

Abstract
Objective
Mutations in patched (PTCH) cause the nevoid basal cell carcinoma syndrome (NBCCS), or Gorlin syndrome. Nevoid basal cell carcinoma syndrome may present with developmental anomalies, including rib and craniofacial abnormalities, and predisposes to several tumor types, including basal cell carcinoma and medulloblastoma. Cleft palate is found in 4% of individuals with nevoid basal cell carcinoma syndrome. Because there might be specific sequence alterations in PTCH that limit expression to orofacial clefting, a genetic study of PTCH was undertaken in cases with cleft lip and/or palate (CL/P) known not to have nevoid basal cell carcinoma syndrome.
Results
Seven new normal variants spread along the entire gene and three missense mutations were found among cases with cleft lip and/or palate. One of these variants (P295S) was not found in any of 1188 control samples. A second variant was found in a case and also in 1 of 1119 controls. The third missense (S827G) was found in 5 of 1369 cases and in 5 of 1104 controls and is likely a rare normal variant. Linkage and linkage desequilibrium also was assessed using normal variants in and adjacent to the PTCH gene in 220 families (1776 individuals), each with two or more individuals with isolated clefting. Although no statistically significant evidence of linkage (multipoint HLOD peak = 2.36) was uncovered, there was borderline evidence of significant transmission distortion for one haplotype of two single nucleotide polymorphisms located within the PTCH gene (p = .08).
Conclusion
Missense mutations in PTCH may be rare causes of isolated cleft lip and/or palate. An as yet unidentified variant near PTCH may act as a modifier of cleft lip and/or palate.
Keywords: cleft lip, cleft palate, PTCH
Patched (PTCH; 9q22.3), the human homolog of the Drosophila segment polarity gene patched, encodes a 12-unit transmembrane protein and is the proposed receptor for the morphogen sonic hedgehog (SHH; Marigo et al., 1996). By binding SHH, PTCH transduces a repressing signal through smoothened (SMO) in a downstream pathway to the nucleus (Taipale et al., 2002). Targets of homologs to hedgehog in vertebrates include wingless type (WNT) and transforming growth factor beta (TGFB) family members and PTCH itself (Bale and Yu, 2001). Loss of SHH signaling in the chick embryonic face causes defects similar to cleft lip and palate and hypotelorism in humans (Hu and Helms, 1999). Haploinsufficiency for SHH can lead to holoprosencephaly and cleft lip and palate in autosomal dominant holoprosencephaly (Muenke and Beachy, 2000).
Mutations in PTCH are associated with a variety of birth defects (Hahn et al., 1996; Goodrich et al., 1997) and are implicated in the development of nevoid basal cell carcinoma syndrome (NBCCS; OMIM 109400), also known as Gorlin syndrome (Gorlin and Goltz, 1960; Johnson et al., 1996). Patients with NBCCS have diverse developmental anomalies, often including rib and craniofacial abnormalities and a mixture of tumor types. Cleft palate is found in 4% of cases (Evans et al., 1993; Kimonis et al., 1997).
Screening of PTCH coding regions revealed a wide spectrum of mutations that are spread throughout the entire gene with no apparent clustering (Wicking et al., 1997). The phenotypic variability has not been correlated with the nature or location of mutations (Bale and Yu, 2001). This suggests that genetic background, environmental effects, or stochastic factors may have an important role in the expression of this syndrome. In addition, recent evidence (Lettice et al., 2002) suggests that long-range regulatory elements also are important in SHH expression, so mutation searches may need to consider elements far beyond traditional gene boundaries. Finally, the signaling pathway that includes PTCH is integrated with cholesterol metabolism, suggesting possible gene and environment interactions with maternal (or fetal) cholesterol levels (Muenke and Beachy, 2000).
Cleft lip and/or palate (CL/P) is a major congenital anomaly with a birth prevalence from 1 in 500 to 1 in 2500 births, depending on geographic, ethnic, and socioeconomic factors (Mossey and Little, 2002). Investigations into the cause of CL/P just have begun to identify a few specific genes that explain a small portion of observed cases (Jezewski et al., 2003; Lidral and Murray, 2004). A few syndromic forms of CL/P can have phenocopies of the isolated forms, including those for muscle segment homeobox homolog 1 (MSX1; van den Boogaard et al., 2000), fibroblast growth factor receptor 1 (FGFR1; Dode et al., 2003), interferon regulatory factor 6 (IRF6; Kondo et al., 2002), and the T-box containing gene TBX22 (Marcano et al., 2004). Because these syndromic forms of clefting include affected individuals who display only an oral cleft, there might be specific sequence alterations that limit expression to a specific organ system in other individuals with nonsyndromic clefting (Stanier and Moore, 2004). To evaluate this possibility, we undertook a genetic study of PTCH in a collection of cases with apparently isolated cleft lip and/or palate known not to have NBCCS. A recent meta-analysis of 13 genome-wide screens for isolated CL/P also suggests that a locus on 9q near the NBCCS gene may be important in the etiology of oral clefts (Marazita et al., 2004).
Methods
Sequencing Analysis
Nonsyndromic cases were identified at the University of Iowa Hospital and Clinics and in the Philippines through Operation Smile (Murray et al., 1997; Romitti et al., 1999). Blood samples for DNA analysis were obtained after signed informed consent following Institutional Review Board (IRB) approval both in the United States (University of Iowa, IRB Committee) and in the Philippines (Hope Foundation, Bacolod City, Negros, Philippines). In this study we sequenced the complete coding sequence and the 23 exon/intron boundaries (Fig. 1), using DNA samples from 90 probands with nonsyndromic cleft lip and palate from Iowa and 90 from the Philippines (Table 1). In the exons where new DNA sequence variations were found, all available family members and an additional 180 cleft cases were analyzed (Table 1).
FIGURE 1.
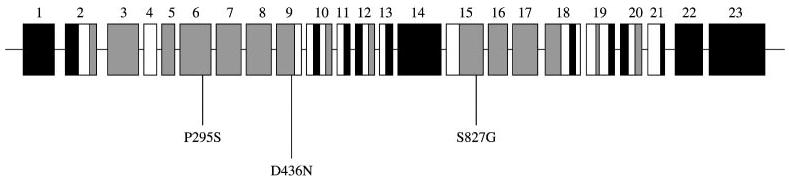
Patched structure. Boxes represent exons and lines represent introns. Black = intracellular domain, white = transmembrane domain, grey = extracellular domain grey. Missense variants are shown.
TABLE 1.
Samples Used in Each Analysis Described
| Analysis | Cases | Controls | Filipino Kindreds (Cases) | |
|---|---|---|---|---|
| Sequencing all 23 exons | 90 nonsyndromic Filipinos | 90 nonsyndromic from Iowa | 92 Filipinos and 95 Europeans | |
| Exons 6, 9, 15 | Additional 180 nonsyndromic Filipinos
Family members of case |
Additional 180 nonsyndromic from Iowa
Family members of case |
||
| Assay of specific missense mutations | 1036 Filipinos | 695 Europeans | 1064 CEPH samples | |
| SNP/microsatellite/linkage data | 1776 (220) | |||
Controls were matched to the population and consisted of convenience samples drawn from the same geographic region and were examined to determine that no cleft was present at birth. DNA sequencing was performed with these controls, a group comprising 95 Europeans and 92 Filipinos (Table 1).
Templates for sequencing were generated by polymerase chain reaction (PCR) in an Applied Biosystems Gene Amp PCR System 9700 (Applied Biosystems, Foster City, CA). The 10-μL reactions contained 1.5 mM Mg2+, 200 μM dNTPs, 0.3 mM each primer, and Bioexact reaction buffer (Bioline USA, Inc., Randolph, MA); 0.25 units of Bioexact and 30 ng to 40 ng DNA were performed in one cycle of 95°C for 5 minutes, 35 cycles of 95°C for 30 seconds, 57°C for 30 seconds, 68°C for 1 minute, and 68°C for 5 minutes. Sequencing then was performed with the DNA sequencing kit, Big Dye™ Terminator Cycle Sequencing (Applied Biosystems, Foster City, CA). The 10-μL sequencing chemistry reactions contained Big Dye Terminator Mix, Big Dye Terminator Buffer reaction, 0.075 mM of the corresponding primer, 1.25 ng per 100 bp of the PCR product, and 5% of DMSO, and were run using at least 35 cycles.
Sequencing reactions were resolved on an ABI Prism 3700 analyzer (Applied Biosystems, Foster City, CA) and analyzed by Polyphred 4.0 (http://droog.mbt.washington.edu/poly_doc40.html) and Consed (http://www.genome.washington.edu/UWGC/analysistools/consed.cfm). The primers used were designed using the primer 3 Web site (http://frodo.wi.mit.edu), and are listed in Appendix A.
PolyPhen Analysis
We assessed the three identified missense mutations with Polymorphism Phenotyping (PolyPhen), a program that predicts the possible impact of an amino acid substitution on the structure and function of a human protein using physical and comparative considerations. (Sunyaev et al., 2001, Ramensky et al., 2002; http://www.bork.embl-heidelberg.de/PolyPhen/).
Missense Mutations Screening
For the three missense mutations identified, allelic discrimination probes were developed using the Taqman Assay System (Ranade et al., 2001). These assays were used to test additional cases and controls in an effort to confirm whether these variants were unique mutations or rare normal single nucleotide polymorphisms (SNPs) if present in both cases and controls. Controls consisted of 1064 members of a human diversity panel from the Centre de Etude du Polymorphisme Humain (CEPH; Cann et al., 2002; Shi et al., 2003), 1036 samples from the Philippines, and 695 samples of European ancestry (435 from Denmark and 260 from Iowa; Table 1).
Gene Bank Accession Data
Nucleotide and amino acid numbering is based on Genbank sequence accession number 59454.
Extended SNP/Microsatellite Analysis
Two SNPs in PTCH, C89T (rs 2297088) and T86C (rs 2236407), were selected from the database based on heterozygosity data available at http://www.appliedbiosystems.com. These SNPs were genotyped in 1776 individuals from 220 Filipino extended kindreds (Table 1; Schultz et al., 2004). Both linkage and association (i.e., transmission distortion) were assessed for the SNPs in the extended kindreds using parametric and nonparametric methods (see below). Association also was assessed for haplotypes formed across the two SNPs. Further, for 1606 Filipino family members who were genotyped for the PTCH SNPs, 17 additional microsatellite markers were available on chromosome 9 (Schultz et al., 2004). Therefore, we also assessed multipoint linkage for chromosome 9, adding the PTCH SNPs to the linkage analysis.
Preliminary Analyses
The inheritance of each marker in all families was assessed with PedCheck (O’Connell and Weeks, 1995) to test for inconsistencies due to nonpaternity or other errors. For the parametric linkage analyses, allele frequencies were estimated from the unaffected founders in the study families. The genetic model parameters were taken from segregation analysis results in a sample of the Filipino families (unpublished results). MEGA2 (Mukhopadhyay et al., 1999, 2001) was used to prepare and to submit analysis files.
Linkage Calculations: LOD Scores
Two-point LOD scores in the extended kindreds were calculated using the Elston-Stewart algorithm (Elston and Stewart, 1971), employing the LINKAGE program with recent updates to speed calculations (Cottingham et al., 1993; Terwilliger and Ott, 1994; O’Connell and Weeks, 1995).
Multipoint LOD Score Calculations
The descent graph method (Sobel and Lange, 1996; Sobel et al., 2001, 2002) implemented in computer program SIM-WALK2 was used for the multipoint LOD score and multi-point heterogeneity LOD score calculations (Smith, 1963).
Model-Free Linkage Calculations
Single-point and multipoint nonparametric linkage calculations were performed using MERLIN (Abecasis et al., 2002).
Allelic Association: TDT Method
The transmission disequilibrium test (TDT), introduced by Spielman et al. (1993), is a powerful family-based method for detecting associations between marker and disease loci in the presence of linkage disequilibrium. Alleles at each marker were tested for association with CL/P using the Family Based Association Test (FBAT; Laird et al, 2000; Rabinowitz and Laird, 2000; Horvath et al., 2001).
Haplotype Transmission Analysis
Haplotype-based transmission disequilibrium statistics were calculated by using the haplotype version of the FBAT (Rabinowitz and Laird, 2000; Horvath et al., 2001). Standard chi-square tests of association were utilized to compare the frequencies of each variant found by sequencing between groups of cases and controls. The Expectation Maximization Algorithm (EM) was used to form the haplotypes and estimate haplotype frequencies, and then transmission distortion was assessed under the null hypothesis of no linkage or no disequilibrium, utilizing the empiric variance-covariance matrix.
Results
Table 2 shows the common polymorphisms identified through DNA sequencing of the cleft cases from Iowa and the Philippines and controls for both populations. The comparison made between controls and cases for both populations gave only two p values lower than .05 (i.e., .04 for T3944C in the Caucasian population and .037 for C32454G in the Filipino population).
TABLE 2.
PTCH Polymorphisms Found (Entries in the Table Are Numbers of Individuals With “Homozygous Common Alele/Heterozygous Rare Allele” 1 rs 574688, 2 rs 2277184, 3 rs 2066830, 4 rs 2236406, 5 rs 2066835, 6 rs 2274690, 7 rs 2236405)
| Filipino Genotype Distributions
|
Caucasian Genotype Distributions
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Filipino Cases | Filipino Controls | p Value* | CARC Cases† | CEPH Controls† | p Value* | Overall Rare Allele Frequency | Status | Change | ||
| T28719C | Intron 5 | 98/33/2 | 76/19/1 | .63 | 70/37/3 | 48/36/2 | .5 | 0.17 | published (51) | |
| G897A | Exon 6 | 85/0/0 | 38/0/0 | ~1.0 | 88/1/0 | 83/0/0 | ~1.0 | 0.001 | new | no |
| C32454G | Intron 11 | 58/24/2 | 56/10/0 | .037 | 29/26/3 | 42/29/3 | .735 | 0.19 | data base (1) | |
| T32497C | 73/10/0 | 66/17/2 | .085 | 59/1/0 | 87/2/1 | .69 | 0.06 | data base (2) | ||
| C1641T | Exon 12 | 118/0/0 | 86/0/0 | ~0.0 | 82/4/0 | 82/3/0 | .71 | 0.01 | data base (3) | no |
| C1665T | 104/14/0 | 78/7/0 | .4 | 69/17/0 | 66/18/1 | .58 | 0.08 | published (52) | no | |
| C39736T | Intron 13 | 63/22/1 | 11/1/00 | .38 | 51/27/3 | 43/23/2 | .97 | 0.17 | new | |
| C2093G | Exon 14 | 80/4/0 | 84/1/0 | .17 | 88/0/0 | 75/0/0 | ~1.0 | 0.0075 | new | P702R |
| A2187G | 81/3/0 | 81/4/0 | .71 | 64/3/0 | 81/3/0 | .78 | 0.02 | published (51) | no | |
| C2210T | 80/4/0 | 82/1/0 | .18 | 88/0/0 | 79/1/0 | .29 | 0.009 | new | A741V | |
| A49783G | Intron 17 | 43/30/2 | 44/33/8 | .21 | 40/37/7 | 37/45/12 | .44 | 0.3 | data base (4) | |
| T2913C | Exon 18 | 63/5/0 | 79/7/1 | .67 | 84/1/0 | 92/0/0 | .3 | 0.02 | new | no |
| T3141G | 57/8/0 | 69/13/2 | .38 | 84/0/0 | 92/1/0 | .34 | 0.04 | data base (5) | no | |
| G55676C | Intron 19 | 75/12/0 | 46/13/0 | .19 | 17/0/0 | 40/0/0 | ~1.0 | 0.06 | data base (6) | |
| C3387T | Exon 20 | 81/5/0 | 53/0/0 | .07 | 53/0/0 | 75/0/0 | ~0.0 | 0.009 | new | no |
| G59656A | Intron 21 | 81/5/0 | 84/3/0 | .46 | 73/1/0 | 80/0/0 | .274 | 0.01 | new | |
| A3583T | Exon 22 | 78/10/0 | 70/18/2 | .095 | 47/0/0 | 87/0/0 | ~1.0 | 0.05 | data base (7) | T1195S |
| T3944C | Exon 23 | 39/32/13 | 105/161/148 | .065 | 11/36/24 | 4/27/35 | .04 | 0.46 | published (53) | L1315P |
CARC = Craniofacial Anomalies Research Center; CEPH = Centre d’Etude du Polymorphisme Humain.
p value from case-control comparison.
Missense Variants
Three new variants were detected, and for those probands, we analyzed all family members available (Figs. 1 and 2).
FIGURE 2.
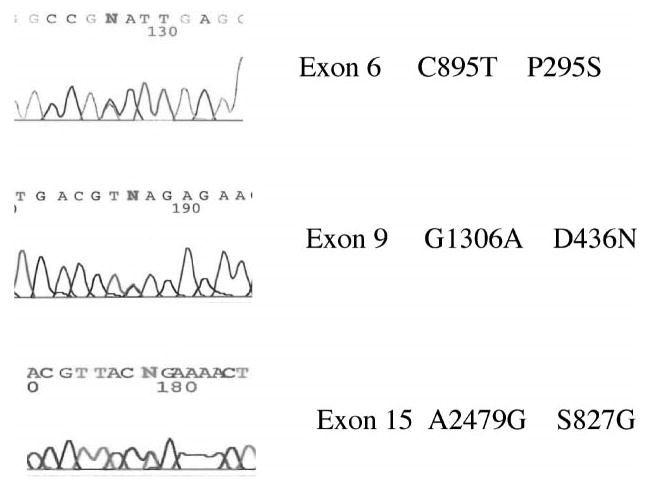
Chromatograms for the missense variants.
In exon 6 (extracellular domain) the variant C895T produces a change from proline to serine (P295S) and was present in an affected Filipino female and also in her two unaffected brothers and father. This variant was not present in controls from the Philippines, Iowa, or the CEPH diversity panel (Table 3). This residue is conserved in mouse, zebra fish, and chicken, but not in Drosophila melanogaster, and was found as a probably damaging variant by PolyPhen.
TABLE 3.
Results of Haplotype Transmission Analysis for PTCH_C89T and PTCH_T86C in 220 Extended Filipino Kindreds
| Haplotype | Allele PTCH_C89T | Allele at PTCH_T86C | Estimated Frequency | HBAT p Value (bi-allelic)* |
|---|---|---|---|---|
| H1 | 1 | 1 | 0.725 | .31 (+) |
| H2 | 2 | 2 | 0.259 | .42 (−) |
| H3 | 2 | 1 | 0.009 | .08 (−) |
| H4 | 1 | 2 | 0.007 | .32 (−) |
HBAT = Haplotype Family Based Association Test; (+) = positive association with the haplotype; (−) = negative association; the multi-allellic p value was .21.
For exon 9, a transition was found in position 1306 (G to A) that results in a substitution from aspartic acid to asparagine (D436N). This residue is located in the transmembrane domain and is conserved in chicken, zebra fish, and mouse. This variation was found in one case from Iowa and in his unaffected mother. This variant was found in 1 of 1119 controls (see Table 4).
TABLE 4.
A: Results From Allelic Discrimination for the Missense Variants Tested in Screening Plates for Cases and CEPH* Diversity Panel as Controls; B: Sequencing Data
| A | |||
|---|---|---|---|
| P295S | D436N | S827G | |
| Cases | 0/1027 | 0/619 | 5/1036 |
| Controls | 0/1064 | 1/1064 | 5/1064 |
| B | |||
| P295S | D436N | S827G | |
|
| |||
| Cases | 1/354 | 1/277 | 1/333 |
| Controls | 0/124 | 0/55 | 0/40 |
CEPH = Centre d’Etude du Polymorphisme Humain.
A third variant found was a transition from A to G in a male with CL/P from the Philippines, but his unaffected mother carried the same allele. This transition was found in the position 2479, exon 15, in an extracellular domain. It produces a change from serine to glycine, at the 827 residue. This variant was found in 5 of 1104 controls.
There was no history or physical evidence for features of Gorlin syndrome in any of the three affected individuals carrying any of these variants or in their available family members. Variants S827G and D436N were predicted as benign by PolyPhen.
The results for the allelic discrimination and the sequencing are shown in Table 4. There were no significant differences in the allele frequency distributions between the case and control groups.
SNP Analysis
There was no evidence of linkage between CL/P and either of these rare SNPs: the maximum 2-point LOD scores were approximately 0.0, and the p values for the single-point Non-Parametric Linkage Analysis (NPL) calculations were .60 for PTCH_C89T and .60 for PTCH_T86C. There was also no statistically significant evidence of transmission distortion for either SNP individually (FBAT p values = .24 for PTCH_C89T and .48 for PTCH_T86C).
Table 3 summarizes the haplotype transmission analyses; there was borderline statistically significant evidence of transmission distortion for PTCH_C89T/PTCH_T86C haplotype 3 (bi-allelic p value = .08), but no overall evidence (multi-allelic p value = .21).
Figure 3 shows the multipoint HLOD plot for chromosome 9, including the two PTCH SNPs along with 25 other markers in 220 Filipino multiplex cleft families (Table 5). The multi-point HLOD peak (HLOD = 2.36, proportion of linked families = 0.10) occurs at PTCH_T86C, approximately cM 103.
FIGURE 3.
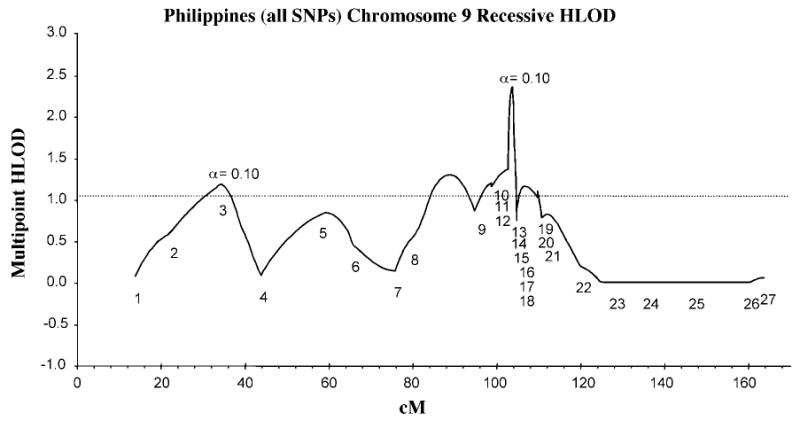
Multipoint HLOD plot for nonsyndromic cleft lip and/or palate versus chromosome 9 markers in 220 Filipino kindreds (see Table 5 for marker names).
TABLE 5.
Marker Guideline for Figure 3
| Marker Number on Plot | Marker Name |
|---|---|
| 1 | D9S2169 |
| 2 | D9S168 |
| 3 | D9S925 |
| 4 | D9S1121 |
| 5 | D9S1118 |
| 6 | D9S301 |
| 7 | D9S922 |
| 8 | D9S1122 |
| 9 | D9S283 |
| 10 | ROR2A113 |
| 11 | ROR2T344 |
| 12 | ROR2A542 |
| 13 | PTCH_C89T |
| 14 | PTCH_T86C |
| 15 | D9S1786 |
| 16 | TGFBA405 |
| 17 | TGFBA396 |
| 18 | TGFBA393 |
| 19 | ZNFC340T |
| 20 | ZNFG256T |
| 21 | D9S938 |
| 22 | D9S930 |
| 23 | D9S934 |
| 24 | D9S1825 |
| 25 | D9S2157 |
| 26 | D9S1826 |
| 27 | D9S1838 |
Discussion
Searches for mutations in candidate genes for cleft lip and palate have met with variable success to date (Murray, 2002; Lidral and Murray, 2004). No mutations were found in searches of the gene causing ectrodactyly, ectodermal dysplasia, and cleft lip and palate syndrome 3, protein 63 (P63; Barrow et al., 2002), and only rare variants of unclear etiologic significance were found in searches of transforming growth factor beta 3 (TGFB3; Lidral et al., 1998) and transforming growth factor alpha (TGFA; Machida et al., 1999). The MSX1 gene has been shown to harbor mutations in about 2% of cases of isolated clefting (Jezewski et al., 2003; Suzuki et al., 2004), as has the T-box 22 (TBX22) gene for a very small percentage of cases of isolated cleft palate (Marcano et al., 2004). Other genes that would make good candidates for at least occasionally having causal mutations and creating phenotypes that could mimic isolated clefting include FGFR1 (Dode et al., 2003), where inactivating mutations cause Kallmann syndrome, and IRF6, in which mutations cause Van der Woude syndrome (Kondo et al., 2002). Recently, IRF6 has been shown to have a common haplotype that increases the risk for isolated CL/P by a factor of about 3 (Zucchero et al., 2004; Scapoli et al., 2005).
In this study, the PTCH gene was examined for mutations in cases of isolated CL/P because about 4% of individuals with NBCCS will have a CL/P. Three new rare missense variants (P295S, D436N, S827G) were detected, which were present in three unrelated pedigrees with isolated CL/P and in unaffected family members. Two of them (P295S and S827G) are located in the predicted extracellular loops of PTCH. These extracellular domains are believed to bind SHH (Marigo et al., 1996). It has been reported that the deletion of one of these loops in mice shows a PTCH protein that did not respond to SHH (Briscoe et al., 2001). We hypothesize that if these variants interfere with the binding of SHH, this signal will not be transmitted to the targets in the nucleus.
The three changes observed in this study involved amino acids that are highly conserved between species and result in amino acid changes: P295S changes a nonpolar for a polar uncharged, D436N changes a negatively charged amino acid for a polar uncharged, and only S827G leads to an unremarkable change to the same type of amino acid. It is important to note, however, that all three of these rare variants occurred in unaffected relatives of the cases, and thus none are completely penetrant.
Only one of the three missense variants (P295S) was not observed in any of more than 1516 controls studied, including 369 specific to the ancestral origin of the case and 1064 from a worldwide ancestral set of samples, making it unlikely that this is a rare normal variant. This variant also was predicted as probably damaging by PolyPhen. Yet S827G was found to be a normal rare variant, present in the same number of controls as cases, although the controls who showed this variant were from China and this population has a higher incidence of CL/P. The D436N variant was found only in 1 of 1119 controls. It remains possible that either of these variants may be coupled to variants in other genes that jointly increase the susceptibility to CL/P.
For the common variants found in exons and in the regulatory regions, further studies in other genes involved in this pathway could disclose variants that would suggest these create hypomorphic alleles or are involved in gene-gene interactions for the cause of clefting. For the many comparisons done in the present study, only two were of borderline significance and therefore seem unlikely to be casually related to the cleft phenotype.
One study of the ligand for PTCH, sonic hedgehog (SHH), did not identify any mutations in SHH in cases of isolated clefting (Orioli et al., 2002), but SHH is known for the presence of distant regulatory elements that are also worth evaluating (Lettice et al., 2002).
Guerrero and Ruiz i Altaba (2003) suggest that there could be other PTCH ligands and that a threshold level of PTCH could be necessary to inhibit the SHH pathway and to initiate apoptosis during development of the neural tube, when PTCH and SHH have been shown to play an essential role in both apoptosis and differentiation. Using this observation, we suggest that PTCH overexpression can induce apoptosis of cells that are involved in the closure of the lip or the palate during development. It also has been suggested that interaction between PTCH domains could affect proliferation and cell death, another mechanism to be examined as a potential cause of clefting, particularly given the role played at the time of palatal fusion involving mesenchymal transformation.
Finally, there was only weak evidence of transmission distortion to suggest that PTCH may act as a modifier of risk to isolated CL/P. Although linkage is found in this location, no common haplotype was identified in this sequencing study of coding regions in PTCH, but a modifier mutation could affect a regulatory element and surrounding PTCH that is outside of its coding sequence. Future studies can search for these elements and also can look for evidence of gene-gene or gene-environment interactions, perhaps of genes involving cholesterol metabolism (Edison and Muenke, 2004).
Data Access
Access to the primary sequence data and SNP genotyping is available on request (J.C.M.).
Acknowledgments
We are especially grateful to the families who participated and many volunteers at Project Hope and Operation Smile. Susie McConnell, Nancy Davin, Cathy Dragan, Diana Caprau, Sandy Daack-Hirsch, Paul Romitti, Alex Vieira, and Sally Santiago provided outstanding technical assistance. Genotyping of anonymous markers on chromosome 9 was provided by a grant from the Center of Inherited Disease Research (CIDR) to Andrew Lidral, M.L.M., and J.C.M. (CIDR is supported by NIH grant N01-HG-65403). This work was supported by DE08559, ES10876, and Fogarty Award DE43TW05503.
This work was supported by DE08559, ES10876, Fogarty Award DE43TW05503, and NIH grant N01-HG-65403 (CIDR).
Appendix A Sequencing Primers
| Exon | Sequence |
|---|---|
| 1B | F 5′-AGGGCGCAGGGTGTGAC-3′ |
| R 5′-CTGCTGCTTTTCCTGGAGAG-3′ | |
| 1–1A | F 5′-CCTCTCCTTAGGCCCTGGT-3′ |
| R 5′-GGGGCTGCAATACAGAAGAG-3′ | |
| 2 | F 5′-CCCATGACGCTCAGATCC-3′ |
| R 5′-GCGCCCAAACAATAAACAAT-3′ | |
| 3 | F 5′-TGGTGAAGTTAACATTGCCATT-3′ |
| R 5′-AGAAACGATAAATCAAGATGAAAA-3′ | |
| 4 and 5 | F 5′-TGCTCGTTTTGACAGATGCT-3′ |
| R 5′-CCCCCGACTATTCACTCAAA-3′ | |
| 6 | F 5′-GAGTCCCAGAACTGCAGCAT-3′ |
| R 5′-CCATAGACAAAGACGATCATGG-3′ | |
| 7 | F 5′-TTTGCCATACACCTCCCATT-3′ |
| R 5′-CCACCAACTCTCTCTGACCA-3′ | |
| 8 | F 5′-AACCATCCTGGTCCCATTTT-3′ |
| R 5′-TCCCAGGATTTTCAATATCAA-3′ | |
| 9 | F 5′-GCCCTGGAATCACGTAGAACT-3′ |
| R 5′-AAGGTAGGCAAACGGCAAA-3′ | |
| 10 | F 5′-GCCCTGGAATCACGTAGAACT-3′ |
| R 5′-AAGGTAGGCAAACGGCAAA-3′ | |
| 11 | F 5′-CTAGGCTTTGGGACGTCAAG-3′ |
| R 5′-TTCCTAAAGGCACCCGAGAT-3′ | |
| 12 | F 5′-TCCCTAATGCCAGCATGATA-3′ |
| R 5′-GGAAAAACTCCAGAAGGGCTTA-3′ | |
| 13 | F 5′-TCACGGTTTCAAATGCTTCA-3′ |
| R 5′-GGGCTGTGAGACTGTTCAGC-3′ | |
| 14 | F 5′-CACAGTGAAAAATGGCAGAATG-3′ |
| R 5′-TGATGAACTCCAAAGGTTCTGTT-3′ | |
| 15 | F 5′-TGGGAGAACAACCCCTACAA-3′ |
| R 5′-AAGTCCATGAAACACGTCAGTG-3′ | |
| 16 | F 5′-GGGACACAGAGGGTGTGTTT-3′ |
| R 5′-TTTCTACCAGCTCCCAGTGC-3′ | |
| 17 | F 5′TGGGATTTTCGACACTTTCAA-3′ |
| R 5′-GTCAACGGATGAAGGCTGTT-3′ | |
| 18 | F 5′-AAAGGCCTGGAGGCTATGAT-3′ |
| R 5′-GGACCTCACCACCTCGAGTA-3′ | |
| 19 | F 5′-AGGCAGTAAAGGCAGTGTCC-3′ |
| R 5′-TGAGGGAAAGGAATCCAGAA-3′ | |
| 20 | F 5′-GACCCAGTGTCATGAAGAGGT-3′ |
| R 5′-CTTGAACTCCTTGACCTTCTGA-3′ | |
| 21 | F 5′-TGAACTGCGGTTGGATAACA-3′ |
| R 5′-CTCTAGCCCTCAAAGCCAGT-3′ | |
| 22 | F 5′-GGGAGGTTAATACGGCACAG-3′ |
| R 5′-CACTACCACGGTGGGAAGAC-3′ | |
| 23 | F 5′-AAACCCAAGGAGGGAAGTGT-3′ |
| R 5′-GAAGCCGTCACAGTGGTGAT-3′ | |
| F 5′-GCATTCTGGCCCTAGCAATA-3′ | |
| R 5′-TGACAAAGCTTGGACACTCA-3′ |
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. MERLIN: rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Bale AE, Yu K. The hedgehog pathway and basal cell carcinomas. Hum Mol Gen. 2001;10:757–762. doi: 10.1093/hmg/10.7.757. [DOI] [PubMed] [Google Scholar]
- Barrow LL, van Bokhoven H, Daack-Hirsch S, Andersen T, van Beersum SEC, Gorlin R, Murray JC. Analysis of the p63 gene in classic EEC syndrome, related syndromes, and non-syndromic orofacial clefts. J Med Genet. 2002;39:559–566. doi: 10.1136/jmg.39.8.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodak N, Queille S, Avril MF, Bouadjar B, Drougard C, Sarasin A, Daya-Grosjean L. High levels of patched gene mutations in basal-cell carcinomas from patients with xeroderma pigmentosum. Proc Natl Acad Sci U S A. 1999;96:5117–5122. doi: 10.1073/pnas.96.9.5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J, Chen Y, Jessell TM, Struthl G. A hedgehog-insensitive form of patched provides evidence for direct long range morphogen activity of sonic hedgehog in the neural tube. Mol Cell. 2001;7:1279–1291. doi: 10.1016/s1097-2765(01)00271-4. [DOI] [PubMed] [Google Scholar]
- Cann HM, de Toma C, Cazes L, Legrand MF, Morel V, Piouffre L, Bodmer J, Bodmer WF, Bonne-Tamir B, Chambón-Thomsen A, Chen Z, Chu J, Carcassi C, Contu L, Du R, Excoffier L, Ferrara GB, Friedlaender JS, Groot H, Gurwitz D, Jenkins T, Herrera RJ, Huang X, Kidd J, Kidd KK, Langaney A, Lin AA, Mehdí SQ, Parham P, Piazza A, Pistillo MP, Qian Y, Shu Q, Xu J, Zhu S, Weber JL, Greely HT, Feldman MW, Thomas G, Dausset J, Cavalli-Sforza LL. A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- Chang-Claude J, Dunning A, Schnitzbauer U, Galmbacher P, Tee L, Wjst M, Chalmers J, Zemzoum I, Harbeck N, Pharoah PD, Hahn H. The patched polymorphism Pro1315Leu (C3944T) may modulate the association between use of oral contraceptives and breast cancer risk. Int J Cancer. 2003;103:779–783. doi: 10.1002/ijc.10889. [DOI] [PubMed] [Google Scholar]
- Cottingham RW, Jr, Idury RM, Schaffer AA. Faster sequential genetic linkage computations. Am J Hum Genet. 1993;53:252–263. [PMC free article] [PubMed] [Google Scholar]
- Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pecheux C, Le Tessier D, Cruaud C, Delpech M, Speleman F, Vermeulen S, Amalfitano A, Bachelot Y, Bouchard P, Cabrol S, Carel JC, Delemarre-van de Waal H, Goulet-Salmon B, Kottler ML, Richard O, Sanchez-Franco F, Saura R, Young J, Petit C, Hardelin JP. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- Dong J, Gailani MR, Pomeroy S, Reardon D, Bale A. Identification of PATCHED mutations in medulloblastomas by direct sequencing. Hum Mut. 2000;16:89–90. doi: 10.1002/1098-1004(200007)16:1<89::AID-HUMU18>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Edison RJ, Muenke M. Central nervous system and limb anomalies in case reports of first-trimester statin exposure. N Engl J Med. 2004;350:1579–1582. doi: 10.1056/NEJM200404083501524. [DOI] [PubMed] [Google Scholar]
- Elston RC, Stewart J. A general model for the genetic analysis of pedigree data. Hum Hered. 1971;21:523–542. doi: 10.1159/000152448. [DOI] [PubMed] [Google Scholar]
- Evans DG, Ladusans EJ, Rimmer S, Thakker N, Farndon PA. Complications of the naevoid basal cell carcinoma syndrome: results of a population-based study. J Med Genet. 1993;30:460–464. doi: 10.1136/jmg.30.6.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Gorlin RJ, Goltz RW. Multiple nevoid basal-cell epithelioma, jaw cysts, and bifid rib: a syndrome. N Engl J Med. 1960;262:908–912. doi: 10.1056/NEJM196005052621803. [DOI] [PubMed] [Google Scholar]
- Guerrero I, Ruiz i Altaba A. Development. Longing for ligand: hedgehog, patched, and cell death. Science. 2003;301:774–776. doi: 10.1126/science.1088625. [DOI] [PubMed] [Google Scholar]
- Hahn H, Christiansen J, Wicking C, Zaphiropoulos PG, Chidambaram A, Gerrard B, Vorechovsky I, Bale AE, Toftgard R, Dean M, Wainwright B. A mammalian patched homolog is expressed in target tissues of sonic hedgehog and maps to a region associated with developmental abnormalities. J Biol Chem. 1996;271:12125–12128. doi: 10.1074/jbc.271.21.12125. [DOI] [PubMed] [Google Scholar]
- Horvath S, Xu X, Laird NM. The family based association test method: strategies for studying general genotype-phenotype associations. Eur J Hum Genet. 2001;9:301–306. doi: 10.1038/sj.ejhg.5200625. [DOI] [PubMed] [Google Scholar]
- Hu D, Helms JA. The role of sonic hedgehog in normal and abnormal craniofacial morphogenesis. Development. 1999;126:4873–4884. doi: 10.1242/dev.126.21.4873. [DOI] [PubMed] [Google Scholar]
- Jezewski PA, Vieira AR, Nishimura C, Ludwig B, Johnson M, O’Brien SE, Daack-Hirsch S, Schultz RE, Weber A, Nepomucena B, Romitti PA, Christensen K, Orioli IM, Castilla EE, Machida J, Natsume N, Murray JC. Complete sequencing shows a role for MSX1 in non-syndromic cleft lip and palate. J Med Genet. 2003;40:399–407. doi: 10.1136/jmg.40.6.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn EH, Myers RM, Cox DR, Epstein EH, Jr, Scott MP. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- Kimonis VE, Goldstein AM, Pastakia B, Yang ML, Kase R, DiGiovanna JJ, Bale AE, Bale SJ. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299–308. [PubMed] [Google Scholar]
- Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S, Sander A, McDonald-McGinn DM, Zackai EH, Lammer EJ, Aylsworth AS, Ardinger HH, Lidral AC, Pober BR, Moreno L, Arcos-Burgos M, Valencia C, Houdayer C, Bahuau M, Moretti-Ferreira D, Richieri-Costa A, Dixon MJ, Murray JC. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19(suppl 1):S36–S42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Lettice LA, Horikoshi T, Heaney SJ, van Baren MJ, van der Linde HC, Breedveld GJ, Joosse M, Akarsu N, Oostra BA, Endo N, Shibata M, Suzuki M, Takahashi E, Shinka T, Nakahori Y, Ayusawa D, Nakabayashi K, Scherer SW, Heutink P, Hill RE, Noji S. Disruption of a long-range CIS-acting regulator for SHH causes preaxial polydactyly. Proc Natl Acad Sci U S A. 2002;99:7548–7553. doi: 10.1073/pnas.112212199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidral AC, Murray JC. Genetics approach to identify disease genes for birth defects with cleft lip/palate as a model. Birth Defects Res Part A Clin Mol Teratol. 2004;70:893–901. doi: 10.1002/bdra.20096. [DOI] [PubMed] [Google Scholar]
- Lidral AC, Romitti PA, Basart AM, Doetschman T, Leysens NJ, Daack-Hirsch S, Semina EV, Johnson LR, Machida J, Burds A, Parnell TJ, Rubenstein JL, Murray JC. Association of MSX1 and TGFB3 with nonsyndromic clefting in humans. Am J Hum Genet. 1998;63:557–568. doi: 10.1086/301956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida J, Yoshiura K, Funkhauser CD, Natsume N, Kawai T, Murray JC. Transforming growth factor-alpha (TGFA): genomic structure, boundary sequences, and mutation analysis in nonsyndromic cleft lip/palate and cleft palate only. Genomics. 1999;61:237–242. doi: 10.1006/geno.1999.5962. [DOI] [PubMed] [Google Scholar]
- Marazita ML, Murray JC, Lidral AC, Arcos-Bargos M, Cooper ME, Goldstein T, Maher BS, Daack-Hirsch S, Schultz R, Mansilla MA, Field LL, Liu YE, Prescott N, Malcolm S, Winter R, Ray A, Moreno L, Valencia C, Neiswanger K, Wyszynski DF, Bailey-Wilson JE, Albacha-Hejazi H, Beaty TH, Mc-Intosh I, Hetmanski JB, Tuncbilek G, Edwards M, Harkin L, Scott R, Rod-dick L. Meta-analysis of 13 genome scans reveals multiple cleft lip/palate genes with novel loci on 9q and 2q. Am J Hum Genet. 2004;75:161–173. doi: 10.1086/422475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcano AC, Doudney K, Braybrook C, Squires R, Patton MA, Lees MM, Richieri-Costa A, Lidral AC, Murray JC, Moore GE, Stanier P. TBX22 mutations are a frequent cause of cleft palate. J Med Genet. 2004;41:68–74. doi: 10.1136/jmg.2003.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marigo V, Scott MP, Johnson RL, Goodrich LV, Tabin CJ. Conservation in the hedgehog signaling: induction of a chicken patched homolog by sonic hedgehog in the developing limb. Development. 1996;122:1225–1233. doi: 10.1242/dev.122.4.1225. [DOI] [PubMed] [Google Scholar]
- Mossey P, Little J. Epidemiology of oral clefts: an international perspective. In: Wyszynski DF, editor. Cleft Lip and Palate: From Origin to Treatment. New York: Oxford University Press; 2002. pp. 127–157. [Google Scholar]
- Muenke M, Beachy PA. Genetics of ventral forebrain development and holoprosencephaly. Curr Opin Genet Dev. 2000;10:262–269. doi: 10.1016/s0959-437x(00)00084-8. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay N, Almasy L, Schroeder M, Mulvihill WP, Weeks DE. MEGA2: a data-handling program for facilitating genetic linkage and association analyses. Am J Hum Genet. 1999;65:A436. doi: 10.1093/bioinformatics/bti364. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay N, Almasy L, Schroeder M, Mulvihill WP, Weeks DE. [Accessed April 18, 2005];MEGA2, Version 2.5. 2001 doi: 10.1093/bioinformatics/bti364. Available at: http://watson.hgen.pitt.edu/mega2.html. [DOI] [PubMed]
- Murray JC. Gene/environment causes of cleft lip and/or palate. Clin Genet. 2002;61:248–256. doi: 10.1034/j.1399-0004.2002.610402.x. [DOI] [PubMed] [Google Scholar]
- Murray JC, Daack-Hirsch S, Buetow KH, Munger R, Espina L, Paglinawan N, Villanueva E, Rary J, Magee K, Magee W. Clinical and epidemiologic studies of cleft lip and palate in the Philippines. Cleft Palate Craniofac J. 1997;34:7–10. doi: 10.1597/1545-1569_1997_034_0007_caesoc_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE. The VITESSE algorithm for rapid exact multilocus linkage analysis via genotype set-recoding and fuzzy inheritance. Nat Genet. 1995;11:402–408. doi: 10.1038/ng1295-402. [DOI] [PubMed] [Google Scholar]
- Orioli IM, Vieira AR, Castilla EE, Ming JE, Muenke M. Mutational analysis of the sonic hedgehog gene in 220 newborns with oral clefts in a South American (ECLAMC) population. Am J Med Genet. 2002;108:12–15. doi: 10.1002/ajmg.10204. [DOI] [PubMed] [Google Scholar]
- Rabinowitz D, Laird N. A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure and arbitrary missing marker information. Hum Hered. 2000;50:211–223. doi: 10.1159/000022918. [DOI] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranade K, Chang MS, Ting CT, Pei D, Hsiao CF, Olivier M, Pesich R, Hebert J, Chen YD, Dzau VJ, Curb D, Olshen R, Risch N, Cox DR, Botstein D. High-throughput genotyping with single nucleotide polymorphisms. Genome Res. 2001;11:262–268. doi: 10.1101/gr.157801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romitti PA, Lidral AC, Munger RG, Daack-Hirsch S, Burns TL, Murray JC. Candidate genes for nonsyndromic cleft lip and palate and maternal cigarette smoking and alcohol consumption: evaluation of genotype-environment interactions from a population-based case-control study of orofacial clefts. Teratology. 1999;59:39–50. doi: 10.1002/(SICI)1096-9926(199901)59:1<39::AID-TERA9>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Scapoli L, Palmieri A, Martinelli M, Pezzetti F, Carinci P, Tognon M, Carinci F. Strong evidence of linkage disequilibrium between polymorphisms at the IRF6 locus and nonsyndromic cleft lip with or without cleft palate, in an Italian population. Am J Hum Genet. 2005;76:180–183. doi: 10.1086/427344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz RE, Cooper ME, Daack-Hirsch S, Shi M, Nepomucena B, Graf KA, O’Brien EK, O’Brien SE, Marazita ML, Murray JC. Targeted scan of fifteen regions for nonsyndromic cleft lip and palate in Filipino families. Am J Med Genet. 2004;125A:17–22. doi: 10.1002/ajmg.a.20424. [DOI] [PubMed] [Google Scholar]
- Shi M, Caprau D, Romitti P, Christensen K, Murray JC. Genotype frequencies and linkage disequilibrium in the CEPH human diversity panel for variants in folate pathway genes MTHFR, MTHFD, MTRR, RFC1, and GCP2. Birth Defects Res Part A Clin Mol Teratol. 2003;67:545–549. doi: 10.1002/bdra.10076. [DOI] [PubMed] [Google Scholar]
- Smith CAB. Testing for heterogeneity of recombination fraction values in human genetics. Ann Hum Genet. 1963;27:175–182. doi: 10.1111/j.1469-1809.1963.tb00210.x. [DOI] [PubMed] [Google Scholar]
- Sobel E, Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker sharing statistics. Am J Hum Genet. 1996;58:1323–1337. [PMC free article] [PubMed] [Google Scholar]
- Sobel E, Papp JC, Lange K. Detection and integration of genotyping errors in statistical genetics. Am J Hum Genet. 2002;70:496–508. doi: 10.1086/338920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel E, Sengul H, Weeks DE. Multipoint estimation of identity-by-descent probabilities at arbitrary positions among marker loci on general pedigrees. Hum Hered. 2001;52:121–131. doi: 10.1159/000053366. [DOI] [PubMed] [Google Scholar]
- Spielman RS, McGinnis RE, Ewens WJ. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM) Am J Hum Genet. 1993;52:506–516. [PMC free article] [PubMed] [Google Scholar]
- Stanier P, Moore GE. Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts. Hum Mol Genet. 2004;13(Spec No 1):R73–R81. doi: 10.1093/hmg/ddh052. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Koch I, Lathe W, III, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Jezewski PA, Machida J, Watanabe Y, Shi M, Cooper ME, Viet LT, Tin NTD, Hai H, Natsume N, Shimozato K, Marazita ML, Murray JC. In a Vietnamese population MSX1 variants contribute to cleft lip and palate. Genet Med. 2004;6:117–125. doi: 10.1097/01.gim.0000127275.52925.05. [DOI] [PubMed] [Google Scholar]
- Taipale J, Cooper MK, Maiti T, Beachy PA. Patched acts catalytically to suppress the activity of smoothened. Nature. 2002;418:892–897. doi: 10.1038/nature00989. [DOI] [PubMed] [Google Scholar]
- Terwilliger JD, Ott J. Handbook of Human Genetic Linkage. Baltimore: Johns Hopkins University Press; 1994. [Google Scholar]
- van den Boogaard MJ, Dorland M, Beemer FA, van Amstel HK. MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet. 2000;24:342–343. doi: 10.1038/74155. [DOI] [PubMed] [Google Scholar]
- Wicking C, Shanley S, Smyth I, Gillies S, Negus K, Graham S, Suthers G, Haites N, Edwards M, Wainwright B, Chenevix-Trench G. Most germ-line mutations in the nevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and no genotype-phenotype correlations are evident. Am J Hum Genet. 1997;60:21–26. [PMC free article] [PubMed] [Google Scholar]
- Zucchero TM, Cooper ME, Maher BS, Daack-Hirsch S, Nepomuceno B, Ribeiro L, Caprau D, Christensen K, Suzuki Y, Machida J, Natsume N, Yoshiura K, Vieira AR, Orioli IM, Castilla EE, Moreno L, Arcos-Burgos M, Lidral AC, Field LL, Liu YE, Ray A, Goldstein TH, Schultz RE, Shi M, Johnson MK, Kondo S, Schutte BC, Marazita ML, Murray JC. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Engl J Med. 2004;351:769–780. doi: 10.1056/NEJMoa032909. [DOI] [PubMed] [Google Scholar]