

Abstract
We report a novel approach to derivatize the primary, secondary, and tertiary hydroxy group(s) of oxysterols with N,N-dimethylglycine (DMG) in the presence of both 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and 4-(N,N-dimethylamino)pyridine to yield their corresponding mono- or di-DMG esters. Eight oxysterols including 7-oxocholesterol, 5α,6α- and 5β,6β-epoxycholesterols, as well as 7α-, 7β-, 24(S)-, 25-, and 27-hydroxycholesterols, were studied. Electrospray ionization tandem mass spectrometric characterization of these singly or doubly protonated derivatives demonstrates the presence of an informative fragmentation pattern for each oxysterol derivative. Potential dissociation pathways for the production of these unique fragmentation patterns are proposed and discussed. Collectively, these informative and unique fragmentation patterns allow rapid and direct discrimination of the identities of 7α-, 7β-, 24(S)-, 25-, and 27-hydroxycholesterol isomers, as well as 5α,6α- and 5β,6β-epoxycholesterol isomers, thereby potentially providing a foundation for quantitative analysis of oxysterols in biological samples in combination with a chromatographic separation.
Oxysterols are 27-carbon oxidation derivatives of cholesterol, which play multiple important roles in biological processes including mediation of inflammatory events in the development of atherosclerotic lesions,1 regulation of cholesterol homeostasis,2 maintenance of neuronal functions,3 and activation of liver X receptors.4 Oxysterols can be produced either by enzymatic or by non-enzymatic oxidation (known as autoxidation) and may also be obtained from dietary sources.5–7
The most common oxysterols are those derived by formation of hydroxyl, oxo, and epoxy moieties. 7α-, 24(S)-, and 27-Hydroxycholesterols are the major products generated by cholesterol hydroxylases.8 7α- and 7β-Hydroxycholesterols are also produced through autoxidation of cholesterol. Whether 7β-hydroxycholesterol (which accounts for less than 10% of 7-hydroxycholesterol) is also the product of an enzymatic action is not well defined.6, 8 7α-Hydroxycholesterol is a central intermediate in the classical pathway of bile acid biosynthesis, while 27-hydroxycholesterol is an intermediate in the alternative pathway for the biosynthesis of bile acid.9, 10 24(S)-Hydroxycholesterol is almost exclusively derived from the central nervous system. Increased mass levels of 24(S)-hydroxycholesterol in plasma is associated with Alzheimer’s disease11, 12 and other neuronal diseases.13
25-Hydroxycholesterol, which inhibits cholesterol biosynthesis14 and low-density lipoprotein receptor expression,6 is present in low abundance in plasma, and arises either through cholesterol autoxidation or through the reaction catalyzed by cholesterol-25-hydroxylase.8 7-Oxocholesterol (or 7-ketocholesterol) is produced not only from the autoxidation of cholesterol, but also from enzymatic dehydrogenation of 7α-hydroxycholesterol.6 7-Oxocholesterol shows cytotoxic activities in endothelial cells, smooth muscle cells, and macrophages.6 The isomeric 5α,6α- and 5β,6β-epoxycholesterols are formed together from cholesterol by many defined oxidants, including air oxidation, and by the actions of soybean lipoxygenases and of liver microsomal lipid peroxidation systems in vitro.15 Both isomers are found in human tissues and foods16, 17 and exhibit cytotoxic effects in in vitro bioassay.17
A variety of analytical methods for the identification and quantitation of oxysterols has been reported, including techniques based on thin-layer chromatography (TLC),18 liquid chromatography (LC),16, 19 and gas chromatography (GC).20–22 Separation of oxysterols from cholesterol by TLC and LC is very laborious and potentially interesting oxysterols could remain undetected by these methods. Isotope dilution GC/mass spectrometry (MS) is currently the most common technique for the analysis of trimethylsilyl derivatives of oxysterols in biological samples.22 However, this approach suffers from being unable to detect thermally unstable oxysterols. As oxysterols are found to participate in an increasing number of cellular events, it is apparent that improved analytical methods are needed for their analysis to understand their roles in the living cells.
Newly developed methods based on atmospheric pressure ionization techniques (e.g., electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI)) alleviate the thermal degradation problems encountered with GC/MS. LC/APCI-MS has been used to determine cholesterol oxidation products without derivatization.23–27 However, unequivocal identification by APCI-MS/MS was impossible because the energy in collision-induced dissociation (CID) used in those studies was not sufficient to break the stable carbon–carbon bonds in sterols.27 The most abundant ions are those resulting from non-specific fragmentation (e.g., loss of water). Although ESI-MS is in general more sensitive than APCI-MS for compounds that carry polar groups and has recently been used for analyses of anabolic steroids,28 oxysterols are not readily detectable by ESI-MS due to their relatively low ionization efficiencies.
Therefore, to resolve this problem, one option is to derivatize these oxysterols with a more polar group than hydroxyl or a charged moiety, thereby converting the hydrophobic oxysterols into compounds that can be efficiently ionized by ESI. Following this line of reasoning, cholesterol has previously been converted into either cholesterol-3-sulfate29 or cholesteryl methoxyacetate30 for analysis by ESI-MS. Oxysterols have been either converted into their corresponding oximes31, 32 or derivatized as Girard P hydra-zones following a two-step procedure.33–35 The derivatives are then analyzed by ESI-MS and/or matrix-assisted laser desorption/ionization (MALDI)-MS at a high sensitivity.
Herein, we report an alternative oxysterol derivatization method in which oxysterols were converted into their N,N-dimethylglycine (DMG) esters in high yield under mild reaction conditions. Moreover, the DMG-oxysterol derivatives are readily ionized by ESI-MS under acidic conditions at a high sensitivity. In the study, we characterized eight common oxysterols (Scheme 1) including 7-oxocholesterol (1a), and 5α,6α- (2a) and 5β,6β-epoxycholesterols (3a), as well as 7α- (4a), 7β- (5a), 24(S)- (6a), 25- (7a), and 27-hydroxycholesterols (8a) as their protonated DMG esters (1b–8b) by tandem mass spectrometric (MS/MS) analysis. Potential fragmentation pathways of these protonated molecules are proposed. It is demonstrated that the [M+H]+ of 7-oxocholesterol (1b), and 5α,6α-(2b) and 5β,6β-epoxycholesterol (3b) derivatives and [M+2H]2+ of 7α- (4b), 7β- (5b), 24(S)- (6b), 25- (7b), and 27-hydroxycholesterol (8b) derivatives yield abundant and informative fragmentation patterns during CID. Collectively, the resultant fragmentation patterns allow one to determine their structures and distinguish the isomers of oxysterols. These fragmentation patterns could also potentially provide a foundation for further quantitative analysis of these oxysterols present in biological samples by using ESI- MS/MS, alone or in combination with chromatographic separation.
Scheme 1.
Chemical structures of oxysterols 1a–8a and their DMG esters 1b–8b.
EXPERIMENTAL
Materials
Oxysterols including 7-oxocholesterol (1a), 5α,6α-epoxycholesterol (2a), 5β,6β-epoxycholesterol (3a), and 7β-hydroxycholesterol (5a) (Scheme 1) were purchased from Sigma-Aldrich (St. Louis, MO, USA); 7α-hydroxycholesterol (4a), 24(S)-hydroxycholesterol (6a), and 25-hydroxycholesterol (7a) were obtained from Steraloids (Newport, RI, USA); and 27-hydroxycholesterol (8a) was supplied by Research Plus (Manasquan, NJ, USA). N,N-Dimethylglycine (DMG), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), and 4-(N,N-dimethylamino)pyridine (DMAP) were purchased from Sigma-Aldrich. Solvents for sample preparation and for MS analysis were obtained from Burdick and Jackson (Honeywell International, Burdick and Jackson, Muskegon, MI, USA). Other chemical reagents were purchased either from Fisher Scientific (Pittsburgh, PA, USA) or from Sigma-Aldrich.
Oxysterol derivatization
Derivatization of oxysterols with DMG was modified from a previously described method in which moisture-sensitive dimethylaminoacetyl chloride or dimethylaminoacetyl imidazolide was employed for alcohol derivatization.36 Specifically, a mixture of DMG (0.5 M) and DAMP (2 M) in CHCl3 (10 μL) and 10 μL of EDC (1 M) in CHCl3 was added to a dried oxysterol sample (10 nmol) in a micro-reaction vessel. The mixture was vortexed for 10 s and incubated at 50°C overnight. After cooling to room temperature, the reaction mixture was extracted with 2 mL of diethyl ether against 1 mL of 0.1 N aqueous ammonia. The aqueous phase was re-extracted with diethyl ether (2 × 2 mL). The combined organic phase was filtered with a 0.2 μm polytetrafluoroethylene syringe filter, and dried under a stream of nitrogen. The residue was dissolved in CHCl3/MeOH/formic acid (50:50:0.1, v/v/v) and the final concentration of oxysterol DMG esters was 0.2 nmol/μL. The solution was stored at −20°C if necessary after gently flushing with nitrogen and used within 1 week after preparation. Before infusion of an oxysterol derivative solution into the ESI source, the prepared solution was diluted with CHCl3/MeOH/formic acid (50:50:0.1, v/v/v) to the desired concentrations as indicated.
ESI-MS/MS analysis of protonated oxysterol DMG esters
A triple quadrupole mass spectrometer (Thermo Electron TSQ Quantum Ultra; Thermo Electron, San Jose, CA, USA) equipped with an ESI source and operated under an Xcalibur software system was utilized in this study. The spray voltage was maintained at +3 kV in the positive ion mode. The offset voltage on the ion transfer capillary was set to −17 V in the positive ion mode. The heater temperature along the ion transfer capillary was maintained at 250°C. The sheath gas (nitrogen) pressure was 2 psi. The diluted lipid extract solution was directly infused into the ESI source at a flow rate of 4 μL/min with a syringe pump (Harvard Apparatus, Holliston, MA, USA). Argon, at a pressure of 1.0 mTorr, was used as the collision gas to obtain product ion mass spectra of selected precursor ions. A collision energy of 24 eV was employed for the production analysis of 7-oxocholesterol, 5α,6α-epoxycholesterol, and 5β,6β- epoxycholesterol DMG esters while a collision energy of 16 eV was used for the analysis of 7α-, 7β-, 24(S)-, 25-, and 27-hydroxycholesterol DMG esters. Typically, a 2-min period of signal averaging in the profile mode was employed for the acquisition of each product ion mass spectrum.
RESULTS AND DISCUSSION
Preparation of dimethylglycine esters of oxysterols
Preparation of DMG esters was reported by reaction of alcohols with moisture-sensitive dimethylaminoacetyl chloride or dimethylaminoacetyl imidazolide.36 Since it is difficult to completely exclude moisture from lipid extracts of biological samples, it is more convenient to employ water-stable reagents. A mixture of DMG, EDC, and DMAP as derivatizing reagents was used in this study. DMG is not soluble in most organic solvents, but it is readily soluble in CHCl3 in the presence of four molar equivalents of DMAP. It was found that a high conversion yield of the derivatization using this new approach was easily obtained. For example, a 100:1 molar ratio of DMG to alcohol could lead to complete conversion to a DMG ester at 50°C overnight. The DMG esters of oxysterols were readily ionized as the protonated species under acidic conditions in the positive ion mode. For example, ESI-MS analysis of the derivatized 5α,6α- epoxycholesterol DMG ester (2b, 0.1 pmol/μL) after direct infusion at a flow rate of 4 μL/min and averaging over a 2-min period displays an abundant ion at m/z 488.3, corresponding to the protonated 5α,6α-epoxycholesterol DMG ester (spectrum not shown). The identity of this protonated species was confirmed by MS/MS (see below).
It was also found that DMG-EDC-DMAP reagents, when they were used to derivatize alcohols, were more reactive than dimethylaminoacetyl imidazolide since the tertiary 25-hydroxyl in 25-hydroxychloesterol (7a) was not esterified with dimethylaminoacetyl imidazolide but was readily derivatized with DMG-EDC-DMAP reagents as demonstrated by mass spectrometric analysis (spectrum not shown). The derivatization of 7-oxocholesterol, 5α,6α-epoxycholesterol or 5β,6β-epoxycholesterol with DMG in the presence of EDC and DMAP produced the corresponding 3β-DMG ester. A bis-DMG ester was obtained from the derivatization of 7α-, 7β-, 24(S)-, 25-, or 27-hydroxycholesterols with DMG in the presence of EDC and DMAP.
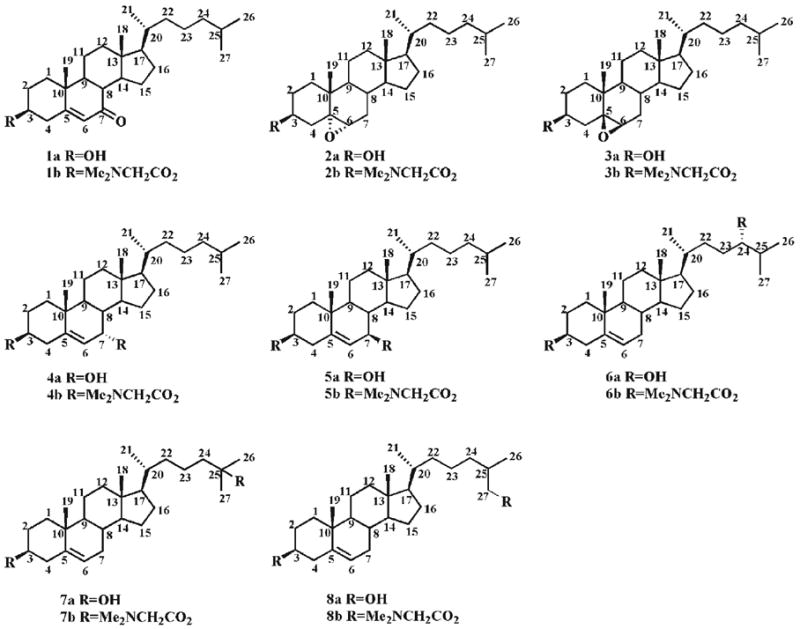
CID spectra and proposed dissociation pathways of protonated mono-DMG esters of oxysterols (i.e., 1b, 2b, and 3b)
The CID spectrum of the protonated DMG ester of 7-oxocholesterol (1b) under low CID energy demonstrates two very abundant product ions at m/z 104.1 and 383.2 (Fig. 1(A)), representing protonated DMG and the fragment resulting from the loss of DMG, respectively. Other low abundance ions are present in the spectrum including the ions at m/z 365.2 and 58.1 (Fig. 1(A)). Potential fragmentation pathways of the protonated 7-oxocholesterol DMG ester are proposed in Scheme 2. Hydrogen migration in the ion at m/z 383.2 may lead to a protonated dienone, which further converts into a protonated trienol via hydrogen rearrangements and the transfer of a proton to the hydroxyl group37 (Scheme 2). The loss of a water molecule from this trienol results in a low abundance ion at m/z 365.2. The neutral loss of acetic acid from the protonated DMG yields iminium at m/z 58.1.
Figure 1.
ESI CID spectra of the protonated DMG esters of 7-oxocholesterol (1b), and 5α,6α- (2b) and 5β,6β-epoxycholesterols (3b). DMG derivatives of 7-oxocholesterol (1a), and 5α,6α- (2a) and 5β,6β-epoxycholesterols (3a) were prepared and analyzed by ESI-MS as described in the Experimental section. CID spectra of the protonated DMG esters of 7-oxocholesterol (A), and 5α,6α- (B) and 5β,6β-epoxycholesterols (C) were acquired with a collision gas (argon) pressure at 1.0 mTorr and a collision energy of 24 eV.
Scheme 2.

Proposed fragmentation pathways of the protonated 7-oxocholesterol DMG ester (1b). rH1,2 represents the rearrangement of a hydrogen atom to an adjacent carbon atom with concurrent α site rearrangement of the charge.28, 37 The ‘-’ symbols (i.e., indicating a loss) and the molecules above the arrows represent the neutral loss of the corresponding fragments.
Since ESI-MS/MS analyses of the protonated 5α,6α-and 5β,6β-epoxycholesterol DMG esters show the matching product ions but in different relative abundance, it is suggested that the fragmentation pathways of these isomers are the same (Scheme 3). The neutral loss of DMG generates a product ion at m/z 385.2, which undergoes a hydrogen shift and the loss of a water molecule to give rise to an ion at m/z 367.2 (Scheme 3). This ion is quite abundant since it is stabilized by two adjacent double bonds. In addition, the signal intensity ratio of the ions at m/z 367.2 and 385.2 observed from 5α,6α-epoxycholesterol DMG ester (2b) is 2.9 while this ratio is 7 from 5β,6β-isomer 3b since the opening of the epoxide is more favorable for the β-isomer than for the α-isomer to release the constraint of the axial C4–C5 bond. This peak signal intensity ratio can be used to distinguish the α- and β-stereoisomers of the derivatized 5,6-epoxycholesterols.
Scheme 3.

Proposed fragmentation pathways of the protonated 5,6-epoxycholesterol DMG esters (2b and 3b). The ‘-’ symbols (i.e., indicating a loss) and the molecules above the arrows represent the neutral loss of the corresponding fragments.
CID spectra and proposed dissociation pathways of the protonated bis-DMG esters of 7α- and 7β-hydroxycholesterols (4b and 5b)
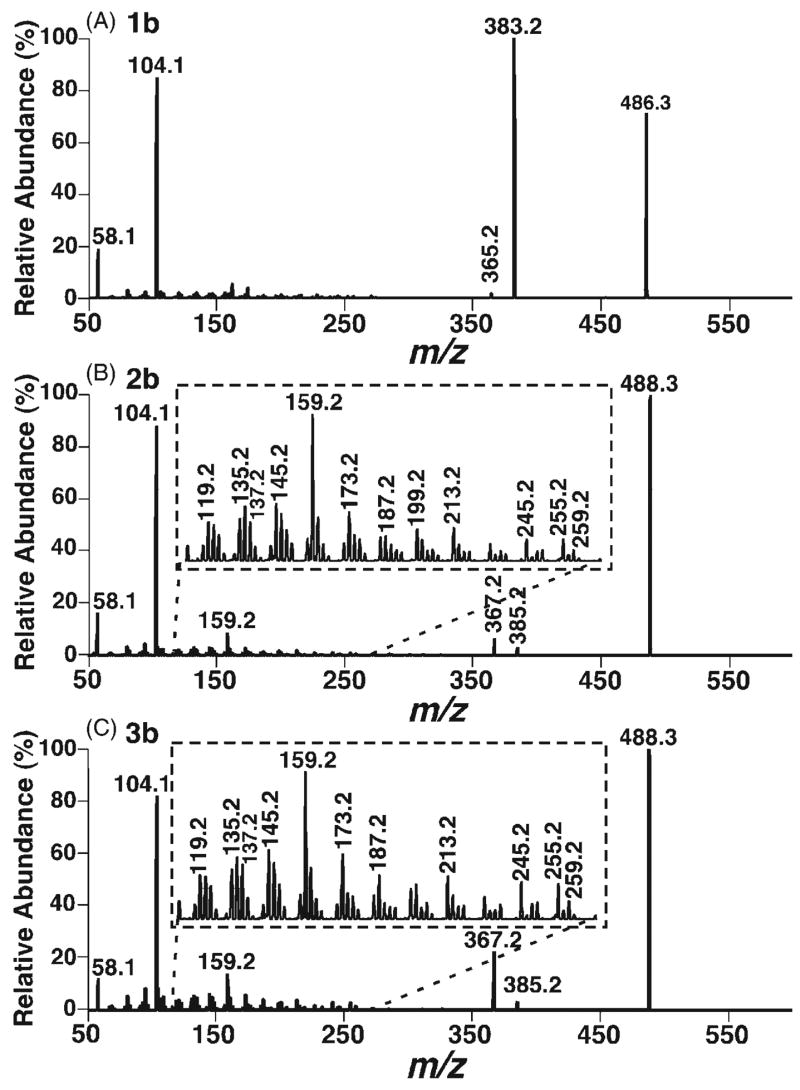
The CID spectra of doubly protonated bis-DMG esters of 7α-and 7β-hydroxycholesterol stereoisomers (4b and 5b) demonstrate informative, abundant product ions (Fig. 2). The product ion spectra show that CID of both doubly charged bis-DMG esters follow identical fragmentation pathways, but that the individual pathways are affected by the stability of the intermediates. Therefore, the product ions from both doubly charged derivatives are identical but possess different abundances (Fig. 2). Dissociation of the [M+2H]2+ ion of either the 7α- or the 7β-hydroxycholesterol DMG ester at m/z 287.2 can involve a combined process (i.e., the loss of DMG, the production of protonated DMG (m/z 104.1), and the production of singly charged ions at m/z 367.2) to yield the ions at m/z 104.1 and 367.2 (Scheme 4(a)). The originally formed m/z 367.2 ions may be composed of an ion with the positive charge at the C7 position, which is stabilized by a resonance structure, and an ion with the positive charge at the C3 position without resonance stabilization. The latter may convert into the resonance-stablilized ion with a positive charge at the C4 or C7 position after hydrogen shifts.
Figure 2.
ESI CID spectra of the doubly protonated DMG esters of 7α- (4b) and 7β-hydroxycholesterols (5b). CID spectra of the doubly protonated DMG esters of 7α- (A) and 7β-hydroxycholesterols (B) were acquired as described in the legend to Fig. 1 except that a collision energy of 16 eV was employed.
Scheme 4.


Proposed fragmentation pathways of the protonated 7-hydroxycholesterol DMG esters (4b and 5b). ‘i’ indicates inductive cleavage;28, 37 rH1,2 represents the rearrangement of a hydrogen atom to an adjacent carbon atom with concurrent α site rearrangement of the charge;28, 37 H1,3 and H1,5 represent 1,3- and 1,5-hydrogen shift, respectively. The ‘-’ symbols and the molecules above the arrows represent the neutral loss of these molecules.
From the m/z 367.2 ion with a positive charge at the C4 position, an ion at m/z 223.2 with its charge stabilized by a resonance structure could be obtained through the loss of the C17 side chain and C18 methyl, the aromatically demethylation of C19 methyl, and the 1,3-hydrogen shift (Scheme 4(b)). The process of the loss of the C17 side chain and C18 methyl has been previously suggested via a combination of a classical 1,4-H2 elimination and a methyl transfer reaction, involving a six-membered ring transition state.38 The m/z 367.2 ion undergoes hydrogen shifts, a retro-cycloaddition reaction,38 and aromatization to yield the ion at m/z 179.1 (Scheme 4(b)). Cleavage of the side chain at the C17–C20 bond in the m/z 367.2 ion with a positive charge at the C7 position via hydrogen shifts and inductive cleavages results in the ion at m/z 255.2 (Scheme 4(c)). Intriguingly, the signal intensity ratios of the pair of product ions at m/z 255.2 and 367.2 show 4-fold differences between these two epimers (i.e. a ratio of 0.44 is present in the CID spectrum of the 7α-isomer 4b whereas a ratio of 1.9 is present in the CID spectrum of the 7β-isomer 5b). Accordingly, these differential signal intensity ratios of the product ions between these epimers can be used as one criterion to discriminate these isomers (Fig. 2).
The signal intensity ratio of the ion pair at m/z 255.2 and 367.2 is influenced by the kinetics of their formation and degradation. Since the structures of the ions at m/z 255.2 and 367.2 resulting from either the 7α-isomer 4b or the 7β-isomer 5b are identical and the m/z 255.2 ion is generated from the m/z 367.2 ion, the kinetics of the formation and degradation of the m/z 255.2 ion from both isomers are identical. However, the ions at m/z 367.2 are comprised of multiple isomeric ions, and only the ion with a positive charge at the C7 position prefers to undergo the pathway to form the m/z 255.2 ion. The difference between the intensity ratios of the pair of ions at m/z 255.2 and 367.2 of the isomers is thus determined by the kinetics of the formation of the m/z 367.2 ion with a positive charge at the C7 position and by its stability. It is not clear yet why the formation of the m/z 367.2 ion with a positive charge at the C7 position in the 7β-isomer 5b is more favorable than in the 7α-isomer 4b.
CID spectrum and proposed dissociation pathways of the protonated bis-DMG ester of 24-hydroxycholesterol (6b)
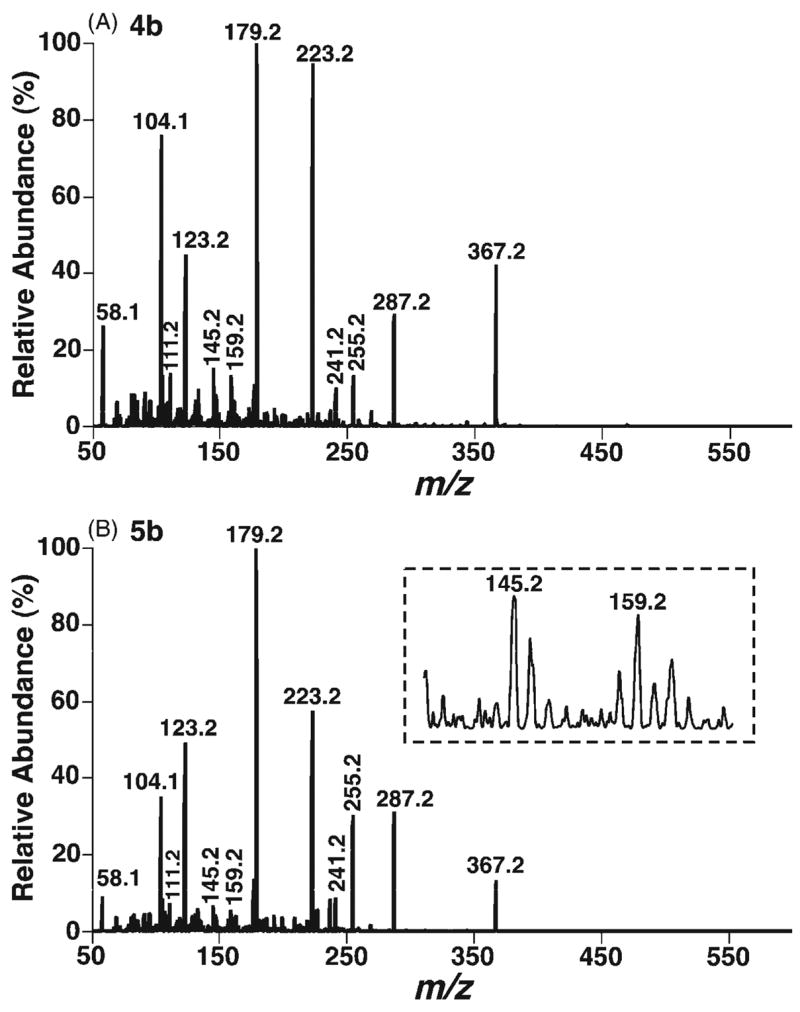
The CID spectrum of the doubly protonated 24-hydroxycholesterol bis-DMG ester (6b) shows a substantially different pattern of ion abundance from those of other bis-DMG esters (Fig. 3(A)). CID of the [M+2H]2+of this DMG ester 6b at m/z 287.2 produces a singly charged ion at m/z 367.2 through the loss of DMG and the production of protonated DMG at m/z 104.1, and alternatively generates a doubly charged ion at m/z 235.7 by the neutral loss of DMG from the C3 position. The positive charge of the ion at m/z 367.2 can be localized at either the C4 position (conjugated with the C5–C6 double bond) or the C24 position. The proposed dissociation of the ion at m/z 367.2 with a positive charge at the C4 position includes fission of the side chain at C17–C20 by hydrogen shifts and inductive cleavages to yield the ions at m/z 111.1 (Scheme 5).
Figure 3.
ESI CID spectra of the doubly protonated DMG esters of 24(S)- (6b), 25-(7b), and 27-hydroxycholesterols (8b). CID spectra of the doubly protonated DMG esters of 24(S)- (A), 25- (B), and 27-hydroxycholesterols (C) were acquired as described in the legend to Fig. 1 except that a collision energy of 16 eV was employed.
Scheme 5.

Proposed fragmentation pathways of 24-hydroxycholesterol DMG ester (6b). ‘i’ indicates inductive cleavage;28, 37 rH1,2 represents the rearrangement of a hydrogen atom to an adjacent carbon atom with concurrent γ site rearrangement of the charge.28, 37 The ‘-’ symbols and the molecules above the arrows represent the neutral loss of these molecules.
CID spectrum and proposed dissociation pathways of the protonated bis-DMG ester of 25-hydroxycholesterol (7b)
The CID spectrum of the [M+2H]2+ ion of 25-hydroxycholesterol DMG ester (7b) at m/z 287.2 demonstrates multiple informative and abundant product ions (Fig. 3(B)). CID of this doubly charged ion under low collision energy yields a singly charged ion at m/z 367.2 by a combination of the loss of DMG with the production of protonated DMG at m/z 104.1. This single positive charge can be located either at the C4 position conjugated with the C5–C6 double bond or at the C25 position. The most abundant product resulting from the doubly protonated 25-hydroxycholesterol bis-DMG ester ion is observed at m/z 223.2, and probably originated from the ion at m/z 367.2 through a similar pathway to that in Scheme 4(b) after hydrogen shifts. Similarly, the ion at m/z 179.2 is probably generated as shown in Scheme 4(b) (Scheme 6).
Scheme 6.
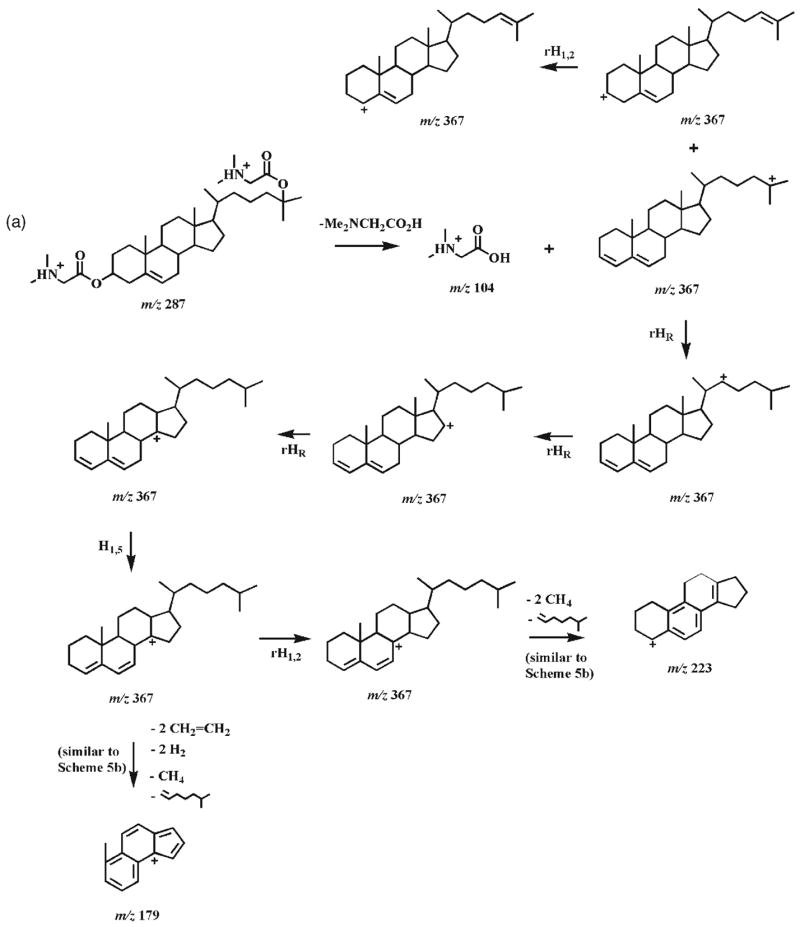
Proposed fragmentation pathways of the protonated 25-hydroxycholesterol DMG ester (7b). rH1,2 represents the rearrangement of a hydrogen atom to an adjacent carbon atom with concurrent α site rearrangement of the charge28, 37 and rHR stands for the rearrangement of a hydrogen atom to a remote site with concurrent γ site rearrangement of the charge.28 H1,3 and H1,5 represent 1,3- and 1,5-hydrogen shift, respectively. The ‘-’ symbols and the molecules above the arrows represent the neutral loss of these molecules.
CID spectrum and proposed dissociation pathways of the protonated bis-DMG ester of 27-hydroxycholesterol (8b)
The CID spectrum of the doubly protonated DMG ester of 27-hydroxycholesterol (8b) has a different appearance from the CID mass spectra of other isomers (Fig. 3). Moderately abundant ions at m/z 362.2, 336.2 and 310.2 are specific to the 27-hydroxycholesterol derivative 8b. There are multiple product ions including those at m/z 470.3, 235.7 and 161.2 present in the product ion mass spectrum of the 27-isomer, which are not present or present at very low signal intensities in that of the 25-isomer. The generation of the ion at m/z 470.3 is postulated to occur by loss of protonated DMG from the C3 position of the [M+2H]2+ ion of the 27-hydroxycholesterol DMG ester, with the single positive charge being located either at the C4 position conjugated with the C5–C6 double bond or at the amino group of DMG. The other postulated fragmentation pathway of the doubly charged 27-hydroxycholesterol DMG ester involves the neutral loss of DMG from the C3 position to yield a stable doubly charged ion at m/z 235.7 (Scheme 7).
Scheme 7.

Proposed fragmentation pathways of the protonated 27-hydroxycholesterol DMG ester (8b). rH1,2 represents the rearrangement of a hydrogen atom to an adjacent carbon atom with concurrent γ site rearrangement of the charge.28, 37.
CONCLUSIONS
A robust and mild derivatization method using DMG, EDC and DMAP has been developed to convert oxysterols (including primary, secondary, and tertiary alcohols) into their DMG esters. The derivatives can be readily ionized as their singly or doubly protonated molecules under acidic conditions. The CID spectra of these protonated DMG esters of oxysterols demonstrate the presence of an informative fragmentation pattern for each oxysterol derivative. Potential dissociation pathways for these unique fragmentation patterns are proposed. These informative fragmentation patterns may allow us rapid and direct differentiation of the identities of 7α-, 7β-, 24(S)-, 25-, and 27-hydroxycholesterols, as well as 5α,6α- and 5β,6β-epoxycholesterol isomers, thereby potentially providing a foundation for quantitative analysis of oxysterols present in biological samples in combination with chromatographic separation.
Acknowledgments
This work was supported by the National Institute on Aging (Grant R01AG23168) and the National Institute of Health (Grants P01HL57278 and P20RR20643), and the Neurosciences Education and Research Foundation.
Footnotes
Contract/grant sponsor: National Institute on Aging; contract/grant number: R01AG23168.
Contract/grant sponsor: The National Institute of Health; contract/grant number: P01HL57278 and P20RR20643.
Contract/grant sponsor: The Neurosciences Education and Research Foundation.
References
- 1.Brown AJ, Jessup W. Atherosclerosis. 1999;142:1. doi: 10.1016/s0021-9150(98)00196-8. [DOI] [PubMed] [Google Scholar]
- 2.Axelson M, Larsson O, Zhang J, Shoda J, Sjovall J. J Lipid Res. 1995;36:290. [PubMed] [Google Scholar]
- 3.Baulieu EE. Recent Prog Horm Res. 1997;52:1. [PubMed] [Google Scholar]
- 4.Bjorkhem I, Meaney S, Diczfalusy U. Curr Opin Lipidol. 2002;13:247. doi: 10.1097/00041433-200206000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Smith LL. Lipids. 1996;31:453. doi: 10.1007/BF02522641. [DOI] [PubMed] [Google Scholar]
- 6.Schroepfer GJ., Jr Physiol Rev. 2000;80:361. doi: 10.1152/physrev.2000.80.1.361. [DOI] [PubMed] [Google Scholar]
- 7.Olkkonen VM, Lehto M. Ann Med. 2004;36:562. doi: 10.1080/07853890410018907. [DOI] [PubMed] [Google Scholar]
- 8.Russell DW. Biochim Biophys Acta. 2000;1529:126. doi: 10.1016/s1388-1981(00)00142-6. [DOI] [PubMed] [Google Scholar]
- 9.Javitt NB. J Lipid Res. 2002;43:665. [PubMed] [Google Scholar]
- 10.Javitt NB. Biochem Biophys Res Commun. 2002;292:1147. doi: 10.1006/bbrc.2001.2013. [DOI] [PubMed] [Google Scholar]
- 11.Lutjohann D, Papassotiropoulos A, Bjorkhem I, Locatelli S, Bagli M, Oehring RD, Schlegel U, Jessen F, Rao ML, von Bergmann K, Heun R. J Lipid Res. 2000;41:195. [PubMed] [Google Scholar]
- 12.Papassotiropoulos A, Lutjohann D, Bagli M, Locatelli S, Jessen F, Rao ML, Maier W, Bjorkhem I, von Bergmann K, Heun R. Neuroreport. 2000;11:1959. doi: 10.1097/00001756-200006260-00030. [DOI] [PubMed] [Google Scholar]
- 13.Leoni V, Masterman T, Patel P, Meaney S, Diczfalusy U, Bjorkhem I. J Lipid Res. 2003;44:793. doi: 10.1194/jlr.M200434-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Nishimura T, Inoue T, Shibata N, Sekine A, Takabe W, Noguchi N, Arai H. Genes Cells. 2005;10:793. doi: 10.1111/j.1365-2443.2005.00879.x. [DOI] [PubMed] [Google Scholar]
- 15.Ansari GA, Smith LL. Methods Enzymol. 1990;186:438. doi: 10.1016/0076-6879(90)86137-k. [DOI] [PubMed] [Google Scholar]
- 16.Csallany AS, Kindom SE, Addis PB, Lee JH. Lipids. 1989;24:645. doi: 10.1007/BF02535082. [DOI] [PubMed] [Google Scholar]
- 17.Bjorkhem I, Diczfalusy U. Arterioscler Thromb Vasc Biol. 2002;22:734. doi: 10.1161/01.atv.0000013312.32196.49. [DOI] [PubMed] [Google Scholar]
- 18.Janoszka B, Tyrpien K, Wielkoszynski T, Dobosz C, Bodzek D, Bodzek P, Olejek A. Chemia Analityczna (Warsaw) 2001;46:11. [Google Scholar]
- 19.Kou IL, Holmes RP. J Chromatogr. 1985;330:339. doi: 10.1016/s0021-9673(01)81991-9. [DOI] [PubMed] [Google Scholar]
- 20.Dionisi F, Golay PA, Aeschlimann JM, Fay LB. J Agric Food Chem. 1998;46:2227. [Google Scholar]
- 21.Sevanian A, Seraglia R, Traldi P, Rossato P, Ursini F, Hodis H. Free Radical Biol Med. 1994;17:397. doi: 10.1016/0891-5849(94)90166-x. [DOI] [PubMed] [Google Scholar]
- 22.Dzeletovic S, Breuer O, Lund E, Diczfalusy U. Anal Biochem. 1995;225:73. doi: 10.1006/abio.1995.1110. [DOI] [PubMed] [Google Scholar]
- 23.Manini P, Andreoli R, Careri M, Elviri L, Musci M. Rapid Commun Mass Spectrom. 1998;12:883. [Google Scholar]
- 24.Razzazi-Fazeli E, Kleineisen S, Luf W. J Chromatogr A. 2000;896:321. doi: 10.1016/s0021-9673(00)00719-6. [DOI] [PubMed] [Google Scholar]
- 25.Bodin K, Diczfalusy U. Eur J Lipid Sci Technol. 2002;104:435. [Google Scholar]
- 26.Burkard I, Rentsch KM, von Eckardstein A. J Lipid Res. 2004;45:776. doi: 10.1194/jlr.D300036-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Raith K, Brenner C, Farwanah H, Muller G, Eder K, Neubert RH. J Chromatogr A. 2005;1067:207. doi: 10.1016/j.chroma.2004.12.053. [DOI] [PubMed] [Google Scholar]
- 28.Guan F, Soma LR, Luo Y, Uboh CE, Peterman S. J Am Soc Mass Spectrom. 2006;17:477. doi: 10.1016/j.jasms.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 29.Sandhoff R, Brugger B, Jeckel D, Lehmann WD, Wieland FT. J Lipid Res. 1999;40:126. [PubMed] [Google Scholar]
- 30.Cheng H, Jiang X, Han X. J Neurochem. 2006 doi: 10.1111/j.1471–4159.2006.04342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu S, Sjovall J, Griffiths WJ. Rapid Commun Mass Spectrom. 2000;14:390. doi: 10.1002/(SICI)1097-0231(20000331)14:6<390::AID-RCM882>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 32.Liu S, Sjovall J, Griffiths WJ. Anal Chem. 2003;75:5835. doi: 10.1021/ac0346297. [DOI] [PubMed] [Google Scholar]
- 33.Griffiths WJ, Liu S, Alvelius G, Sjovall J. Rapid Commun Mass Spectrom. 2003;17:924. doi: 10.1002/rcm.1002. [DOI] [PubMed] [Google Scholar]
- 34.Griffiths WJ, Wang Y, Alvelius G, Liu S, Bodin K, Sjovall J. J Am Soc Mass Spectrom. 2006;17:341. doi: 10.1016/j.jasms.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Hornshaw M, Alvelius G, Bodin K, Liu S, Sjovall J, Griffiths WJ. Anal Chem. 2006;78:164. doi: 10.1021/ac051461b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson DW. J Mass Spectrom. 2001;36:277. doi: 10.1002/jms.125. [DOI] [PubMed] [Google Scholar]
- 37.McLafferty FW, Turecek F. Interpretation of Mass Spectra. 4. University Science Books; Sausalito, California: 1993. [Google Scholar]
- 38.Griffiths WJ. Mass Spectrom Rev. 2003;22:81. doi: 10.1002/mas.10046. [DOI] [PubMed] [Google Scholar]