

Plusbacin A3 is a lipodepsipeptide isolated from a fermentation broth of Pseudomonas sp. PB-6250 obtained from a soil sample collected in the Okinawa Prefecture, Japan (Figure 1).1 The structure of plusbacin A3 was established in 1992. It is a member of a family of lipodepsipeptide natural products that differ either in the structure of their respective fatty acid side chains or in substitution of L-proline for 3-hydroxy-L-proline residues.2
Figure 1.

Structure of plusbacin A3 and its retrosynthetic analysis.
In a recent evaluation, plusbacin A3 displayed strong antibiotic activity against methicillin-resistant Staphylococcus aureus and VanA-type vancomycin-resistant enterococci with minimum inhibitory concentration (MIC) values from 0.78 to 3.13 μg/mL.3 Plusbacin A3 also inhibited incorporation of N-acetylglucosamine into staphylococcal cell wall peptidoglycan with a 50% inhibitory concentration (IC50) that was close to its MIC value. Like vancomycin, plusbacin A3 was found to inhibit nascent peptidoglycan formation; however, unlike vancomycin, plusbacin was also found to inhibit the formation of the lipid intermediates utilized in bacterial cell wall biosynthesis. Interestingly, the activity of plusbacin A3 was not antagonized by the presence of N-acetyl-L-Lys-D-Ala-D-Ala, a tripeptide mimic of the binding domain for vancomycin,3 suggesting that if plusbacin A3 achieves its biological activity through binding to the lipid intermediates or to nascent peptidoglycan it does so at a site on these precursors that is not utilized by vancomycin itself. As a result, plusbacin A3 possesses significant promise for use in the treatment of vancomycin-resistant infections.
The amino acid sequences of the plusbacins were established through Edman degradation of their deacylated products and supported by mass spectrometric studies. Degradation experiments also suggested a lactone linkage between an L-threo-β -hydroxyaspartic acid residue and a 3-hydroxy fatty acid subunit. In the case of plusbacin A3, the fatty acid component is reported to be 3-hydroxyisopentadecanoic acid, although the stereochemical configuration at the hydroxyl stereocenter has yet to be assigned.
Plusbacin A3 also has several non-proteinogenic amino acids embedded in its peptide backbone. In addition to the L-threo-β -hydroxyaspartic acid residue mentioned above, other non-natural amino acids contained in plusbacin A3 include D-threo-β -hydroxyaspartic acid, D-allo-threonine, and trans-3-hydroxy-L-proline. The presence of these non-natural amino acids, coupled with the base sensitivity of the lactone linkage, renders plusbacin A3 a challenging target for total synthesis.
Our retrosynthetic analysis for plusbacin A3 with our selected disconnections is presented in Figure 1. We chose to divide the target molecule into four fragments of approximately equal complexity. This was done in order to provide a measure of flexibility over the amide bond to be made in the final macrolactamization step since there was no solution conformation data available to guide our selection. Our analysis was further guided by a hypothesis that the hydroxyproline residues located at each end of plusbacin A3 may enforce a β-turn type conformation that could influence the intervening residues to adopt a conformation that would facilitate the final macrocyclization event.
Upon closer examination of the structure of plusbacin A3, one is drawn to the possibility that the fatty acid side chain and the arginine residue may play important roles in its biological activity. For example, the fatty acid side chain may be important for membrane localization, while the terminal guanidine group of the arginine residue could be important for binding interactions with diphosphate or carboxylate groups present in the lipid intermediates and/or nascent peptidoglycan. Thus, we wished to devise a synthetic strategy that would easily accommodate variations in the lipophilic side chain and arginine/ornithine residues in order to gain a greater insight into the roles each may have in the biological activity of the plusbacins.
The preparation of fragments 2–4 is presented in Scheme 1. Dipeptide 2 was prepared via coupling of Boc-D-Ser(OBn)– OH with the allyl ester obtained from commercially available trans-3-hydroxy-L-proline (EDCI, DIEA, HOAt, 89%). The C-terminal allyl ester was then cleanly removed (PdCl2(PPh3)3, Ph3SiH, CH2Cl2, 85%) under standard deprotection conditions to provide the C-terminal acid 2 in high overall yield.
Scheme 1.
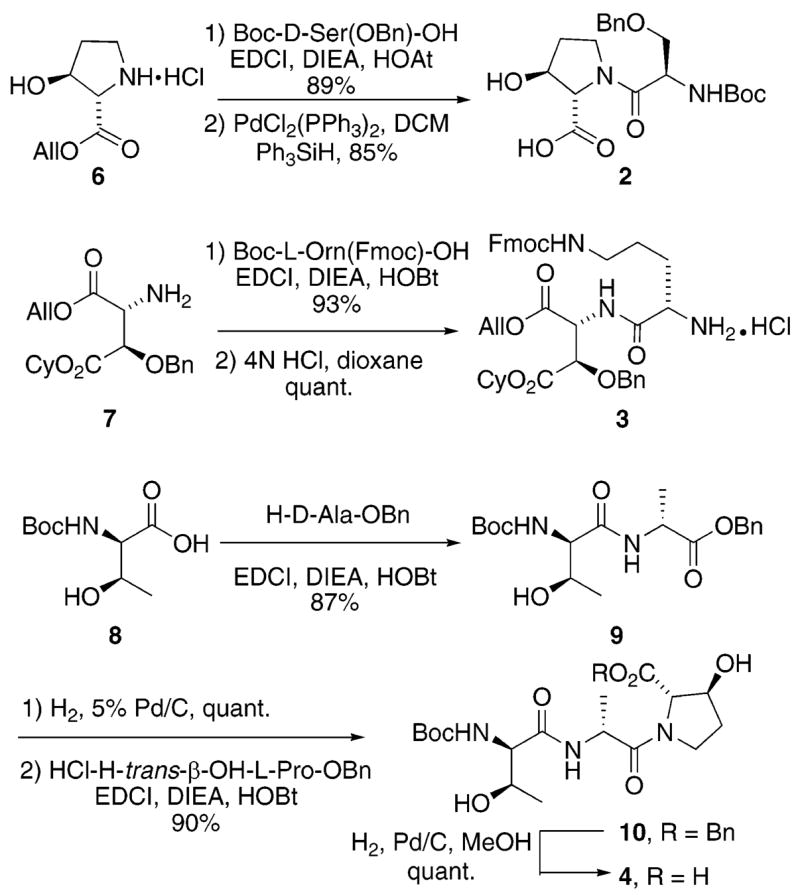
Synthesis of Fragments 2–4
Synthesis of fragment 3 required an independent synthesis of an orthogonally protected D-threo-hydroxyaspartic acid derivative 7. This compound was prepared in eight steps from the Garner’s aldehyde derived from Boc-L-Ser.4 Coupling of this precursor with Boc-L-Orn(Fmoc) – OH (EDCI, DIEA, HOAt, 93%) provided the corresponding dipeptide. Cleavage of the N-terminal Boc protective group (4 N HCl, dioxane, quantitative yield) provided the N-terminal amine 3 as its hydrochloride salt.
The synthesis of tripeptide 4 proceeded from Boc-protected D-allo-threonine that was prepared according to the Elliot protocol.5 Coupling with H-D-Ala–OBn (EDCI, DIEA, HOBt, 87%) provided dipeptide 9 in high yield. Hydrogenolysis of the C-terminal benzyl ester (H2, 5% Pd/C, quantitative yield) followed by coupling of the C-terminal carboxyl group with HCl•H-trans-β -OH-L-Pro–OBn (EDCI, DIEA, HOBt, 90%) provided tripeptide 10. Hydrogenolytic cleavage of the C-terminal benzyl ester (H2, Pd/C, MeOH, quantitative yield) cleanly provided the target tripeptide 4.
Our synthetic route to fragment 5 is provided in Scheme 2. Given the uncertainty regarding the stereochemistry at the lactone stereocenter, we desired a synthetic method that would allow access to a suitably protected β-hydroxyester derivative (e.g., 12) in both enantiomeric forms. The alkene functional group would provide a handle for an olefin cross metathesis reaction that could be used for incorporation of the remainder of the isohexadecanoic acid side chain, as well as numerous additional side chain variations.
Scheme 2.
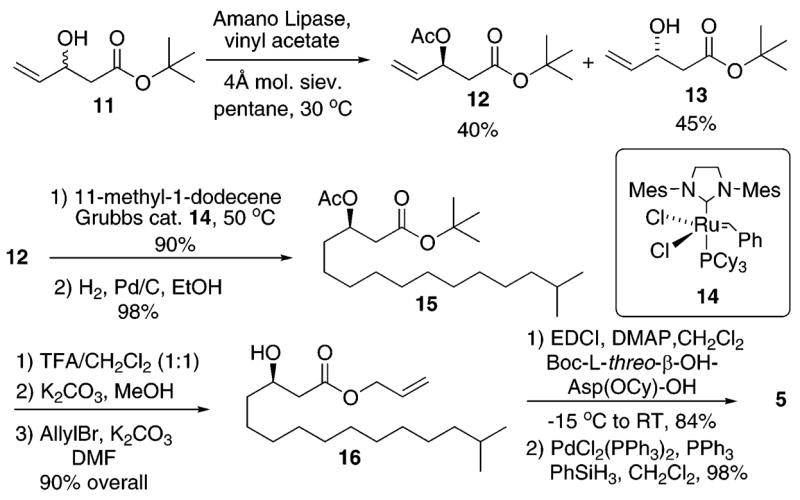
Synthesis of Fragment 5
Our sequence began with a lipase-mediated kinetic resolution of racemic β-hydroxyester 11. The kinetic resolution utilized the Amano PS lipase6–8 and provided enantioenriched β-acetoxyester (S)-12 and (R)-allylic alcohol 13 in good yields and with very high enantiomeric purities as determined by chiral HPLC analysis.9 An alkene cross metathesis reaction between (S)-12 and 11-methyl-1-dodecene employing the Grubbs second generation catalyst 1410 followed by reduction of the alkene intermediate (H2, Pd/C, MeOH) provided β-hydroxyester 15 in good overall yield. Cleavage of the tert-butyl ester (TFA/CH2Cl2) and esterification of the free carboxylic acid (allyl-Br, K2CO3, DMF) provided the allyl-protected β-hydroxyester 16 in 90% overall yield. Finally, coupling of Boc-L-threo-β -OH–Asp(OCy) – OH (EDCI, DMAP, CH2Cl2, 84%) and allyl ester deprotection (PdCl2(PPh3)2, PPh3, PhSiH3, CH2Cl2, 98%) cleanly provided the target fragment 5.
With the four target fragments in hand, the stage was now set for their assembly and elaboration into plusbacin A3. Our fragment assembly began with coupling of fragments 3 and 4. The coupling reaction (EDCI, HOBt, DIEA, THF) cleanly provided pentapeptide 17 in 91% yield. Removal of the N-terminal Boc protective group followed by coupling with carboxylic acid 5 (EDCI, HOBt, THF, 83%) provided ester 18. It is noteworthy that no β-elimination of the chemically sensitive β-acyloxy substituent was observed during the activation/coupling sequence. Boc cleavage (4 N HCl, dioxane, quantitative) and coupling of fragment 2 (EDCI, HOBt, DIEA, DMF, 72%) provided the fully assembled linear lipodepsipeptide precursor 19 for the final macrocyclization reaction.
Our choice for coupling of the N-terminal D-serine residue with the C-terminal β-hydroxyaspartic acid residue was driven by the efficiency of the macrocyclization of the simplified model substrate 24 shown in eq 1. The linear precursor was assembled using standard protection/deprotection and activation/coupling sequences in analogy to those utilized for the assembly of depsipeptide 18. Non-hydroxylated amino acid residues were incorporated into the peptide backbone of the model substrate, and a β-alanine subunit was introduced in place of the depsipeptide linkage. Cyclization of 24 to 25 was cleanly (72% for R = Bn; 65% for R = Ph) with EDCI/HOAt activation.

To set the stage for the final macrocyclization (Scheme 3), the C-terminal carboxyl group was unmasked (PdCl2(PPh3)3, Ph3SiH, CH2Cl2, 95%). Cleavage of the N-terminal Boc protective group (4 N HCl, dioxane, quantitative) provided the amino acid cyclization precursor 21. Carboxyl activation followed by cyclization (EDCI, HOBt, DMF, 48 h) provided the cyclic depsipeptide 22 in 72% yield. Finally, cleavage of the side chain Fmoc protective group (5% piperidine/DMF, quantitative yield) provided depsipeptide 23. Guanidinylation (N,N′-diBoc-N″-triflylguanidine, DIEA, DMF, 70%) followed by cleavage of the cyclohexyl esters (HF, anisole) provided target compound, plusbacin A3, possessing the (R)-configuration at the lactone stereocenter.
Scheme 3. Synthesis of Plusbacin A3—The Final Stagesa.

a (a) EDCI, HOBt, DIEA, THF, 91%; (b) 4 N HCl/dioxane, quantitative; then EDCI, HOBt, 5, DIEA, THF, 83%; (c) 4 N HCl, dioxane, quantitative; (d) EDCI, HOBt, 2, DIEA, DMF, 72%; (e) PdCl2(PPh3)2, Ph3SiH, CH2Cl2, 95%; (f) 4 N HCl, dioxane, quantitative; (g) EDCI, HOBt, 0 °C, DMF, 48 h, 72%; (h) 5% piperidine/DMF, quantitative; (i) N,N′-diBoc-N″-triflylguanidine, DIEA, DMF, 70%; (j) HF, anisole, 20% after HPLC purification.
Our synthetic sample of plusbacin A3 was then compared with an authentic sample of the natural product provided by the Shionogi corporation and a synthetic sample of the plusbacin A3 diastereomer that incorporated the (S)-3-hydroxyisopentanoic acid fragment in the peptide backbone.11 The synthetic substance containing the (R)-3-hydroxyisopentanoic acid residue in the peptide backbone was identical in all respects to an authentic sample of the natural product, thus establishing that the 3-hydroxyisopentanoic acid residue in plusbacin A3 has the (R)-configuration.
In summary, we have described the first total synthesis of plusbacin A3, a promising depsipeptide antibiotic activity against vancomycin-resistant organisms. Future studies are aimed at the study of the structure and function of this very promising antibacterial agent. The results of these studies will be reported in due course.
Supplementary Material
Acknowledgments
Financial support provided by the National Institutes of Health (AI 059327), The Hellman Foundation, and the Regents of the University of California is gratefully acknowledged. M.S.V. also thanks Eli Lilly and Company for support in the form of an Eli Lilly and Company New Faculty Award. We thank the Shionogi Corporation for a gift of plusbacin A3.
Footnotes
Supporting Information Available: Experimental details and spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Shoji J, Hinoo H, Katayama T, Matsumoto K, Tanimoto T, Hattori T, Higashiyama I, Miwa H, Motokawa K, Yoshida T. J Antibiot. 1992;45 doi: 10.7164/antibiotics.45.817. [DOI] [PubMed] [Google Scholar]
- 2.Shoji J, Hinoo H, Katayama T, Nakagawa Y, Ikenishi Y, Iwatani K, Yoshida T. J Antibiot. 1992;45:824–831. doi: 10.7164/antibiotics.45.824. [DOI] [PubMed] [Google Scholar]
- 3.Maki H, Miura K, Yamano Y. Antimicrob Agents Chemother. 2001;45:1823–1827. doi: 10.1128/AAC.45.6.1823-1827.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wohlrab, A.; VanNieuwenhze, M. S. Scheme provided in Supporting Information.
- 5.Elliott DF. J Chem Soc, Chem Commun. 1949:589–594. [Google Scholar]
- 6.Takahata H, Uchida Y, Momose T. Tetrahedron Lett. 1992;33:3331–3332. [Google Scholar]
- 7.Tan CH, Stork T, Feeder N, Holmes AB. Tetrahedron Lett. 1999;40:1397–1400. [Google Scholar]
- 8.Vrielynck S, Vandewalle M. Tetrahedron Lett. 1995;36:9023–9026. [Google Scholar]
- 9.Chiral HPLC analysis of the respective p-nitrobenzyl ether derivatives revealed the enantiomeric purity of each allylic alcohol to be >99%.
- 10.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 11.The requisite fragment containing the (S)-3-hydroxyisopentanoic acid subunit was prepared in six steps from allylic alcohol 13 via the sequence (no acetate deprotection necessary) outlined in Scheme 2.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.