

Abstract
Cardiac injury is a major complication for oxidative stress-generating anticancer agents exemplified by Adriamycin (ADR). Recently, several histone deacetylase inhibitors (HDACIs) including phenylbutyrate (PBA) have shown promise in the treatment of cancer with little known toxicity to normal tissues. PBA has been shown to protect against oxidative stress in normal tissues. Here, we examined whether PBA might protect heart against ADR toxicity in a mouse model. The mice were i.p. injected with ADR (20 mg/kg). PBA (400 mg/kg/day) was i.p. injected one day before and daily after the ADR injection for two days. We found that PBA significantly decreased the ADR-associated elevation of serum lactase dehydrogenase (LDH) and creatine kinase (CK) activities, and diminished ADR-induced ultrastructual damages of cardiac tissue by more than 70%. Importantly, PBA completely rescued ADR-caused reduction of cardiac functions exemplified by ejection fraction and fraction shortening, and increased cardiac MnSOD protein and activity. Our results reveal a previously unrecognized role of HDACIs in protecting against ADR-induced cardiac injury, and suggest that PBA may exert its cardioprotective effect, in part, by the increase of MnSOD. Thus, combining HDACIs with ADR could add a new mechanism to fight cancer while simultaneously decrease ADR-induced cardiotoxicity.
Keywords: sodium phenylbutyrate, Adriamycin, oxidative stress, histone deacetylase inhibitor, heart, mitochondria, antioxidant
Introduction
Adriamycin (ADR) is a potent anticancer drug that is used in treating both haematological and solid tumors [1]. However, severe cardiomyopathy and heart failure have been observed in ADR-treated cancer patients [2], which limits the clinical dosage of ADR in cancer treatments, i.e. 450 mg/m2 body surface [1]. The anticancer activity and cardiotoxicity of ADR are mediated via different mechanisms [3]. DNA intercalation and DNA topoisomerase II inhibition underscore ADR anticancer activity [4]. Oxidative stress is generally held as the mediating mechanism in the multiple biological processes leading to ADR cardiotoxicity, e.g. redox mediated superoxide radical production [5,6], tissue-specific mitochondrial DNA damage [7], and disturbances of calcium [8,9] or iron homeostasis [10,11]. Structurally, ADR is a quinone, which can generate a large amount of O2•- via a redox cycling reaction catalyzed by several endogenous reductases [5] and endothelial isoform of nitric oxide synthase [6]. O2•- in turn gives rise to a variety of more active reactive oxygen species (ROS), including H2O2, •OH and ONOO-., which trigger further oxidation of bio-molecules [12]. Despite its side effects, ADR remains as an important component in most chemotherapeutic regimens, due to its efficacy in treating a broad-spectrum of cancers. Numerous research have focused on prevention of ADR-induced cardiac injuries [2]. Co-administration of cardioprotective agents, which do not attenuate the anticancer activity of ADR, is one approach [13-17].
Acetylation homeostasis is a major epigenetic mechanism in cancer development and heart dysfunction [18-20], which is tightly regulated by the opposing histone acetyltransferases (HATs) and histone deacetylases (HDACs) [21,22]. Acetylation of histones opens chromatin structure for gene transcription; and acetylation also stabilizes and activates transcription factors for an increased activation of target genes [21]. Acetylation is negatively regulated by HDACs. In mammals, there are four classes of HDACs (class I, IIa, IIb, III and IV) categorized based on homology to yeast HDACs [23]. Acetylation homeostasis can be easily modulated by the ever-growing members of HDAC inhibitors (HDACIs) [21,24], which currently categorized into six structurally distinct groups: short-chain fatty acids, hydroxamates, cyclic tetrapeptides, benzamides, electrophilic ketone, and miscellaneous [23,25].
Phenylbutyric acid (PBA) is a short chain fatty acid that has been clinically tested as an anticancer drug [26]. Towards the normal tissues it not only shows little toxicity, but also provides protections against various stimuli [27-32]. The anticancer activity of PBA is generally attributed to its function as an HDACI [23,26]. However, multiple activities can be assigned to its protection of normal tissues, such as the activity of an HDACI [28], of a chemical chaperone [27,29,30], and of an ammonia sink [31,32].
Based on the previous reports that PBA protects normal tissues against oxidative stress [27,28], we envisioned a possibility that PBA might protect heart from ADR injury. In this study, we investigated the effects of PBA on acute ADR cardiotoxicity in wild type C57BL/6 mice, by echocardiographic characterization, ultrastructural pathology analysis, and measurement of serum lactate dehydrogenase (LDH) and creatine kinase (CK) activities. We also examined the protein and activity levels of the primary antioxidant enzyme, manganese superoxide dismutase (MnSOD), in PBA-treated cardiac tissues.
Materials and methods
Reagents
Regents of the highest grade available were purchased from Sigma (St. Louis, MO, USA), unless otherwise specified. Adriamycin (doxorubicin hydrochloride) was purchased from Pharmacia, Inc. (Kalamazoo, MI, USA). PBA was obtained from Scandinavian Formulas Inc. (Sellesville, PA, USA).
Mice and treatment
Wild-type male mice of inbred strain C57BL/6, 8-12 weeks of age and 22-28 grams of body weight, were randomly divided into four groups, saline, ADR, PBA, and ADR+PBA, and were treated according to Scheme 1. Further characterizations were performed after the treatments. Animal experiments were performed in accordance with NIH policies on the humane care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee, University of Kentucky.
Scheme 1.

Treatment schedule. C57BL/6 mice were divided into four groups: saline, ADR, PBA, and ADR+PBA, and treated with i.p. injection ADR (20 mg/kg) or PBA (400 mg/kg/day). The same volume of saline was used in control treatment.
Echocardiography
Five mice were used in the saline and PBA treatment and seven were used ADR and PBA+ADR treatment. After the treatments, the mice being anesthetized with 2% isoflurane, and echocardiographic images were taken with a Vevo 660 high-resolution Imaging system (VisualSonic, Toronto, Ontario, Canada) equipped with a high-frequency 30 MHz probe. Short axis motion mode (M-mode) images were recorded at the papillary muscle level for cardiac function analysis.
Ultrastructural examination of cardiac tissues
Ultrastructural injury in cardiac tissues was evaluated by electron microscopic analysis on four mice from each treatment groups, using methods described by Yen et al. [33] with minor modifications. Heart tissue was cut into 1 mm3 pieces and fixed in fullstrength Karnovsky′s fixative [4% paraformaldehyde and 5% glutaraldehyde in 0.1 mol/L sodium phosphate buffer (pH 7.4)] for 4 to 5 hours, and then postfixed in Caulfield′s osmium tetroxide with sucrose for 30 to 60 minutes at 4°C. Tissue was dehydrated in graded ethanol series and 100% propylene oxide and embedded in Embed 812. Thin sections were cut with an LKB ultramicrotome (Ultratome NOVA, LKB 2188, Bromma, Sweden) and transferred to copper grids. The grids were stained with lead citrate and uranylacetate, and observed with a Hitachi H-600 electron microscope. Systematic random sampling was achieved by scanning the grid at low magnification so that cell injury was not apparent. Grids were scanned continuously in equal spaces from top to bottom and left to right. Thirty cardiomyocytes were photographed and analyzed for each mouse. Quantification of mitochondrial damage was performed following the strict criteria for mitochondrial injury and image analysis techniques as previously described [34]. Investigators performing morphometry were blinded as to the categories of the samples.
Western blot
To analyze MnSOD protein, heart tissue homogenates were separated by SDS-PAGE and electro-transferred to nitrocellulose membranes. MnSOD and GAPDH (loading control) was first probed with anti-MnSOD (1: 20,000) and anti-GAPDH (1:5,000) (Upstate Biotechnology, Lake Placid, NY, USA), and then hybridized with horseradish peroxidase-conjugated anti-rabbit IgG (1:2,000) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The signals were developed with an enhanced chemiluminescence (ECL Plus) detection system (Amersham Pharmacia Biotech, Piscataway, N.J.), recorded with a fluorescent imager, Storm 860 (Molecular Dynamics, Sunnyvale, CA, USA), and quantified with Quantity One (Bio-Rad Laboratories, Hercules, CA, USA).
MnSOD activity
Manganese superoxide dismutase (MnSOD) activity was measured by a modified nitroblue tetrazolium (NBT) assay, which is based on the competition scavenging of superoxide radicals, generated by xanthine/xanthine oxidase, between SOD and NBT [35]. Briefly, 20 μL of different amounts of total protein from heart tissue homogenate were added to the 160 μL of reaction buffer (50 mM potassium phosphate, pH 7.8, 1 mM diethylenetriaminepenta acetic (DETAPAC), 10 U/ml CAT, 56 μM NBT, 0.1 mM xanthine) in the presence of 5 mM NaCN, a copper-zinc-SOD inhibitor. The rate of reaction was followed for 2 min at 560 nm immediately after adding 20 μL of properly diluted xanthine oxidase. One unit of MnSOD is defined as the amount of protein providing 50% inhibition of NBT reduction.
Serum LDH and CK activity
Within 2 h after blood being collected into serum separator tubes (Becton Dickinson, Rutherford, NJ, USA), serum was obtained by centrifugation at 6,000 g for 6 min. Serum CK and LDH activity in serum was analyzed as previously described [33].
Statistics
Analysis of variance was performed for multiple samples, and statistical significance was determined by using ANOVA. P value < 0.05 was considered to be statistically significant. All data were presented as mean ± SEM.
Results
Echocardiography
Noninvasive and sensitive, echocardiography is used clinically for detecting ADR-induced cardiomypathy [2]. In order to determine whether PBA treatment has any effect on cardiac functions, we performed echocardiographic measurements of mouse left ventricles (Fig. 1A), after the mice received different treatments (Scheme 1). Ejection fraction and fraction shortening were calculated. Prior to receiving any treatment, the mice did not show any difference in the basal values for both parameters (data not shown). After the treatments, the changes of ejection fraction and fraction shortening were normalized as the percentage of the animals’ own basal values (Fig. 1B and C). Compared with the saline control, PBA alone did not significantly change any cardiac function parameters (Fig. 1B and C), indicating PBA had no cardiac toxicity. In contrast, ADR alone significantly decreased both ejection fraction and fraction shortening (Fig. 1B and C). In the combined treatment of PBA and ADR, PBA completely rescued ejection fraction and fraction shortening to the levels of the saline control.
Figure 1.

Chemical structures of Adriamycin (ADR) and sodium phenylbutyrate (PBA).
Ultrastructural pathology
Electron micrographs showed that heart tissues from PBA-treated mice displayed similar ultrastructural features as those from saline-treated mice, demonstrating normal cardiac muscle with numerous mitochondria (M), prominent myofilaments (Myo) and lipid (Lip) (Fig. 2A), confirming the results of echocardiographic analysis that PBA had no cardiac toxicity (Fig. 1). Profound damages were observed in the heart tissues of ADR-treated mice, especially in mitochondria, e.g. mitochondrial vacuolization (V), the presence of myelin figures in mitochondria (solid arrow), disruption of mitochondrial membrane (D), and degeneration of mitochondria (open arrow) (Fig. 2A). Mice that received the combined treatment of PBA and ADR showed much less ultrastructural damages in their heart tissues (Fig. 2). Quantitative analysis of mitochondrial and cellular damages in cardiomyocytes confirmed the visual observation (Fig. 2B). Mice treated with saline and PBA alone showed minimal damages, while ADR-treated mice the highest level on damages. When PBA and ADR were combined, PBA decreased ADR-induced mitochondrial and total cellular ultrastructural damages by ∼ 75% and ∼ 70%, respectively (Fig. 2B). Electron micrographic analysis clearly demonstrated the efficacy of PBA in the reduction of ADR-induced cardiomyocyte injury, revealing the ultrastructural basis for a mechanism by which PBA may rescue cardiac functions upon ADR treatment (Fig. 1).
Figure 2.
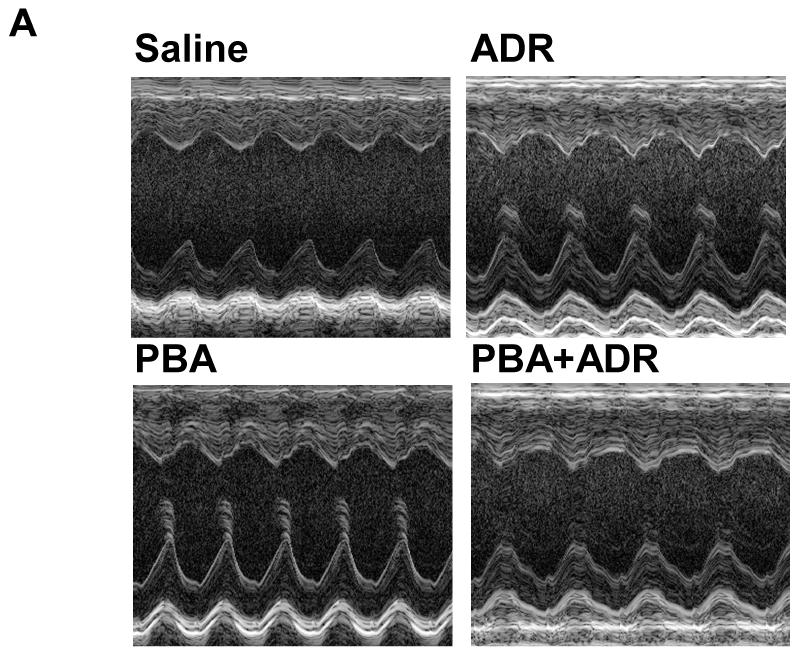


Echocardiograhic analysis of cardiac function. The mice were treated with saline (N = 5), ADR (N = 7), PBA (N = 5), and PBA+ADR (N = 7), respectively. Echocardiographic images were taken after the mice being anesthetized with 2% isoflurane. Short axis motion mode (M-mode) images were recorded at the papillary muscle level for cardiac functional analysis (A). Changes of ejection fraction (B) and fraction shortening (C) were plotted as percentage changes of the basal line values. Data were represented as mean ± SEM * P < 0.05 compared with saline, PBA, and PBA+ADR.
MnSOD
Electron micrographic analysis revealed that PBA rescued severe structural damages of the cardiac mitochondria manifested in the ADR treatment (Fig. 2). Our previous study has shown that induction of MnSOD, the primary antioxidant enzyme residing in mitochondrion, mediates the cardioprotective mechanism of Tamoxifen, an anticancer drug used in breast cancer treatment [17]. Using heterozygous MnSOD knockout (MnSOD+/-) and human MnSOD transgenic mice, we have also demonstrated that increase of MnSOD activity alone leads to protection against ADR cardiotoxicity [17,33,36]. To explore whether MnSOD played a role in the observed cardioprotective effects of PBA, we examined MnSOD levels in the heart samples after mice were treated with ADR or/and PBA. Compared with the saline control, ADR or PBA increased MnSOD both at protein and activity levels (Fig. 3A and B), and combination of PBA and ADR achieved higher increases of MnSOD protein and activity than ADR alone (Fig. 3A and B). There existed a strong linear correlation (R2 = 0.98, P < 0.05) between MnSOD protein and activity levels (Fig. 3C), indicating that the increased MnSOD protein was functionally active.
Figure 3.


PBA protects heart from ADR injuries. (A) Representative electron micrographs (× 11, 800) of mouse cardiomyocytes after the wide type C57BL/6 mice were treated with saline (a), ADR 20 kg/kg (b), PBA 400 mg/kg/day (c), or PBA+ADR (d), respectively, and each treatment group contained four mice. Mice treated with saline and PBA show normal ultrastructural features of cardiac muscle with numerous mitochondria (M), prominent myofilaments (Myo) and lipid (Lip). Mice treated with ADR show profound mitochondrial damage with pathological features such as mitochondrial vacuolization (V), the presence of myelin figures (solid arrow), degeneration of mitochondria (D), and disruption of mitochondrial membrane (open arrow). Mice treated with PBA+ADR show significantly less injuries than mice treated with ADR. (B) Quantitative analysis of damaged area in cardiomyocytes. * P < 0.05 compared with saline control. # P < 0.05 compared with PBA+ADR. + P < 0.05 compared with PBA.
Mitochondrial injury = mitochondrial damage/total mitochondrial area
Cellular injury = total cellular damage (mitochondria + cytoplasm)/total cellular area excluding nuclei.
Serum LDH and CK
Even though not as specific as echocardiographic and ultrastructural pathological analysis, the activities of serum LDH and CK have been widely used clinically as parameters for the diagnosis of cardiac diseases. We tested whether serum LDH and CK could be used as a quick and easy way to characterize the cardioprotective effects of PBA. As shown in Table 1, compared with the saline control, PBA treatment alone did not change the serum level of LDH, but slightly increased the serum level of CK, even though not significantly different from the saline treatment (Tab. 1). ADR alone increased the serum LDH and CK levels by 12 and 38 folds over the saline control, respectively (Tab. 1). The serum LDH and CK in the combined treatments of PBA and ADR were greatly decreased to values that were not significant higher than that of the saline control (Tab. 1).
Table 1.
Serum levels of LDH and CK
| Saline | ADR | PBA | PBA+ADR | |
|---|---|---|---|---|
| LDH activity (μmole/L) | 195 ± 46 | 2335 ± 522* | 263 ± 46 | 615 ± 96 |
| N | 7 | 8 | 7 | 8 |
| CK activity (IU/L) | 419 ± 66 | 16022 ± 3217** | 2315 ± 695 | 4792 ± 889 |
| N | 5 | 8 | 7 | 8 |
Three measurements were performed for each sample.
P < 0.001 compared to saline, PBA and PBA-ADR.
P < 0.001 compared to saline, PBA and PBA-ADR.
There are no significant differences among saline, PBA and PBA-ADR (P > 0.05).
Discussion
We investigated effects of PBA on ADR cardiotoxicity in wild type C57BL/6 mice. Our cardiac functional study showed that PBA improved cardiac functions in ADR-treated mice (Figure 1). Consistent with the functional results, PBA attenuated ADR-induced cardiac unltrastructural damages, decreasing cardiac mitochondrial and total cellular damages by about 75% and 70%, respectively (Fig. 2). Cardiac mitochondria, which occupy about 40% of total intracellular volume of cardiomyocytes [37], are the most affected organelle in ADR-induced oxidative stress [7]. It has been reported that mitochondrial defects leads to cardiomyopathy and heart failure [37], suggesting mitochondrial damages may play a pivotal role in ADR-induced cardiomyopathy and heart failure. We have previously demonstrated that modulation of MnSOD, the primary antioxidant enzyme that resides only in mitochondria, protects against ADR cardiotoxicity [17,33,36]. Consistent with the previous results, our current result demonstrated that PBA increased MnSOD at both protein and activity levels (Fig. 3). Using the human MnSOD transgenic mice model, we have shown that a two-fold increase of MnSOD protein and activity alone leads to a protection against ADR cardiotoxicity [17,33,36]. Interestingly, in the current study there was a two-fold increase of MnSOD protein and activity where PBA demonstrated a protection against ADR cardiac toxicity (Fig. 3), suggesting that MnSOD might play an important mechanistic role in the observed PBA cardio-protective effects. We also showed that PBA greatly decreased the ADR associated elevations of serum LDH and CK activities (Tab. 1), two non-specific but widely used cardiac injury markers. Taken together, our data indicated that PBA protected against ADR cardiotoxicity.
PBA has multiple biological activities, including as HDAC inhibition [28], chemical chaperoning upon endoplasmic reticulum (ER) stress [38,39], ammonia scavenging in urea cycle dysfunction [31,32]. Urea cycle dysfunction in heart has never been reported after decades of intensive research of ADR cardiotoxicity. ER stress has been reported to provide unexpectedly beneficial protection towards ADR toxicity [40]. Thus, it seemed more likely that the cardioprotective effects of PBA observed in our experiment was mediated through HDAC inhibition.
Upon the ADR-induced oxidative insult, transcription of protective genes [41,42] requires the acetylation of histones (H2A,H2B, H3, and H4) and transcription factors, such as RelA, Sp1, and p53 [22,24,43,44]. HDAC inhibitors (HDACIs) may sustain the activation of target genes via inhibition of HDACs [21,24]. More importantly, in cardiac cells, ADR leads to the degradation of p300, a major nuclear histone acetyltransferase, via a p38-mediated phosphorylation-dependent pathway [45,46]; in such a scenario, inhibition of HDACs may attenuate the decrease of acetylation levels due to the destruction of p300.
Even though our current data did not delineate the mechanism by which PBA induced MnSOD, the discovery of Maehara et al provides some helpful insights [24]. They have shown that, in a mouse myoblast cell line (C2C12), HDAC1 (a member of class I HDACs) executes a localized deacetylation of H3 and H4 in the proximal promoter region of MnSOD gene, suppressing MnSOD transcription; and this suppression can be removed by a class I HDACs specific inhibitor, trichostatin A (TSA) , resulting in activation of MnSOD transcription [24]. PBA is also a class I HDAC specific HDACI [23,47]. Thus, PBA should be able to induce MnSOD by inhibiting HDAC1, which might explain the increase of MnSOD in the observed PBA protection against ADR cardiac toxicity.
In current studies of ADR cardiotoxicity, the single high dose model [33,34,48,49] and the low dose chronic model [50,51] are two widely used dosage models, which both provided valuable biological insights in of ADR-induced cardiac injury. For the single high dose model, the dosage, 20 mg/kg, is equivalent to a high dose single injection in cancer patients, such as patients with small cell lung cancer [52]. For the low dose chronic model, a small dose of ADR (e.g. 2 mg/kg) was periodically given to experimental animals over a period of several weeks until the total dosage reaches an equivalent dosage received by patients (e.g. 2,520 mg/70 kg man) [50]. In the current study, we applied the single high dose model (20 mg/kg) and studied that PBA protected heart from acute ADR toxicity. Rosenoff et al. reported that subacute cardiomyopathy occurred four days after a single high dosage of ADR, which is similar to the accumulative ADR-induced cardiomyopathy noted in human [48]. Similarly, our previous and current results revealed mitochondrial ultrastructural injury after a single high dose of ADR (20mg/kg) [33]. Thus, the single high dose model is biologically relevant and serves as a valuable tool to study fatal ADR cardiotoxicity.
In summary, our data showed PBA provided protection against ADR cardiotoxicity, indicating that combining HDACIs with ADR could decrease ADR-induced cardiotoxicity in treatment of cancer. PBA greatly decreased the ADR-associated elevation of serum LDH and CK, ADR-mediated mitochondrial and total cellular ultrastructural damages, completely rescued ADR-caused reduction of ejection fraction and fraction shortening, and increased MnSOD protein and activity in the heart tissues, suggesting that PBA may exert an antioxidant function, via modulation of MnSOD, to protect cardiac tissue against ADR-induced injury.
Figure 4.



PBA treatment increased MnSOD. The mice were treated with saline, ADR, PBA, and PBA+ADR. (A) Western blot analysis showed protein level changes of MnSOD. Data were plotted as mean ± SEM. * P < 0.05 compared to the saline treatment. # P < 0.05 compared to the ADR treatment. (B) Activity level changes were revealed by MnSOD activity assay. Data were plotted as mean ± SEM. * P < 0.05 compared to the saline treatment. # P < 0.05 compared to the ADR treatment. + P < 0.01 compared to the PBA treatment. (C) Correlation between MnSOD protein and activity. Data plotted were the mean values from Western blot analysis and activity assay.
Acknowledgments
This work is supported by NIH grants CA 59797 and CA 94853.
Abbreviations
- ADR
Adriamycin
- HAT
Histone acetyltransferase
- HDAC
Histone deacetylase
- HDACI
HDAC inhibitor
- PBA
Phenylbutyric acid
- LDH
Lactate dehydrogenase
- CK
Creatine kinase
- MnSOD
Manganese superoxide dismutase
- DETAPAC
Diethylenetriaminepenta acetic
- TSA
Trichostatin A
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Horan PG, McMullin MF, McKeown PP. Anthracycline cardiotoxicity. Eur Heart J. 2006;27:1137–1138. doi: 10.1093/eurheartj/ehi702. [DOI] [PubMed] [Google Scholar]
- [2].Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- [3].Wang SW, Konorev EA, Kotamraju S, Joseph J, Kalivendi S, Kalyanaraman B. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms - intermediacy of h2o2- and p53-dependent pathways. Journal Of Biological Chemistry. 2004;279:25535–25543. doi: 10.1074/jbc.M400944200. [DOI] [PubMed] [Google Scholar]
- [4].Darpa P, Liu LF. Topoisomerase-targeting antitumor drugs. Biochimica Et Biophysica Acta. 1989;989:163–177. doi: 10.1016/0304-419x(89)90041-3. [DOI] [PubMed] [Google Scholar]
- [5].Doroshow JH. Effect of anthracycline antibiotics on oxygen radical formation in rat-heart. Cancer Res. 1983;43:460–472. [PubMed] [Google Scholar]
- [6].Vasquezvivar J, Martasek P, Hogg N, Masters BSS, Pritchard KA, Kalyanaraman B. Endothelial nitric oxide synthase-dependent superoxide generation from adriamycin. Biochemistry. 1997;36:11293–11297. doi: 10.1021/bi971475e. [DOI] [PubMed] [Google Scholar]
- [7].Lebrecht D, Kokkori A, Ketelsen UP, Setzer B, Walker UA. Tissue-specific mtdna lesions and radical-associated mitochondrial dysfunction in human hearts exposed to doxorubicin. Journal of Pathology. 2005;207:436–444. doi: 10.1002/path.1863. [DOI] [PubMed] [Google Scholar]
- [8].Kusuoka H, Futaki S, Koretsune Y, Kitabatake A, Suga H, Kamada T, et al. Alterations of intracellular calcium homeostasis and myocardial energetics in acute adriamycin-induced heart-failure. Journal of Cardiovascular Pharmacology. 1991;18:437–444. doi: 10.1097/00005344-199109000-00017. [DOI] [PubMed] [Google Scholar]
- [9].Solem LE, Henry TR, Wallace KB. Disruption of mitochondrial calcium homeostasis following chronic doxorubicin administration. Toxicology and Applied Pharmacology. 1994;129:214–222. doi: 10.1006/taap.1994.1246. [DOI] [PubMed] [Google Scholar]
- [10].Minotti G, Cairo G, Monti E. Role of iron in anthracycline cardiotoxicity: New tunes for an old song? Faseb Journal. 1999;13:594. (vol 13, pg 199, 1999) [PubMed] [Google Scholar]
- [11].Kotamraju S, Chitambar CR, Kalivendi SV, Joseph J, Kalyanaraman B. Transferrin receptor-dependent iron uptake is responsible for doxorubicin-mediated apoptosis in endothelial cells - role of oxidant-induced iron signaling in apoptosis. Journal of Biological Chemistry. 2002;277:17179–17187. doi: 10.1074/jbc.M111604200. [DOI] [PubMed] [Google Scholar]
- [12].Chen Y, Daosukho C, Opii WO, Turner DM, Pierce WM, Klein JB, et al. Redox proteomic identification of oxidized cardiac proteins in adriamycin-treated mice. Free Radical Biology and Medicine. 2006;41:1470–1477. doi: 10.1016/j.freeradbiomed.2006.08.006. [DOI] [PubMed] [Google Scholar]
- [13].Singal PK, Siveskiiliskovic N, Hill M, Thomas TP, Li TM. Combination therapy with probucol prevents adriamycin-induced cardiomyopathy. Journal Of Molecular And Cellular Cardiology. 1995;27:1055–1063. doi: 10.1016/0022-2828(95)90074-8. [DOI] [PubMed] [Google Scholar]
- [14].Seifert CF, Nesser ME, Thompson DF. Dexrazoxane in the prevention of doxorubicin-induced cardiotoxicity. Annals Of Pharmacotherapy. 1994;28:1063–1072. doi: 10.1177/106002809402800912. [DOI] [PubMed] [Google Scholar]
- [15].Chua CC, Liu X, Gao J, Hamdy RC, Chua BHL. Multiple actions of pifithrin-{alpha} on doxorubicin-induced apoptosis in rat myoblastic h9c2 cells. Am J Physiol Heart Circ Physiol. 2006;290:H2606–2613. doi: 10.1152/ajpheart.01138.2005. [DOI] [PubMed] [Google Scholar]
- [16].Li K, Sung RYT, Huang WZ, Yang M, Pong NH, Lee SM, et al. Thrombopoietin protects against in vitro and in vivo cardiotoxicity induced by doxorubicin. Circulation. 2006;113:2211–2220. doi: 10.1161/CIRCULATIONAHA.105.560250. [DOI] [PubMed] [Google Scholar]
- [17].Daosukho C, Ittarat W, Lin SM, Sawyer DB, Kiningham K, Lien YC, et al. Induction of manganese superoxide dismutase (mnsod) mediates cardioprotective effect of tamoxifen (tam) Journal Of Molecular And Cellular Cardiology. 2005;39:792–803. doi: 10.1016/j.yjmcc.2005.07.011. [DOI] [PubMed] [Google Scholar]
- [18].Lund AH, van Lohuizen M. Epigenetics and cancer. Genes Dev. 2004;18:2315–2335. doi: 10.1101/gad.1232504. [DOI] [PubMed] [Google Scholar]
- [19].Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- [20].McKinsey TA, Olson EN. Toward transcriptional therapies for the failing heart: Chemical screens to modulate genes. J. Clin. Invest. 2005;115:538–546. doi: 10.1172/JCI24144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: Importance in inflammatory lung diseases. European Respiratory Journal. 2005;25:552–563. doi: 10.1183/09031936.05.00117504. [DOI] [PubMed] [Google Scholar]
- [22].Saha RN, Pahan K. Hats and hdacs in neurodegeneration: A tale of disconcerted acetylation homeostasis. Cell Death And Differentiation. 2006;13:539–550. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- [24].Maehara K, Uekawa N, Isobe KI. Effects of histone acetylation on transcriptional regulation of manganese superoxide dismutase gene. Biochemical And Biophysical Research Communications. 2002;295:187–192. doi: 10.1016/S0006-291X(02)00646-0. [DOI] [PubMed] [Google Scholar]
- [25].Liu T, Kuljaca S, Tee A, Marshall GM. Histone deacetylase inhibitors: Multifunctional anticancer agents. Cancer Treatment Reviews. 2006;32:157–165. doi: 10.1016/j.ctrv.2005.12.006. [DOI] [PubMed] [Google Scholar]
- [26].Carducci MA, Gilbert J, Bowling MK, Noe D, Eisenberger MA, Sinibaldi V, et al. A phase i clinical and pharmacological evaluation of sodium phenylbutyrate on an 120-h infusion schedule. Clin Cancer Res. 2001;7:3047–3055. [PubMed] [Google Scholar]
- [27].Qi X, Hosoi T, Okuma Y, Kaneko M, Nomura Y. Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Molecular Pharmacology. 2004;66:899–908. doi: 10.1124/mol.104.001339. [DOI] [PubMed] [Google Scholar]
- [28].Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. Journal Of Neurochemistry. 2005;93:1087–1098. doi: 10.1111/j.1471-4159.2005.03077.x. [DOI] [PubMed] [Google Scholar]
- [29].Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce er stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu XL, Done SC, Yan K, Kilpelainen P, Pikkarainen T, Tryggvason K. Defective trafficking of nephrin missense mutants rescued by a chemical chaperone. Journal Of The American Society Of Nephrology. 2004;15:1731–1738. doi: 10.1097/01.asn.0000129826.28932.fd. [DOI] [PubMed] [Google Scholar]
- [31].Scaglia F, Carter S, O′Brien WE, Lee B. Effect of alternative pathway therapy on branched chain amino acid metabolism in urea cycle disorder patients. Molecular Genetics And Metabolism. 2004;81:S79–S85. doi: 10.1016/j.ymgme.2003.11.017. [DOI] [PubMed] [Google Scholar]
- [32].Maestri NE, Brusilow SW, Clissold DB, Bassett SS. Long-term treatment of girls with ornithine, transcarbamylase deficiency. New England Journal Of Medicine. 1996;335:855–859. doi: 10.1056/NEJM199609193351204. [DOI] [PubMed] [Google Scholar]
- [33].Yen HC, Oberley TD, Vichitbandha S, Ho YS, Stclair DK. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. Journal of Clinical Investigation. 1996;98:1253–1260. doi: 10.1172/JCI118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chaiswing L, Cole M, St. Clair D, Ittarat W, Szweda L, Oberley T. Oxidative damage precedes nitrative damage in adriamycin-induced cardiac mitochondrial injury. Toxicologic Pathology. 2004;32:536–547. doi: 10.1080/01926230490502601. [DOI] [PubMed] [Google Scholar]
- [35].Spitz DR, Oberley LW. An assay for superoxide dismutase activity in mammalian tissue homogenates. Analytical Biochemistry. 1989;179:8–18. doi: 10.1016/0003-2697(89)90192-9. [DOI] [PubMed] [Google Scholar]
- [36].Yen HC, Oberley TD, Gairola CG, Szweda LI, St Clair DK. Manganese superoxide dismutase protects mitochondrial complex i against adriamycin-induced cardiomyopathy in transgenic mice. Archives of Biochemistry and Biophysics. 1999;362:59–66. doi: 10.1006/abbi.1998.1011. [DOI] [PubMed] [Google Scholar]
- [37].Goffart S, von Kleist-Retzow JC, Wiesner RJ. Regulation of mitochondrial proliferation in the heart: Power-plant failure contributes to cardiac failure in hypertrophy. Cardiovascular Research. 2004;64:198–207. doi: 10.1016/j.cardiores.2004.06.030. [DOI] [PubMed] [Google Scholar]
- [38].Kubota K, Niinuma Y, Kaneko M, Okuma Y, Sugai M, Omura T, et al. Suppressive effects of 4-phenylbutyrate on the aggregation of pael receptors and endoplasmic reticulum stress. Journal Of Neurochemistry. 2006;97:1259–1268. doi: 10.1111/j.1471-4159.2006.03782.x. [DOI] [PubMed] [Google Scholar]
- [39].Vilatoba M, Eckstein C, Bilbao G, Smyth CA, Jenkins S, Thompson JA, et al. Sodium 4-phenylbutyrate protects against liver ischemia reperfusion injury by inhibition of endoplasmic reticulum-stress mediated apoptosis. Surgery. 2005;138:342–351. doi: 10.1016/j.surg.2005.04.019. [DOI] [PubMed] [Google Scholar]
- [40].Kim SJ, Park KM, Kim N, Yeom YI. Doxorubicin prevents endoplasmic reticulum stress-induced apoptosis. Biochemical and Biophysical Research Communications. 2006;339:463–468. doi: 10.1016/j.bbrc.2005.11.040. [DOI] [PubMed] [Google Scholar]
- [41].Brown HR, Ni H, Benavides G, Yoon L, Hyder K, Giridhar J, et al. Correlation of simultaneous differential gene expression in the blood and heart with known mechanisms of adriamycin-induced cardiomyopathy in the rat. Toxicologic Pathology. 2002;30:452–469. doi: 10.1080/01926230290105604. [DOI] [PubMed] [Google Scholar]
- [42].Lien YC, Noel T, Liu H, Stromberg AJ, Chen KC, St Clair DK. Phospholipase c-delta 1 is a critical target for tumor necrosis factor receptor-mediated protection against adriamycin-induced cardiac injury. Cancer Res. 2006;66:4329–4338. doi: 10.1158/0008-5472.CAN-05-3424. [DOI] [PubMed] [Google Scholar]
- [43].Rahman I, Marwick J, Kirkham P. Redox modulation of chromatin remodeling: Impact on histone acetylation and deacetylation, nf-kappa b and pro-inflammatory gene expression. Biochemical Pharmacology. 2004;68:1255–1267. doi: 10.1016/j.bcp.2004.05.042. [DOI] [PubMed] [Google Scholar]
- [44].Adcock IM, Cosio B, Tsaprouni L, Barnes PJ, Ito K. Redox regulation of histone deacetylases and glucocorticoid-mediated inhibition of the inflammatory response. Antioxidants & Redox Signaling. 2005;7:144–152. doi: 10.1089/ars.2005.7.144. [DOI] [PubMed] [Google Scholar]
- [45].Poizat C, Sartorelli V, Chung G, Kloner RA, Kedes L. Proteasome-mediated degradation of the coactivator p300 impairs cardiac transcription. Molecular And Cellular Biology. 2000;20:8643–8654. doi: 10.1128/mcb.20.23.8643-8654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Poizat C, Puri PL, Bai Y, Kedes L. Phosphorylation-dependent degradation of p300 by doxorubicin-activated p38 mitogen-activated protein kinase in cardiac cells. Molecular And Cellular Biology. 2005;25:2673–2687. doi: 10.1128/MCB.25.7.2673-2687.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. Journal Of Clinical Oncology. 2005;23:3971–3993. doi: 10.1200/JCO.2005.16.600. [DOI] [PubMed] [Google Scholar]
- [48].Rosenoff SH, Olson HM, Young DM, Bostick F, Young RC. Adriamycin-induced cardiac damage in mouse - small-animal model of cardiotoxicity. Journal of the National Cancer Institute. 1975;55:191–194. doi: 10.1093/jnci/55.1.191. [DOI] [PubMed] [Google Scholar]
- [49].Lien YC, Lin SM, Nithipongvanitch R, Oberley TD, Noel T, Zhao Q, et al. Tumor necrosis factor receptor deficiency exacerbated adriamycin-induced cardiomyocytes apoptosis: An insight into the fas connection. Molecular Cancer Therapeutics. 2006;5:261–269. doi: 10.1158/1535-7163.MCT-05-0390. [DOI] [PubMed] [Google Scholar]
- [50].Yi XM, Bekeredjian R, Defilippis NJ, Siddiquee Z, Fernandez E, Shohet RV. Transcriptional analysis of doxorubicin-induced cardiotoxicity. American Journal of Physiology-Heart and Circulatory Physiology. 2006;290:H1098–H1102. doi: 10.1152/ajpheart.00832.2005. [DOI] [PubMed] [Google Scholar]
- [51].Lebrecht D, Setzer B, Ketelsen UP, Haberstroh J, Walker UA. Time-dependent and tissue-specific accumulation of mtdna and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation. 2003;108:2423–2429. doi: 10.1161/01.CIR.0000093196.59829.DF. [DOI] [PubMed] [Google Scholar]
- [52].Piscitelli SC, Rodvold KA, Rushing DA, Tewksbury DA. Pharmacokinetics and pharmacodynamics of doxorubicin in patients with small-cell lung cancer. Clinical Pharmacology & Therapeutics. 1993;53:555–561. doi: 10.1038/clpt.1993.69. [DOI] [PubMed] [Google Scholar]