

Abstract
Antibiotic resistance has become a significant public health concern. Antibiotics that belong to new structural classes and manifest their biological activity via novel mechanisms are urgently needed. Lysobactin, a depsipeptide antibiotic has displayed very strong antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA) as well as vancomycin-resistant enterococci (VRE) with minimum inhibitory concentrations (MICs) ranging from 0.39 to 0.78 μg/mL. The MIC values against VRE were more than 50-fold lower than those reported for vancomycin itself. Lysobactin was found to inhibit nascent peptidoglycan formation; however, this activity was not antagonized in the presence of N-acyl-L-Lys-D-Ala-D-Ala, the binding domain on the cell wall precursors that is utilized by vancomycin. Thus, lysobactin represents a promising agent for the treatment bacterial infections due to resistant pathogens. We describe a convergent synthesis of lysobactin that relies upon a highly efficient macrocyclization reaction to assemble the 28-membered cyclic depsipeptide. This synthesis provides the foundation for further study of the mode of action utilized by lysobactin and its analogues.
Introduction
Inhibition of bacterial cell wall (peptidoglycan) biosynthesis is a mechanism exploited by many commonly used antibiotics, including the β-lactams and glycopeptides.1 The peptidoglycan biosynthetic pathway has proven to be a very popular target for the development of chemotherapeutic agents for the treatment of bacterial infections.1 One very important attribute of this biosynthetic pathway as a target for antibacterial agents is that it is unique to bacterial cells; there is no mammalian counterpart. Thus, an agent that inhibits any step in this pathway could be expected to show selective toxicity toward bacterial cells, compromise the integrity of the bacterial cell wall, and result in cell death.2 The historical popularity of the β-lactam antibiotics is in part due to the fact that their molecular target is displayed on the cell surface, thus cellular penetration is not required to achieve their antibiotic effect. The glycopeptide antibiotics (e.g., vancomycin), the antibiotic of last resort for treatment of serious Gram-positive infections, also target a reaction in the cell wall biosynthetic pathway that occurs on the cell surface.
Bacterial resistance to these agents, as well as other classes of antibacterials, has created an urgent need for new chemotherapeutic agents with novel modes of action.3 According to a recent estimate, 20% of people admitted to hospitals have or will develop an infection, and 70% of the bacteria that give rise to these infections are resistant to one or more of the common antimicrobial agents used to fight infections.4 Antibiotics that belong to new structural classes or that function via novel mechanisms are less likely to show cross-resistance with existing antibiotics. Unfortunately, in the period between 1962 and 2000, only one such drug (linezolid) was introduced clinically; all other antibiotics that entered the market during this period were analogues of existing drugs.5 Thus, antibiotics belonging to new structural classes with novel modes of action are urgently needed.
Lysobactin 1 is a cyclic depsipeptide antibiotic produced by a species of Lysobacter (ATCC53042) that was isolated by a group at the Squibb Institute.6, 7 The structure of lysobactin (Figure 1) was determined through a series of chemical and enzymatic transformations and subsequent spectroscopic analysis.8 The core depsipeptide is a 28-membered macrocycle composed of nine amino acid residues with an ester linkage between the hydroxyl group of a β-hydroxyphenylalanine residue and the carboxyl group of a serine residue. A L-Leu-D-Leu dipeptide side chain is appended to the N-terminus of the core depsipeptide. Of the nine amino acids comprising the core structure of lysobactin, five are nonproteinogenic, including four that are β-hydroxylated.6, 8 Three of these residues are β-hydroxylated variants of proteinogenic amino acids (Phe, Leu, and Asn), while the fourth is L-allo threonine (aThr).
Figure 1.

Structure and retrosynthetic analysis of lysobactin.
In work that just preceded the disclosure of lysobactin, Shionogi and Company reported the structure of katanosin B, a cyclic depsipeptide antibiotic that was isolated from a producing organism (PBJ-5356) related to the genus Cytophaga.9 Interestingly, the structure reported for katanosin B was identical to that reported for lysobactin, save for the absolute configuration of the allo threonine residue. In their initial isolation paper, the Shionogi group assigned the allo-threonine residue the D-absolute configuration10 via a protocol that entailed L-leucylation of the amino acid followed by HPLC comparison to reference standards. In a subsequent report, the absolute configuration of the allo-threonine residue katanosin B was changed from D to L.11 Thus, katanosin B and lysobactin have identical structures.
In a recent evaluation, lysobactin displayed very strong antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA) as well as vancomycin-resistant enterococci (VRE) with minimum inhibitory concentrations (MICs) ranging from 0.39 to 0.78 μg/mL.12 The MIC values against VRE were more than 50-fold lower than those reported for vancomycin itself. Lysobactin inhibited the incorporation of isotopically labeled N-acetylglucosamine (GlcNAc) into staphylococcal cell wall peptidoglycan with a 50% inhibitory concentration (IC50 = 0.8 μg/mL) very close to its MIC.12 Lysobactin was also found to inhibit nascent peptidoglycan formation; however, unlike vancomycin, its activity was not antagonized in the presence of N-acetyl-L-Lys-D-Ala-D-Ala.12 Thus, if lysobactin achieves its inhibition of the peptidoglycan biosynthetic machinery through binding to the lipid intermediates (Figure 2) or nascent peptidoglycan, it does so at a site on these precursors that is not utilized by vancomycin itself.
Figure 2.

The latter stages of the peptidoglycan biosynthetic pathway are illustrated in Figure 2. The lipid I intermediate 4 is anchored to the cytoplasmic surface of the cell membrane by a lipid head group where it participates in a glycosyl transfer reaction mediated by MurG. This reaction adds an N-acetylglucosamine residue and provides lipid II 5, the final monomeric intermediate utilized in the peptidoglycan biosynthetic pathway. Lipid II is then translocated to the extracellular surface of the cell membrane where it is a substrate for the transglycosylation and transpeptidation reactions. The transglycosylation utilizes the lipid II intermediate in a polymerization reaction that provides the carbohydrate backbone of peptidoglycan. The transpeptidation reaction results in the installation of a cross-link between peptide chains displayed on adjacent glycan strands.2
Although the mode of action of lysobactin is not precisely understood, the available data suggest that its antibacterial activity may be due to inhibition of the transglycosylation reaction and/or steps in the biosynthetic pathway that precede it (e.g., the MurG reaction). This activity may be the result of binding to the lipid intermediates, a notion supported by antagonism of its antibacterial activity by a cell wall-membrane particulate.12 In this respect, its mode of action would be quite similar to that utilized by ramoplanin, a cyclic depsipeptide antibiotic currently in phase III trials for the treatment of VRE infections.13 As a result of its impressive antibiotic activity, lysobactin possesses significant promise for the treatment of infections owing to vancomycin-resistant organisms.
Several efforts have been directed toward the synthesis of various fragments of the lysobactin core structure.14–16 In addition, a solid-phase synthesis of a structurally simplified lysobactin analogue17 has been reported. Recently, a group at Bayer AG reported the total synthesis and X-ray crystal structure of lysobactin.18 This disclosure has prompted us to report our effort that has culminated in the total synthesis of lysobactin.19
Our retrosynthetic analysis for lysobactin is presented in Figure 1. We chose to construct the depsipeptide macrocycle via an intramolecular coupling between L-HyAsn5 and Gly.6 Coupling reactions employing activated esters of glycine residues are generally very efficient. In addition, since glycine lacks an α-substituent, any possibility of epimerization during the course of the activation/coupling sequence would be avoided. The linear depsipeptide would be assembled via coupling reaction between Leu12 and D-Arg11 on the basis of the literature precedent that suggests amide bond formation reactions between D and L coupling partners generally proceed more efficiently than those employing L,L or D,D coupling partners.20, 21, 22 Thus our target fragments for the assembly of lysobactin were depsipeptide 2, incorporating the L-Leu-D-Leu dipeptide side chain, and tetrapeptide 3.
Ideally, we would have preferred to introduce the dipeptide side chain onto an N-terminal amino group at a late stage of the synthesis since this is an obvious site for future modification. This would require generation of a free amino group (e.g., as in 6) that would be poised to undergo O,N-acyl migration; a reaction frequently observed upon N-deprotection of linear peptides containing N-terminal serine and threonine residues bearing O-acyl protective groups. Adding to our concern, we noted during the earlier work on the structure determination of lysobactin, that two cycles of the Edman degradation reaction provided clean phenylthiohydantion products for cleavage of the first two N-terminal amino acid residues (D-Leu and L-Leu).8 During the third cycle, however, only a portion of the didesleucyl peptide reacted with phenyl isothiocyanate suggesting that structural constraints provided by the macrocycle may at most only partially suppress the propensity for O,N-acyl migration upon generation of a free N-terminal amino group8 (Figure 3).
Figure 3.
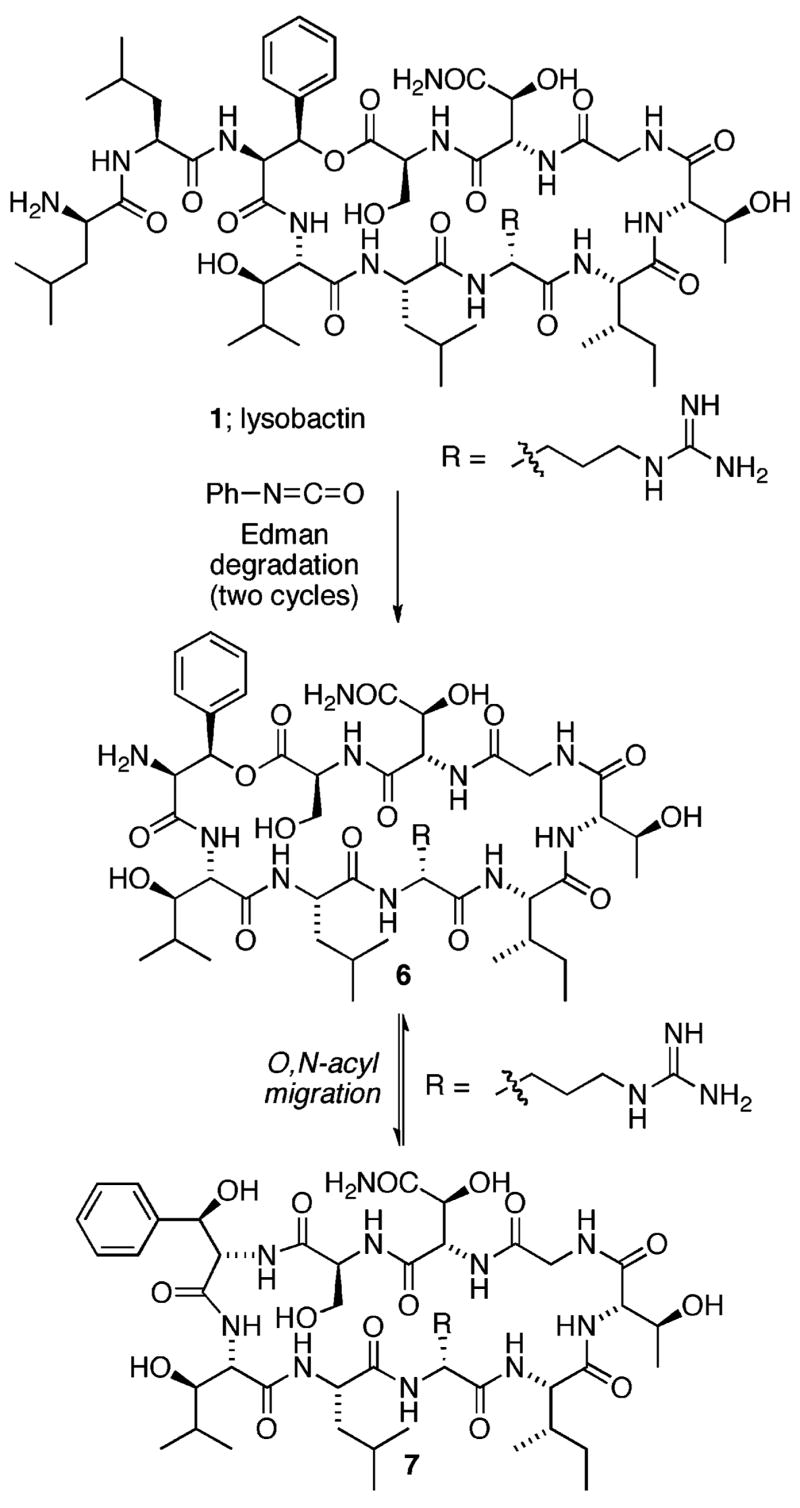
Possible O,N-acyl migration of lysobactin core depsipeptide during Edman degradation.
Results and Discussion
Fragment Synthesis
Our synthesis of fragment 2 (Scheme 1) began with the methyl ester of β-hydroxyphenylalanine 8, protected as the 2-(trimethylsilyl)ethyl carbamate (Teoc), that was readily available via Sharpless asymmetric aminohydroxylation of (E)-methyl cinnamate.23 Cleavage of the Teoc protecting group was efficiently achieved upon exposure of 8 to TFA in dichloromethane and provided the free amine 9. Coupling of 9 with Boc-D-Leu-L-Leu-OH was achieved through carboxyl activation with 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT)24, 25 and provided tripeptide 10 in 80% overall yield. DEPBT was the reagent of choice for all peptide coupling reactions on the basis of its demonstrated ability to suppress racemization at the adjacent α-stereocenter of activated carboxylates.24, 26
Scheme 1.
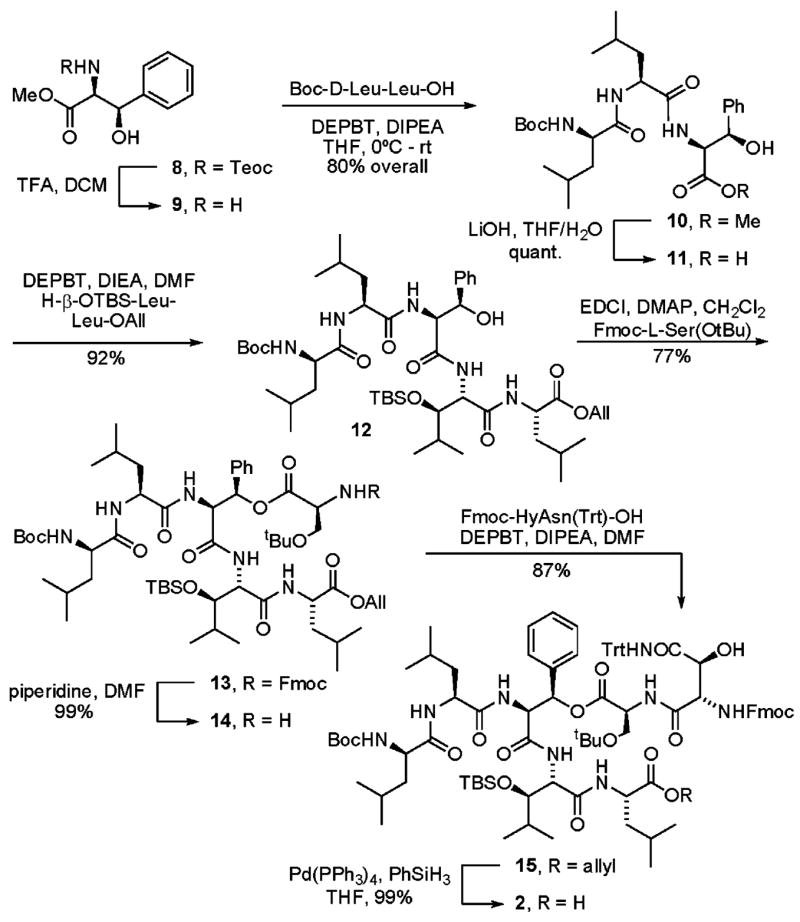
Synthesis of Fragment 2
With tripeptide 10 in hand, we now needed to install a dipeptide fragment at its carboxy terminus and acylate its β-hydoxyl group. In his seminal report of the synthesis of ramoplanin A2 and the ramoplanose aglycon, Boger noted that carboxylate activation of substrates containing β-acyloxy substituents typically resulted in preferential β-elimination, irrespective of the reagents used for carboxyl activation.27, 28 Given this precedent, we chose to install the dipeptide at the carboxy terminus of the tripeptide prior to introduction of the backbone ester. Thus, hydrolysis of methyl ester 10 (LiOH, THF/H2O, quantitative) followed by coupling with H-β-OTBS-Leu-Leu-OAll (DEPBT, DIPEA, DMF, 92%) provided pentapeptide 12. Installation of the backbone ester was achieved through coupling of 12 with Fmoc-L-Ser-OtBu (EDCI, DMAP, CH2Cl2) and provided depsipeptide 13 in 77% yield. The N-terminal Fmoc protective group was cleanly removed (piperidine, DMF, 99%) and the resulting free amine was coupled with Fmoc-HyAsn-(Trt)-OH29 (DEPBT, DIPEA, DMF, 87%) to provide 15. Finally, cleavage of the C-terminal allyl ester (Pd(PPh3)4, PhSiH3, THF, 99%) provided fragment 2.
Our synthesis of fragment 3 (Scheme 2) began with D-allo-Thr-OH 16 that was easily prepared from D-threonine via the Elliott protocol.30 Conversion of the carboxylic acid to its methyl ester (SOCl2, MeOH) followed by coupling with Boc-L-Ile-OH (DEPBT, DIPEA, THF) provided dipeptide methyl ester 17 in 92% overall yield. Hydrolysis of the methyl ester (LiOH, THF/H2O, quantitative) followed by coupling with H-Gly-OAll (DEPBT, DIPEA, THF) provided tripeptide 19 in 95% yield. Removal of the Boc protective group (4N HCl, dioxane) followed by coupling with Fmoc-D-Arg(Boc)2-OH (DEPBT, DIPEA, THF) provided tetrapeptide 20 in 96% overall yield. A final Fmoc cleavage (10% piperidine, DMF, 98%) provided fragment 3.
Scheme 2.
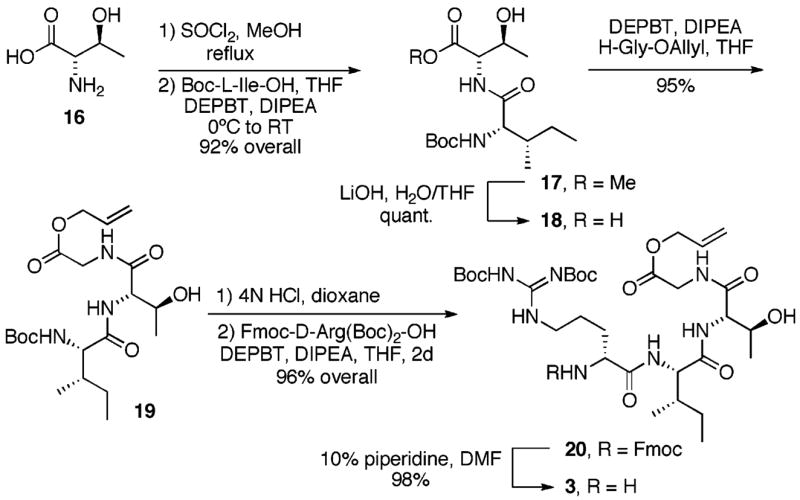
Synthesis of Fragment 3
Macrocyclization and End Game
The final stages of our lysobactin synthesis began with coupling of the C-terminal carboxyl group of 2 with the N-terminal amino group of 3 (Scheme 3). Coupling of carboxylic acid 2 with the free amine 3 (DEPBT, DIEA, DMF, 0°C to room temp) provided depsipeptide 21 in 94% yield. Unmasking of the C-terminal carboxyl group was readily achieved through treatment of 21 with Pd(PPh3)4 and phenylsilane in DMF and provided the free carboxylic acid 22 in 93% yield. The N-terminal Fmoc protective group was cleanly removed upon exposure to piperidine in DMF. Macrocyclization was very efficiently achieved by exposure of the amino acid intermediate to DEPBT (DIPEA, DMF) and provided cyclic depsipeptide 23 in 83% overall yield. A final global deprotection (TFA/H2O, 95:5) provided lysobactin 1 in 33% isolated yield after reverse-phase HPLC purification. The synthetic material was identical in all respects to an authentic sample kindly provided by Shionogi and Company.
Scheme 3.

Final Assembly of Lysobaction
Conclusion
In summary, we have developed a convergent synthesis of the depsipeptide antibiotic, lysobactin. Key to the success of our synthetic route was a two-step deprotection/coupling sequence that provided a 28-membered cyclic depsipeptide in 83% yield. The efficiency of this transformation is noteworthy when viewed in the context of previously reported results for depsipeptide macrocyclization.31, 32 The relatively unhindered structure of the glycine electrophile is one factor that may contribute to the efficiency of the macrocyclization. A second factor may involve preorganization of the cyclization precursor resulting in more favorable cyclization kinetics.33 This attribute has been used to great advantage in the synthesis of ramoplanin A2,13, 27, 34 tyrocidine A,35 and gramicidin S36 and is believed to be due to a β-sheet structure, or minimally a structural element that enforces a β-turn, adopted by the cyclization precursors that entropically enhances the rate of head-to-tail cyclization.
Lysobactin is a promising depsipeptide antibiotic with activity against vancomycin-resistant organisms. Although little is known regarding the mode of action utilized by lysobactin, the available data implicate a target in the bacterial cell wall biosynthetic pathway possibly involving binding/sequestration of the lipid intermediate(s).12 Future work will focus on lucidating the molecular target(s) of lysobactin, identification of the key structural attributes responsible for its biological activity, and structural studies of the complexes lysobactin forms with the lipid intermediates. In addition, spectroscopic studies designed to probe the role that substrate preorganization may play in the overall efficiency of the lysobactin macrocyclization are currently in progress. Results from these studies will be presented in due course.
Supplementary Material
Acknowledgments
Financial support provided by the National Institutes of Health (Grant AI 059327), The Hellman Foundation, and the Regents of the University of California, is gratefully acknowledged. M.S.V. thanks Eli Lilly and Company for support in the form of an Eli Lilly and Company New Faculty Award. A.G.M. thanks the University of California at San Diego for support in the form of GAANN Fellowship. We thank Shionogi and Company for kindly providing a sample of lysobactin (katanosin B).
Footnotes
Supporting Information Available: Experimental procedures and spectroscopic data for compounds 1–3, 10–15, and 17–23. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gale EF, Cundliffe E, Reynolds PE, Richmond MH, Waring MJ. The Molecular Basis of Antibiotic Action. 2. Wiley-Interscience; New York: 1981. [Google Scholar]
- 2.Bugg TDH, Walsh CT. Nat Prod Rep. 1992:199–215. doi: 10.1039/np9920900199. [DOI] [PubMed] [Google Scholar]
- 3.Chu DTW, Plattner JJ, Katz L. J Med Chem. 1996;39:3853–3874. doi: 10.1021/jm960294s. [DOI] [PubMed] [Google Scholar]
- 4.Coates A, Hu Y, Bax R, Page C. Nat Rev Drug Discovery. 2002;1:895–910. doi: 10.1038/nrd940. [DOI] [PubMed] [Google Scholar]
- 5.Walsh C. Nat Rev Microbiol. 2003;1:65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- 6.O’Sullivan J, McCullough JE, Tymiak AA, Kirsch DR, Trejo WH, Principe PA. J Antibiot. 1988;41:1740–1744. doi: 10.7164/antibiotics.41.1740. [DOI] [PubMed] [Google Scholar]
- 7.Bonner DP, O’Sullivan JO, Tanaka SK, Clark JM, Whitney RR. J Antibiot. 1988;41:1745–1751. doi: 10.7164/antibiotics.41.1745. [DOI] [PubMed] [Google Scholar]
- 8.Tymiak AA, McCormick TJ, Unger SE. J Org Chem. 1989;54:1149–1157. [Google Scholar]
- 9.Shoji J, Hinoo H, Matsumoto K, Hattori T, Yoshida T, Matsuura S, Kondo E. J Antibiot. 1988;41:713–718. doi: 10.7164/antibiotics.41.713. [DOI] [PubMed] [Google Scholar]
- 10.Kato T, Hinoo H, Terui Y, Kikuchi J, Shoji J. J Antibiot. 1988;41:719–725. doi: 10.7164/antibiotics.41.719. [DOI] [PubMed] [Google Scholar]
- 11.Kato T, Hinoo H, Terui Y, Kikuchi J, Shoji J. J Antibiot. 1989;42:C-2. [Google Scholar]
- 12.Maki H, Miura K, Yamano Y. Antimicrob Agents Chemother. 2001;45:1823–1827. doi: 10.1128/AAC.45.6.1823-1827.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker S, Chen L, Hu Y, Rew Y, Shin D, Boger DL. Chem Rev. 2005;105:449–475. doi: 10.1021/cr030106n. [DOI] [PubMed] [Google Scholar]
- 14.Armaroli S, Cardillo G, Gentilucci L, Gianotti M, Tolomelli A. Org Lett. 2000;2:1105–1107. doi: 10.1021/ol005659o. [DOI] [PubMed] [Google Scholar]
- 15.Cardillo G, Gentilucci L, Gianotti M, Tolomelli A. Eur J Org Chem. 2000:2489–2494. [Google Scholar]
- 16.Palomo C, Oiarbide M, Ganboa I, Miranda JI. Tetrahedron Lett. 2001;42:8955–8957. [Google Scholar]
- 17.Egner BJ, Bradley M. Tetrahedron. 1997;53:14021–14030. [Google Scholar]
- 18.von Nussbaum F, Anlauf S, Benet-Bucholz J, Häbich D, Köbberling J, Musza L, Telser J, Rübsamen-Waigmann H, Brunner NA. Angew Chem, Int Ed Engl. 2007;46:2039–2042. doi: 10.1002/anie.200604232. [DOI] [PubMed] [Google Scholar]
- 19.Initial disclosure of the work presented in this manuscript: Guzman-Martinez A, Lamer R, VanNieuwenhze MS. 232nd ACS National Meeting; San Francisco, CA. September 10–14, 2006.
- 20.Rich DH, Bhatnagar P, Mathiaparanam P, Grant JA, Tam JP. J Org Chem. 1978;43:296–302. doi: 10.1021/jo00396a027. [DOI] [PubMed] [Google Scholar]
- 21.Brady SF, Varga SL, Freidinger RM, Schwenk DA, Mendlowski M, Holly FW, Veber DF. J Org Chem. 1979;44:3101–3105. [Google Scholar]
- 22.Birch D, Hill RR, Jeffs GE, North M. Chem Commun. 1999:941–942. [Google Scholar]
- 23.Tao B, Schlingloff G, Sharpless KB. Tetrahedron Lett. 1998;39:2507–2510. [Google Scholar]
- 24.Li H, Jiang X, Ye Y, Fan C, Romoff T, Goodman M. Org Lett. 1999;1:91–93. doi: 10.1021/ol990573k. [DOI] [PubMed] [Google Scholar]
- 25.Fan CX, Hao XL, Ye YH. Synth Commun. 1996;26:1455–1457. [Google Scholar]
- 26.Ye YH, Li H, Jiang X. Biopolymers. 2005;80:172–178. doi: 10.1002/bip.20201. [DOI] [PubMed] [Google Scholar]
- 27.Jiang W, Wanner J, Lee RJ, Bounaud PY, Boger DL. J Am Chem Soc. 2002;124:5288–5290. doi: 10.1021/ja020237q. [DOI] [PubMed] [Google Scholar]
- 28.Carboxyl activation with DEPBT was the only exception to this general trend.
- 29.Guzman-Martinez, A.; VanNieuwenhze, M. S. Unpublished results.
- 30.Elliott DF. J Chem Soc, Chem Commun. 1949:589–594. [Google Scholar]
- 31.Davies JS. J Peptide Res. 2003;9:471–501. doi: 10.1002/psc.491. [DOI] [PubMed] [Google Scholar]
- 32.Hamada Y, Shioiri T. Chem Rev. 2005;105:4441–4482. doi: 10.1021/cr0406312. [DOI] [PubMed] [Google Scholar]
- 33.Blankenstein J, Zhu J. Eur J Org Chem. 2005:1949–1964. [Google Scholar]
- 34.Jiang W, Wanner J, Lee RJ, Bounaud PY, Boger DL. J Am hem Soc. 2003;125:1877–1887. doi: 10.1021/ja0212314. [DOI] [PubMed] [Google Scholar]
- 35.Bu X, Wu X, Xie G, Guo Z. Org Lett. 2002;4:2893–2895. doi: 10.1021/ol0263191. [DOI] [PubMed] [Google Scholar]
- 36.Bu X, Wu X, Ng NLJ, Mak CK, Qin C, Guo Z. J Org Chem. 2004;69:2681–2685. doi: 10.1021/jo035712x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.