

Summary
The halogenation of thousands of natural products occurs during biosynthesis and often confers important functional properties. While haloperoxidases had been the default paradigm for enzymatic incorporation of halogens, via X+ equivalents into organic scaffolds, a combination of microbial genome sequencing, enzymatic studies and structural biology have provided deep new insights into enzymatic transfer of halide equivalents in three oxidation states. These are: (1) the halide ions (X−) abundant in nature, (2) halogen atoms (X•), and (3) the X+ equivalents. The mechanism of halogen incorporation is tailored to the electronic demands of specific substrates and involves enzymes with distinct redox coenzyme requirements.
Introduction
Almost five thousand natural products that contain one or more carbon-halogen bonds have been isolated [1]. The great majority of halogenated metabolites are from prokaryotes and single cell eukaryotes but the tri-iodo (T3) and tetra-iodo (T4) forms of thyronine, the master homeostatic thyroid hormone, remind us of the long reach of halogenation biology [2]. Medicinal chemists have used regio- and stereoselective halogenation of drug candidates to optimize a variety of molecular properties including dipole moment and pKa, to control pharmacokinetics and tissue distribution, and to block or redirect metabolism. Undoubtedly Nature is using equivalent logic in the enzymatic tailoring of natural product scaffolds by halogenation. For example, three common antibiotics, chlortetracycline [3], chloramphenicol [4], and vancomycin [5] are all chlorinated. In vancomycin, the chlorination affects atropisomer distribution and is required to achieve clinically active conformation [6]. Dictyostelium uses chlorinated signaling small molecules [7], and bacteria make antifungal agents with chlorinated heterocyclic units [8].
Dramatic advances in deciphering the logic of halogenation enzymes have occurred in the recent past in part through bacterial genomic and bioinformatic analyses which allow identification of two new classes of halogenases, the flavin-dependent and mononuclear nonheme iron families, collocated with nonribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) biosynthetic gene clusters [9–21]. Complementary studies of purified proteins in each class have allowed codification of each class as O2-consuming halogenases [22,23] and have led to mechanistic and structural studies that have uncovered the molecular logic of catalytic oxidative halogenation during biosynthesis [24–28].
While most of the enzymatic halogenation reactions are oxidative, recently a new non-enzymatic non-oxidative strategy was elucidated, believed to be responsible for the halogenation of enediyne-derived macrolides isolated from marine actinomycetes [29]. This finding represents an important addition to the substrate diversity of halogenated molecules in nature.
Scope of halogenation reactions in biological molecules
A large variety of aromatic and aliphatic carbon centers are halogenated during natural product biosynthesis, with over 95% of the cases involving chloride or bromide [30]. These include chlorination at positions 4, 5, 6 and 7 of tryptophan-derived rings, chlorination of tyrosines at the ortho position and mono- and di-chlorination of pyrroles [3,31]. In marine organisms where bromide is in higher concentration than in fresh water there is comparable bromination of aromatic and heteroaromatic molecules as well as bromination of isoprenoid metabolites [32,33]. Halogenations occur adjacent to carbonyl groups, as in the dichloroacetyl moiety of chloramphenicol [4]. Several amino acid side chains in nonribosomal peptides are chlorinated at the γ or δ carbons. For example, the barbamide series of marine metabolites exibit -CHCl2 and -CCl3 substituents derived from a methyl group in leucine [34]. Most remarkable are natural product functional groups such as vinyl chlorides and bromides, alkynyl chlorides and bromides, and bromoallenes [20,35–37]. Iodination is relatively rare with approximately 120 known examples and organofluorination is even less prevalent with only 30 fluorinated natural products known to date [30]. While the paucity of organoiodides may reflect the low natural abundance of iodide, the scarcity of fluorinated metabolites is most likely due to a different, nonoxidative mechanism for fluoride incorporation compared to the oxidative halogenation enzymology open to chloride, bromide, and iodide.
Halogenases vs haloperoxidases
From the original discovery of a fungal chloroperoxidase in the 1960s the paradigm of H2O2 and chloride ion giving an Fe-OCl equivalent in a heme protein active site was the knowledge base for chlorination, bromination, and iodination enzymology [38]. The finding that bromoperoxidases from marine algae are vanadium-containing enzymes, using a V-OBr brominating species for bromoterpene biosynthesis expanded the scope of H2O2-dependent biological halogenation machinery [32]. The view that haloperoxidases were the full story changed with the genetic demonstration that the chlorination step in chlortetracycline biosynthesis was encoded by a flavoprotein homolog [9]. Bioinformatic analysis of the biosynthetic gene clusters for other polyketides and nonribosomal peptides identified additional flavin-based halogenases. Biochemical characterization of some representative members has demonstrated that they use O2 to form a flavin-OOH intermediate which oxidizes halide ions [25,31,39]. Given that flavoprotein oxygenases are not powerful enough to hydroxylate unactivated aliphatic carbons, it was anticipated that flavoprotein halogenases would similarly not be up to the task for such halogenations. Indeed, bioinformatic analysis of NRPS gene clusters for syringomycin, barbamide, and coronatine indicated the absence of flavoenzymes. Instead, researchers identified mononuclear nonheme iron enzymes of the O2 and α-ketoglutarate-dependent family that constitute a novel types of halogenases [15,17–19]. The sections that follow categorize the scope of substrates halogenated by whether the enzymes use X−, X•, or X+ as proximal halogen donors, titrated to the electronic demand of the cosubstrates. This in turn determines which cofactors are required to affect catalysis.
Halogenation via X−
The best example for use of ground state halide ions as nucleophiles in C-X bond formation has been the bacterial enzyme fluorinase [40]. The enzyme has two obvious requirements for enabling catalysis. First, it must provide a route to desolvation of F− so that this electronegative anion can function as nucleophile. A serine side chain in the active site is proposed to provide an alternate hydrogen bond to assist solvation [41]. Second, there must be an electrophilic carbon site in an organic cosubstrate for a halide ion to attack. This is provided by S-adenosylmethionine (SAM) where the 5′carbon of the ribose moiety is electrophilic and the methionine serves as a good leaving group (Figure 1a) as 5-F-adenosine is formed. Subsequent metabolism generates fluoroacetaldehyde, a precursor to fluroacetate and 4-fluorothreonine. The Streptomyces cattleya fluorinase will use Cl− slowly as alternate nucleophile [42].
Figure 1.

Halogenations with halide anions. (A) Halide ions as nucleophiles toward carbon electrophiles: Streptomyces cattleya fluorinase [40]. (B) Non-enzymatic halogenation via trapping of p-benzyne biradical intermediate in Bergman cyclization of marine enediynes [29].
Recently, a non-enzymatic pathway for the incorporation of halide anions into organic substrates has been postulated for the biosynthesis of halogenated polycyclic nuclei of sporolides A and B, and cyanosporosides A and B, isolated from marine actinomycetes Salinispora tropica and Salinispora pacifica, respectively [43,44] (Figure 1b). These molecules are proposed to be biosynthetically derived from an enediyne PKS [44,45], the presence of which was recently confirmed by genome sequencing of S. tropica [46]. The unique halogenation event is postulated to occur during the cycloaromatization of the enediyne unit. The feasibility of this proposal has been demonstrated in a model system, where it was found that slight heating of the enediyne cyclodeca-1,5-diyn-3-ene, in the presence of lithium chloride, bromide or iodide and a weak acid, is converted to 1-halotetrahydronaphthalene [29]. The kinetic parameters of the reaction are consistent with rate-limiting formation of p-benzyne intermediate. Trapping of this intermediate by halide nucleophile and the protonation of the resulting aryl anion leads to the formation of monohalogenated products (Figure 1b).
In general enzymatic halogenation via X− attack on electrophilic carbon sites is rare. Fluoride is restricted to this nonoxidative route because its high electronegativity makes it resistant to oxidative chemistry. Chloride, bromide and iodide could be incorporated this way but thus far only few metabolites – SAM and p-benzyne biradical intermediates –were demonstrated to be viable substrates.
Halogenation via X+
The more common mechanism for enzymatic halogenation has been oxidative conversion of X− to enzyme-bound –OX, where the hypohalites act as delivery agents for “X+” equivalents. These are the now-classic cases of the heme-iron haloperoxidases and the vanadyl bromoperoxidases [32,47]. The oxygen-based oxidant cosubstrate is hydrogen peroxide as the enzyme nomenclature reflects. The metal-OOH species proceed to form metal-OX forms as proximal halogenating agents [48–50]. Halogenation reactions with OX− equivalents require the reacting carbon center in the substrate to have the opposite polarity from the case described above. Now the reacting carbon site must be electron rich. Thus halogenation adjacent to phenolic oxygens, and bromination of pyrroles are typical outcomes (Figure 2a) [8,32,51]. Similarly π-electons in isoprenoid metabolites can yield cyclic bromonium ions as evidenced by the conversion of nerolidol to snyderol regioisomers by a vanadium-dependent bromoperoxidase [33] (Figure 2a). Finally, the bromoallene in the natural product laurallene is derived via bromonium-ion promoted cyclization of prelaureatin catalyzed by a bromoperoxidase (Figure 2a)[52].
Figure 2.

Hypohalites as electrophilic halogens toward carbon nucleophiles. (A) Marine haloperoxidases [32,33,50]. (B) Halogenation of tryptophan by flavin-dependent halogenases.[24,54–57] PrnA – Trp-7 halogenase in pyrrolnitrin biosynthesis (Pseudomonas fluorescens); RebH- Trp-7 halogenase in rebeccamycin biosynthesis (Lechevalieria aerocolonigenes); ThaI- Trp-6 halogenase in thienodolin biosynthesis (Streptomyces albogriseolus); PyrH- Trp-5 halogenase in pyrroindomycin biosynthesis (Streptomyces rugosporus). (C) Novel halogenated metabolites discovered by flavin halogenase genetic screen [59].
A second class of enzymes generating –OX equivalents are the flavin-dependent halogenases found in PKS and NRPS biosynthetic clusters [31,39]. In this case molecular oxygen, not hydrogen peroxide is the oxidizing cosubstrate. The dihydroflavin (FADH2) oxidation state is the reactive form of the coenzyme and, by one electron pathways and radical recombination, yields the prototypic FAD-4a-OOH that is typical in all flavoproteins that react with O2. At this point a chloride (or bromide) ion in the active site fragments the O-O single bond to generate HO-Cl [25,27]. While this could be the proximal donor of “Cl+”, the FAD cofactor is positioned 10 Å away from the bound substrate and it is likely that the ε-NH2 of an active site lysine, conserved in all flavin-dependent halogenases, intervenes and yields a chloramine [24,53]. The –NH–Cl species would have higher kinetic stability than HOCl and allow for regiospecific placement of the “Cl+” equivalent near the bound substrate, thus explaining the ability of three different flavoenzyme halogenases to act as 5-,6-, or 7-chlorotryptophan forming enzymes (Figure 2b) [54–57]. As in the haloperoxidases that generate “X+” equivalents, the substrates for O2- and FADH2-dependent halogenases are electron rich scaffolds such as phenols, indoles, and pyrroles, which provide the nucleophilic character on the reactive carbon in halogenation transition states [39]. While some members of this halogenase class work on small molecules such as free tryptophan [54,56], others halogenate polyketide and nonribosomal peptide intermediates, tethered as S-pantetheinyl thioesters to carrier protein domains, being built on enzymatic assembly lines [58].
Recently, a systematic genetic profiling approach has been applied to the discovery of new halogenated metabolites [59]. Conserved primers that target flavin-dependent halogenases have been used in a genomic screening of a large library of actinomycete strains. This strategy led to the discovery of novel halogenating enzymes in 20% of screened genomes. Subsequent analysis of the genetic content of a gene upstream and a gene downstream of the corresponding halogenase gene allowed for the prediction of the compound class putatively produced by each of the strains. Products of two of the gene clusters harboring halogenases were isolated and structurally characterized, resulting in two novel chlorinated metabolites, glycopeptide antibiotic CB2364-I and a novel polycyclic xanthone CBS40 (Figure 2c). These findings demonstrate that screening of large strain collections with halogenase probes is a valuable approach not only for genome-guided discovery of novel halogenated compounds, but also allows for the rapid pre-selection of strains that may produce therapeutically useful metabolites.
Halogenation via X•
The discovery of mononuclear iron enzymes using O2 and α-ketoglutarate to effect chlorination rather than hydroxylation of unactivated methyl groups in substrates has been made recently [21,23,60,61]. The first example was the enzyme SyrB2, which generates a 4-chloro-L-threonine residue incorporated into the framework of the nonribosomal lipopeptidolactone syringomycin produced by Pseudomonas syringae (Figure 3a)[23]. This initial discovery has been followed up by characterization of comparable NRPS-associated nonheme iron halogenases active in the biosyntheses of coronatine, barbamide, and dichloroaminobutyrate [21,60,61]. These chloromethyl products can in turn in some cases undergo enzyme-mediated intramolecular elimination to the cyclopropane ring as it is the case in coronamic acid biosynthesis (Figure 3a )[60]. This may be a likely route to the methylcyclopropane ring in the cyanobacterial metabolite curacin [16].
Figure 3.

Halide atom incorporation by mononuclear nonheme iron halogenases. (A) Chlorination in syringomycin and coronamic acid biosyntheses [23,60]. (B) Mononuclear nonheme iron dioxygenases vs mononuclear nonheme iron halogenases [28,63]. (C) Mononuclear nonheme iron halogenases utilize high-valent iron(IV)-oxo intermediates to activate the substrate via hydrogen atom abstraction [28].
The amino acid moiety to be chlorinated is presented to a halogenase active site as an S-pantetheinyl carrier protein. X-ray analysis of SyrB2 revealed that the normal two His, one Asp/Glu “facial triad” ligand set found in related dioxygenases [62] is replaced by a two His set with chloride ion replacing the Asp/Glu due to a mutation of that residue to Ala (Figure 3b) [26]. The reaction path for both hydroxylases and halogenases in this nonheme iron enzyme family proceeds through comparable FeIV=O intermediates that can abstract an H• from the substrate methyl group [28,63]. In hydroxylases there is an OH• rebound while for halogenases Cl•\ rebound is proposed (Figure 3c) to capture a transient primary radical on the substrate. Stopped flow absorbance and rapid freeze quench Mössbauer spectroscopies have allowed for trapping of the high valent oxo iron species in both hydroxylases and halogenases of this class [28,63], confirming the conserved mechanism of hydrogen-atom abstraction by these enzymes.
Conclusions and Unsolved Problems
The combination of microbial genome sequencing, bioinformatic analysis, halogenase purification and mechanistic study, mechanistic organic chemistry and structural biology efforts in the past five years have dramatically changed the paradigms for how halogens are incorporated into natural products. Three new classes of enzymes, fluorinase [40], FADH2- and O2-dependent halogenases [22,24,25,27,39,54,56,58], and nonheme FeII α-ketoglutarate- and O2-dependent halogenases [21,23,28,60], have been characterized. These findings have led to a codification of the kinds of halogenating enzymes and their associated coenzymes that are matched to substrate electronic demand in C-X bond formation. In addition, a nonenzymatic nucleophilic addition of halides has recently been elucidated in a model system [29].
There are still halogenated functional groups in natural products whose origin is as yet unclear. These include the vinyl chloride and alkynyl bromide groups in jamaicamide (Figure 4) [20]. The vinyl chloride group could arise by a convergence of the logic and enzymatic machinery of mononuclear nonheme iron chlorination and the formation of Δ2-isopentenyl-S-carrier proteins-bound intermediates, found in bacillaene [64,65], curacin [66] and myxovirescin A [67] gene clusters. The bromoalkyne origin is mysterious although perhaps it could arise from double hydrogen bromide elimination of a terminal tribromohexanoyl group.
Figure 4.
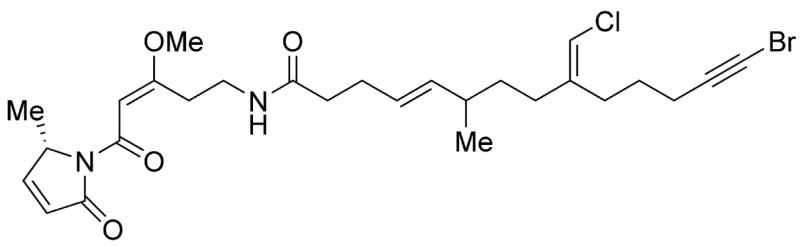
Jamaicamide A, halogenated natural product with unsolved carbon-halogen bond-forming mechanisms [20].
Acknowledgments
We thank Dr. Christopher S. Neumann for careful proofreading of the review. This work was supported in part by NIH grants GM 20011 and GM 49338 to C.T.W. and the Damon Runyon Cancer Research Foundation Fellowship to D.G.F (DRG-1893-05)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Recommended Reading
• of special interest
•• of outstanding interest
- 1.Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Nature’s inventory of halogenation catalysts: oxidative strategies predominate. Chem Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. • The review provides a comprehensive insight into the biological halogenation catalysts. [DOI] [PubMed] [Google Scholar]
- 2.Morrison M, Schonbaum GR. Peroxidase-catalyzed halogenation. Annu Rev Biochem. 1976;45:861–888. doi: 10.1146/annurev.bi.45.070176.004241. [DOI] [PubMed] [Google Scholar]
- 3.van Pee KH. Biosynthesis of halogenated metabolites by bacteria. Annu Rev Microbiol. 1996;50:375–399. doi: 10.1146/annurev.micro.50.1.375. [DOI] [PubMed] [Google Scholar]
- 4.Piraee M, White RL, Vining LC. Biosynthesis of the dichloroacetyl component of chloramphenicol in Streptomyces venezuelae ISP5230: genes required for halogenation. Microbiology. 2004;150:85–94. doi: 10.1099/mic.0.26319-0. [DOI] [PubMed] [Google Scholar]
- 5.Kahne D, Leimkuhler C, Lu W, Walsh C. Glycopeptide and lipoglycopeptide antibiotics. Chem Rev. 2005;105:425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 6.Harris C, Kannan R, Kopecka H, Harris T. The role of chlorine substituents in the antibiotic vancomycin: preparation and characterization of mono- and didechlorovancomycin. J Am Chem Soc. 1985;107:6652–6658. [Google Scholar]
- 7.Kay RR. The biosynthesis of differentiation-inducing factor, a chlorinated signal molecule regulating Dictyostelium development. J Biol Chem. 1998;273:2669–2675. doi: 10.1074/jbc.273.5.2669. [DOI] [PubMed] [Google Scholar]
- 8.Gribble GW. Natural organohalogens - occurrence, sources, quantities, natural function, and benefits. Euro Chlor Science Dossier. 2004:1–77. [Google Scholar]
- 9.Dairi T, Nakano T, Mizukami T, Aisaka K, Hasegawa M, Katsumata R. Conserved organization of genes for biosynthesis of chlortetracycline in Streptomyces strains. Biosci Biotechnol Biochem. 1995;59:1360–1361. doi: 10.1271/bbb.59.1360. [DOI] [PubMed] [Google Scholar]
- 10.Hammer PE, Hill DS, Lam ST, Van Pee KH, Ligon JM. Four genes from Pseudomonas fluorescens that encode the biosynthesis of pyrrolnitrin. Appl Environ Microbiol. 1997;63:2147–2154. doi: 10.1128/aem.63.6.2147-2154.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pelzer S, Sussmuth R, Heckmann D, Recktenwald J, Huber P, Jung G, Wohlleben W. Identification and analysis of the balhimycin biosynthetic gene cluster and its use for manipulating glycopeptide biosynthesis in Amycolatopsis mediterranei DSM5908. Antimicrob Agents Chemother. 1999;43:1565–1573. doi: 10.1128/aac.43.7.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Wageningen AM, Kirkpatrick PN, Williams DH, Harris BR, Kershaw JK, Lennard NJ, Jones M, Jones SJ, Solenberg PJ. Sequencing and analysis of genes involved in the biosynthesis of a vancomycin group antibiotic. Chem Biol. 1998;5:155–162. doi: 10.1016/s1074-5521(98)90060-6. [DOI] [PubMed] [Google Scholar]
- 13.Nowak-Thompson B, Chaney N, Wing JS, Gould SJ, Loper JE. Characterization of the pyoluteorin biosynthetic gene cluster of Pseudomonas fluorescens Pf-5. J Bacteriol. 1999;181:2166–2174. doi: 10.1128/jb.181.7.2166-2174.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang JH, Quigley NB, Gross DC. Analysis of the syrB and syrC genes of Pseudomonas syringae pv. syringae indicates that syringomycin is synthesized by a thiotemplate mechanism. J Bacteriol. 1995;177:4009–4020. doi: 10.1128/jb.177.14.4009-4020.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guenzi E, Galli G, Grgurina I, Gross DC, Grandi G. Characterization of the syringomycin synthetase gene cluster. A link between prokaryotic and eukaryotic peptide synthetases. J Biol Chem. 1998;273:32857–32863. doi: 10.1074/jbc.273.49.32857. [DOI] [PubMed] [Google Scholar]
- 16.Chang Z, Sitachitta N, Rossi JV, Roberts MA, Flatt PM, Jia J, Sherman DH, Gerwick WH. Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine cyanobacterium Lyngbya majuscula. J Nat Prod. 2004;67:1356–1367. doi: 10.1021/np0499261. [DOI] [PubMed] [Google Scholar]
- 17.Chang Z, Flatt P, Gerwick WH, Nguyen VA, Willis CL, Sherman DH. The barbamide biosynthetic gene cluster: a novel marine cyanobacterial system of mixed polyketide synthase (PKS)-non-ribosomal peptide synthetase (NRPS) origin involving an unusual trichloroleucyl starter unit. Gene. 2002;296:235–247. doi: 10.1016/s0378-1119(02)00860-0. [DOI] [PubMed] [Google Scholar]
- 18.Buell CR, Joardar V, Lindeberg M, Selengut J, Paulsen IT, Gwinn ML, Dodson RJ, Deboy RT, Durkin AS, Kolonay JF, et al. The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc Natl Acad Sci U S A. 2003;100:10181–10186. doi: 10.1073/pnas.1731982100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ullrich M, Guenzi AC, Mitchell RE, Bender CL. Cloning and expression of genes required for coronamic acid (2-ethyl-1-aminocyclopropane 1-carboxylic acid), an intermediate in the biosynthesis of the phytotoxin coronatine. Appl Environ Microbiol. 1994;60:2890–2897. doi: 10.1128/aem.60.8.2890-2897.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 21.Ueki M, Galoniæ DP, Vaillancourt FH, Garneau-Tsodikova S, Yeh E, Vosburg DA, Schroeder FC, Osada H, Walsh CT. Enzymatic generation of the antimetabolite γ, γ-dichloroaminobutyrate by NRPS and mononuclear iron halogenase action in a streptomycete. Chem Biol. 2006;13:1183–1191. doi: 10.1016/j.chembiol.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 22.Keller S, Wage T, Hohaus K, Holzer M, Eichhorn E, van Pee KH. Purification and partial characterization of tryptophan 7-halogenase (PrnA) from Pseudomonas fluorescens. Angew Chem Int Ed Engl. 2000;39:2300–2302. doi: 10.1002/1521-3773(20000703)39:13<2300::aid-anie2300>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 23.Vaillancourt FH, Yin J, Walsh CT. SyrB2 in syringomycin E biosynthesis is a nonheme FeII α-ketoglutarate- and O2-dependent halogenase. Proc Natl Acad Sci USA. 2005;102:10111–10116. doi: 10.1073/pnas.0504412102. •• SyrB2 is the first in vitro reconstituted mononuclear nonheme iron halogenase. Herein cofactor requirements of this enzyme are described in detail. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeh E, Blasiak LC, Koglin A, Drennan CL, Walsh CT. Chlorination by a long-lived intermediate in the mechanism of flavin-dependent halogenases. Biochemistry. 2007;46:1284–1292. doi: 10.1021/bi0621213. •• Characterization of a kinetically competent, long-lived intermediate in flavin-dependent halogenase RebH is described. It is postulated that this intermediate is the chloramine species obtained in the reaction of HOCl with an active site lysine residue. [DOI] [PubMed] [Google Scholar]
- 25.Yeh E, Cole LJ, Barr EW, Bollinger JM, Jr, Ballou DP, Walsh CT. Flavin redox chemistry precedes substrate chlorination during the reaction of the flavin-dependent halogenase RebH. Biochemistry. 2006;45:7904–7912. doi: 10.1021/bi060607d. • Authors have carried out kinetic characterization of RebH-catalyzed chlorination. [DOI] [PubMed] [Google Scholar]
- 26.Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL. Crystal structure of the non-haem iron halogenase SyrB2 in syringomycin biosynthesis. Nature. 2006;440:368–371. doi: 10.1038/nature04544. •• The crystal structure of nonheme iron halogenase SyrB2 revealed that the active site iron in Fe(II) state is coordinated by two protein-derived histidine residues and a chlorine atom, thus distinguishing halogenases from related dioxygenases. [DOI] [PubMed] [Google Scholar]
- 27.Dong C, Flecks S, Unversucht S, Haupt C, van Pee KH, Naismith JH. Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science. 2005;309:2216–2219. doi: 10.1126/science.1116510. •• Crystal structure of flavin halogenase PrnA lead to the hypothesis that HOCl is generated during the flavin halogenases catalysis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galonic DP, Barr EW, Walsh CT, Bollinger JM, Jr, Krebs C. Two interconverting Fe(IV) intermediates in aliphatic chlorination by the halogenase CytC3. Nat Chem Biol. 2007;3:113–116. doi: 10.1038/nchembio856. •• Stopped-flow absorption and Mössbauer spectroscopies were used to elucidate mechanism of aliphatic halogenation by mononuclear nonheme iron halogenase CytC3. The presence of two interconverting Fe(IV)-oxo intermediates distinguishes aliphatic halogenases from mononuclear nonheme iron dioxygenases. [DOI] [PubMed] [Google Scholar]
- 29.Perrin CL, Rodgers BL, O’Connor JM. Nucleophilic addition to a p-benzyne derived from an enediyne: a new mechanism for halide incorporation into biomolecules. J Am Chem Soc. 2007;129:4795–4799. doi: 10.1021/ja070023e. •• The authors describe a model study of non-enzymatic non-oxidative halogenation of p-benzyne biradical intermediate in Bergman cyclization of an enediyne. This mode of halogenation is postulated to account for the formation of several monochlorinated marine macrolides. [DOI] [PubMed] [Google Scholar]
- 30.Gribble GW. Natural organohalogens: a new frontier for medicinal agents? J Chem Ed. 2004;81:1441–1449. [Google Scholar]
- 31.van Pee KH. Microbial biosynthesis of halometabolites. Arch Microbiol. 2001;175:250–258. doi: 10.1007/s002030100263. [DOI] [PubMed] [Google Scholar]
- 32.Butler A, Walker JV. Marine haloperoxidases. Chem Rev. 1993;93:1937–1944. [Google Scholar]
- 33.Carter-Franklin JN, Butler A. Vanadium bromoperoxidase-catalyzed biosynthesis of halogenated marine natural products. J Am Chem Soc. 2004;126:15060–15066. doi: 10.1021/ja047925p. [DOI] [PubMed] [Google Scholar]
- 34.Orjala J, Gerwick WH. Barbamide, a chlorinated metabolite with molluscicidal activity from the Caribbean cyanobacterium Lyngbya majuscula. J Nat Prod. 1996;59:427–430. doi: 10.1021/np960085a. [DOI] [PubMed] [Google Scholar]
- 35.Blunt JW, Copp BR, Hu WP, Munro MH, Northcote PT, Prinsep MR. Marine natural products. Nat Prod Rep. 2007;24:31–86. doi: 10.1039/b603047p. [DOI] [PubMed] [Google Scholar]
- 36.Gribble GW. Naturally occurring organohalogen compounds. Acc Chem Res. 1998;31:141–152. [Google Scholar]
- 37.Williamson RT, Pal Singh I, Gerwick WH. Taveuniamides: new chlorinated toxins from a mixed assemblage of marine cyanobacteria. Tetrahedron. 2004;60:7025–7033. [Google Scholar]
- 38.Hager LP, Morris DR, Brown FS, Eberwein H. Chloroperoxidase. II Utilization of halogen anions. J Biol Chem. 1966;241:1769–1777. [PubMed] [Google Scholar]
- 39.van Pee KH, Patallo EP. Flavin-dependent halogenases involved in secondary metabolism in bacteria. Appl Microbiol Biotechnol. 2006;70:631–641. doi: 10.1007/s00253-005-0232-2. [DOI] [PubMed] [Google Scholar]
- 40.O’Hagan D, Schaffrath C, Cobb SL, Hamilton JT, Murphy CD. Biochemistry: biosynthesis of an organofluorine molecule. Nature. 2002;416:279. doi: 10.1038/416279a. [DOI] [PubMed] [Google Scholar]
- 41.Dong C, Huang F, Deng H, Schaffrath C, Spencer JB, O’Hagan D, Naismith JH. Crystal structure and mechanism of a bacterial fluorinating enzyme. Nature. 2004;427:561–565. doi: 10.1038/nature02280. [DOI] [PubMed] [Google Scholar]
- 42.Deng H, Cobb SL, McEwan AR, McGlinchey RP, Naismith JH, O’Hagan D, Robinson DA, Spencer JB. The fluorinase from Streptomyces cattleya is also a chlorinase. Angew Chem Int Ed Engl. 2006;45:759–762. doi: 10.1002/anie.200503582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buchanan GO, Williams PG, Feling RH, Kauffman CA, Jensen PR, Fenical W. Sporolides A and B: structurally unprecedented halogenated macrolides from the marine actinomycete Salinispora tropica. Org Lett. 2005;7:2731–2734. doi: 10.1021/ol050901i. [DOI] [PubMed] [Google Scholar]
- 44.Oh DC, Williams PG, Kauffman CA, Jensen PR, Fenical W. Cyanosporasides A and B, chloro- and cyano-cyclopenta[a]indene glycosides from the marine actinomycete “Salinispora pacifica”. Org Lett. 2006;8:1021–1024. doi: 10.1021/ol052686b. [DOI] [PubMed] [Google Scholar]
- 45.Fenical W, Jensen PR. Developing a new resource for drug discovery: marine actinomycete bacteria. Nat Chem Biol. 2006;2:666–673. doi: 10.1038/nchembio841. [DOI] [PubMed] [Google Scholar]
- 46.Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc Natl Acad Sci U S A. 2007;104:10376–10381. doi: 10.1073/pnas.0700962104. •• Genome sequencing of marine actinomycete Salinispora tropica provides an insight into biosynthesis of a number of secondary metabolites, including anticancer agent salinosporamide A and enediyne-derived chlorinated compounds sporolides A and B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beckwith JR, Clark R, Hager LP. Biological chlorination. VII Studies on the biosynthesis of caldariomycin. J Biol Chem. 1963;238:3086–3090. [PubMed] [Google Scholar]
- 48.Wagenknecht HA, Woggon WD. Identification of intermediates in the catalytic cycle of chloroperoxidase. Chem Biol. 1997;4:367–372. doi: 10.1016/s1074-5521(97)90127-7. [DOI] [PubMed] [Google Scholar]
- 49.Soedjak HS, Walker JV, Butler A. Inhibition and inactivation of vanadium bromoperoxidase by the substrate hydrogen peroxide and further mechanistic studies. Biochemistry. 1995;34:12689–12696. doi: 10.1021/bi00039a027. [DOI] [PubMed] [Google Scholar]
- 50.Butler A, Carter-Franklin JN. The role of vanadium bromoperoxidase in the biosynthesis of halogenated marine natural products. Nat Prod Rep. 2004;21:180–188. doi: 10.1039/b302337k. [DOI] [PubMed] [Google Scholar]
- 51.Gribble GW. Naturally occuring halogenated pyrroles and indoles. Prog Heterocycl Chem. 2003;15:58–74. [Google Scholar]
- 52.Ishihara J, Shimada Y, Kanoh N, Takasugi Y, Fukuzawa A, Murai A. Conversion of prelaureatin into laurallene, a bromo-allene compound, by enzymatic and chemical bromo-etherification reactions. Tetrahedron. 1997;53:8371–8382. [Google Scholar]
- 53.Naismith JH. Inferring the chemical mechanism from structures of enzymes. Chem Soc Rev. 2006;35:763–770. doi: 10.1039/b602313d. [DOI] [PubMed] [Google Scholar]
- 54.Yeh E, Garneau S, Walsh CT. Robust in vitro activity of RebF and RebH, a two-component reductase/halogenase, generating 7-chlorotryptophan during rebeccamycin biosynthesis. Proc Natl Acad Sci U S A. 2005;102:3960–3965. doi: 10.1073/pnas.0500755102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zehner S, Kotzsch A, Bister B, Sussmuth RD, Mendez C, Salas JA, van Pee KH. A regioselective tryptophan 5-halogenase is involved in pyrroindomycin biosynthesis in Streptomyces rugosporus LL-42D005. Chem Biol. 2005;12:445–452. doi: 10.1016/j.chembiol.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 56.Hohaus K, Altmann A, Burd W, Fischer I, Hammer PE, Hill DS, Ligon JM, van Pee K-H. NADH-Dependent halogenases are more likely to be involved in halometabolite biosynthesis than haloperoxidases. Angew Chem Int Ed Engl. 1997;36:2012–2013. [Google Scholar]
- 57.Kling E, Schmid C, Unversucht S, Wage T, Zehner S, van Pee K-H. In: Enzymatic Incorporation of Halogen Atoms into Natural Compounds. Wohlleben W, Spellig T, Muller-Tiemann B, editors. Berlin Heidelberg New York: Springer; 2005. [DOI] [PubMed] [Google Scholar]
- 58.Dorrestein PC, Yeh E, Garneau-Tsodikova S, Kelleher NL, Walsh CT. Dichlorination of a pyrrolyl-S-carrier protein by FADH2-dependent halogenase PltA during pyoluteorin biosynthesis. Proc Natl Acad Sci U S A. 2005;102:13843–13848. doi: 10.1073/pnas.0506964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hornung A, Bertazzo M, Dziarnowski A, Schneider K, Welzel K, Wohlert SE, Holzenkampfer M, Nicholson GJ, Bechthold A, Sussmuth RD, et al. A genomic screening approach to the structure guided identification of drug candidates from natural sources. Chembiochem. 2007;8:757–766. doi: 10.1002/cbic.200600375. •• Genome-guided discovery of novel halogenated natural products by screening of a large strain collection with primers targeting flavin halogenases is described. [DOI] [PubMed] [Google Scholar]
- 60.Vaillancourt FH, Yeh E, Vosburg DA, O’Connor SE, Walsh CT. Cryptic chlorination by a non-haem iron enzyme during cyclopropyl amino acid biosynthesis. Nature. 2005;436:1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 61.Galoniæ DP, Vaillancourt FH, Walsh CT. Halogenation of unactivated carbon centers in natural product biosynthesis: trichlorination of leucine during barbamide biosynthesis. J Am Chem Soc. 2006;128:3900–3901. doi: 10.1021/ja060151n. [DOI] [PubMed] [Google Scholar]
- 62.Costas M, Mehn MP, Jensen MP, Que L., Jr Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 63.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: a high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 64.Calderone CT, Kowtoniuk WE, Kelleher NL, Walsh CT, Dorrestein PC. Convergence of isoprene and polyketide biosynthetic machinery: isoprenyl-S-carrier proteins in the pksX pathway of Bacillus subtilis. Proc Natl Acad Sci U S A. 2006;103:8977–8982. doi: 10.1073/pnas.0603148103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Butcher RA, Schroeder FC, Fischbach MA, Straight PD, Kolter R, Walsh CT, Clardy J. The identification of bacillaene, the product of the PksX megacomplex in Bacillus subtilis. Proc Natl Acad Sci U S A. 2007;104:1506–1509. doi: 10.1073/pnas.0610503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gu L, Jia J, Liu H, Hakansson K, Gerwick WH, Sherman DH. Metabolic coupling of dehydration and decarboxylation in the curacin a pathway: functional identification of a mechanistically diverse enzyme pair. J Am Chem Soc. 2006;128:9014–9015. doi: 10.1021/ja0626382. [DOI] [PubMed] [Google Scholar]
- 67.Simunovic V, Zapp J, Rachid S, Krug D, Meiser P, Muller R. Myxovirescin A biosynthesis is directed by hybrid polyketide synthases/nonribosomal peptide synthetase, 3-hydroxy-3-methylglutaryl-CoA synthases, and trans-acting acyltransferases. Chembiochem. 2006;7:1206–1220. doi: 10.1002/cbic.200600075. [DOI] [PubMed] [Google Scholar]