

Abstract
The levels of soluble beta amyloid (Aβ) are correlated with symptom severity in Alzheimer’s disease. Soluble Aβ has been shown to disrupt synaptic function and it has been proposed that accumulation of soluble Aβ triggers synapse loss over the course of the disease. Numerous studies indicate that soluble Aβ has multiple targets, one of which appears to be the nicotinic acetylcholine receptor, particularly for Aβ concentrations of pM-nM. Moreover, pM-nM soluble Aβ was found to increase presynaptic Ca2+ levels, suggesting that it may have an impact on neurotransmitter release. In the present study, soluble Aβ was perfused into mouse prefrontal cortex and the effect on the release of dopamine outflow via microdialysis was assessed. In the presence of tetrodotoxin, Aβ1-42 at 100nM evoked the release of dopamine to ∼170% of basal levels. The Aβ1-42-evoked dopamine release was sensitive to antagonists of α7 nicotinic receptors and was absent in mice harboring a null mutation for the α7 nicotinic subunit, but was intact in mice harboring a null mutation for the β2 nicotinic subunit. The control peptide Aβ40-1 was without effect. In contrast, Aβ1-42 at 1-10pM caused a profound but slowly developing decrease in dopamine outflow. These results suggest that Aβ alters dopamine release in mouse prefrontal cortex, perhaps involving distinct targets as it accumulates during Alzheimer’s disease and leading to disruption of synaptic signaling.
Section: Disease-Related Neuroscience
Keywords: Prefrontal Cortex, Beta Amyloid, Dopamine Release, In Vivo Microdialysis
1. Introduction
One of the major pathological entities in Alzheimer’s disease (AD) is beta amyloid (Aβ). Aβ refers to a collection of peptides of 38-43 amino acids in length, which are derived from the amyloid precursor protein (APP) by sequential proteolytic cleavage (reviewed in Walsh and Selkoe, 2007). The predominant forms of Aβ found in AD are Aβ1-40 and Aβ1-42, with their levels progressively increasing over the course of the disease (Ingelsson et al., 2004). Though Aβ notably accumulates in plaques, a significant portion exists in soluble forms, mainly as oligomers (or ADDLs; Lambert et al., 1998), and it has been proposed that increased levels of soluble Aβ oligomers in the early stages of AD cause synaptic dysfunction (Hardy and Selkoe, 2002; Selkoe and Schenk, 2003) and, later, synapse loss (Lue et al., 1999). The Aβ1-42 form, though less abundant than the Aβ1-40 form, has the highest propensity to form toxic oligomers (Walsh and Selkoe, 2007) and correlates with disease onset (Kumar-Singh et al., 2006). In addition, aggregated and fibrillar forms of Aβ also accumulate and hence different forms of Aβ may have different effects in the brain, which, together with neurofibrillary pathology, likely includes synaptic dysfunction, synapse loss, oxidative damage, inflammation and neuron loss (Selkoe, 2002; Mattson, 2004).
The accumulation of Aβ in AD is selective, with particularly high concentrations found in frontal cortex, entorhinal cortex, hippocampus, amygdala and parietal association cortex (Braak and Braak, 1991). Moreover, the accumulation of Aβ is largely linked to its release from synaptic nerve endings, as evidenced by its loss from terminal innervation sites of lesioned pathways in APP transgenic mice (Lazarov et al., 2002; Sheng et al., 2002). In addition, soluble Aβ oligomers, particularly Aβ1-42, has been shown to disrupt synaptic plasticity, specifically LTP in the hippocampus (Walsh et al., 2005). On the other hand, it appears that oligomerization of Aβ starts intraneuronally (Oddo et al., 2006). The extent to which Aβ increases or decreases synaptic activity, and its site(s) of action, remains to be fully elucidated.
Previous work has demonstrated a potent action of soluble Aβ on nicotinic acetylcholine receptors (nAChRs), including both antagonist (Pettit et al., 2001; Liu et al., 2001; Grassi et al., 2003; Wu et al, 2004) and agonist (Dineley et al., 2001; Dougherty et al., 2003; Fu et al., 2003) effects. We have shown that pM-nM Aβ1-42 induces increased [Ca2+]i in isolated presynaptic terminals from rat cortex and hippocampus in a manner susceptible to partial antagonism by classical nAChR antagonists, such as α-bungarotoxin and dihydro-β-erythroidine. Moreover, prior activation of presynaptic nAChRs attenuated subsequent responses to Aβ1-42. However, there remains some reservation regarding the actual target for soluble Aβ, particularly the role of nAChRs, and whether activation of presynaptic nAChRs leads to altered synaptic transmission. Here, we employed preparations from mice harboring null mutations for either the α7 subunit or the β2 subunit of the two major nAChR subtypes present in brain, namely the α-bungarotoxin-sensitive and high affinity subtypes, respectively (Role and Berg, 1996; Zoli et al., 1998) to examine the effect of Aβ on the release of dopamine in the prefrontal cortex of the intact brain. Dopamine (DA) is a prominent player in the functioning of the prefrontal cortex (Arnsten and Li, 2005) and alterations in its release by Aβ could lead to altered prefrontal cortical function.
2. Results
2.1 Nicotine and β-amyloid evoke the release of dopamine in mouse prefrontal cortex
The release of dopamine in prefrontal cortex was measured in freely moving mice via in vivo microdialysis (Fig. S1) in the presence of TTX in order to isolate presynaptic regulation from cellular effects. Nicotine (1μM) in the presence of TTX evoked an increase in DA outflow (∼300% over basal) and this increase was blocked by prior treatment with a nAChR antagonist (Fig. 2A). These results are generally consistent with previous reports of nicotine-evoked DA release in rodent prefrontal cortex slices (Rao et al., 2003; Cao et al., 2005). Perfusion with soluble 100nM of Aβ1-42 (Fig. 1) also evoked the release of DA (∼170% over basal; p<0.05) in a manner sensitive to nAChR antagonists (Fig. 2B). (Perfusion of Aβ using microdialysis has been previously characterized (Parks et al., 2001; Trabace et al., 2007).) Perfusion with 100nM of the soluble core amyloid fragment Aβ12-28 (Wang et al., 2000a; Dougherty et al., 2003) also evoked the release of DA (Fig. 2C), but the response was rather variable. Curiously, perfusion with 1-10pM Aβ1-42 did not evoke an increase in DA outflow, but, rather, caused a slowly developing, profound decrease in DA outflow (Fig. 2D), down to just detectable levels (fg/μl DA in perfusate). (With shorter time of perfusion of 100pM or 100nM Aβ, the DA outflow was decreased at later time points after initially evoking an increase (see Fig. 3); longer times of perfusion with Aβ likely did not allow the peptide concentration to fall to pM over the course of the recording.) The decrease in DA release was not significantly affected by nAChR antagonist (not shown). Perfusion with the control peptide, “Aβ40-1”, was without effect (Fig. 2E).
Fig. 2.

Nicotine and β-amyloid evoke increased DA outlflow in prefrontal cortex. (A) Effect of perfusing 1μM nicotine by “reverse” dialysis into prefrontal cortex of freely moving C57B1/6 mice on the release of DA was assessed via in vivo microdialysis in the presence of TTX. The perfusate was switched to aCSF containing 1uM TTX (as control), TTX plus 1uM nicotine or TTX plus 1uM nicotine plus 2mM MLA for 5min, after which the perfusate was switched by to aCSF containing TTX alone. Under the time frame used for drug perfusion (1-3h), TTX alone did not significantly affect DA outflow. After extended perfusion (4-6h), the DA outflow dropped substantially (not shown). MLA was used a general nAChR antagonist, as its use at 2mM will block both α7 and non-α7 nAChRs (Ward et al., 1990; Mogg et al., 2002). (B) Perfusion with 100nM Aβ1-42 in aCSF in TTX for 20min in the absence or presence of BgTx (1μM) or MLA (1μM), also delivered for 20min. BgTx was delivered via a second, open cannula, implanted next to and slightly above the microdialysis probe. (C) Perfusion with 100nM Aβ12-28 in TTX, a highly soluble core fragment which strongly competes for Aβ1-42 interaction with α7 nAChRs (Wang et al., 2000a). (D) Perfusion with various concentrations of Aβ1-42 in TTX. (E) Perfusion with the peptide “Aβ40-41” in TTX, which served as a control “reverse” peptide having no effect on presynaptic Ca2+ (Dougherty et al., 2003). (Lack of effect of “Aβ42-1” on presynaptic Ca2+ has also been observed (Mehta et al., manuscript in preparation).) Dopamine (DA) content in the fractions (preloaded with perchloric acid) was determined via HPLC with an electrochemical detector, as described in the Experimental Procedures. Data are presented as averages ± s.e.m (4-6 replicates each). Mean basal values for DA: 0.5-1pg/ul, adjusted for recovery (4-10%); *p<0.05 relative to baseline.
Fig. 1.

Microdialysis perfusion of β-amyloid. β-amyloid perfused into surrounding tissue via the microdialysis probe (*blank areas along right side of micrographs) placed in the prefrontal cortex for 30 min was detected via immunocytochemistry, as described in Experimental Procedures, using an anti-Aβ monoclonal antibody (right) in comparison to a control section incubated with fluorescein-conjugated secondary antibody alone (left). Images of 40μm sections around the microdialysis probe (*) were taken using confocal microscopy. Scale bar = 12 μm. Posthoc staining with Hoescht did not reveal any gross alteration in the tissue following Aβ perfusion (not shown).
Fig. 3.
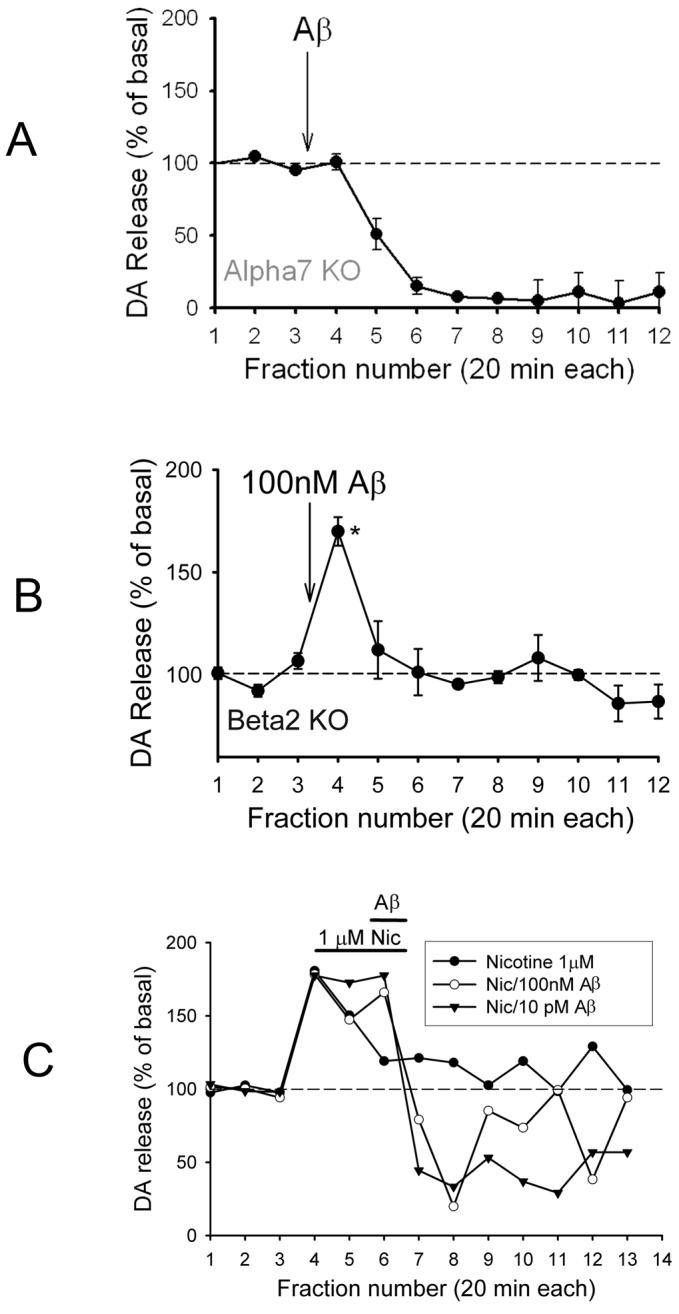
β-amyloid-evoked DA outflow in prefrontal cortex of mice harboring null mutations of nAChR subunits. (A) Perfusion with 100pM or 100nM Aβ1-42 for 5min into prefrontal cortex of α7 nAChR null-mutant mice (Alpha7 KO). (B) Perfusion with 100nM Aβ1-42 into prefrontal cortex of β2 nAChR null-mutant mice (Beta2 KO). Shorter perfusion with Aβ in A and B was performed in order to reveal possible slowly developing inhibition of DA outflow resulting from the fall-off in Aβ following perfusion. (C) Perfusion with 1μM nicotine for 30min, then 100nM or 10pM Aβ1-42 with nicotine for an additional 20min. Experiments were performed as described in the legend to Fig. 2. Data are presented as averages ± s.e.m; n=3-5 replicates each, except in C where only averages are shown for clarity (n=3). Mean basal values for DA: 0.5-1pg/ul, adjusted for recovery (8-23%); *p<0.05 relative to baseline.
2.2 β-amyloid evoked release of dopamine is mediated by α7* nAChRs
The pharmacological assessment of the Aβ-evoked DA release in prefrontal cortex indicated involvement of α7* nAChRs, based on inhibition by α-bungarotoxin (Fig. 2B). To ascertain definitely the role of particular nAChR subtypes, the Aβ-evoked DA release was examined in mice harboring null mutations for either the α7 subunit or β2 subunit of nAChRs. The DA outflow evoked by 100nM Aβ1-42 was absent in α7 null-mutant mice, whereas the inhibition of DA release remained (Fig. 3A). By contrast, DA release evoked by 100nM Aβ1-42 was intact in β2 null-mutant mice (Fig. 3B). Prior treatment with nicotine attenuated the subsequent Aβ-evoked DA outflow, but did not affect the slowly developing decrease in DA release (Fig. 3C). These results indicate that the transient increase in DA outflow evoked by Aβ was mediated by activation of α7* nAChRs.
3. Discussion
It has been proposed that under normal conditions soluble Aβ has a physiological function (Kamenetz et al., 2003; Wilquet and De Strooper, 2004; Pearson and Peers, 2006). As Aβ accumulates during the course of AD, however, it likely disrupts neuronal signaling in a variety ways, dependent upon its actual form (monomeric, small or large oligomeric, aggregated, protofibrillar, fibrillar) (eg. Ye et al., 2004; Bell et al., 2004). That accumulation of soluble Aβ is relevant to AD is supported by the strong correlation of soluble Aβ levels with neuropathology in AD patients (Lue et al., 1999; McLean et al., 1999). It was previously demonstrated that acute application of relatively low concentrations (pM-nM) soluble Aβ can induce increases in presynaptic Ca2+ level in a manner that appears to involve nAChRs (Dougherty et al., 2003). Freshly made Aβ solutions at pM to nM final concentrations will result largely in the formation of small oligomers (Klein, 2002; Chromy et al., 2003; Gong et al., 2003; Stine et al., 2003; Bell et al., 2004). In the present study, acute application of soluble Aβ into mouse prefrontal cortex was shown to increase transiently the release of DA, measured as overflow via microdialysis in the presence of TTX, whereas control peptides were without effect. A core fragment comprising residues 12-28 of Aβ (see Wang et al., 2000a) also stimulated DA release but the effect was variable.
The stimulatory effect of Aβ occurred through an action on presynaptic α7* nAChRs, as based on the block of the stimulatory effect by α-bungarotoxin (Fig. 2B) and the loss of the stimulatory effect in α7 null mutant mice (Fig. 3A). In addition, very low relative concentrations (pM) of soluble Aβ caused a slowly developing, long-lived depression in DA in the prefrontal cortex. In view of the lack of this inhibited phase of DA release in β2 null mice (Fig. 3B) or mice treated with a high concentration of a nicotinic antagonist (MLA in Fig.2B), the possibility exists that the slowly developing depression of DA outflow involves β2 containing nAChRs. Interestingly, the apparent EC50 for the stimulation of presynaptic Ca2+ by Aβ was in the pM range (Dougherty et al., 2003), at which only the inhibited phase of DA release was evident (Fig. 2D). Thus, pM Aβ via presynaptic nAChRs may evoke a sustained but low level increase in presynaptic Ca2+, leading to synaptic depression. On the other hand, nicotine perfusion did not evoke this inhibited phase of DA outflow under any condition, nor did nicotine alter the inhibited phase. Thus, alternative targets may exist. Based on previous studies of the effects of soluble Aβ on synaptic plasticity, possible additional candidate targets might include metabotropic glutamate receptors (Wang et al., 2004) and the nitric oxide pathway (Puzzo et al., 2005; Trabace et al., 2007). Whether the profound decrease in DA in response to very low concentrations of Aβ represents synaptic depression also remains to be uncovered. Previous evidence has implicated Aβ in synaptic depression under a negative feedback system for activity-dependent APP processing in rat hippocampus in a manner dependent upon glutamatergic receptors (Kamenetz et al., 2003). In separate studies, depression of synaptic transmission by Aβ or Aβ fragments (eg. Aβ25-35) has been linked to inhibition of voltage-gated Ca2+ channels (Ashenafi et al., 2005; Santos-Torres et al., 2007) and reduction of AMPA receptor currents (Hsieh et al., 2006; Shemer et al., 2006; Ting et al., 2007). Furthermore, as soluble Aβ levels rise in AD, stimulation of DA release might also lead to synaptic depression by depletion over the long run, dependent on the cholinergic activity at the presynaptic nAChRs as well as nerve activity (Dougherty et al., 2003; Kamenetz et al., 2003). It remains to be elucidated whether these effects are pathological or are part of a physiological action of Aβ. It is very likely that this distinction depends critically on the concentration of soluble Aβ.
The question arises as to the impact of chronic Aβ on presynaptic nAChRs. It was previously reported that α7* nAChRs are up-regulated in a mouse model for AD, Tg2576, at 9 months of age and then decline thereafter (Jones et al., 2006; however see Oddo and LaFerla, 2006). In preliminary experiments using a model AD mouse at 9 months of age, the DA outflow in response to Aβ was substantially increased (∼3-fold) as compared to age-matched controls (Wu and Nichols, unpublished observations). The extent to which alteration of DA outflow in response to Aβ reflects up-regulation of nAChRs would need to be determined. The findings presented here would be consistent with the possibility that Aβ, shown to act as a ligand for α7* nAChRs, induces up-regulation when chronically present. In AD, and in older AD mice, the α7* nAChRs decrease (Guan et al., 2000; Oddo et al., 2005), suggesting that the system ultimately undergoes degeneration, due most likely to synapse and cell loss.
The prefrontal cortex is implicated in higher executive functions (eg. attention, planning, impulse control), with a prominent role for DA (Arnsten and Li, 2005). The dopaminergic pathway, in turn, is modulated by, among others, nicotine (George et al., 2000). Interestingly, high levels of DA, released, for example, in response to stress, worsen prefrontal cortex function, as gauged by attentional performance (Granon et al., 2000). Thus, normal tonic levels of DA in prefrontal cortex likely act to enhance synaptic function (eg. Matsuda et al., 2006). On the other hand, excessive DA release will disrupt prefrontal cortex synaptic function. The results of the present study suggest that the induction of DA release by Aβ may alter synaptic function in prefrontal cortex.
4. Experimental Procedures
4.1 Animal Surgery
Adult male C57BL/6J mice (Jackson), C57B1/6 mice harboring a null mutation for the nAChR α7 subunit (Orr-Urtreger et al., 1997) or C57B1/6 mice harboring a null mutation for the nAChR β2 subunit (Picciotto et al., 1995) were deeply anesthetized with 60mg/kg i.p. pentobarbital (Henry Schein). Under sterile conditions, the anesthetized animals were placed into a Kopf rodent stereotaxic apparatus designed for use with mice and a guide cannula (CMA7) for the microdialysis probe was inserted into the prefrontal cortex, using 10° angle, at 2.0mm anterior to bregma, 1.0mm lateral and -3.0mm ventral from dura (Ihalainen et al., 1999; Paxinos and Franklin, 2001). The probe position and size were chosen to minimize overflow from other structures. (The position of the probe was verified at the end of every experiment (example shown in supplemental Fig. S1).) In the case of perfusion with α-bungarotoxin (Calbiochem), a second, open cannula was cemented next to and slightly above the microdialysis probe cannula, implanting both during surgery, as the toxin will not pass through the microdialysis probe membrane. (In preliminary experiments, perfusion of β-amyloid via the second cannula was also performed, but yielded equivalent results to those obtained wherein β-amyloid was delivered via the microdialysis probe (unpublished findings).) Animals were kept on an isothermal pad (Deltaphase) during recovery, while bupivacaine (0.5% solution; Henry Schein) was applied liberally to the surgical site during and after recovery. The surgical procedure followed a protocol approved by the Drexel University College of Medicine Institutional Animal Care and Use Committee. A colony of mice harboring a null mutation for the nAChR α7 subunit was established from heterozygous breeders obtained courtesy of Dr. Michael Marks, University of Colorado. A colony of mice harboring a null mutation for the nAChR β2 subunit was established from heterozygous breeders obtained courtesy of Dr. Marina Picciotto, Yale University. Routine genotyping of offspring was performed to identify homozygous null mutants (α7-/- and β2-/-).
4.2 In Vivo Microdialysis
Between 18-24h after surgery, the holder insert in the cannula was removed and the microdialysis probe (CMA11, membrane length: 1 mm) was inserted. An output dialysis line from a microdialysis pump (CMA/102) was attached via a freely rotating swivel assembly (BAS) to the microdialysis probe of cannulated mice. The mice were maintained in a plastic cage where they could freely move and have access to food and water during the course of the experiment. Perfusion with sterile artificial cerebrospinal fluid (aCSF) composed of 145mM NaCl, 2.8mM KCl, 1.2mM CaCl2, 1.2mM MgCl2, 0.25mM ascorbate, 5mM glucose plus 20 mM Na-phosphate, pH 7.4 (He and Shippenburg, 2000) was commenced at 1 μl/min. After 1-2h, sampling of the perfusate was started, collecting fractions into perchloric acid in tubes in a microfraction collector (CMA/142), typically every 20min. After collecting three fractions to determine basal outflow, perfusion with 1 μM tetrodotoxin (TTX; Calbiochem) in sterile aCSF was initiated 10 min prior to perfusion with stimulatory agents (in TTX/aCSF). Even though nM TTX is sufficient to block action potentials in isolated preparations, the use of 1 μM TTX is necessary to ensure full block (Boehnke and Rasmusson, 2001) without noted risk to the mice. Where antagonists were used, they were included during the TTX preincubation period. β-amyloid peptides (Bachem) were solubilized in aCSF immediately before use, as described previously (Dougherty et al., 2003). Fractions were analyzed on an ESA Coulochem II HPLC equipped with a 3 μm-particle MD150X2 column (mobile phase: ESA MD-TM (acetonitrile: octanesulfonate/EDTA/TEA/Na•phosphate in HPLC-grade water, pH 3) at 0.5ml/min) and a high-sensitivity electrochemical detector (guard cell: 350mV; analytical cell: 300mV detection potential; 0.05-0.1 pg/μl sensitivity for dopamine). Dopamine (DA) standards were injected into the HPLC before assaying the dialysates in order to determine DA retention time. Mean basal levels of DA (pg/μl) were determined for the first 3 fractions (basal) based on regression analysis of the areas under the DA peaks relative to DA standards, as adjusted for probe recovery (Wang et al., 2000b; Knobelman et al., 2001). Release of DA in response to applied agents is expressed as a percentage of the basal level in order to normalize responses across animals.
4.3 Immunostaining
To assess perfusion of Aβ into the prefrontal cortical tissue surrounding the probe, microdialysis with 100nM Aβ1-42 was performed as described in the previous section for 30 min. The probe and guide cannula were immediately removed and India ink was quickly injected into the hole formerly occupied by the cannula to mark the probe site. The mouse was euthanized and the intact brain was removed to an ice-cold glass plate. The area of interest was sliced out, using the dye as a guide. The brain area was then fixed in 4% paraformaldehyde in 0.1 M sodium phosphate-buffered saline (PBS) for 2h. The slice was then transferred to 20% sucrose in PBS for cryoprotection for 15h at 4°C, sectioned on a freezing microtome at 40μm and mounted on glass slides. The sections were incubated or not (control) for 40h at 4°C in anti-β amyloid monoclonal antibody (6E10; Sigma) at 1:400 dilution in 0.1 M PBS containing 4% bovine serum albumin and 0.3% Triton X-100 (PBS-BSA-T). The sections were then washed three times in PBS for 10min each, incubated with FITC-conjugated affinity purified goat anti-mouse IgG (1:200; Jackson ImmunoResearch) in PBS-BSA-T for 2 h, washed with PBS and finally coverslipped, sealing with nail polish. Sections were imaged via confocal microscopy. Sections incubated without primary antibody (control) were imaged first, to set range for the baseline “black-level” on the confocal imaging system for background staining due to the secondary antibody alone (see Wu et al., 2006).
4.4 Statistics
Most experiments were replicated 3-6 times. Where indicated, the significance of the difference between mean values was determined by one-way ANOVA followed by Scheffé’s F test. Differences were considered significant when p was minimally <0.05.
Supplementary Material
Fig. S1. Microdialysis probe track visualized in C57BL/6 mouse brain after perfusing briefly with tracking dye, verifying the probe placement into prefrontal cortex at the end of a typical experiment. The front of the brain was sectioned back to the prefrontal area to reveal the track.
Acknowledgements
The work was supported by grants from the NIH (AG21586) and the State of Pennsylvania Tobacco Formula Funds. We thank Ms. Catherine Choi and Ms. Tejal Mehta for help in establishing the colonies of transgenic mice.
Abbreviations
- Aβ
β-amyloid
- aCSF
artificial cerebrospinal fluid
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- BgTx
α-bungarotoxin
- DA
dopamine
- DHBE
dihydro-β-erythroidine
- HBS
HEPES-buffered saline
- MLA
methyllycaconitine
- nAChRs
nicotinic acetylcholine receptors
- 3xTg-AD
triple-transgenic AD mice
- TTX
tetrodotoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnsten AFT, Li B.-Mi. Neurobiology of executive functions: catecholamine influences on prefrontal cortical functions. Biol. Psychiatry. 2005;57:1377–1384. doi: 10.1016/j.biopsych.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Ashenafi S, Fuente A, Criado JM, Riolobos AS, Heredia M, Yajeya J. β-amyloid peptide25-35 depresses excitatory synaptic transmission in the rat basolateral amygdale “in vitro”. Neurobiol. Aging. 2005;26:419–428. doi: 10.1016/j.neurobiolaging.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Bell KA, O’Riordan KJ, Sweatt JD, Dineley KT. MAPK recruitment by β-amyloid in organotypic hippocampal slice cultures depends on physical state and exposure time. J. Neurochem. 2004;91:349–361. doi: 10.1111/j.1471-4159.2004.02722.x. [DOI] [PubMed] [Google Scholar]
- Boehnke SE, Rasmusson DD. Time course and effective spread of lidocaine and tetrodotoxin delivered via microdialysis: an electrophysiological study in cerebral cortex. J. Neurosci. Meth. 2001;105:133–141. doi: 10.1016/s0165-0270(00)00348-4. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Cao Y-J, Surowy CS, Puttfarcken PS. Different nicotinic acetylcholine receptors subtypes mediating striatal and prefrontal cortical [3H]dopamie release. Neuropharmacol. 2005;48:72–79. doi: 10.1016/j.neuropharm.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL. Self-assembly of Aβ1-42 into globular neurotoxins. Biochem. 2003;42:12749–12760. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Hsiao Ashe K, Sweatt JD. β-amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer’s disease. J. Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JJ, Wu J, Nichols RA. β-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J. Neurosci. 2003;23:6740–6747. doi: 10.1523/JNEUROSCI.23-17-06740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, Jhamandas JH. β-amyloid peptide activates non-α7 nicotinic acetylcholine receptors in rat basal forebrain neurons. J. Neurophysiol. 2003;90:3130–3136. doi: 10.1152/jn.00616.2003. [DOI] [PubMed] [Google Scholar]
- George TP, Verrico CD, Picciotto MR, Roth RH. Nicotinic modulation of mesoprefrontal dopamine neurons: pharmacologic and neuroanatomic characterization. J. Pharmacol. Exp. Therap. 2000;295:58–66. [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer’s disease-affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl Acad. Sci. USA. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granon S, Passetti F, Thomas KL, Dalley JW, Everitt BJ, Robbins TW. Enhanced and impaired attentional performance after infusion of D1 dopaminergic receptor agents into rat prefrontal cortex. J. Neurosci. 2000;20:1208–1215. doi: 10.1523/JNEUROSCI.20-03-01208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi F, Palma E, Tonini R, Amici M, Ballivet M, Eusebi F. Amyloid β1-42 peptice alters the gating of human and mouse α-bungarotoxin-sensitive nicotinic receptors. J. Physiol. 2003;547:147–157. doi: 10.1113/jphysiol.2002.035436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z-Z, Zhang X, Ravid R, Nordberg A. Decreased protein levels of nicotinic receptor subunits in the hippocampus and temporal cortex of patients with Alzheimer’s disease. J. Neurochem. 2000;74:237–243. doi: 10.1046/j.1471-4159.2000.0740237.x. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- He M, Shippenberg TS. Strain differences in basal and cocaine-evoked dopamine dynamics in mouse striatum. J. Pharmacol. Exp. Therap. 2000;294:121–127. [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihalainen JA, Rieffinen P, Feenstra MGP. Comparison of dopamine and noradrenaline release in mouse prefrontal cortex, striatum and hippocampus using microdialysis. Neurosci. Letts. 1999;277:71–74. doi: 10.1016/s0304-3940(99)00840-x. [DOI] [PubMed] [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Aβ accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- Jones IW, Westmacott A, Chan E, Jones RW, Dineley K, O’Neill MJ, Wonnacott S. α7 nicotinic acetylcholine receptor expression in Alzheimer’s disease: receptor densities in brain regions of the APP(SWE) mouse model and in human peripheral blood lymphocytes. J. Mol. Neurosci. 2006;30:83–84. doi: 10.1385/JMN:30:1:83. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Klein WL. Aβ toxicity in Alzheimer’s disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem. Int. 2002;41:345–352. doi: 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- Knobelman DA, Hen R, Lucki I. Genetic regulation of extracellular serotonin by 5-hydroxytryptamine1A and 5-hydroxytryptamine1B autoreceptors in different brain regions of the mouse. J. Pharmacol. Exp. Therap. 2001;298:1083–1091. [PubMed] [Google Scholar]
- Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, Cruts M, Dermaut B, Wang R, Van Broeckhoven C. Mean age-of-onset of familial Alzheimer disease caused by presenilin mutations correlates with both increased Aβ42 and decrease Aβ40. Human Mutat. 2006;27:686–695. doi: 10.1002/humu.20336. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Violoa KL, Wals P, Zhang C, Finch CE, Krafft CA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl Acad. Sci. USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released β-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–9793. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q-S, Kawai H, Berg DK. β-amyloid peptide blocks the response of α7-containing nicotinic receptors on hippocampal neurons. Proc. Natl Acad. Sci. USA. 2001;98:4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue L-F, Kuo Y-M, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda Y, Marzo A, Otani S. The presence of background dopamine signal converts long-term synaptic depression to potentiation in rat prefrontal cortex. J. Neurosci. 2006;26:4803–4810. doi: 10.1523/JNEUROSCI.5312-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, Wonnacott S. Methyllycaconitine is a potent antagonist of α-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. J. Pharmacol. Exp. Therap. 2002;301:197–204. doi: 10.1124/jpet.302.1.197. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Green KN, Liang K, Tran L, Chen Y, Leslie FM, LaFerla FM. Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA. 2005;102:3046–3051. doi: 10.1073/pnas.0408500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, Laferla FM. Temporal profile of amyloid-β (Aβ) oligomerization in an in vivo model of Alzheimer’s disease. J. Biol. Chem. 2006;281:1599–1604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- Oddo S, LaFerla FM. The role of nicotinic acetylcholine receptors in Alzheimer’s disease. J, Physiol, Paris. 2006;99:172–179. doi: 10.1016/j.jphysparis.2005.12.080. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Göldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, Dani JA, Patrick JW, Beaudet AL. Mice deficient in the α7 neuronal nicotinic acetylcholine receptor lack α-bungarotoxin binding sites and hippocampal fast nicotinic currents. J. Neurosci. 1997;17:9165–9171. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks JK, Smith TS, Trimmer PA, Bennett JP, Jr., Parker WD., Jr. Neurotoxic Aβ peptides increase oxidative stress in vivo through NMDA-receptor and nitric oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J. Neurochem. 2001;76:1050–1056. doi: 10.1046/j.1471-4159.2001.00112.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. 2nd Edition Academic Press; San Diego: 2001. [Google Scholar]
- Pearson HA, Peers C. Physiological roles for amyloid β peptides. J. Physiol. 2006;575:5–10. doi: 10.1113/jphysiol.2006.111203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit DL, Shao Z, Yakel J. β-amyloid1-42 peptide directly modulates nicotinic receptors in the rat hippocampal slice. J. Neurosci. 2001;21:RC120. doi: 10.1523/JNEUROSCI.21-01-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Léna C, Bessis A, Lallemand Y, Le Novère, Vincent P, Pich EM, Brûlet P, Changeux J-P. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–67. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Vitolo O, Trinchese F, Jacob JP, Palmeri A, Arancio O. Amyloid-β peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. J. Neurosci. 2005;25:6887–6897. doi: 10.1523/JNEUROSCI.5291-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao TS, Correa LD, Adams P, Santori EM, Sacaan AI. Pharmacological characterization of dopamine, norepinephrine and serotonin release in the rat prefrontal cortex by neuronal nicotinic acetylcholine receptor agonist. Brain Res. 2003;990:203–208. doi: 10.1016/s0006-8993(03)03532-7. [DOI] [PubMed] [Google Scholar]
- Role LW, Berg DK. Nicotinic receptors in the development and modulation of CNS synapses. Neuron. 1996;16:1077–1085. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- Santos-Torres J, Fuente A, Criado JM, Riobolos AS, Heredia M, Yajeya J. Glutamatergic synaptic depression by synthetic amyloid β-peptide in the medial septum. J. Neurosci. Res. 2007;85:634–648. doi: 10.1002/jnr.21150. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D. Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu. Rev. Pharmacol. Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- Shemer I, Holmgren C, Min R, Fulop L, Zilberter M, Sousa KM, Farkas T, Hartig W, Penke B, Burnashev N, Tanila H, Zilberter Y, Harkany T. Non-fibrillar β-amyloid abates spike-timing-dependent synaptic potentiation at excitatory synapses in layer 2/3 of the neocortex by targeting postsynaptic AMPA receptors. Eur. J. Neurosci. 2006;23:2035–2047. doi: 10.1111/j.1460-9568.2006.04733.x. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Price DL, Koliatsos VE. Disruption of corticocortical connections ameliorates amyloid burden in terminal fields in a transgenic model of Aβ amyloidosis. J Neurosci. 2002;22:9794–9799. doi: 10.1523/JNEUROSCI.22-22-09794.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine WB, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J. Biol. Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- Ting JT, Kelley BG, Lambert TJ, Cook DG, Sullivan JM. Amyloid precursor protein overexpression depresses excitatory transmission. Proc. Natl Acad. Sci. U.S.A. 2007;104:353–358. doi: 10.1073/pnas.0608807104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabace L, Kendrick KM, Catriganano S, Colaianna M, De Giogi A, Schiavone S, Lanni C, Cuomo V, Govoni S. Soluble amyloid beta1-42 reduces dopamine levels in prefrontal cortex: relationship to nitric oxide. Neurosci. 2007;147:652–663. doi: 10.1016/j.neuroscience.2007.04.056. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Aβ - a decade of discovery. J. Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, Agnaf OE, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid β-peptide (Aβ) fibrillogenesis block oligomerization of natural Aβ and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-Y, Lee DHS, D’Andrea MR, Peterson PA, Shank RP, Reitz AB. β-amyloid1-42 binds to α7 nicotinic acetylcholine receptor with high affinity: implications for Alzheimer’s disease. J. Biol. Chem. 2000a;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kikuchi T, Sakai M, Wu JL, Sata K, Okumura F. Age-related modifications of effects of ketamine and propofol on rat hippocampal acetylcholine release studied by in vivo microdialysis. Acta Anaesthiol. Scand. 2000b;44:112–117. doi: 10.1034/j.1399-6576.2000.440120.x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan M, Selkoe D, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid β-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J. Neurosci. 2004;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JM, Cockcroft VB, Lunt GG, Smilie FS, Wonnacott S. Methyllycaconitine: a selective probe for neuronal α-bungarotoxin binding sites. FEBS Lett. 1990;270:45–48. doi: 10.1016/0014-5793(90)81231-c. [DOI] [PubMed] [Google Scholar]
- Wilquet V, De Strooper B. Amyloid-beta precursor protein processing in neurodegeneration. Curr. Op. Neurobiol. 2004;14:582–588. doi: 10.1016/j.conb.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Wu J, Kuo Y-P, George AA, Xu L, Hu J, Lukas RJ. β-amyloid directly inhibits human α7β2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J. Biol. Chem. 2004;279:37842–37851. doi: 10.1074/jbc.M400335200. [DOI] [PubMed] [Google Scholar]
- Wu J, Dougherty JJ, Nichols RA. Dopamine receptor regulation of Ca2+ levels in individual isolated nerve terminals from rat striatum: comparison of presynaptic D1-like and D2-like receptors. J. Neurochem. 1999;98:481–494. doi: 10.1111/j.1471-4159.2006.03901.x. [DOI] [PubMed] [Google Scholar]
- Ye C, Walsh DM, Selkoe DJ, Hartley DM. Amyloid β-protein induced electrophysiological changes are dependent on aggregation state: N-methyl-D-aspartate (NMDA) versus non-NMDA receptor/channel activation. Neurosci. Letts. 2004;366:320–325. doi: 10.1016/j.neulet.2004.05.060. [DOI] [PubMed] [Google Scholar]
- Zoli M, Léna C, Picciotto MR, Changeux J-P. Identification of four classes of brain nicotinic receptors using β2 mutant mice. J. Neurosci. 1998;18:4461–4472. doi: 10.1523/JNEUROSCI.18-12-04461.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Microdialysis probe track visualized in C57BL/6 mouse brain after perfusing briefly with tracking dye, verifying the probe placement into prefrontal cortex at the end of a typical experiment. The front of the brain was sectioned back to the prefrontal area to reveal the track.