

Summary
The maturation of dendritic cells (DCs) following exposure to microbial products or inflammatory mediators plays a critical role in initiating the immune response. We now find that maturation can also occur under steady state conditions, triggered by alterations in E-cadherin-mediated DC-DC adhesion. Selective disruption of these interactions induces the typical features of DC maturation including the upregulation of costimulatory molecules, MHC class II, and chemokine receptors. These events were triggered at least in part by activation of the β-catenin pathway. However, unlike maturation induced by microbial products, E-cadherin-stimulated DCs failed to release immunostimulatory cytokines, exhibiting an entirely different transcriptional profile. As a result, E-cadherin-stimulated DCs elicited an entirely different T cell response in vivo, generating T cells with a regulatory as opposed to an effector phenotype. These DCs induced tolerance in vivo and may thus contribute to the elusive steady state “tolerogenic DCs”.
Introduction
Dendritic cells (DCs) reside at the interface of innate and adaptive immunity. As the sentinels of the immune system, immature DCs are distributed in peripheral tissues where they continuously sample the environment by endocytosis (Banchereau and Steinman, 1998). Upon encountering pathogens or a variety of pro-inflammatory mediators, DCs commence a complex and heterogeneous transformation process termed “maturation”, which greatly enhances their capacity for antigen processing and presentation. Maturation may occur prior to, during, or after migration to secondary lymphoid organs where the DCs serve to prime naïve T cells (Banchereau and Steinman, 1998). The general features of DC maturation are well understood (Mellman and Steinman, 2001) and involve the translocation of MHC class II molecules (MHCII) from lysosomal compartments to the plasma membrane, the upregulation of costimulatory molecules such as CD80 and CD86, the activation of lysosomal antigen processing, and the release of a host of immunostimulatory cytokines (Trombetta and Mellman, 2005). There is also a marked increase in the expression of lymphoid chemokine receptors such as CCR7, required for directed migration of DCs to lymph nodes (Randolph et al., 2005). Maturation is most often thought of as being triggered by activation of one or more Toll-like receptors (TLRs), although a variety of pro-inflammatory mediators and T cell products can also induce DCs to mature (Mellman and Steinman, 2001; Trombetta and Mellman, 2005).
Although the phenotypic correlates of DC maturation are clear, their relationship to DC function is complex. For example, depending on the type of microbial stimulus, DCs can prime qualitatively different types of effector T cell responses (Lanzavecchia and Sallusto, 2001). In addition, DCs play a role in maintaining tolerance to self proteins (Steinman et al., 2003). Precisely how DCs accomplish this latter task is unclear, but is thought to involve ingestion of apoptotic cells in peripheral tissues and the presentation of captured self antigens in lymph nodes in a fashion that results in transient stimulation and death of autoreactive T cells (Steinman et al., 2003; Steinman et al., 2000). The maturation state, origin, and phenotype of these “tolerogenic DCs” remain poorly understood.
Recent work has suggested that the features associated with DC maturation can be quite variable. For example, DC maturation and migration to lymph nodes can be independently regulated (Geissmann et al., 2002; Verbovetski et al., 2002), although the underlying mechanisms have not been elucidated. In DCs lacking the TLR adaptor MyD88, the phenotypic maturation of DCs can occur without inflammatory cytokine production (Kaisho et al., 2001). Such DCs cannot activate naïve CD4 T cells in vivo suggesting that this phenotype, should it occur physiologically, might play a role in tolerance (Pasare and Medzhitov, 2004). Indeed, DCs matured by inflammatory cytokines in the absence of TLR agonists may not be able to prime CD4 T cell immunity completely (Lutz and Schuler, 2002; Sporri and Reis e Sousa, 2005).
Can DCs initiate maturation in the absence of inflammatory or microbial stimuli? DCs of the skin, particularly epidermal Langerhans cells (LCs), present an intriguing example. LCs form networks anchored to neighboring keratinocytes via E-cadherin, a component of epithelial cell junctions that is also expressed by LCs (Jakob et al., 1999; Tang et al., 1993). Although these networks are quite stable, LCs appear to traffic to lymph nodes, with their rate of emigration being enhanced by UV exposure or mechanical trauma (Jakob et al., 2001; Merad et al., 2002). How this occurs is unknown, but seems likely to require the disruption of E-cadherin interactions. In epithelial cells, E-cadherin forms a complex with members of the catenin family, which control interactions with the actin cytoskeleton and (after translocation to the nucleus) act as cofactors for TCF/LEF transcriptional activators (Vasioukhin and Fuchs, 2001). Given these functions, the amount of free cytosolic catenins, especially β-catenin, is carefully regulated. Under resting conditions, the bulk of β-catenin is sequestered to the E-cadherin cytoplasmic domain, with the cytosolic pool further attenuated by its phosphorylation by glycogen synthase kinase 3β (GSK3β) and subsequent proteasomal degradation (Nelson and Nusse, 2004; Staal and Clevers, 2005). Activation of Wnt signaling activates TCF/LEF-dependent transcription by increasing free β-catenin due in part to an inhibition of GSK3β.
In LCs, it is unclear whether E-cadherin-mediated cell-cell adhesion is linked to activation of β-catenin signaling, although early work demonstrated that disruption of LC-LC interactions in vitro could trigger phenotypic maturation (Jakob and Udey, 1998; Riedl et al., 2000a; Riedl et al., 2000b). We find that the E-cadherin/β-catenin expression is not limited to LCs, and that loss of E-cadherin adhesion triggers a functionally distinct pathway of maturation that appears more closely linked to maintaining tolerance than to initiating immunity.
Results
Disruption of E-cadherin-mediated contacts induce DC maturation
It has long been observed in primary bone marrow-derived cultures that differentiating DCs form clusters and exhibit spontaneous maturation when the clusters are inadvertently disaggregated (Pierre et al., 1997). Since several components of tight and adherence junctions have been observed in murine DCs (at the mRNA level) (Rescigno et al., 2001), it was conceivable that loss of E-cadherin contacts induced the maturation of bone marrow-derived DCs (BMDCs) as previously suggested for LCs (Riedl et al., 2000a). Indeed, flow cytometry revealed that CD11c+ BMDCs exhibited high amounts of surface E-cadherin after 5-6 days in culture, which remained high even after stimulation with the TLR4 agonist LPS (Suppl. Fig. 1A). Importantly, E-cadherin was responsible for maintaining the cell clusters in these cultures, as they could be dissociated by addition of an inhibitory E-cadherin mAb, as shown previously for LCs (Suppl. Fig. 1B). Physical disruption of the BMDC clusters (accomplished by passing them over magnetic columns), however, triggered all of the morphological features of DC maturation (Mellman and Steinman, 2001). These include the redistribution of MHC class II molecules from lysosomes to the cell surface (Fig. 1A), the down regulation of macropinocytosis (not shown), the upregulation of costimulatory molecules and ability to present peptide to antigen-specific T cells (Suppl. Fig. 1C). Maturation was due at least in part to the loss of E-cadherin contacts, since it could be prevented by adding the E-cadherin blocking antibody either before or during cluster disruption (CD) (Fig. 1B). Interestingly, an Fab fragment failed to inhibit CD-induced maturation (Suppl. Fig. 1D), suggesting that clustering of the surface E-cadherin by bivalent IgG may be required for the inhibition. Maturation was not blocked using non-specific mAb or isotype-matched mAb against the DC integrin CD11b (Fig. 1B), nor did anti-E-cadherin inhibit maturation induced by LPS (see below). Analogous results were obtained using human CD34+ derived LCs (not shown).
Figure 1. Disruption of E-cadherin-mediated clusters results in DC maturation.
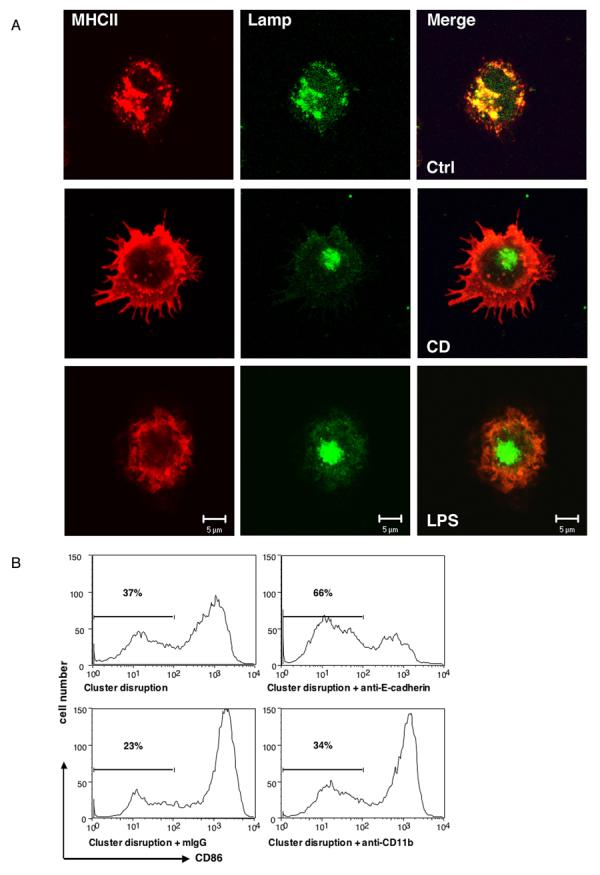
(Panel A) DCs matured after cluster disruption (CD) exhibited similar mophological changes as induced by LPS. DCs matured by CD or LPS were labeled for MHC II (red) and the lysosomal marker Lamp 2 (green). (Panel B) Anti-E-cadherin antibodies can block DC maturation induced by CD. BMDCs were prepared as described and CD11c+ DCs were purified at day 6 and replated at 5 × 105 cells/ml. Treatment with an anti-E-cadherin mAb (Sigma) but not isotype-matched anti-CD11b mAb or mouse IgG inhibited the upregulation of CD86.
Disruption of E-cadherin-mediated adhesion activates a β-catenin-TCF/LEF signaling pathway independent of TLR signaling
DC maturation is exceedingly sensitive to minute amounts of contaminating LPS. To determine if contaminating LPS contributed to the E-cadherin-induced maturation, we compared the signaling events induced by CD to those induced by LPS stimulation of TLR4. TLR signaling is well known to be associated with the activation of NF-κB and p38 MAPK (Barton and Medzhitov, 2003; Takeda and Akira, 2004). As expected, LPS induced a strong activation of both signaling pathways, as revealed by the phosphorylation of IκBαand p38 MAPK (Fig. 2A). In contrast, neither p38 MAPK nor IκBαwas detectably phosphorylated following CD (Fig. 2A). In addition, TLR4−/− DCs, which do not respond to LPS, exhibited robust maturation following CD (Suppl. Fig. 2A), further demonstrating that CD signals maturation independently by a mechanism that is distinct from that due to TLR agonists.
Figure 2. Disruption of the E-cadherin-mediated adhesion activates a distinct β-catenin/TCF signaling pathway independent of TLR signaling.
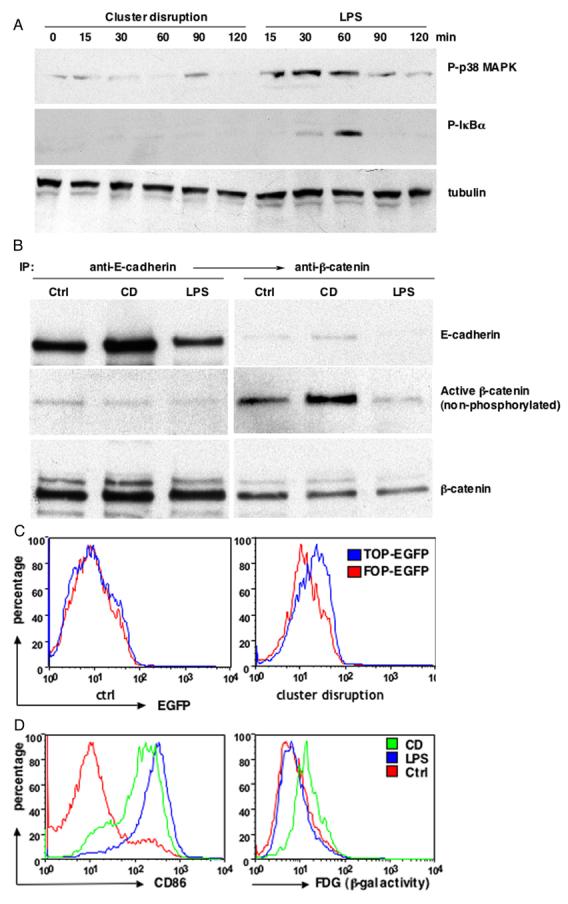
(Panel A) CD did not activate NF-κB and p38 MAPK signaling pathways. Cell lysates from different treatments were analyzed by immunoblotting with anti-phospho-p38 MAPK Ab (top), phosphorylation-specific Ab against IκBα (middle) and anti-tubulin Ab (bottom). (Panel B) CD resulted in activation of β-catenin. BMDCs were either treated with LPS or CD and cell lysates from CD11c+ DCs were subject to sequential immunoprecipitation with antibodies against E-cadherin and β-catenin, followed by immunoblotting with antibodies against E-cadherin (top), active β-catenin (middle) and total β-catenin (bottom). (Panel C) CD resulted in β-catenin/TCF mediated transcription. BMDC cultures were transfected with pLTRH1 containing the TOP-EGFP or FOP-EGFP at day 2 and transfected cells were purified with magnetic columns at day 6. EGFP was measured on CD11c+ DCs immediately after purification (control) or 48 hr later (CD) by FACS.
(Panel D) CD but not LPS treatment led to transactivation of TOPgal reporter. BMDCs from transgenic TOPGAL reporter mice were matured by LPS or CD, β-galactosidase activity was measured by flow cytometry using fluorescein di-β-D-galactosidase (FDG) as a substrate.
The fact that alterations in E-cadherin interactions induced DC maturation raised the possibility that maturation involved the activation of β-catenin. We therefore used a monoclonal antibody that specifically recognizes a non-phosphorylated form of β-catenin induced after canonical Wnt signaling (van Noort et al., 2002). As shown in Figure 2B, CD caused the accumulation of non-phosphorylated β-catenin relative to control or LPS-treated DCs. The active β-catenin was apparently cytosolic as it was not co-precipitated with anti-E-cadherin (Fig. 2B).
We next asked whether CD could drive activation of the β-catenin-dependent transcriptional activators TCF/LEF. This was investigated using two different reporter systems. First, DCs were transduced with retroviruses encoding EGFP under the control of an optimal TCF promoter (TOP-EGFP) or with a construct containing inactive mutant promoter (FOP-EGFP) (Korinek et al., 1997). EGFP production was monitored by flow cytometry after CD. Untreated control DCs expressed similar amounts of EGFP (Fig. 2C, left). 48 hr after CD treatment, however, there was a significant increase in EGFP expression in the TOP-EGFP transfected DCs compared to the FOP-EGFP expressing DCs (Fig. 2C, right), indicating that cluster disruption activated TCF/LEF-dependent transcription. Although the signal obtained using the EGFP reporter was less than that obtained after Wnt activation using a luciferase assay with the same reporter system, unlike luciferase, EGFP signals are not amplified. Indeed, a similarly modest EGFP increase was observed in MDCK cells expressing the same constructs following Wnt activation by lithium treatment (which inhibits GSK3β) (Suppl. Fig. 2B). A >10 fold increase was generated using the luciferase assay in MDCK cells under the same conditions (Lyons et al., 2004). In our experiments, it was necessary to use an EGFP reporter to identify the small fraction (<10%) of productively infected DCs by flow cytometry.
To overcome the quantitative limitation of retrovirus approach, we next took advantage of transgenic mice that uniformly express the TOPGAL reporter (DasGupta and Fuchs, 1999). CD as well as LPS treatment resulted in strong maturation with DCs prepared from transgenic mice (Fig. 2D, left). However, only cluster disruption produced a significant increase in β-galactosidase activity, indicative of TCF/LEF activation (Fig. 2D, right).
Together, the reporter assays provide direct evidence that disruption of E-cadherin-mediated adhesion activates the β-catenin-TCF/LEF signaling pathway in DCs.
Activation of β-catenin signaling plays a role in DC maturation
To determine if activation of β-catenin signaling pathway might actually contribute to DC maturation, we first used a pharmacologic approach. SB216763 is a selective inhibitor of GSK3β(Coghlan et al., 2000), the kinase whose phosphorylation of β-catenin marks it for degradation by the proteasome. As expected, treatment of immature cells with SB216763 resulted in a dose-dependent accumulation of β-catenin in the cytosol in both murine (Fig. 3A) and human DCs (not shown), as assayed by cell fractionation and Western blot.
Figure 3.
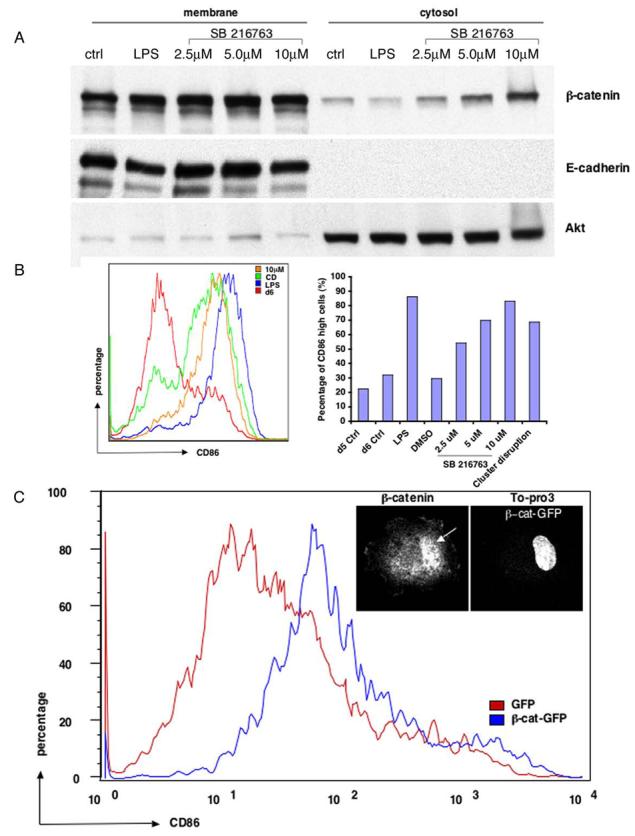
Activation of β-catenin signaling pathway induces DC maturation (Panel A) Dose-dependent accumulation of cytosolic β-catenin after treatment with GSK3β inhibitor SB216763. BMDCs were treated with either LPS or different doses of SB216763. CD11c+ DCs were then fractionated into membrane and cytosolic fractions, followed by immunoblotting with antibodies against β-catenin (top) and E-cadherin (middle). Akt was probed as a loading control (bottom). (Panel B) Inhibition of GSK3β results in DC maturation. CD11c+ DCs after different stimuli were subject to FACS analysis. The left histogram overlay shows a representative FACS profile of CD86 expression for each condition, with SB216763 at 10 μm. CD86high cells represent mature DCs on the right. (Panel C) Expression of β-catenin enhanced spontaneous DC maturation. BMDC cultures were transfected either with GFP or β-catenin-GFP and were subject to FACS analysis for CD86 expression at day 6. Expression of β-catenin-GFP but not GFP induced CD86 upregulation, although not as strongly as after CD or drug treatment. Insert: β-catenin translocates to the nucleus. 12 hr after CD of DCs expressing β-catenin-GFP, cells were fixed, labeled with a β-catenin antibody and the DNA dye TO-Pro3, and imaged by confocal microscopy. β-catenin was clearly translocated into the nucleus (arrow).
SB216763 not only stabilized β-catenin, but also was a potent inducer of DC maturation. When the GSK3β inhibitor was added to immature DCs, the mature population (defined as the percentage of cells expressing high CD86) increased in a dose-dependent manner (Fig. 3B). Indeed, at 10 μM, SB216763 was nearly as effective as LPS at triggering DC maturation, and slightly more effective than CD. Similar results were obtained for human CD34+-derived DCs (not shown). By immunofluorescence, it was clear that DCs treated with the inhibitor assumed the classical mature DC phenotype, with MHC class II molecules redistributing from lysosomes to the plasma membrane (not shown; see Fig. 1A).
Although these results suggested that activation of β-catenin by either CD or inhibition of GSK3β led to DC maturation, both treatments may have relevant downstream targets other than β-catenin. Therefore, we asked if a selective increase in β-catenin could induce DC maturation. A recombinant retrovirus was used to express β-catenin-GFP. As shown in Figure 3C, expression of β-catenin-GFP, but not of GFP alone, resulted in a significant increase in the fraction of CD86-high mature DCs. As expected if β-catenin were responsible for activation, the expressed GFP construct was at least partly localized to the nucleus (Fig. 3C inset, arrow). Similar results were obtained using a virus encoding a stabilized mutant β-catenin lacking the GSK3βphosphorylation sites (GSK*β-catenin-GFP), although expression of a GSK*β-catenin-GFP also lacking its C-terminal transcriptional transactivation domain (Votin et al., 2005) failed to induce DC maturation (Suppl. Fig. 2C), suggesting that the β-catenin-induced DC maturation is dependent on TCF/LEF-mediated transcription activity. Although the extent of CD86 upregulation due to β-catenin expression was not as great as found for CD or SB216763 treatment, the GFP tag may have interfered with β-catenin, or the concentrations of β-catenin achieved may have been too low for optimal maturation. Alternatively, β-catenin may work synergistically with other components that may be targets of GSK3β or E-cadherin activation.
Taken together, these results strongly suggest that activation of the β-catenin signaling pathway, by CD, inhibition of GSK3β, or expression of exogenous β-catenin, can at least in part induce a mature DC phenotype.
DCs matured by cluster disruption exhibit a distinct transcriptional profile
We performed a genome-wide microarray analysis to study the expression profiles of DCs matured by CD as opposed to a conventional TLR agonist (E. coli, which stimulates multiple TLRs). RNA was isolated from human CD34+ DCs at various times after stimulation and used to probe Affymetrix U95Av2 chips. >700 genes were found differentially regulated upon maturation by either CD or bacterial stimulation (Fig. 4A and Suppl. Fig. 3). Cluster analysis revealed that following an early phase (1-3 hr) of similarity, expression profiles exhibited by the two sets of DCs diverged significantly at later time points (>6 hr) (Fig. 4A). A large number of transcripts were markedly upregulated (red) in the bacteria-stimulated set that remained relatively unchanged or actually decreased (blue) in the cluster-disrupted set. There were some transcripts upregulated in cluster-disrupted cells, however, with at least some of these increases prevented by including anti-E-cadherin mAb under conditions that blocked maturation. Clearly, the transcriptional events induced by alteration of E-cadherin-mediated adhesion were quite distinct from those induced by TLR activation.
Figure 4. CD-matured human DCs failed to produce inflammatory cytokines.
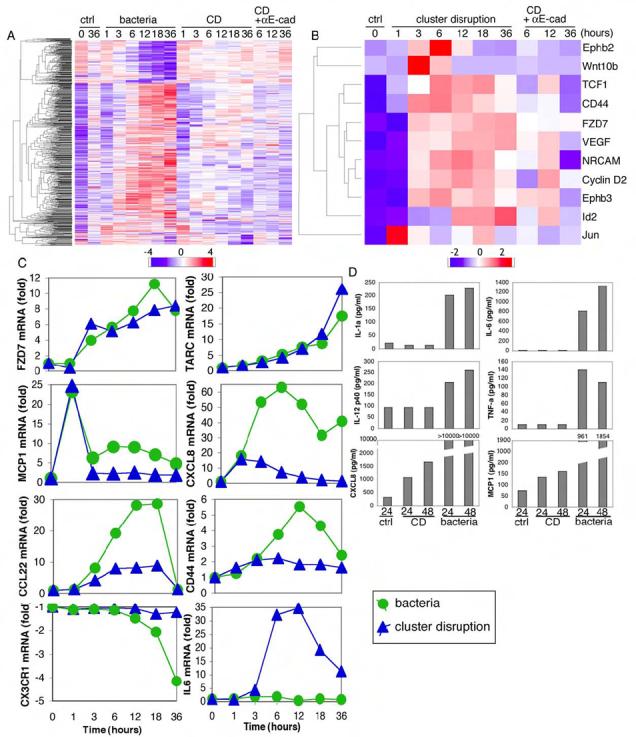
(Panel A) More than 700 genes were differentially regulated upon maturation by either CD or bacterial stimulation. Heatmap was generated as detailed in the Experimental Procedures. (Panel B) CD led to upregulation of 10 direct β-catenin/TCF target genes. Target genes were selected according to R. Nusse and colleagues and heatmap was created as described in Experimental Procedures. Wnt10b was not a target gene but was included for comparison. (Panel C) Representative gene expression profiles were plotted from the microarray data. (Panel D) Human CD34+ DCs matured by CD did not produce inflammatory cytokines. Luminex assays for multiple cytokines and chemokines were performed on supernatants from CD or bacteria-matured DCs. One of two independent experiments is shown.
We next asked if any targets of β-catenin-induced transcription were upregulated in DCs stimulated by CD. Guided by the gene set compiled for various cell types by R. Nusse and colleagues (http://www.stanford.edu/~rnusse/pathways/targets.html), we identified increases in at least 10 β-catenin-TCF/LEF targets, including: Ephb2, TCF1, CD44, FZD7 (Frizzled homolog 7; a component of the Wnt pathway), VEGF, cyclin D2, Ephb3, and Id2 (Fig. 4B). Each of these inductions occurred after a lag of 1-3 hr and each was partially inhibited by anti-E-cadherin mAb (Fig. 4B), suggesting that CD activated at least some elements of the β-catenin signaling pathway.
We then quantified the extent to which selected transcripts were upregulated by CD vs. TLR stimulation (Fig. 4C). Several were enhanced by both stimuli including FZD7 as well as the chemokines TARC and MCP-1. The chemokines IL-8 (CXCL8) and CCL22 as well as the adhesion protein CD44 were also enhanced by both stimuli, albeit to a greater extent by bacteria. In contrast, a cluster of genes including chemokine receptor CX3CR1 was strongly downregulated by TLR signaling but not by CD (Fig. 4C, lower left). Few if any genes were upregulated to a greater extent by CD than by TLR stimulation (with TARC and Wnt10b as potential exceptions [not shown]). Most striking, however, was the fact that genes encoding inflammatory cytokines, such as IL-6, were dramatically upregulated by TLR stimulation, but not at all by CD (Fig. 4C, lower right).
DCs matured by cluster disruption neither produce nor secrete inflammatory cytokines
As expected, treatment of human DCs with bacteria greatly enhanced release of the inflammatory cytokines IL-1α, IL-6, TNF-α, and IL-12 p40 24-48 hr after stimulation, as determined by Luminex assay (Fig. 4D). DCs matured by CD, however, failed to secrete any of these cytokines above background levels. Consistent with the array results, the release of several chemokines including CXCL8, MCP-1 and MIP-1α were enhanced after CD, albeit in amounts far lower than DCs matured by bacteria (Fig. 4D and data not shown). Thus, activation via E-cadherin upregulated the chemokine pathway without inducing inflammatory cytokines in human CD34+-derived DCs.
Similar results were obtained for murine BMDCs. By RT-PCR, CD did not induce significant increases in transcription of genes encoding TNF-α, IL-6, IL-1β, or IL-12p40. In contrast, LPS treatment resulted in dramatic (if sometimes transient) increases in each of these inflammatory markers (Fig. 5A).
Figure 5. CD-matured murine BMDCs upregulated CCR7 without inflammatory cytokine induction.
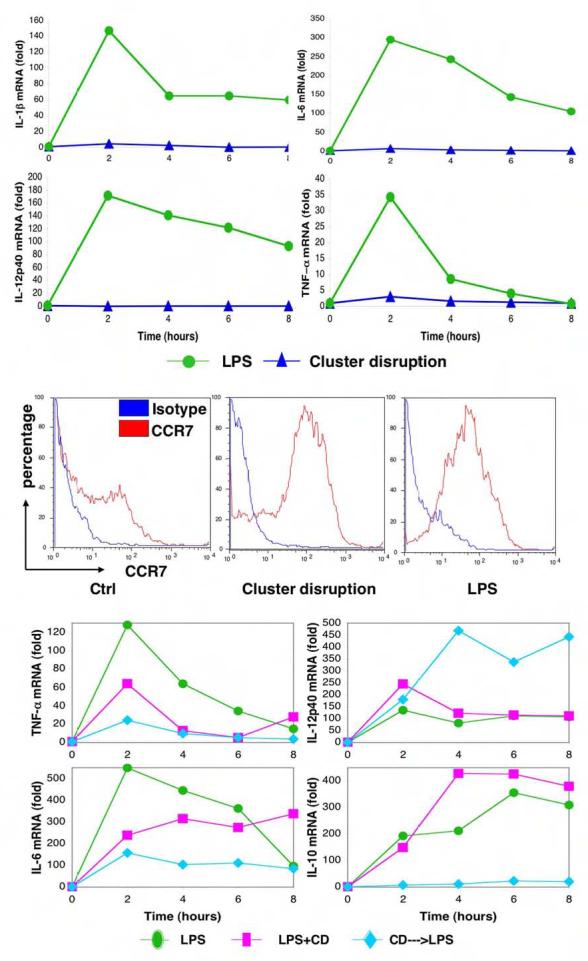
(Panel A) CD-matured murine BMDCs did not induce inflammatory cytokines IL-1β, IL-6, IL-12p40 and TNFα. Real-time RT-PCRs were performed on total RNA isolated from DCs treated with either LPS or CD for the indicated times, the expression of each gene then was normalized to β-actin expression. (Panel B) CD-matured BMDCs express elevated level of surface CCR7. DCs untreated or matured by either CD or LPS were subjected to FACS analysis. (Panel C) Addition of LPS after cluster disruption synergistically enhances or inhibits cytokine production. Real-time RT-PCRs were performed and analyzed as described in Panel A. Cluster-disrupted DCs were stimulated with LPS simultaneously (CD+LPS) or LPS was added 14-18 hr afterwards for the indicated times (CD--->LPS). Results from one of three different sets of samples are shown.
CD treatment of BMDCs did lead to upregulation of CCR7, both at the mRNA level (not shown) and on the plasma membrane (Fig. 5B). CCR7 is a chemokine receptor important for the migration of DCs from the periphery to T cell areas of lymph nodes (Randolph et al., 2005), so that in principle, activation of the E-cadherin pathway in vivo would result in cells capable of migrating to secondary lymphoid organs. These results suggested that the loss of E-cadherin-mediated adhesion might provide a spatial cue for the generation of mature, migratory DCs but without the ability to induce T cell immunity.
To determine if the two maturation signals synergize or compete with each other, we next examined the cytokine profiles of DCs matured by LPS alone or by both CD and LPS. In general, addition of LPS at the time of CD yielded a phenotype more similar to LPS alone than to CD alone (TNF-α, IL-6, IL-10, and IL12 p40; Fig. 5C). This finding suggested that the TLR signal was dominant to the E-cadherin-induced signal, at least when presented simultaneously. A rather different set of results was obtained if LPS was added to DCs that had been matured by CD for over 12 hr beforehand. LPS could no longer induce the transcription of IL-10 mRNA, and only partly induced IL-6 and TNF-α transcription. Transcription of IL-12p40 and IL-1β, on the other hand, was more effectively induced (Fig. 5C and not shown). Thus, although LPS could enhance cytokine secretion if provided during the CD step, prior E-cadherin activation either prevented or enhanced (in the case of IL-12) the LPS effects.
This prompted us to investigate whether different maturation signals regulated the ability to activate naïve T cells. Fixed DCs that had been pulsed with OVA protein and matured by different treatments were tested for their ability to activate OVA-specific OTII CD4 and OTI CD8 T cells in vitro. DCs matured by CD elicited both CD4 and CD8 T cell responses, while LPS-matured DCs were able to stimulate only CD4 T cells (Suppl. Fig. 4). Although this observation was made previously (Delamarre et al., 2003), the current data allow a direct, quantitative comparison of both maturation signals. Indeed, sequential treatment by CD followed by LPS caused a rather substantial synergistic increase in cross-presentation to OT1 cells. These data emphasize that cluster disruption matures DCs through a mechanism distinct from TLR signaling. For both CD8 and CD4 responses, the addition of LPS to CD-matured DCs greatly enhanced the extent of T cell activation (Suppl Fig. 4), consistent with the synergy observed in cytokine production. Thus, E-cadherin-induced maturation program could modulate subsequent LPS stimulation to enhance T cell response.
DCs matured by E-cadherin vs. TLR activation elicit distinct T cell responses in vivo
Since DCs matured by CD alone could efficiently present antigen but did not produce inflammatory cytokines, they might not be immunogenic in vivo. We thus compared the ability of DCs matured by CD or CD followed by LPS to elicit T cell responses in mice. Both types of mature DCs were incubated in vitro with OVA-derived peptides and then injected into C57B/6 mice. Consistent with our in vitro results, immunization with either population of mature DCs led to the proliferation of adoptively transferred CD4 and CD8 T cells transgenic for OVA-specific T cell receptors (not shown). Thus, the injected DCs could encounter and stimulate naïve, but antigen-specific T cells in vivo.
We next asked if the DCs could actually stimulate immunity: can they prime naïve CD4 T cells to become IFN-γ-producing effectors in vivo? DCs were matured by CD alone or sequentially by CD and LPS, loaded with OVA peptide 323-339, and injected three times over a one week period (see Methods). Three days after the last injection, splenocytes were isolated and restimulated in vitro with the OVA peptide. While sequentially matured DCs led to a dramatic increase in the production of IFN-γduring this recall response, DCs matured by CD alone failed to prime CD4 T cells to produce IFN-γ but did generate high levels of IL10 (Fig. 6A) together with other cytokines (not shown) consistent with type I regulatory T cells (O'Garra and Vieira, 2004). Furthermore, while both DCs expanded the overall population of activated CD4+CD25+ T cells to similar extents (not shown), only immunization with DCs that had been matured by CD and LPS generated IFN-γ-producing CD4 T cells in peripheral lymph nodes (Fig. 6B, middle). In stark contrast, DCs matured by CD induced only IL10-producing T cells (Fig. 6B, top). These cells also were found to exhibit elevated amounts of the transcription factor Foxp3, another hallmark of regulatory T cells (AJ and IM, unpublished observations). Finally, DCs matured by CD failed to generate a significant antibody response as compared to DCs matured by both CD and LPS (AJ and IM, unpublished observations). Thus, DCs matured by CD alone were not immunogenic and instead induced the production of IL10-producing, putative regulatory T cells.
Figure 6. DCs matured by CD alone generated IL10-producing CD4 T cells leading to tolerance and protection from EAE in vivo.
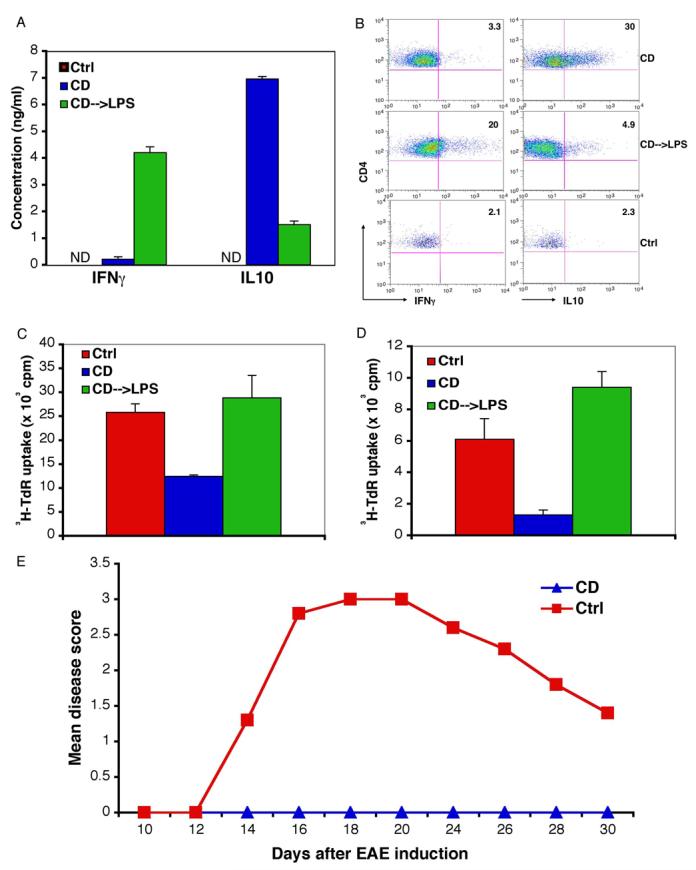
(Panel A) Immunization with DCs matured by CD alone induced T cells that produced IL10 instead of IFN-γ. CD11c+ BMDCs were purified at day 6-7 of culture, pulsed with OVA peptide 323-339 (10 μg/ml) for 2 hr and washed extensively before resuspension in PBS. 1-2.5 × 106 DCs were injected intravenously into C57BL/6 mice at day 0, 2 and 4. Splenocytes (1 × 106 cells/well) were prepared at day 7 and stimulated with antigens for 3 days. The supernatants were collected and cytokines were measured with the Luminex assays. (Panel B) DC matured by CD generated IL10-producing CD4 T cells instead of IFN-γ-producing effector cells. BFA (5 μg/ml) was added for 6 hr at the end of 2-3 day restimulation, splenocytes were then stained for cytokines and analyzed by FACS. The numbers indicate the percentage of IFN-γ–or IL10-positive cells of gated CD4+ CD25+ cells. Results are representative of 4 similar experiments, each consisted of two mice for CD and CD—>LPS treatments. (Panel C) Peripheral tolerance induced by treatment with DCs matured by CD alone. Mice were injected 3 times as described in Panel A and challenged 3 days after the last injection with 200 μg OTII peptide in CFA. 5 × 105 cells from pooled draining lymph nodes 3 days later were cultured with 2 × 105 mitomycin C-treated splenocytes in the presence of OTII peptide and T cell proliferation was measured by 3H-thymidine incorporation.
(Panel D) T cells from mice receiving CD-matured DCs showed diminished response to challenge with OTII peptide. Mice were injected 3 times with OVA peptide-pulsed DCs after adoptive transfer of naïve OTII CD4 T cells, 200 μg OTII peptide in CFA was injected subcutaneously 7 days later. 2 × 105 CD4+ cells from pooled draining lymph nodes 3 days after the challenge were cultured with 1 × 105 mitomycin C-treated splenocytes in the presence of OTII peptide; T cell proliferation was measured by 3H-thymidine incorporation. Similar results were obtained when the mice were injected once with DCs.
(Panel E) Immunization with DCs matured by CD alone protect mice from EAE. DCs were purified as described and cultured for 12-16h before being pulsed with MOG peptide for 2h, washed extensively and 2-3 × 106 cells were injected three times (at days 0, 2, 4) intravenously into 4-5 mice per group. EAE was induced 5, 7 or 9 days after the last injection and the mice were observed for paralysis. Induction of EAE on day 7 was shown.
E-cadherin-matured DCs induce peripheral T cell tolerance in vivo and protect against EAE
To determine directly if DCs matured by loss of E-cadherin adhesion could induce peripheral tolerance, mice were injected with ova-loaded DCs as above, and then challenged with the OVA OTII peptide (in complete Freunds adjuvant) 3-7 days after the last DC injection. T cells were isolated from draining lymph nodes and then assayed for their ability to proliferate in response to OVA in vitro. As shown in Figure 6C, T cells from mice immunized with CD-matured DCs proliferated poorly as compared to T cells from untreated control mice or mice immunized with DCs matured by LPS and CD. Thus, that DCs matured by CD alone diminished the number or activity of ova-responsive T cells, indicating that tolerance to OVA had been induced.
To confirm specifically that CD4 T cell tolerance had been obtained, we transferred transgenic OTII CD4+ cells into the mice 24 hr prior to DC injection, and challenged with 200 μg OTII peptide as above. CD4+ T cells from draining lymph nodes of mice immunized with DCs matured by CD alone again proliferated far less well than CD4+ cells from untreated mice or mice immunized with DCs matured by LPS and CD (Fig. 6D).
As an independent measure of tolerance, we asked if injection of E-cadherin-matured DCs could prevent the induction of experimental autoimmune encephalitis (EAE) in mice, induced by subcutaneous injection of an encephalitogenic myelin-oligodendricyte glycoprotein (MOG) peptide. As shown in Figure 6E, three prior injections of MOG peptide-pulsed, CD-matured DCs could completely protect against the neuropathology associated with EAE, suggesting that the DCs had induced tolerance to the peptide. These results were qualitatively similar to previous work showing that EAE could be prevented by targeting MOG peptide to DCs in vivo using an anti-DC antibody-MOG conjugate (in the absence of adjuvant) or by adoptive transfer of MOG peptide-loaded DCs matured in vitro by TNFα(Hawiger et al., 2004; Menges et al., 2002). In both cases, the mechanism of protection was associated with the induction of T cell tolerance, as we demonstrated for OVA (Fig. 6C,D). Taken together, these results strongly suggest that maturation induced by the loss of E-cadherin-mediated adhesion produces DCs that exhibit a functionally tolerogenic phenotype.
Discussion
One of the most intriguing specializations of DCs is the process of maturation (Trombetta and Mellman, 2005). The term was originally used to describe the acquisition of antigen-presenting activity by murine epidermal LCs after isolation from skin (Romani et al., 2003). More recently, maturation has come to describe a panoply of functional and morphological transformations triggered by TLR ligands, microbial products, inflammatory cytokines, or T cell surface proteins (eg CD40 ligand) (Mellman and Steinman, 2001). We can now add another mediator of DC maturation to this list, the activation following alterations in E-cadherin-mediated cell adhesion. Although we have not completely defined the biochemical features or physiological significance of this new pathway, we have established its functional consequences in vitro and in vivo and found them to be strikingly different from virtually all other pathways of DC maturation.
E-cadherin-induced maturation is unique on several accounts. First, it is not triggered by components associated with the inflammatory response or microbial invasion. The E-cadherin/ β-catenin pathway is best known for its role in the function of epithelial tissues and in organogenesis. In these examples, the sequestration of β-catenin by cadherins helps to regulate Wnt signaling and thereby cell proliferation and morphogenesis (Nelson and Nusse, 2004). In addition, induced alterations in homotypic cadherin interactions may play a direct role in triggering β-catenin signaling in endothelial cells during angiogenesis (Dejana, 2004). Although we cannot conclude that alterations in E-cadherin-mediated adhesion act alone (eg in the absence of a Wnt signal) or functions exclusively by activating the β-catenin pathway, our data do strongly suggest that β-catenin signaling is at least partly involved.
Second, unlike other signals studied thus far, induction of maturation by E-cadherin can occur under steady state conditions. LCs and possibly all other DCs that reside in peripheral tissues interact with neighboring cells by adhesion mediated via E-cadherin or related adhesion molecules. Disruption of these interactions, which must occur concomitant with tissue emigration, would lead to the activation of DC maturation even in the absence of infection or inflammation. That there is continuous traffic of DCs from tissues is clear, but it is unclear what triggers the loss of adhesion. As monomers, cadherin interactions are relatively low affinity, with adhesive strength reflecting the contribution of multiple cadherins at contact sites. Cadherin-mediated adhesions are also dependent on links to the actin cytoskeleton via α-catenin, which binds to the cadherin cytoplasmic domain via β-catenin, and via p120ctn (an effector of Rho GTPase activity) (Perez-Moreno and Fuchs, 2006). Thus, any physical disruption (analogous to that performed in vitro) that dissociates even a few E-cadherin interactions may be sufficient to reduce the strength of a given contact to induce maturation and migration. The mild mechanical trauma that occurs continuously in the skin might serve this purpose, and has already been associated with LC traffic to lymph nodes (Jakob et al., 2001). Similarly, in motile organs such as the gut, E-cadherin has been implicated in the anchoring of DCs in the mucosa (Rescigno et al., 2001); mild trauma may contribute as well. Alternatively, the steady state production of local agonists (including Wnt) may stochastically signal the dissociation of β-catenin following E-cadherin phosphorylation, weakening adhesive contacts (Dejana, 2004). Agonists might also elevate β-catenin levels by inhibiting GSK3β, since multiple signaling pathways activate kinases such as Akt that phosphorylate GSK3β (Doble and Woodgett, 2003).
Finally, and most strikingly, the E-cadherin-mediated pathway produces DCs that contains the phenotypic hallmarks of mature DCs but without the production inflammatory cytokines Thus, despite redistributing MHC class II molecules from lysosomal compartments, down regulating endocytosis, up regulating costimulatory molecules and chemokine receptors, and enhancing antigen processing activity, E-cadherin-induced DCs would not be expected to produce a sustained immune response. Such a phenotype might well be associated with the induction of peripheral tolerance in vivo (Pasare and Medzhitov, 2004); tolerance is a function associated with the constitutive traffic of otherwise unstimulated DCs from the periphery to lymph nodes (Steinman et al., 2003). Indeed, while DCs matured upon alteration of E-cadherin-mediated adhesion expanded the activated CD4 T cells as well as DCs matured by LPS, they failed to prime them to become IFN-γ–producing effectors, instead, they developed into IL10-producing cells with characteristics of regulatory T cells, resulting in peripheral CD4 T cell tolerance. Stimulation of the E-cadherin pathway may thus represent a signal for generating tolerogenic DCs under steady state conditions.
Signaling and DC function. Although it was established >10 years ago that LCs expressed E-cadherin (Tang et al., 1993) and that E-cadherin interactions may regulate LC maturation (Jakob and Udey, 1998; Riedl et al., 2000a; Riedl et al., 2000b), the underlying mechanism was not explored. E-cadherin in mouse and human DCs forms a complex with β-catenin and p120catenin at the plasma membrane (not shown). Conceivably, upon cluster disruption, some β-catenin is discharged from the plasma membrane with a fraction translocating to the nucleus. Stabilization of cytosolic β-catenin by inactivating GSK3β (which phosphorylates β-catenin for proteasome destruction) also induced DC maturation as did, albeit to a lesser extent, transfection of immature DCs with β-catenin cDNA. Maturation by CD activated β-catenin-TCF/LEF-mediated transcription (mouse DCs) and enhanced the expression of genes associated with β-catenin-TCF/LEF-mediated transcriptional events (human DCs). Although our data clearly established that the alteration of E-cadherin-mediated adhesion alone could lead to activation of β-catenin/TCF pathway, it remains possible that Wnt signaling also contributes. This might occur following CD, perhaps by the attendant shear force and signaling pathways possibly associated with primary cilia expressed by virtually all cells, events reported to initiate Wnt signaling in other systems (Norvell et al., 2004; Simons et al., 2005).
The ability of the GSK3β inhibitor SB216763 to induce maturation was striking. While it is possible this effect was limited to the dramatic increase in free β-catenin, GSK3β also has other targets. For example, by inhibiting GSK3β-mediated phosphorylation of NFAT, this transcriptional activator would be more readily translocated into the nucleus, possibly synergyzing with β-catenin (Crabtree and Olson, 2002). Other potential mediators (eg Hedgehog, p53, c-myc) might also be affected (Doble and Woodgett, 2003). However, the ability of transfected β-catenin cDNA to affect at least a partial maturation response strongly suggests that SB216763-induced maturation was at least partly due to a direct effect on β-catenin. SB216763 also induced the formation of the same array of chemokines as did CD (eg IP10, MCP-1, MIP-α, CXCL8) and failed to induce the production of inflammatory cytokines. Interestingly, inhibition of GSK3β in human monocytes was also found to downregulate inflammatory cytokine production while enhancing anti-inflammatory cytokine production by differentially regulating NF-κB and CREB (Martin et al., 2005). Possible off target effects of the drug must be considered, but in the context of other data implicating the β-catenin system, it seems most likely that inhibition of GSK3β was at least in part involved.
There are only a few signaling pathways in DCs that to date have been found to yield a similar maturation phenotype to CD or GSK3β inhibition. One pathway results from ligation of the orphan plasma membrane receptor TREM-2 (Bouchon et al., 2001). TREM-2 signals through the ITAM-bearing adaptor DAP-12 to moderately upregulate CD40, CD86 and MHC II, strongly upregulates CCR7, and fails to induce production of inflammatory cytokines or NF-κB and p38 MAP kinase activation (Bouchon et al., 2001). Although the function of TREM-2 or the identity of its ligand are unknown, DAP12−/− mice exhibited accumulation of DCs in muco-cutaneous epithelia as if the emigration from tissues was inhibited (Tomasello et al., 2000). Another pathway is induced by TSLP (thymic stromal lymphopoietin) (Watanabe et al., 2005). Although first described as a product of inflammed epithelia, TSLP is also produced by Hassall's corpuscles in the thymus, a site of Treg formation. Interestingly, TSLP induces phenotypic maturation but not the release of inflammatory cytokines such as IL12; the DCs so produced can generate CD4+CD25+ Tregs in vitro.
Physiological role of E-cadherin-induced DC maturation. Although the observation that E-cadherin/ β-catenin signaling is likely to play a dramatic regulatory role in DCs is noteworthy, it will next be important to determine the in vivo significance of this pathway. In principle, this might be accomplished using targeted deletions of essential components in the pathway.
The failure of DCs matured in vitro by loss of E-cadherin adhesion to produce inflammatory cytokines suggests that these cells are involved in peripheral tolerance. In support of this possibility are both our own in vivo experiments and recent observations concerning MyD88−/− DCs. Despite exhibiting a typical mature phenotype following LPS treatment, MyD88−/− DCs fail to produce inflammatory cytokines and also fail to activate naïve CD4 T cells in vivo, due to the suppressive effect of Treg's (Pasare and Medzhitov, 2004). Thus, like MyD88-deficient DCs, E-cadherin activation of normal DCs activates those features of the maturation pathway required for efficient antigen processing and presentation, yet there is a failure of cytokine production. Adoptive transfer of these DCs into naïve mice produces IL10-secreting T cells, which have been associated with tolerance in other systems (Menges et al., 2002; O'Garra and Vieira, 2004). By this general definition, we too found that tolerance was induced, as antigen-specific CD4 T cells activated by CD-matured DCs proliferated far more weakly than those from controls or mice injected with DCs matured by CD and LPS together. Further evidence that CD-matured DCs could induce tolerance was demonstrated by the fact that prior transfer of MOG peptide-loaded DCs could protect mice against developing EAE after subsequent injection of free MOG peptide.
What is known about “tolerogenic” DCs in vivo? It has been elegantly demonstrated that targeting antigens to DCs using antibody to DEC-205 in the absence of overt inflammatory or immunostimulatory mediators led to tolerance, while further maturation by CD40 ligand (CD40L) resulted in immunity (Bonifaz et al., 2002; Hawiger et al., 2001). The steady state DCs present antigens efficiently to drive T cell proliferation, which were then deleted (Hawiger et al., 2001). These “tolerogenic” DCs were phenotypically mature, with their expression of CD40 and CD86 being upregulated only slightly upon further stimulation by CD40L, similar to our observations. More strikingly, while immunization with E-cadherin-matured DCs generated IL-10-producing Tregs and peripheral tolerance, treating these matured DCs with LPS led to strong immunity. Since we could detect the expression of E-cadherin in primary DCs from lymph nodes and peripheral tissues (AJ and IM, unpublished), it is possible that at least some of the DCs in lymphoid organs at steady state had been “matured” by alterations in adhesion. It is also of interest to note that a heterotypic ligand for E-cadherin, CD103, is expressed by some DCs and T cells that play an important role in regulating tolerance vs inflammation in the gut (Annacker et al., 2005).
The events induced by alterations in E-cadherin adhesion may represent one of several programs that work in various combinations to achieve DC maturation. It seems possible that the adhesion-triggered program induces a variety of organizational changes that enable phenotypic maturation: mobilization of MHC class II to the surface, extension of dendritic processes, induction of costimulatory molecules, upregulation of antigen processing activity, etc. By this view, other programs induced by microbial or inflammatory stimuli would add the production of critical cytokines required for the induction of T cell immunity. Alterations in adhesion can thus be seen as producing phenotypically mature tolerogenic DCs at the steady state, while microbial stimuli yield functionally mature immunogenic DCs under conditions of inflammation. There is clearly cross-talk between the various programs. Many microbial or inflammatory stimuli can induce both phenotypic and functional maturation, at least in vitro, in the absence of alterations in adhesion. Maturation of DCs by pathogens (eg E. coli) does seem to stimulate many of the same transcripts as does CD (including TCF/LEF targets), while LPS alone (which stimulates only a single TLR, unlike E. coli which stimulates several) fails to produce active β-catenin (Fig. 2B and 2D). Activation of Akt by TLR-induced maturation may phosphorylate GSK3β thereby helping to activate the β-catenin pathway, even in the absence of adhesion alterations. The two classes of maturation programs are also hierarchically arranged, since microbial stimuli can always induce cytokine secretion even if added to DCs already matured by alterations in E-cadherin adhesion. While much remains to be determined about the biochemistry and function of these maturation programs, our data demonstrate that the E-cadherin system -- despite not being specific to the immune system -- is likely to have an important effect on the ability of DCs to control one of the most finely tuned and complex aspects of the immune response.
Experimental Procedures
Reagents and Antibodies Rat anti-E-cadherin, mouse anti-β-catenin, and mouse anti-p120 catenin were purchased from BD Transduction Laboratories (Lexingon, CA). Both mouse anti-E-cadherin (HECD-1 and SHE78-7) and rat anti-E-cadherin monoclonal antibodies were obtained from Zymed (San Francisco, CA). Rat monoclonal anti-E-cadherin and rabbit polyclonal β-catenin were obtained from Sigma-Aldrich. Secondary antibodies for immunofluorescence and FluroReporterR lacZ flow cytometry kit were from Molecular Probes (Eugene, Oregon). Anti-active β-catenin antibody was purchased from Upstate Biotechnology (Lake Placid, NY). FACS antibodies: CD86, CD11c, human CD4, mouse CD4, CD8, TCR Vα2, TCR Vβ5, CD25 and MHCII I-Ab were from Pharmingen (San Diego, CA); CD25, IL2, IFN-γ, FOXP3, and IL10 were from eBioscience (San Diego, CA). Antibodies against phospho-specific p38 and IKBα were from Cell Signaling Technology Inc (Beverly, MA). SB 216763 was purchased from Tocris Cookson Inc. (Ellisville, MO). A PE-Rat anti-mouse CCR7 antibody was purchased from Biolegend (San Diego, CA). Brefeldin A (BFA) was purchased from Epicentre. Murine β-catenin-GFP, GSK*β-catenin-GFP and C-del-GSK*β-catenin-GFP were kindly provided by Dr. James Nelson (Stanford University).
Mice C57BL/6 mice were purchased from Charles Rivers Laboratories and and CD45.1 C57BL/6, C57BL/10ScCr (TLR4−/−), OT I and OT II TCR transgenic mice were purchased from Jackson Laboratories. The transgenic TOPGAL reporter mice were kindly provided by Dr. E. Fuchs (DasGupta and Fuchs, 1999) (Rockefeller University).
Flow cytometry assays Cells were stained for 30 min on ice with primary antibody and if necessary secondary antibody, washed, and then evaluated on a FACSCalibur™ (Becton Dickinson). A fluoReporterR LacZ flow cytometry kit was used to measure β-galatosidase activity for BMDCs from TOPgal transgenic mice following the manufacturer's recommendations. For intracellular staining, splenocytes (1 × 106 cell/well) were incubated with BFA (5 μg/ml) for the last 6 hours of their in vitro restimulation and surface staining and intracellular staining were performed with BD cytofix/cytoperm™ plus kit with manufacturer's protocol.
Cell Culture and purification of DC, OT-I and OT-II T cells Mouse BM-derived CD11c+ DCs were isolated using anti-CD11c-conjugated beads and columns (Miltenyi Biotech) according to the manufacturer's protocol. The resulted CD11c+ DCs were then resuspended in the same culture media at 5-10 × 105 cells/ml; these cells were in single cell suspension and thus referred to as cluster-disrupted cells. CD4 or CD8 T cells were isolated with CD4 and CD8 T cells isolation kits (Miltenyi Biotech) from lymph nodes according to manufacture's protocol. For proliferation assays, cells were labeled with CFSE (Molecular Probes) at 5 μM at 37°C for 10 min and washed extensively before injection or plating.
Cell fractionation, immunoprecipitation and Western blotting Cells were homogenized in hypotonic buffer (10mM HEPES-KOH pH 7.4, 1.5mM MgCl2, 10mM KCl, 0.5mM DTT) with proteinase and phosphatase inhibitors by passing through 30 gauge needles for 10 times. Postnuclear fractions were centrifuged at 45,000 rpm for 45 min to separate membrane and cytosol fractions. Total cell lysates were obtained with 1% NP-40 lysis buffer (1% Nonidet P-40, 150mM NaCl, 20mM Tris, and ImM EDTA (pH 7.5)) supplemented with proteinase and phosphatase inhibitors. For immunoprecipitation, cells were lysed in 1% digitonin and supernatants were then incubated with antibodies and protein G-sepharose at 4°C with constant rotation for 2-4 hr, captured immune complexes were subjected to SDS-PAGE and Western blotting analysis.
Antigen presentation assay For peptide antigen, CD11c+ DCs after different treatments were incubated with OVA-peptide (323-339) at 37°C for 2-3 hr. The cells were then extensively washed with serum free RPMI, fixed with 1% PFA for 10 min and extensively washed before addition of 1 × 105 CD4 T cells freshly purified from OT II lymph nodes. For protein antigen, cells were pulsed with OVA protein (grade VI; Sigma-Aldrich or Worthington) for 2 hr at 37°C before different treatment. CD11c+ DCs were then fixed with 1% PFA before addition to either CD4 or CD8 T cells from OT-II and OT-I mouse lymph nodes, respectively. Supernatants were taken out after 24 hr incubation at 37°C and frozen at -70°C overnight before ELISA assay for IL-2.
Retrovirus generation and transfection of DCs pEGFP-β-catenin and EGFP were cloned into pLZRS and then transfected into Φx-ecotropic cells using Fugene 6 (Roche). Retrovirus was generated in Φx-ecotropic cells and subsequently used to transfect DCs (Chow et al., 2002). TOP-EGFP and FOP-EGFP, constructed from pTOP/FOPFlash (with minimal c-fos promoter from Dr. H Clevers) were kindly provided by Dr. A. Sartorelli (Yale University). Top/Fop-EGFP were then cloned into the retroviral vector LTRH1 provided by Dr. R. Medzhitov (Yale University) (Barton and Medzhitov, 2002).
Immunofluorescence microscopy Cells were fixed in 4% PFA, permeabilized in RPMI with 10% goat serum and 0.25% saponin, followed by 30 min each with primary antibody and secondary antibody with appropriate Alexa® Fluors (Molecular Probes, Inc.) before mounted in Prolong Gold solution. Confocal microscopy was performed using a laser scanning microscope (LSM 510; Carl Zeiss MicroImaging, Inc.), 40x water immersion lens (n = 1.5), at 25°C. Images were processed using Adobe Photoshop® (Adobe Systems, Inc.) version 7.0 software. Microarrays and data analysis Total RNA from human CD34+ DCs was isolated using RNeasy kits (Qiagen), followed by cDNA synthesis (5 μg RNA per sample) using the SuperScript system (Invitrogen). Samples were then cleaned, prepared and hybridized to Affymetrix (Santa Clara, CA) Human Genome U95Av2 arrays (representing approximately 8500 genes) according to manufacturer's protocol. Raw data correction and normalization was performed using Affymetrix Microarray Suite 5.0 for background and PM/MM corrections. The probe set based summary data were then log transformed and normalized for probe set intensity-dependent biases. Loess normalization of M vs. A relationship for all chip-pairs was performed. We considered a gene to be regulated by a treatment only if its expression intensity was increased or reduced by at least 3 fold compared to the intensity measured at time 0. For clustering and heatmap generation, log ratios of expression intensities were standardized within each gene, thereby transforming the distribution of log ratios into one with mean at 0 and standard derivation of 1. Heatmap-associated color scale bars visualize the scaling relationship between color intensities and corresponding standardized log ratio values: up-regulated genes are shown in red while down-regulated genes are shown in blue. Clustering of experimental samples based on transcriptional profiles was carried out using an agglomerative hierarchical clustering algorithm. Log ratio values of all regulated genes were used to construct feature vectors for each sample. A dissimilarity measurement between samples was computed as a Euclidean distance between feature vectors. Cluster dissimilarities were computed using group average method. Unless otherwise specified, all data analysis procedures were implemented using S-Plus software (Insightful Corp.). Cytokine and chemokine multiplex analysis The levels of cytokines and chemokines were measured with Luminex™ suspension array technology. Supernatants were collected and frozen at −80°C. For human CD34+ -derived DCs, cell culture supernatants were then analyzed using the Beadlyte cytokine assay kit (Upstate) with manufacturer's protocol. For mouse cytokines, supernatants were analyzed using the Bio-Plex cytokine assay kit and supporting reagents (BioRad) following manufacturer's procedures.
Real-time RT-PCR Total RNA was isolated from differently treated human or murine cells with the RNAeasy kit from Invitrogen according to manufacturer's recommendation. Quantitative real-time RT-PCR was carried out with the DNA Engine Opticon® 2 real time detection system (MJ Research Inc.) and SYBR Green system (Stategene), and data were normalized by the level of β-actin expression in each individual sample.
Adoptive transfer with DCs and T cells and in vitro restimulation and proliferation For in vivo proliferation assays, 0.5-1.5 × 106 labeled or unlabeled purified T cells were injected intravenously into the lateral tail vein of mice, 24 hr later 1 × 106 DCs pulsed with proper antigens were injected intravenously into the same mice. Adoptive transferred T cells were analyzed for their proliferation 3, 5 or 7 days after the last injection. For in vivo CD4 T cell priming, BMDCs were prepared as described and pulsed with OVA peptide 323-339 (10 μg/ml) after maturation treatment for 2 hr at 37°C and then washed extensively. 1-2.5 × 106 matured DCs were then injected intravenously at day 0, 2 and 4 and spleen cells were restimulated at day 7 with 10 μg/ml OVA peptide 323-339. Cell supernatants were taken after 72 hr and cytokines were measured as described. For in vitro proliferation assay of draining LNs, mice were immunized at 4 places on the back with 200 μg OVA peptide in CFA and total cells or CD4 from the pooled LNs were incubated with mitomycin C-treated splenocytes with 10 μg/ml OVA peptide, proliferation of T cells was measured by incorporation of 3H-thymidine during the last 16 hr of the 72 hr incubation. For intracellular staining, BFA (5 μg/ml) was added for the last 6 hr of the in vitro restimulation and the cells were fixed and permeabilized using BD cytofix/cytoperm™ plus kit.
EAE Induction EAE was induced by subcutaneous injections of 300 μg of MOG35–55 peptide in CFA (Difco, Detroit, MI) with 500 μg of Mycobacterium tuberculosis on days 0 and 7, supplemented by intraperitoneal injections of 500 ng of pertussis toxin (List Biological, Campbell, CA) on days 0 and 2. The mice were observed and scored on a scale of 0 to 5 with gradations of 0.5 for intermediate scores: 0, no clinical signs; 1, loss of tail tone; 2, wobbly gait; 3, hind limb paralysis; 4, hind and fore limb paralysis; 5, death.
Supplementary Material
Acknowledgements
We thank Chunmei Fu, Hongmei Li, Horace Rhee and members of the Mellarren group for technical help and discussion. We are grateful to Xiaobing Qian and Yi Zhou for help, and Dr. Lynda Bennett for validation in the microarray work. The use of real-time RT-PCR was supported by NIH grant AR46032. A.J. was supported by a Cancer Research Institute fellowship. This work was supported by the NIH (R37AI34098 and U19AI057234), the Sandler Family Foundation, and by the Ludwig Institute for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Annacker O, Coombes JL, Malmstrom V, Uhlig HH, Bourne T, Johansson-Lindbom B, Agace WW, Parker CM, Powrie F. Essential role for CD103 in the T cell-mediatedregulation of experimental colitis. J Exp Med. 2005;202:1051–1061. doi: 10.1084/jem.20040662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Barton GM, Medzhitov R. Retroviral delivery of small interfering RNA into primarycells. Proc Natl Acad Sci U S A. 2002;99:14943–14945. doi: 10.1073/pnas.242594499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leadsto antigen presentation on major histocompatibility complex class I products and peripheral CD8+ Tcell tolerance. J Exp Med. 2002;196:1627–1638. doi: 10.1084/jem.20021598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchon A, Hernandez-Munain C, Cella M, Colonna M. A DAP12-mediatedpathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. JExp Med. 2001;194:1111–1122. doi: 10.1084/jem.194.8.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow A, Toomre D, Garrett W, Mellman I. Dendritic cell maturation triggersretrograde MHC class II transport from lysosomes to the plasma membrane. Nature. 2002;418:988–994. doi: 10.1038/nature01006. [DOI] [PubMed] [Google Scholar]
- Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, et al. Selective small molecule inhibitors ofglycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol. 2000;7:793–803. doi: 10.1016/s1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexesduring hair follicle development and differentiation. Development. 1999;126:4557–4568. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- Delamarre L, Holcombe H, Mellman I. Presentation of exogenous antigens on majorhistocompatibility complex (MHC) class I and MHC class II molecules is differentially regulatedduring dendritic cell maturation. J Exp Med. 2003;198:111–122. doi: 10.1084/jem.20021542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J CellSci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Dieu-Nosjean MC, Dezutter C, Valladeau J, Kayal S, Leborgne M, Brousse N, Saeland S, Davoust J. Accumulation of immature Langerhans cells in human lymphnodes draining chronically inflamed skin. J Exp Med. 2002;196:417–430. doi: 10.1084/jem.20020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, Ravetch JV, Steinman RM, Nussenzweig MC. Dendritic cells induce peripheral T cell unresponsiveness understeady state conditions in vivo. J Exp Med. 2001;194:769–779. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawiger D, Masilamani RF, Bettelli E, Kuchroo VK, Nussenzweig MC. Immunological unresponsiveness characterized by increased expression of CD5 on peripheral T cellsinduced by dendritic cells in vivo. Immunity. 2004;20:695–705. doi: 10.1016/j.immuni.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Jakob T, Brown MJ, Udey MC. Characterization of E-cadherin-containing junctionsinvolving skin- derived dendritic cells. J Invest Dermatol. 1999;112:102–108. doi: 10.1046/j.1523-1747.1999.00475.x. [DOI] [PubMed] [Google Scholar]
- Jakob T, Ring J, Udey MC. Multistep navigation of Langerhans/dendritic cells in andout of the skin. J Allergy Clin Immunol. 2001;108:688–696. doi: 10.1067/mai.2001.118797. [DOI] [PubMed] [Google Scholar]
- Jakob T, Udey MC. Regulation of E-cadherin-mediated adhesion in Langerhans cell-like dendritic cells by inflammatory mediators that mobilize Langerhans cells in vivo. J Immunol. 1998;160:4067–4073. [PubMed] [Google Scholar]
- Kaisho T, Takeuchi O, Kawai T, Hoshino K, Akira S. Endotoxin-induced maturationof MyD88-deficient dendritic cells. J Immunol. 2001;166:5688–5694. doi: 10.4049/jimmunol.166.9.5688. [DOI] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Lanzavecchia A, Sallusto F. Antigen decoding by T lymphocytes: from synapses to fatedetermination. Nat Immunol. 2001;2:487–492. doi: 10.1038/88678. [DOI] [PubMed] [Google Scholar]
- Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells: whichsignals induce tolerance or immunity? Trends Immunol. 2002;23:445–449. doi: 10.1016/s1471-4906(02)02281-0. [DOI] [PubMed] [Google Scholar]
- Lyons JP, Mueller UW, Ji H, Everett C, Fang X, Hsieh JC, Barth AM, McCrea PD. Wnt-4 activates the canonical beta-catenin-mediated Wnt pathway and binds Frizzled-6CRD: functional implications of Wnt/beta-catenin activity in kidney epithelial cells. Exp Cell Res. 2004;298:369–387. doi: 10.1016/j.yexcr.2004.04.036. [DOI] [PubMed] [Google Scholar]
- Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokineproduction is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigenprocessing machines. Cell. 2001;106:255–258. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- Menges M, Rossner S, Voigtlander C, Schindler H, Kukutsch NA, Bogdan C, Erb K, Schuler G, Lutz MB. Repetitive injections of dendritic cells matured with tumornecrosis factor alpha induce antigen-specific protection of mice from autoimmunity. J Exp Med. 2002;195:15–21. doi: 10.1084/jem.20011341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M, Manz MG, Karsunky H, Wagers A, Peters W, Charo I, Weissman IL, Cyster JG, Engleman EG. Langerhans cells renew in the skin throughout life under steady-stateconditions. Nat Immunol. 2002;3:1135–1141. doi: 10.1038/ni852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norvell SM, Alvarez M, Bidwell JP, Pavalko FM. Fluid shear stress induces beta-catenin signaling in osteoblasts. Calcif Tissue Int. 2004;75:396–404. doi: 10.1007/s00223-004-0213-y. [DOI] [PubMed] [Google Scholar]
- O'Garra A, Vieira P. Regulatory T cells and mechanisms of immune system control. Nat Med. 2004;10:801–805. doi: 10.1038/nm0804-801. [DOI] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Toll-dependent control mechanisms of CD4 T cell activation. Immunity. 2004;21:733–741. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Perez-Moreno M, Fuchs E. Catenins: keeping cells from getting their signals crossed. Dev Cell. 2006;11:601–612. doi: 10.1016/j.devcel.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre P, Turley SJ, Gatti E, Hull M, Meltzer J, Mirza A, Inaba K, Steinman RM, Mellman I. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature. 1997;388:787–792. doi: 10.1038/42039. [DOI] [PubMed] [Google Scholar]
- Randolph GJ, Sanchez-Schmitz G, Angeli V. Factors and signals that govern themigration of dendritic cells via lymphatics: recent advances. Springer Semin Immunopathol. 2005;26:273–287. doi: 10.1007/s00281-004-0168-0. [DOI] [PubMed] [Google Scholar]
- Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl JP, Ricciardi-Castagnoli P. Dendritic cells express tight junction proteinsand penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- Riedl E, Stockl J, Majdic O, Scheinecker C, Knapp W, Strobl H. Ligation of E-cadherin on in vitro-generated immature Langerhans-type dendritic cells inhibits their maturation. Blood. 2000a;96:4276–4284. [PubMed] [Google Scholar]
- Riedl E, Stockl J, Majdic O, Scheinecker C, Rappersberger K, Knapp W, Strobl H. Functional involvement of E-cadherin in TGF-beta 1-induced cell cluster formation of invitro developing human Langerhans-type dendritic cells. J Immunol. 2000b;165:1381–1386. doi: 10.4049/jimmunol.165.3.1381. [DOI] [PubMed] [Google Scholar]
- Romani N, Holzmann S, Tripp CH, Koch F, Stoitzner P. Langerhans cells -dendritic cells of the epidermis. Apmis. 2003;111:725–740. doi: 10.1034/j.1600-0463.2003.11107805.x. [DOI] [PubMed] [Google Scholar]
- Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, et al. Inversin, the gene product mutated in nephronophthisistype II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporri R, Reis e Sousa C. Inflammatory mediators are insufficient for full dendritic cellactivation and promote expansion of CD4+ T cell populations lacking helper function. Nat Immunol. 2005;6:163–170. doi: 10.1038/ni1162. [DOI] [PubMed] [Google Scholar]
- Staal FJ, Clevers HC. WNT signalling and haematopoiesis: a WNT-WNT situation. Nat Rev Immunol. 2005;5:21–30. doi: 10.1038/nri1529. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. AnnuRev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance bydendritic cells that have captured apoptotic cells. J Exp Med. 2000;191:411–416. doi: 10.1084/jem.191.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Tang A, Amagai M, Granger LG, Stanley JR, Udey MC. Adhesion of epidermalLangerhans cells to keratinocytes mediated by E- cadherin. Nature. 1993;361:82–85. doi: 10.1038/361082a0. [DOI] [PubMed] [Google Scholar]
- Tomasello E, Desmoulins PO, Chemin K, Guia S, Cremer H, Ortaldo J, Love P, Kaiserlian D, Vivier E. Combined natural killer cell and dendritic cell functional deficiency inKARAP/DAP12 loss-of-function mutant mice. Immunity. 2000;13:355–364. doi: 10.1016/s1074-7613(00)00035-2. [DOI] [PubMed] [Google Scholar]
- Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- van Noort M, Meeldijk J, van der Zee R, Destree O, Clevers H. Wnt signalingcontrols the phosphorylation status of beta-catenin. J Biol Chem. 2002;277:17901–17905. doi: 10.1074/jbc.M111635200. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V, Fuchs E. Actin dynamics and cell-cell adhesion in epithelia. Curr OpinCell Biol. 2001;13:76–84. doi: 10.1016/s0955-0674(00)00177-0. [DOI] [PubMed] [Google Scholar]
- Verbovetski I, Bychkov H, Trahtemberg U, Shapira I, Hareuveni M, Ben-Tal O, Kutikov I, Gill O, Mevorach D. Opsonization of apoptotic cells by autologous iC3b facilitatesclearance by immature dendritic cells, down-regulates DR and CD86, and up-regulates CCchemokine receptor 7. J Exp Med. 2002;196:1553–1561. doi: 10.1084/jem.20020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votin V, Nelson WJ, Barth AI. Neurite outgrowth involves adenomatous polyposiscoli protein and beta-catenin. J Cell Sci. 2005;118:5699–5708. doi: 10.1242/jcs.02679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Wang YH, Lee HK, Ito T, Wang YH, Cao W, Liu YJ. Hassall'scorpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature. 2005;436:1181–1185. doi: 10.1038/nature03886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.