trends were observed at other ages. Sulfatides, which were reported to differ between apoE3 and apoE4 mice (Han et al. 2003), did not differ significantly in LDM domains between apoE3/APP and apoE4/APP mice.
Table 1.
Lipid analysis of the LDM from apoE3/APP and apoE4/APP mice brainsa
| ApoE type | Age | Cerebrosides | Sulfatides | Ceramides | Phosphatidic acids | Phosphatidylglycerols | Phosphatidylserines | Phosphatidylinositols |
|---|---|---|---|---|---|---|---|---|
| ApoE3-LDM | 12 months | 203.62 ± 12.69 | 108.17 ± 5.54 | 9.06 ± 0.58 | 1.10 ± 0.06 | 10.18 ± 0.53 | 228.03 ± 7.22 | 26.46 ± 2.45 |
| 15 months | 205.57 ± 17.99 | 113.03 ± 5.57 | 8.37 ± 0.42 | 1.06 ± 0.07 | 10.49 ± 0.73 | 225.12 ± 13.44 | 27.85 ± 5.46 | |
| ApoE4-LDM | 12 months | 210.33 ± 23.61 | 110.05 ± 4.63 | 8.62 ± 0.85 | 1.10 ± 0.07 | 8.82 ± 1.69 | 217.44 ± 13.43 | 19.19 ± 0.07* |
| 15 months | 206.17 ± 27.67 | 110.11 ± 6.55 | 8.44 ± 0.31 | 1.09 ± 0.04 | 8.51 ± 0.53* | 216.06 ± 7.83 | 20.82 ± 1.15 | |
| ApoE type | Age | Phosphatidylcholine | Phosphatidylethanolamine | Sphingomyelins | Free cholesterol | Total cholesterol | Free fatty acid | |
|
| ||||||||
| ApoE3-LDM | 12 months | 782.86 ± 25.75 | 1060.44 ± 32.33 | 45.75 ± 1.60 | 826.94 ± 34.14 | 827.15 ± 61.89 | 290.71 ± 14.72 | |
| 15 months | 757.92 ± 24.67 | 1037.16 ± 21.38 | 38.81 ± 3.08 | 832.19 ± 135.14 | 835.84 ± 50.10 | 330.03 ± 14.79 | ||
| ApoE4-LDM | 12 months | 909.32 ± 53.93* | 1130.96 ± 58.01 | 52.11 ± 4.66 | 951.13 ± 108.97 | 913.69 ± 76.66 | 317.37 ± 13.39 | |
| 15 months | 896.58 ± 44.10* | 1117.12 ± 45.57 | 51.18 ± 1.16* | 879.18 ± 86.74 | 834.56 ± 108.48 | 302.58 ± 8.88 | ||
Data are expressed in nmol/mg of protein and represent means ± SD (n = 5 at 12 months of age; n = 6 at 15 months of age).
p < 0.01 compared with the apoE3/APP mice at the same age (Student’s t-test).
Discussion
Our previous observations have indicated that apoE4-carrying brains are associated with an earlier onset, but not an increased extent of Aβ deposition (Morishima-Kawashima et al. 2000). An increased number of Aβ40-positive plaques apparently specific for apoE4 carriers (Gearing et al. 1996) can be explained by longer presence of senile plaques: it is likely that Aβ40 deposition continues to increase, whereas Aβ42 deposition levels off (Morishima-Kawashima et al. 2000). In contrast, the data are conflicting as to whether there are apoE isoform-specific effects on Aβ deposition in mouse models (Holtzman et al. 2000; Carter et al. 2001; Lesuisse et al. 2001; Mann et al. 2004; Yao et al. 2004; Fryer et al. 2005). In most reports, the mice examined had abnormally high levels of apoE expression or its ectopic expression, or different genetic backgrounds. Exogenous and endogenous apoE could interfere or interact with each other in an unpredictable manner, which may obscure the human apoE-specific effects. We used mice that express human apoE3 or apoE4 alone under the control of the endogenous mouse regulatory elements, which provides a clearer model to observe the effects of human apoE3 and apoE4.
In apoE-null mice, Aβ deposition is delayed and its tissue distribution is altered markedly (Bales et al. 1997; Irizarry et al. 2000). The Aβ concentration in LDM domains is significantly lower in these mice than in wild-type mice expressing mouse apoE (Fagan et al. 2002), indicating that apoE has a significant role in Aβ localization to LDM domains. Our study shows that the Aβ40 concentration in apoE3/APP mice is slightly, but significantly higher than in apoE4/APP mice. Neither the concentration nor the proportion of Aβ42 in LDM domains differed between apoE3/APP and apoE4/APP mice at any age (Figs 1 and 2). Because these data represent monomeric Aβ, it is possible that the apoE genotype affects the concentration of oligomeric Aβ, which might be more relevant to Aβ deposition but unquantifiable by ELISA. However, western blotting detected no dimeric or oligomeric Aβ, except for one apoE3/APP mouse (Fig. 4). This agrees with an immunoelectron microscopic observation on the Tg2576 mouse brain (Kokubo et al. 2005a), which showed that oligomeric Aβ is localized to cellular processes, especially axon terminals, but not to lipid rafts (LDM domains). These results indicate that Aβ localization to LDM domains might be mediated in part through apoE, irrespective of its isoform, and that, in this aspect, there are no isoform-specific effects in apoE KI and apoE/APP mice.
The Aβ42 concentration in LDM domains increased in an age-dependent manner and thus the Aβ42 proportion was higher in the older mice, independent of apoE genotype (Fig. 1). These data imply that Aβ42 becomes more concentrated in LDM domains during aging. Lipid rafts have high specific activity of γ-secretase (Wahrle et al. 2002; Wada et al. unpublished observation), and thus the increasing steady-state concentration of Aβ42 in LDM domains may even be aided by subtly lower rate of secretion or degradation, which may also be associated with aging. The concentrated Aβ42 would then acquire a high aggregation potential and favor the accumulation in LDM domains, as cholesterol and sphingolipids enriched in LDM domains bind Aβ and alter its conformation (Avdulov et al. 1997; Choo-Smith and Surewicz 1997; Kakio et al. 2001). Membrane-bound Aβ has been observed in LDM domains within senile plaques at the early stage of Aβ deposition in Tg2576 mice (Kokubo et al. 2005b). This supports the assumption that Aβ42 in LDM domains can trigger the initial phase of Aβ deposition in the brain. ApoE isoforms could exert their effects on the Aβ fibrillogenesis, both in vivo and in vitro (Wisniewski et al. 1994; Holtzman et al. 2000), which might become overt in the later phase when Aβ forms fibrils in the parenchyma. It is possible that, because we studied the mice either prior to Aβ deposition or at the very early stage of Aβ deposition, no isoform-specific effects were observed. However, this is unlikely because our previous observations using ELISA showed that apoE4 carriers have a significantly earlier initial Aβ accumulation phase, which can be detected only by ELISA and represents probably non-fibrillar Aβ aggregates and the membrane-bound form (Funato et al. 1998; Morishima-Kawashima et al. 2000; Yamaguchi et al. 2000).
It is also possible that the isoform-specific effects on the Aβ accumulation in the brain cannot be recapitulated in our mouse model. Human apoE might interact with its downstream mouse molecules in a different way in our model than in humans. ApoE3 and apoE4 could interact differently with human apoE receptors than with mouse apoE receptors. These subtle differences in Aβ clearance would increase the Aβ42 concentration, leading to earlier Aβ accumulation in apoE4 carriers; but these effects might not be generated in the apoE KI model mice.
The apoE isoforms did not affect the cholesterol concentration in LDM domains, which is consistent with previous reports (Han et al. 2003; Mann et al. 2004; Fryer et al. 2005; Igbavboa et al. 2005). In addition consistent is the observation that the cholesterol concentration in the synaptosomal LDM domains does not differ significantly between apoE3 and apoE4 KI mice (Igbavboa et al. 2005). However, the local cholesterol concentration in the membrane could be altered in an apoE isoform-specific manner. The same research group showed that the amount of cholesterol is higher in the exofacial leaflet and lower in the cytofacial leaflet of synaptic plasma membranes from apoE4 mice than from apoE3 mice (Hayashi et al. 2002).
Apolipoprotein E isoform-specific alterations occurred in some other lipids (see Table 1). These include lower concentrations of phosphatidylglycerol and phosphatidylinositol, and higher concentrations of phosphatidylcholine and sphingomyelin in apoE4 mice than in apoE3 mice of the same age. These observed changes in apoE4 LDM domains are similar to those in the exofacial leaflet of the synaptic plasma membrane (Wood et al. 2002). Thus, apoE isoforms modulate the lipid composition in both the plasma and brain LDM domains.
We used ESI/MS to compare the lipid composition of LDM domains from the prefrontal cortices of non-demented human subjects with high Aβ concentrations (n = 3; 1300, 1700, 2200 pmol of Aβ42/g wet tissue) and those with low Aβ concentrations (n = 3; <5 pmol of total Aβ/g wet tissue). There were non-significant trends toward decreasing concentrations of phosphatidylglycerol, phosphatidylinositol, phosphatidylcholine, phosphatidylethanolamine, and sphingomyelin in the Aβ-accumulating cortices compared with the Aβ-non-accumulating cortices (p > 0.05, Student’s t-test), but no difference in cholesterol concentration (see Table S1). At present, a correlation between the concentrations of certain lipids and Aβ accumulation in the brain has not been confirmed. Based on these findings, we believe it unlikely that small differences in the lipid concentrations brought about by apoE4 isoform is a major factor in accelerated Aβ deposition in the brain of human apoE4 carriers.
In summary, apoE isoforms appear to slightly modulate the Aβ concentration and lipid composition in LDM domains, but these alterations may not be relevant to an accelerated Aβ deposition observed in the brain of human apoE4 carriers. It is likely that an accelerated Aβ deposition in the presence of apoE4 isoform is mediated through mechanisms other than LDM domains. Alternatively, the isoform-specific effects may require a particular human molecule that interacts with apoE in an isoform-specific manner.
Supplementary Material
Supplementary material
The following material is available for this paper online.
Table S1 Lipid analysis of the LDM from human brains.
This material is available as part of the online article from http://www.blackwell-synergy.com
Acknowledgments
We thank Dr Karen Hsiao Ashe and the Mayo Foundation for Medical Education and Research for the Tg2576 APP transgenic mouse line. We also thank Dr Satoko Wada-Kakuda for her helpful work. This work was supported in part by a Grant-in-aid for Scientific Research on Priority Areas – Advanced Brain Science Project – from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (to YI), and an NIA grant (AG23168 to XH).
Abbreviations used
- Aβ
amyloid β-protein
- AD
Alzheimer’s disease
- apoE
apolipoprotein E
- APP
β-amyloid precursor protein
- ELISA
enzyme-linked immunosorbent assay
- ESI–MS
electrospray ionization–mass spectrometry
- KI
knock-in
- LDM
low-density membrane
- MES
2-morpholinoethanesulfonic acid
- SDS
sodium dodecyl sulfate
- TS
Tris-saline
References
- Avdulov NA, Chochina SV, Igbavboa U, Warden CS, Vassiliev AV, Wood WG. Lipid binding to amyloid β-peptide aggregates: preferential binding of cholesterol as compared with phosphatidylcholine and fatty acids. J Neurochem. 1997;69:1746–1752. doi: 10.1046/j.1471-4159.1997.69041746.x. [DOI] [PubMed] [Google Scholar]
- Bales KR, Verina T, Dodel RC, et al. Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition. Nat Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- Carter DB, Dunn E, McKinley DD, Stratman NC, Boyle TP, Kuiper SL, Oostveen JA, Weaver RJ, Boller JA, Gurney ME. Human apolipoprotein E4 accelerates β-amyloid deposition in APPsw transgenic mouse brain. Ann Neurol. 2001;50:468–475. doi: 10.1002/ana.1134. [DOI] [PubMed] [Google Scholar]
- Choo-Smith LP, Surewicz WK. The interaction between Alzheimer amyloid β (1–40) peptide and ganglioside GM1-containing membranes. FEBS Lett. 1997;402:95–98. doi: 10.1016/s0014-5793(96)01504-9. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Enya M, Morishima-Kawashima M, Yoshimura M, et al. Appearance of sodium dodecyl sulfate-stable amyloid β-protein (Aβ) dimer in the cortex during aging. Am J Pathol. 1999;154:271–279. doi: 10.1016/s0002-9440(10)65273-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine ApoE markedly alters Aβ metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2002;9:305–318. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- Fassbender K, Simons M, Bergmann C, et al. Simvastatin strongly reduces concentrations of Alzheimer’s disease β-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98:5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frears ER, Stephens DJ, Walters CE, Davies H, Austen BM. The role of cholesterol in the biosynthesis of β-amyloid. Neuroreport. 1999;10:1699–1705. doi: 10.1097/00001756-199906030-00014. [DOI] [PubMed] [Google Scholar]
- Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM. Human apolipoprotein E4 alters the amyloid-β 40:42 proportion and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci. 2005;25:2803–2810. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato H, Yoshimura M, Kusui K, Tamaoka A, Ishikawa K, Ohkoshi N, Namekata K, Okeda R, Ihara Y. Quantitation of amyloid β-protein (Aβ) in the cortex during aging and in Alzheimer’s disease. Am J Pathol. 1998;152:1633–1640. [PMC free article] [PubMed] [Google Scholar]
- Gearing M, Mori H, Mirra SS. Aβ-peptide length and apolipoprotein E genotype in Alzheimer’s disease. Ann Neurol. 1996;39:395–399. doi: 10.1002/ana.410390320. [DOI] [PubMed] [Google Scholar]
- Hamanaka H, Katoh-Fukui Y, Suzuki K, et al. Altered cholesterol metabolism in human apolipoprotein E4 knock-in mice. Hum Mol Genet. 2000;9:353–361. doi: 10.1093/hmg/9.3.353. [DOI] [PubMed] [Google Scholar]
- Han X. The role of apolipoprotein E in lipid metabolism in the central nervous system. Cell Mol Life Sci. 2004;61:1896–1906. doi: 10.1007/s00018-004-4009-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Gross RW. Shotgun lipidomics: Electrospray ionization mass spectroscopic analysis and quantitation of the cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom Rev. 2005a;24:367–412. doi: 10.1002/mas.20023. [DOI] [PubMed] [Google Scholar]
- Han X, Gross RW. Shotgun lipidomics: multi-dimensional mass spectrometric analysis of cellular lipidomes. Expert Rev Proteomics. 2005b;2:253–264. doi: 10.1586/14789450.2.2.253. [DOI] [PubMed] [Google Scholar]
- Han X, Cheng H, Fryer JD, Fagan AM, Holtzman DM. Novel role for apolipoprotein E in the central nervous system. Modulation of sulfatide content. J Biol Chem. 2003;278:8043–8051. doi: 10.1074/jbc.M212340200. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Igbavboa U, Hamanaka H, Kobayashi M, Fujita SC, Wood WG, Yanagisawa K. Cholesterol is increased in the exofacial leaflet of synaptic plasma membranes of human apolipoprotein E4 knock-in mice. Neuroreport. 2002;13:383–386. doi: 10.1097/00001756-200203250-00004. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Fagan AM. Potential role of apoE in structural plasticity in the nervous system; implications for disorders of the central nervous system. Trends Cardiovasc Med. 1998;8:250–255. doi: 10.1016/s1050-1738(98)00017-6. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, Chang LK, Sun Y, Paul SM. Expression of human apolipoprotein E reduces amyloid-β deposition in a mouse model of Alzheimer’s disease. J Clin Invest. 1999;103:R15–R21. doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, et al. Apolipoprotein E isoform-specific amyloid deposition and neuritic degeneproportionn in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Igbavboa U, Eckert GP, Malo TM, et al. Murine synaptosomal lipid raft protein and lipid composition are altered by expression of human apoE 3 and 4 and by increasing age. J Neurol Sci. 2005;229–230:225–232. doi: 10.1016/j.jns.2004.11.037. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Rebeck GW, Cheung B, Bales K, Paul SM, Holzman D, Hyman BT. Modulation of Aβ deposition in APP transgenic mice by an apolipoprotein E null background. Ann N Y Acad Sci. 2000;920:171–178. doi: 10.1111/j.1749-6632.2000.tb06919.x. [DOI] [PubMed] [Google Scholar]
- Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000;356:1627–1631. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- Kakio A, Nishimoto SI, Yanagisawa K, Kozutsumi Y, Matsuzaki K. Cholesterol-dependent formation of GM1 ganglioside-bound amyloid β-protein, an endogenous seed for Alzheimer amyloid. J Biol Chem. 2001;276:24985–24990. doi: 10.1074/jbc.M100252200. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG. Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2004;24:3801–3809. doi: 10.1523/JNEUROSCI.5543-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubo H, Kayed R, Glabe CG, Saido TC, Iwata N, Helms JB, Yamaguchi H. Oligomeric proteins ultrastructurally localize to cell processes, especially to axon terminals with higher density, but not to lipid rafts in Tg2576 mouse brain. Brain Res. 2005a;1045:224–228. doi: 10.1016/j.brainres.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Kokubo H, Saido TC, Iwata N, Helms JB, Shinohara R, Yamaguchi H. Part of membrane-bound Aβ exists in rafts within senile plaques in Tg2576 mouse brain. Neurobiol Aging. 2005b;26:409–418. doi: 10.1016/j.neurobiolaging.2004.04.008. [DOI] [PubMed] [Google Scholar]
- LaDu MJ, Reardon C, Van Eldik L, Fagan AM, Bu G, Holtzman D, Getz GS. Lipoproteins in the central nervous system. Ann N Y Acad Sci. 2000;903:167–175. doi: 10.1111/j.1749-6632.2000.tb06365.x. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT, Jr, Kosik KS. A detergent-insoluble membrane compartment contains Aβ in vivo. Nat Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- Lehman EJ, Kulnane LS, Gao Y, Petriello MC, Pimpis KM, Younkin L, Dolios G, Wang R, Younkin SG, Lamb BT. Genetic background regulates β-amyloid precursor protein processing and β-amyloid deposition in the mouse. Hum Mol Genet. 2003;12:2949–2956. doi: 10.1093/hmg/ddg322. [DOI] [PubMed] [Google Scholar]
- Lesuisse C, Xu G, Anderson J, et al. Hyper-expression of human apolipoprotein E4 in astroglia and neurons does not enhance amyloid deposition in transgenic mice. Hum Mol Genet. 2001;10:2525–2537. doi: 10.1093/hmg/10.22.2525. [DOI] [PubMed] [Google Scholar]
- Li G, Higdon R, Kukull WA, et al. Statin therapy and risk of dementia in the elderly: a community-based prospective cohort study. Neurology. 2004;63:1624–1628. doi: 10.1212/01.wnl.0000142963.90204.58. [DOI] [PubMed] [Google Scholar]
- Mann KM, Thorngate FE, Katoh-Fukui Y, Hamanaka H, Williams DL, Fujita S, Lamb BT. Independent effects of APOE on cholesterol metabolism and brain Aβ concentrations in an Alzheimer disease mouse model. Hum Mol Genet. 2004;13:1959–1968. doi: 10.1093/hmg/ddh199. [DOI] [PubMed] [Google Scholar]
- Mori T, Kobayashi M, Town T, Fujita SC, Asano T. Increased vulnerability to focal ischemic brain injury in human apolipoprotein E4 knock-in mice. J Neuropathol Exp Neurol. 2003;62:280–291. doi: 10.1093/jnen/62.3.280. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Ihara Y. The presence of a-myloid β-protein in the detergent-insoluble membrane compartment of human neuroblastoma cells. Biochemistry. 1998;37:15247–15253. doi: 10.1021/bi981843u. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Oshima N, Ogata H, Yamaguchi H, Yoshimura M, Sugihara S, lhara Y. Effect of apolipoprotein E allele ε4 on the initial phase of amyloid β-protein accumulation in the human brain. Am J Pathol. 2000;157:2093–2099. doi: 10.1016/s0002-9440(10)64847-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima N, Morishima-Kawashima M, Yamaguchi H, Yoshimura M, Sugihara S, Khan K, Games D, Schenk D, Ihara Y. Accumulation of amyloid β-protein in the low-density membrane domain accurately reflects the extent of β-amyloid deposition in the brain. Am J Pathol. 2001;158:2209–2218. doi: 10.1016/s0002-9440(10)64693-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Morishima-Kawashima M, Sato T, Mitsumori R, Ihara Y. Distinct mechanisms by mutant presenilin 1 and 2 leading to increased intracellular levels of amyloid β-protein 42 in Chinese hamster ovary cells. Biochemistry. 2003;42:1042–1052. doi: 10.1021/bi0267590. [DOI] [PubMed] [Google Scholar]
- Ravona-Springer R, Davidson M, Noy S. The role of cardiovascular risk factors in Alzheimer’s disease. CNS Spectr. 2003;8:824–833. doi: 10.1017/s109285290001926x. [DOI] [PubMed] [Google Scholar]
- Rea TD, Breitner JC, Psaty BM, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Arch Neurol. 2005;62:1047–1051. doi: 10.1001/archneur.62.7.1047. [DOI] [PubMed] [Google Scholar]
- Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000;7:321–331. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- Sawamura N, Morishima-Kawashima M, Waki H, et al. Mutant presenilin 2 transgenic mice. A large increase in the concentrations of Aβ42 is presumably associated with the low density membrane domain that contains decreased concentrations of blycero-phospholipids and sphingomyelin. J Biol Chem. 2000;275:27901–27908. doi: 10.1074/jbc.M004308200. [DOI] [PubMed] [Google Scholar]
- Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD. Increased amyloid β-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer’s disease. Proc Natl Acad Sci USA. 1993;90:9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons. Proc Natl Acad Sci USA. 1998;95:6460–6464. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoog I, Nilsson L, Palmertz B, Andreasson LA, Svanborg A. A population-based study of dementia in 85-year-olds. N Engl J Med. 1993;328:153–158. doi: 10.1056/NEJM199301213280301. [DOI] [PubMed] [Google Scholar]
- Sparks DL, Scheff SW, Hunsaker JC, III, Liu H, Landers T, Gross DR. Induction of Alzheimer-like β-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp Neurol. 1994;126:88–94. doi: 10.1006/exnr.1994.1044. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T, Younkin LH, Younkin SG, Golde TE. Cholesterol-dependent γ-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol Dis. 2002;9:11–23. doi: 10.1006/nbdi.2001.0470. [DOI] [PubMed] [Google Scholar]
- Walker LC, Pahnke J, Madauss M, Vogelgesang S, Pahnke A, Herbst EW, Stausske D, Walther R, Kessler C, Warzok RW. Apolipoprotein E4 promotes the early deposition of Aβ42 and then Aβ40 in the elderly. Acta Neuropathol (Berl) 2000;100:36–42. doi: 10.1007/s004010051190. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Weisgraber KH. Apolipoprotein E: structure-function relationships. Adv Protein Chem. 1994;45:249–302. doi: 10.1016/s0065-3233(08)60642-7. [DOI] [PubMed] [Google Scholar]
- Weisgraber KH, Mahley RW. Human apolipoprotein E: the Alzheimer’s disease connection. FASEB J. 1996;10:1485–1494. doi: 10.1096/fasebj.10.13.8940294. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Castano EM, Golabek A, Vogel T, Frangione B. Acceleration of Alzheimer’s fibril formation by apolipoprotein E in vitro. Am J Pathol. 1994;145:1030–1035. [PMC free article] [PubMed] [Google Scholar]
- Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- Wong GT. Speed congenics: applications for transgenic and knock-out mouse strains. Neuropeptides. 2002;36:230–236. doi: 10.1054/npep.2002.0905. [DOI] [PubMed] [Google Scholar]
- Wood WG, Schroeder F, Igbavboa U, Avdulov NA, Chochina SV. Brain membrane cholesterol domains, aging and amyloid β-peptides. Neurobiol Aging. 2002;23:685–694. doi: 10.1016/s0197-4580(02)00018-0. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Maat-Schieman ML, van Duinen SG, Prins FA, Neeskens P, Natte R, Roos RA. Amyloid β protein (Aβ) starts to deposit as plasma membrane-bound form in diffuse plaques of brains from hereditary cerebral hemorrhage with amyloidosis-Dutch type, Alzheimer disease and nondemented aged subjects. J Neuropathol Exp Neurol. 2000;59:723–732. doi: 10.1093/jnen/59.8.723. [DOI] [PubMed] [Google Scholar]
- Yao J, Petanceska SS, Montine TJ, et al. Aging, gender and APOE isotype modulate metabolism of Alzheimer’s Aβ peptides and F-isoprostanes in the absence of detectable amyloid deposits. J Neurochem. 2004;90:1011–1018. doi: 10.1111/j.1471-4159.2004.02532.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
The following material is available for this paper online.
Table S1 Lipid analysis of the LDM from human brains.
This material is available as part of the online article from http://www.blackwell-synergy.com