

Summary
Trafficking of AMPA receptors (AMPARs) is regulated by specific interactions of the subunit intracellular C-terminal domains (CTDs) with other proteins, but the mechanisms involved in this process are still unclear. We have found that the GluR1 CTD binds to cGMP-dependent protein kinase II (cGKII) adjacent to the kinase catalytic site. Binding of GluR1 is increased when cGKII is activated by cGMP. cGKII and GluR1 form a complex in the brain, and cGKII in this complex phosphorylates GluR1 at S845, a site also phosphorylated by PKA. Activation of cGKII by cGMP increases the surface expression of AMPARs at extrasynaptic sites. Inhibition of cGKII activity blocks the surface increase of GluR1 during chemLTP, and reduces LTP in the hippocampal slice. This work identifies a novel pathway, downstream from the NMDA receptor (NMDAR) and nitric oxide (NO), which stimulates GluR1 accumulation in the plasma membrane and plays an important role in synaptic plasticity.
Introduction
AMPARs are ionotropic glutamate receptors that mediate rapid excitatory transmission in the mammalian brain. They are hetero-tetrameric cation channels comprised of a combinatorial assembly of four subunits, GluR1-GluR4 (GluRA-D) (Hollmann and Heinemann, 1994). Regulated trafficking of AMPARs has emerged as an important mechanism that underlies the activity-dependent modification of synaptic strength. Delivery of AMPARs to the postsynaptic membrane leads to long-term potentiation (LTP), whereas removal of these receptors leads to long-term depression (LTD) (Barry and Ziff, 2002; Bredt and Nicoll, 2003; Malinow and Malenka, 2002; Sheng and Lee, 2001; Song and Huganir, 2002). Both of these forms of synaptic plasticity are influenced by NMDAR activity (Bliss and Collingridge, 1993; Malenka and Bear, 2004). Regulation of AMPAR synaptic insertion is determined by the receptor subunit composition. While synaptic activity drives GluR1-containing receptors to the synapse, thus enhancing transmission, AMPARs lacking GluR1, such as GluR2/3 heteromers, constitutively cycle in and out of the synapse, in an activity-independent manner, entering and leaving sites initially occupied by GluR1-containing receptors. This distinction in subunit trafficking is determined by the subunit intracellular CTDs (Passafaro et al., 2001; Shi et al., 2001).
Several lines of evidence indicate that GluR1 has an important role in LTP. GluR1 is delivered to the synapse during LTP (Hayashi et al., 2000), adult GluR1 -/- mice do not express LTP in CA3 to CA1 synapses (Zamanillo et al., 1999), and LTP is deficient in mice with knockin mutations in the GluR1 PKA and CaMKII phosphorylation sites (Lee et al., 2003). The molecular mechanisms that regulate GluR1 synaptic delivery during LTP are complex, and involve interactions of the GluR1 CTD with scaffolding proteins, such as protein 4.1N and SAP97 (Leonard et al., 1998; Shen et al., 2000), and a series of phosphorylation steps at several Ser residues on the GluR1 CTD (Boehm and Malinow, 2005). The CTD of GluR1 is phosphorylated at S831 by both CaMKII and PKC (Barria et al., 1997; Mammen et al., 1997; Roche et al., 1996), at S845 by PKA (Roche et al., 1996), and at S818 by PKC (Boehm et al., 2006). While CaMKII drives GluR1 to the synapse and may contribute to induction of LTP, a mutation of GluR1 S831 that prevents phosphorylation by CaMKII does not prevent synaptic delivery of the receptor by active CaMKII or LTP (Hayashi et al., 2000), suggesting that CaMKII acts on a different target to induce GluR1 synaptic delivery. Interestingly, mutagenesis of S845 of GluR1 showed that phosphorylation of this site is required, although not sufficient, for GluR1 synaptic insertion during LTP (Esteban et al., 2003). Phosphorylation of S845 by PKA has also been shown to increase the delivery of AMPARs to extrasynaptic sites, and to prime the receptor for synaptic insertion (Oh et al., 2006; Sun et al., 2005).
Many studies have suggested that the diffusible second messenger, nitric oxide (NO), contributes to the mechanism of LTP (Bon et al., 1992; Bon and Garthwaite, 2003; Haley et al., 1992; O'Dell et al., 1994; Schuman and Madison, 1991; Zhuo et al., 1993), and LTP in the CA1 region of hippocampus is reduced in double knockout mice lacking both endothelial and neuronal NO synthase (eNOS and nNOS, respectively) (Son et al., 1996), the two major NO-producing enzymes in the brain. Although many studies have addressed the role of NO in LTP, the molecular mechanisms underlying the NO regulation of synaptic plasticity are still elusive. NO activates soluble guanylate cyclase (sGC), which induces the formation of cGMP, and one cGMP target is the cGMP-dependent kinases (cGKs). There are two cGK isoforms, cGKI and cGKII. While cGKI is cytosolic and in the brain is preferentially enriched in the cerebellum, cGKII is located in cellular membranes and is widely distributed in the brain (Francis and Corbin, 1999).
Here we report that cGKII binds the GluR1 CTD in a cGMP-dependent manner, and that in this complex, cGKII can phosphorylate S845 of GluR1 and increase GluR1 levels in the plasma membrane. Glycine-induced chemLTP promotes cGKII interaction with GluR1 and phosphorylation of S845. Expression of a cGKII inhibitor peptide in cultured hippocampal neurons blocks the increase of surface GluR1 and of mEPSCs frequency and amplitude after chemLTP, and when expressed in the mouse hippocampus, reduces LTP in the hippocampal slice. This function of cGKII provides a new mechanism for increasing the levels of GluR1 in the plasma membrane that complements the PKA-induced GluR1 surface increase. Because the NMDAR regulates NO production by nNOS, and hence controls cGMP levels and cGKII activity, this pathway provides a novel mechanism for NMDAR and NO control of GluR1 accumulation in the plasma membrane.
Results
Interaction of the GluR1 CTD with cGKII
Using the GluR1 CTD as bait, we screened 2 × 106 cDNAs for GluR1 CTD interactors by the Sos recruitment system, which detects protein-protein interactions in the yeast cytoplasm (Aronheim et al., 1997). We obtained three independent positive transformants, which all encoded the region between aa 400-762 of cGKII. cGKII is comprised of three functional domains, an N-terminal regulatory domain, a cGMP-binding domain that contains two tandem cGMP-binding sites, and a catalytic domain (Pfeifer et al., 1999) (Fig. 1B). The N-terminal domain contains a dimerization domain and an autoinhibitory (AI) domain, which includes autophosphorylation sites. The clones encoded the second cGMP-binding domain plus the catalytic domain, located at the cGKII C-terminus (Fig. 1B). In the Sos recruitment assay, neither deletion of the last 8 aa of GluR1, which contain the PDZ ligand and is required for interaction with SAP97 (Leonard et al., 1998) (Fig. 1Ai), nor deletion of the region from the membrane-spanning domain to aa 839, a region required for protein 4.1N interaction (Shen et al., 2000) (Fig. 1Ai), blocked the aa 400-762 cGKII interaction (Fig. 1Aii). We conclude that aa 839-881 of the GluR1 CTD, a region not known to bind other proteins, is important for the interaction with cGKII.
Figure 1. Interaction between cGKII and GluR1.

(A) Yeast two-hybrid analysis of the GluR1 CTD and the cGKII catalytic domain interaction. (Ai) Schematic of the GluR1 CTD and the regions required for binding with protein 4.1N and SAP97. (Aii) The GluR1 CTD and deletion mutants were screened for interaction with the aa 400-762 fragment of cGKII (cGKII aa 400–762) by the Sos recruitment system as described in Experimental Procedures.
(B) Schematic of cGKII fragments and interaction with the GluR1 CTD in the mammalian recruitment assay (See Figure S1).
(C) Interaction of the GluR1 CTD with full-length cGKII, and stimulation by cGMP. Lysates of 293T cells expressing wt cGKII were incubated with GST-GluR1C. 293T cells were either treated with 8-Br-cGMP (50 μM) for 10 min, or left untreated (control), before lysis. The supernatant (SN) and the pellet (P) fractions were fractionated by SDS PAGE. The P fraction contains bound proteins and the SN, unbound proteins. The whole P and approximately 10% of the SN were fractionated in the gel. Proteins were detected by IB. Bottom panel shows the Coomassie blue staining of GST species and their degradation products, which are commonly seen.
(D) Specificity of the GluR1 CTD and cGKII interaction. Lysates from 293T cells expressing wt cGKII were incubated with GST, GST-GluR1C and GST-GluR2C. 293T cells were treated with 8-Br-cGMP (50 μM) for 10 min before lysis. Bound proteins were detected by IB. Bottom panel shows the Coomassie blue staining of GST species.
(E) GST pulldown experiment. Lysates from 293T cells expressing cGKII were incubated with GST alone or with GST-GluR1C deletion mutants. 293T cells were treated with 8-Br-cGMP (50 μM) for 10 min before lysis. Bound proteins were detected by IB. A 23 aa region between aa 850 and aa 873 in the GluR1 CTD is necessary and sufficient for binding with cGKII.
Using a mammalian recruitment system in 293T cells, we confirmed that the GluR1 CTD fused to GFP (GluR1C-GFP) bound the plasma membrane-anchored cGKII catalytic domain (cGKII aa 400-762) (Fig. S1) and found that the C-terminal 173 aa of cGKII, which contain a segment of the catalytic domain, are required for interaction with GluR1 (Fig. S1A and B).
Regulation of GluR1 CTD binding to cGKII by cGMP
In the cGKII inactive, autoinhibited state, the cGKII AI domain binds the substrate-docking domain at the catalytic site (Fig. 1B). Activation by cGMP unfolds the AI domain and exposes the C-terminal catalytic domain (Wall et al., 2003; Zhao et al., 1997). We speculated that in the full-length cGKII, the AI domain-catalytic domain interaction could block GluR1 binding to the catalytic domain. If so, displacement of the AI following cGMP activation of cGKII could facilitate GluR1 interaction with the full-length kinase. In a GST pulldown assay, the GluR1 CTD fused to GST (GST-GluR1C) pulled down full-length cGKII from lysates of both control and 8-Br-cGMP treated 293T cells expressing full-length cGKII (Fig. 1C). However, strikingly, treatment of 293T cells with 8-Br-cGMP greatly increased the interaction with cGKII (Fig. 1C). As controls, neither GST alone nor the GluR2 CTD fused to GST (GST-GluR2C) pulled down cGKII, even after 8-Br-cGMP treatment (Fig. 1D). These results confirm that the GluR1 CTD interacts with full-length cGKII, and that kinase activation increases the interaction.
We used GST GluR1 CTD deletion mutant pulldowns to map the cGKII-interacting region within the CTD (Fig. S2). While certain CTD mutants bound cGKII with intermediate affinity (Fig. S2), the GluR1 CTD 23 aa sequence between aa 850 and aa 873 was necessary and sufficient for binding cGKII, although binding was decreased compared to wt (Fig. 1E). The CTD mutation R837A also decreased CTD binding to cGKII, and the mutation R837AE completely disrupted the binding (Fig. 1E), suggesting that R837 is critical for the cGKII-GluR1 interaction.
cGKII cofractionates and colocalizes with GluR1 and binds GluR1 in vivo
Myristoylation of the Gly2 residue targets cGKII to the plasma membrane (Vaandrager et al., 1996). In assays of the cGKII brain subcellular distribution, cGKII was present in the synaptosome and postsynaptic density (PSD) fractions, but was most abundant in the lipid raft fraction of the synaptosomes (Fig. 2A). GluR1 was also in the lipid raft fraction (Fig. 2A), as previously shown (Hering et al., 2003), and thus in the same cellular compartment as cGKII.
Figure 2. cGKII interacts with GluR1 in the brain and in cortical cultures, colocalizes with GluR1 in cultured hippocampal neurons, and phosphorylates GluR1 S845 in vitro and in vivo.

(A) Subcellular fractionation of adult rat brains. Equal amounts of rat brain fractions were analyzed by IB and probed as indicated. P2, crude membranes; PSD, postsynaptic density.
(B) Hippocampal neuron stained for endogenous MAP2 (blue), cGKII (red) and synaptophysin (green). Scale bar, 20 μm. Arrows indicate cGKII and synaptophysin colocalization.
(C) Endogenous cGKII (green) and GluR1 (red) staining in hippocampal neurons. Scale bar, 5 μm. Arrows indicate cGKII and GluR1 colocalization.
(D) cGKII associates with GluR1 in rat brain tissue. Rat brain synaptosomal lysates were precipitated with a control IgG, an anti-cGKII Ab or an anti-GluR1 Ab. Bound proteins were detected by IB.
(E) 8-Br-cGMP increases the interaction of cGKII with GluR1 in cortical cultures. Cells were treated with 8-Br-cGMP, or saline (control). Cellular lysates were precipitated with an anti-cGKII Ab, and bound proteins were detected by IB.
(F) CoIP performed as in (E). Glycine and the NO donor NOR-3 increase the interaction of GluR1 with cGKII.
(G) In vitro phosphorylation of the GluR1 CTD by cGKII. GST, GST-GluR1C and GST-GluR2C were incubated with the indicated purified kinases and [γ-32P] ATP, resolved by SDS-PAGE, and analyzed by autoradiography. (i) Phosphorylation of GST-GluR1C by cGKII. The arrows indicate autophosphorylated cGKII. The lower cGKII band is a degradation product of the purified cGKII used in these assays. (ii) Phosphorylation of GST-GluR1C by PKA.
(H) Phosphorylated GST-GluR1C pulls down autophosphorylated cGKII. (i) GST-GluR1C was incubated with purified cGKII in the presence of [γ-32P] ATP. After incubation, protein complexes containing GST-R1C were analyzed by pulldown and autoradiography. The arrows indicate autophosphorylated cGKII. The lower cGKII band is a degradation product of the purified cGKII used in these assays. (ii) Same as in (i), but the reaction was incubated with unlabeled ATP. Proteins were detected by IB. The same two bands seen with labeled ATP in (i) were seen after IB with an anti-cGKII Ab.
(I) In vitro phosphorylation of GST-GluR1C at S845 by cGKII and PKA. Reactions were performed as in (A) but with unlabeled ATP. Levels of S845-PO4 and of total GluR1C were determined by SDS-PAGE and IB using phospho-specific and total GluR1 Abs, respectively.
(J) GluR1 phosphorylation at S845 in cortical cultures is increased by stimulation of the NMDA-NO-cGKII pathway. Cortical cultures were treated with glycine, the NO donor NOR-3, 8-Br-cGMP or with saline (control). Inhibitors used as indicated were: AP5, NMDAR antagonist, and KT5823, cGK inhibitor. Cultures were preincubated for 30 min with okadaic acid (1 μM) to inhibit phosphatase activity. Total amount of GluR1 was assayed with an anti-GluR1 Ab.
(K) Quantitation of the cGKII phosphorylation of GluR1 S845 from several experiments as the one shown in (D). Phospho-specific signal was normalized to the total amount of GluR1, and final values were normalized to control. *p<0.05, n=4.
In cultured hippocampal neurons, cGKII was punctate in the somatic cytoplasm and in neurites (Fig. 2B). cGKII puncta colocalized with or were adjacent to synaptophysin puncta, suggesting a synaptic location. GluR1 also displayed puncta that in many cases colocalized with the cGKII puncta (Fig. 2C), confirming GluR1 and cGKII presence in the same cellular compartment. Furthermore, an antiserum to cGKII, but not a control IgG, coprecipitated GluR1 with cGKII from brain synaptosomal lysate (Fig. 2D), confirming the cGKII-GluR1 interaction in the brain synaptosomal compartment, which is a synaptic and perisynaptic fraction. Significantly, treatment of cultured neurons with 8-Br-cGMP (500 μM; 5 min) increased the coprecipitation of GluR1 with cGKII, confirming that cGMP increases their binding (Fig. 2E). In a chemical form of LTP (chemLTP), short application of glycine at high concentration selectively activates synaptic NMDA receptors (Lu et al., 2001). Ca++ influx through the NMDAR activates nNOS, which increases NO, which activates the cGMP-producing enzyme, sGC (Bredt and Snyder, 1989; Garthwaite et al., 1988). Glycine application (200 μM; 3 min) increased the interaction of cGKII with GluR1, as did the NO donor, NOR-3 (10 μM; 10 min) (Fig. 2F). This confirms that NMDAR activation, NO or cGMP, all can increase the interaction of GluR1 with cGKII.
cGKII phosphorylates GluR1 at S845 in vitro and in vivo
GluR1 CTD S818, S831 and S845 are major phosphorylation sites (Boehm et al., 2006; Roche et al., 1996). When incubated with GST-GluR1C and [γ-32P] ATP, purified cGKII robustly phosphorylated GST-GluR1C (Fig. 2Gi). This was specific since cGKII phosphorylated neither GST alone nor GST-GluR2C (Fig. 2Gi). As expected, PKA also phosphorylated GST-GluR1C (Fig. 2Gii).
We next tested whether autophosphorylated cGKII could physically interact with GST-GluR1C as well as phosphorylate it (Fig. 2H). When we incubated GST-GluR1C and cGKII with [γ-32P] ATP, 32P labeled cGKII was pulled down by GST-GluR1C, which was also 32P labeled. Thus, cGKII not only phosphorylates GluR1, but activated autophosphorylated cGKII can physically interact with the phosphorylated GluR1 CTD (Fig. 2H).
Among the protein kinases, cGKs (cGKI and cGKII) are most closely related to PKA, and cGKs frequently recognize the canonical PKA phosphorylation site motif (Wang and Robinson, 1997). Also, both kinase types share substrates in the brain, with DARPP-32 one example (Hemmings et al., 1984). Since PKA phosphorylates S845 of GluR1, we assayed cGKII phosphorylation of this site. Notably, incubation of GST-GluR1C with either cGKII or PKA increased the signal for S845-PO4 detected by immunoblotting (IB) with a phosphopeptide-specific Ab to S845-PO4 (Fig. 2I). Thus, in vitro, cGKII phosphorylates GluR1 at S845. Furthermore, in cultured neurons, glycine, the NO donor NOR-3, and 8-Br-cGMP, all increased S845 phosphorylation (Fig. 2J and K). Pretreating with the NMDAR blocker, AP5 (25 μM; 20 min), or with the cGKII inhibitor, KT5823 (2 μM; 30 min), before glycine application, abolished the increase of S845 phosphorylation, confirming that glycine acts through the NMDAR and requires cGKII activity (Fig. 2J and K). However, AP5 pretreament did not prevent the effects of NOR-3 on S845 phosphorylation, although KT5823 reduced the increase of S845 phosphorylation after NOR-3 treatment almost to control levels (Fig. 2J and K). We conclude that cGKII phosphorylates GluR1 at S845 in vitro, and that activation of cGKII, which is under the regulation of the NMDAR and NO, leads to S845 phosphorylation in neurons.
cGKII activation increases surface GluR1
Phosphorylation of S845 by PKA is sufficient to increase GluR1 at the plasma membrane (Oh et al., 2006). When we expressed epitope-tagged GluR1 (HAGluR1) from a viral vector in cultured hippocampal neurons (Fig. 3A), a 5 min 8-Br-cGMP (500 μM) treatment caused a significant increase in the surface/total ratio of HAGluR1, compared to control cells (control: 1±0.14; 8-Br-cGMP: 2.18±0.26; values normalized to control, p<0.01) (Fig. 3Ai and Aii) that was significantly blocked by preincubation with KT5823 (2 μM, 30 min) (1.26±0.22) (Fig. 3Ai and Aii), suggesting that cGKII mediates a cGMP-dependent accumulation of GluR1 at the plasma membrane (Fig. 3A). In contrast, surface/total ratio of HAGluR1-S845A, which cannot be phosphorylated at S845, was not significantly increased by 8-Br-cGMP compared to untreated HAGluR1-S845A (untreated HAGluR1-S845A: 0.72±0.11, HAGluR1-S845A + 8-Br-cGMP: 0.99±0.07; p>0.05) or to untreated wt (1±0.09, p>0.05) (Fig. 3Bi and Bii). Similar results were obtained when neurons were infected with wt HA-GluR1 or HAGluR1-S845A and treated with the NO donor NOR-3 (10 μM, 10 min) [HAGluR1 wt + saline (control): 1±0.1, HAGluR1 wt + NOR-3: 4.6±0.9, p<0.005; and HAGluR1-S845A + saline (control): 1±0.1, HAGluR1-S845A + NOR-3: 0.9±0.1, p>0.05] (Fig. 3C). These results indicate that phosphorylation of S845 is required for the cGMP- and the NO-dependent GluR1 surface increase.
Figure 3. Effects of cGKII activation on GluR1 trafficking.

(Ai) Hippocampal neurons expressing HAGluR1 and treated for 5 min with saline (control), 8-Br-cGMP, or 8-Br-cGMP following pretreatment with the cGK inhibitor KT5823. Neurons were stained live for surface HA (red), followed by fixation, permeabilization and staining for total HA (green). Scale bar, 20 μm. (Aii) Quantitation of surface/total ratio of HAGluR1 on neurons from experiments as the one shown in (Ai). Bar graph shows mean±SEM; n=35 for control, 30 for 8-Br-cGMP and 20 for 8-Br-cGMP+KT5823; **p<0.01.
(Bi) Representative images of hippocampal neurons expressing HAGluR1 or HAGluR1-S845A and treated for 5 min with saline (control) or 8-Br-cGMP. Staining was done as in (Ai). Scale bar, 20 μm. (Bii) Quantitation of surface/total ratio of HAGluR1 and HAGluR1-S845A on neurons from experiments as the one shown in (Bi). Bar graph shows mean±SEM; n=33 for HAGluR1 wt, 20 for HAGluR1-S845A and 20 for HAGluR1-S845A+8-Br-cGMP; NS = non-significant.
(C) Quantitation of surface/total ratio of HAGluR1 wt and HAGluR1-S845A on neurons treated for 10 min with saline (control) or NOR-3. Bar graph shows mean±SEM; n=20 for HAGluR1 wt control, 18 for HAGluR1 wt + NOR-3, 22 for HAGluR1-S845A control and 24 for HAGluR1-S845A + NOR-3; **p<0.01.
(D) Images of surface GluR1 (red) and SV2 (green) staining in hippocampal neurons treated with saline (control), glycine for chemLTP, 8-Br-cAMP or 8-Br-cGMP. Neurons were stained live for surface GluR1, and after fixation and permeabilization, for SV2. Scale bar, 2 μm.
(E) Quantitation of surface levels of GluR1 on neurons from experiments as the one shown in (D). Bar graph shows mean±SEM; n=18 for control, 15 for glycine, 16 for 8-Br-cAMP and 19 for 8-Br-cGMP; ***p<0.001.
(F) Quantitation of synaptic GluR1 on neurons from experiments similar to those of (D). Synaptic GluR1 was expressed as the intensity of the GluR1 signal that overlapped with SV2 divided by the total intensity of the GluR1 signal. Bar graph shows mean±SEM; n = same as in (B); ***p<0.001.
(G) Examples of mEPSCs before (-5 min), and 5 and 25 min after the application of glycine.
(H) Application of glycine (n=6, filled red circles), but neither 8-Br-cGMP (n=6, open green squares), nor 8-Br-cAMP (n=6, open black squares), enhanced mEPSC amplitude in cultured hippocampal neurons. Controls were performed with vehicle (n=5, open red circles). The thick line represents the duration of glycine, 8-Br-cGMP or 8-Br-cAMP application and error bars represent SEM.
PKA increases surface GluR1 but only at extrasynaptic sites (Oh et al., 2006; Sun et al., 2005). To determine if cGKII acted similarly, we assayed GluR1 surface location following 8-Br-cGMP application, with chemLTP, which increases GluR1 synaptic insertion (Lu et al., 2001), as a control (Fig. 3D). We measured synaptic GluR1 by quantifying through immune fluorescence labeling, the intensity of the GluR1 fluorescent signal that overlaps with the signal of SV2, a synaptic marker, divided by the total intensity of the GluR1 signal (see Experimental Procedures). In control conditions, 34±2% of surface GluR1 overlapped with the synaptic marker SV2 (Fig. 3D and F). Although both 8-Br-cAMP (500 μM) and 8-Br-cGMP increased surface GluR1 (Fig. 3D and E), neither treatment significantly changed the amount of synaptic GluR1, measured as the overlap with SV2 (40±2% of GluR1 overlapped with SV2 in cells treated with 8-Br-cAMP, p>0.05, compared to control, and 39±2% in cells treated with 8-Br-cGMP, p>0.05, compared to control) (Fig. 3F). Induction of chemLTP by glycine increased surface GluR1 relative to the control (control: 1±0.15, glycine: 2.7±0.44, p<0.001) (Fig. 3E). However, as opposed to 8-Br-cAMP and 8-Br-cGMP treatment, chemLTP also significantly increased the overlap of GluR1 with SV2 relative to control (70±3%, p<0.001), as expected (Fig. 3D and F). These results suggest that while glycine, cGMP and cAMP all increase GluR1 surface levels, only with chemLTP is synaptic GluR1 increased.
To validate these results using electrophysiology, we recorded miniature excitatory postsynaptic currents (mEPSCs) on the cultured hippocampal neurons (Fig. 3G). Consistent with the earlier study (Lu et al., 2001), application of glycine (200 μM, 3 min) produced an immediate and long-lasting increase in mEPSC amplitude (131.6±12.3% of baseline at 25 min after glycine application, n=6) (Fig. 3G and H). However, unlike glycine, neither 8-Br-cGMP (500 μM) nor 8-Br-cAMP (500 μM) enhanced mEPSC amplitude (93.3±16.3% of baseline at 25 min after 8-Br-cGMP application, n=6; 78.6±10.1% of baseline at 25 min after 8-Br-cAMP application, n=6) (Fig. 3H), confirming that while 8-Br-cGMP or 8-Br-cAMP increase surface GluR1 (Fig. 3D), they are not sufficient to induce GluR1 synaptic insertion (Fig. 3H).
cGKII action is independent from PKA
Our results suggest that cGKII and PKA share a common mechanism to increase GluR1 in the plasma membrane. To rule out the possibility of cross talk between the cyclic nucleotide pathways, in which cGMP activates PKA or cAMP activates cGKII, we tested the effects of pharmacological inhibitors specific for cGKII and PKA (Fig. 4). 8-Br-cGMP increased the surface level of endogenous GluR1 approximately 3.5 fold (value of 3.48±0.30, treated cells; 1±0.15, control; p<0.001) (Fig. 4Ai and Aii). The effects of 8-Br-cGMP were reduced almost to control levels by the cGKII-specific inhibitor KT5823 (GluR1 surface level of 1.60±0.21), but not by the PKA-inhibitor KT5720 (2 μM, 30 min) (3.47±0.51, p<0.001), (Fig. 4Ai and Aii). Similarly, the increase in surface AMPARs by 8-Br-cAMP (2.82±0.38; control values same as above; p<0.001) was inhibited by the PKA inhibitor KT5720 (1.32±0.24; p>0.05, compared to control) (Fig. 4Bi and Bii), while the cGKII inhibitor KT5823, although it slightly reduced the cAMP-dependent increase of surface GluR1, still left significant higher surface levels of surface receptors compared to the control (1.94±0.22; p<0.01) (Fig. 4Bi and Bii).
Figure 4. Trafficking of GluR1 by cGMP and cAMP.
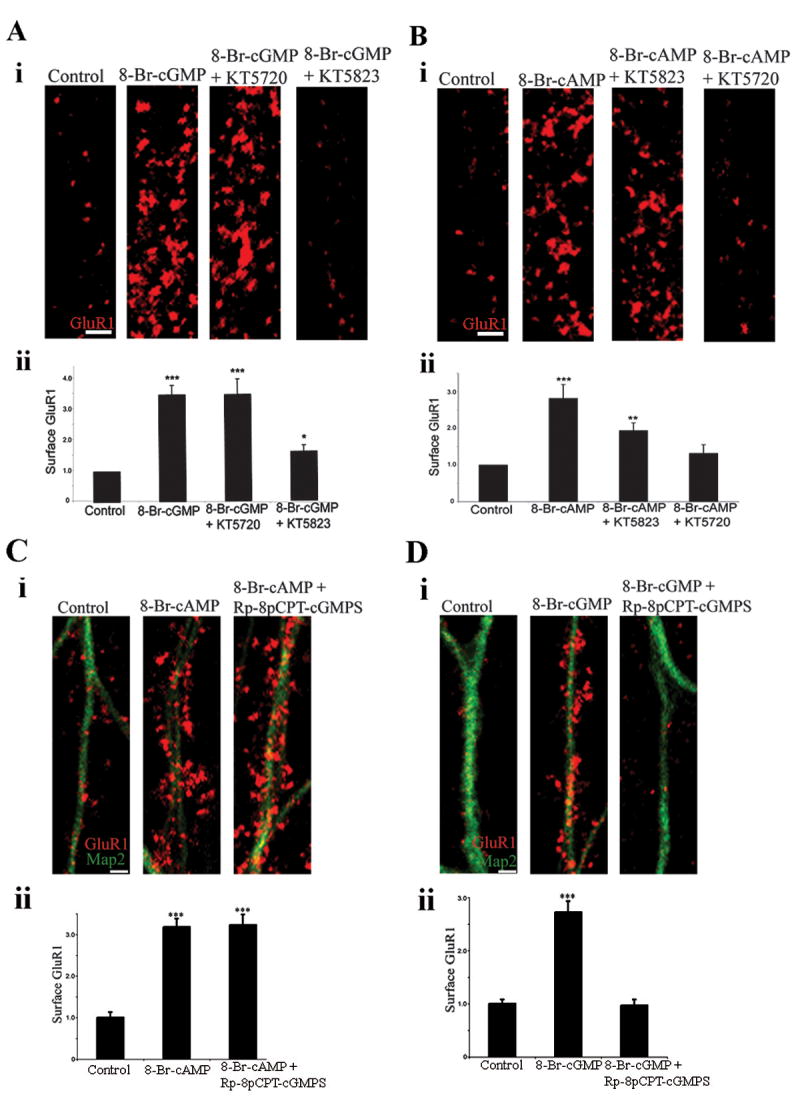
(Ai) Images of surface GluR1 in neurons treated for 5 min with saline (control), 8-Br-cGMP, 8-Br-cGMP following pretreatment with the PKA inhibitor KT5720, or 8-Br-cGMP following pretreatment with the cGK inhibitor KT5823. Neurons were then stained live for surface GluR1. Scale bar, 2 μm. (Aii) Quantitation of surface levels of GluR1 on neurons from experiments as the ones shown in (Ai). Bar graph shows mean±SEM; n=20 for all experimental groups; *p<0.05, ***p<0.001.
(Bi) Images of surface GluR1 in neurons treated for 5 min with saline (control), 8-Br-cAMP, 8-Br-cAMP following pretreatment with the cGK inhibitor KT5823, or 8-Br-cAMP following pretreatment with the PKA inhibitor KT5720. Staining was done as in (A). Scale bar, 2 μm. (Bii) Quantitation of surface levels of GluR1 on neurons from experiments as the ones shown in (Bi). Bar graph shows mean±SEM; n=20 for control, 14 for 8-Br-cAMP, and 16 for 8-Br-cAMP+KT2823 and 8-Br-cAMP+KT5720; **p<0.005, ***p<0.001.
(Ci) Images of surface GluR1 (red) in neurons treated for 5 min with saline (control), 8-Br-cAMP or 8-Br-cAMP following pretreatment with the cGKII inhibitor Rp-8-pCPT-cGMPS. Neurons were then stained live for surface GluR1, followed by fixation, permeabilization and staining for MAP2 (green). Scale bar, 2 μm. (Cii) Quantitation of surface levels of GluR1 on neurons from experiments as the one shown shown in (Ci). Bar graph shows mean±SEM; n=30 for control, 30 for 8-Br-cAMP and 27 for 8-Br-cAMP+Rp-8-pCPT-cGMPS. ***p<0.001.
(Di) Images of surface GluR1 (red) in neurons treated for 5 min with saline (control), 8-Br-cGMP or 8-Br-cGMP following pretreatment with the cGKII inhibitor Rp-8-pCPT-cGMPS. Neurons were then stained live for surface GluR1, followed by fixation, permeabilization and staining for MAP2 (green). Scale bar, 2 μm. (Dii) Quantitation of surface levels of GluR1 on neurons from experiments as the one shown in (Di). Bar graph shows mean±SEM; n=29 for control, 27 for 8-Br-cGMP and 26 for 8-Br-cGMP+Rp-8-pCPT-cGMPS. ***p<0.001.
We confirmed these results with Rp-8-pCPT-cGMPS, a more potent and very highly cell permeable cGKII inhibitor (Butt et al., 1994), which blocks nucleotide binding to the cGMP-binding site of cGKII, while KT5823 inhibits ATP binding to the catalytic domain (Kase et al., 1987). Rp-8-pCPT-cGMPS (10 μM, 30 min) did not reduce surface GluR1 following 8-Br-cAMP application (1±0.13, control; 3.18±0.2, 8-Br-cAMP;3.23±0.25, 8-Br-cAMP+Rp-8-pCPT-cGMPS) (Fig. 4Ci and Cii), however it completely prevented the 8-Br-cGMP increase of surface GluR1 (1±0.08, control; 2.72±0.21, 8-Br-cGMP; 0.97±0.11, 8-Br-cGMP+Rp-8-pCPT-cGMPS) (Fig. 4Di and Dii). We conclude that 8-Br-cGMP acts through cGKII, that its effects are independent from PKA, and that each kinase can act independently to increase the levels of GluR1 in the plasma membrane.
Different molecular mechanisms mediate GluR1 phosphorylation by PKA and cGKII
The GluR1 carboxyl terminus binds SAP97, a PDZ scaffolding protein that binds the A kinase anchoring protein, AKAP79, which binds and tethers PKA (Colledge and Scott, 1999). Phosphorylation of GluR1 by PKA requires this complex since disruption of the SAP97-GluR1 interaction decreased PKA phosphorylation of S845 (Colledge et al., 2000). Because cGKII directly binds GluR1, we hypothesized that its structural requirements for phosphorylation of GluR1 will differ. Both 8-Br-cAMP and 8-Br-cGMP significantly increased the phosphorylation of HAGluR1 wt in neuronal cultures, as expected (Fig. 5Ai and Aii). However, 8-Br-cAMP did not increase S845 phosphorylation of HAGluR1Δ7, a mutant that lacks the last 7 amino acids of GluR1 and does not bind the SAP97-AKAP-PKA complex, although 8-Br-cGMP still induced a significant increase of HAGluR1Δ7 S845 phosphorylation (Fig. 5Bi and Bii). Thus cGKII does not require the GluR1 PDZ ligand. Likewise, while 8-Br-cAMP did not change the surface/total ratio of HAGluR1Δ7 (ratio of 1±0.1 for untreated HAGluR1Δ7 and 1.1±0.1 for HAGluR1Δ7+8-Br-cAMP, p>0.05), 8-Br-cGMP did increase the HAGluR1Δ7 surface/total ratio significantly (1.43±0.1, p<0.001, compared to untreated) (Fig. 5C). Thus, PKA but not cGKII requires the GluR1 PDZ ligand to regulate GluR1 trafficking, as expected from the structural interactions. Moreover, in the absence of PKA regulation of GluR1, cGKII regulation of GluR1 is not significantly affected.
Figure 5. Different molecular mechanisms mediate phosphorylation and trafficking of GluR1 by PKA and cGKII.

(Ai) Lysates from neurons infected with HAGluR1 wt were precipitated with an anti-HA Ab. The IPs were probed with the indicated Abs. NTD = N terminal domain. (Aii) Quantitation of S845 phosphorylation from several experiments as the one shown in (Ai). Phospho-specific signal was normalized to the total amount of GluR1, and final values were normalized to control. ***p<0.001, n=3.
(Bi) Lysates from neurons infected with HAGluR1Δ7 were precipitated with an anti-HA Ab. The IPs were probed with the indicated Abs. Note that HAGluR1Δ7 is not recognized by the GluR1 CTD Ab, since this Ab recognizes a sequence present in the C terminus of GluR1, which has been deleted in HAGluR1Δ7 (see text). (Bii) Quantitation of S845 phosphorylation from several experiments as the one shown in (Bi). Phospho-specific signal was normalized to the total amount of GluR1, and final values were normalized to control. ***p<0.001, n=3.
(C) Hippocampal neurons expressing HAGluR1Δ7 and treated for 5 min with saline (control), 8-Br-cAMP, or 8-Br-cGMP. Neurons were stained live for surface HA (red), followed by fixation, permeabilization and staining for total HA (green). Scale bar, 20 μm; lower panel, 2 μm. The right lower panel shows the quantitation of surface/total ratio of HAGluR1. Bar graph shows mean±SEM; n=35 for control, 30 for 8-Br-cAMP and 20 for 8-Br-cGMP + KT5823; ***p<0.001.
(Di) In vitro phosphorylation of GST-GluR1C wt, GST-GluR1C R837A and GST-GluR1C R837E at S845 by cGKII. Levels of S845-PO4 and of total GluR1C were determined by SDS-PAGE and IB using phospho-specific and total GluR1 Abs, respectively. (Dii) Quantitation of S845 phosphorylation from several experiments as the one shown in (Di). Phospho-specific signal was normalized to the total amount of GluR1, and final values were normalized to control. *p<0.05, **p<0.005, n=3.
(E) Hippocampal neurons expressing HAGluR1 wt, HAGluR1 R837A and HAGluR1 R837E. Neurons were stained live for surface HA (red), followed by fixation, permeabilization and staining for total HA (green). Scale bar, 20 μm; lower panel, 2 μm.
(F) Quantitation of surface/total ratio of HAGluR1 on neurons from experiments as the ones shown in (E). Bar graph shows mean±SEM; n=18 for HAGluR1 wt, 20 for HAGluR1 R837A and 16 for HAGluR1 R837E; **p<0.01, ***p<0.001.
(G) Quantitation of surface/total ratio of HAGluR1 R837A on neurons treated with saline (control), treated with 8-Br-cGMP or with 8-Br-cAMP. Bar graph shows mean±SEM; n=18 for control, 20 for 8-Br-cGMP and 8-Br-cAMP; ***p<0.001.
(H) Quantitation of surface/total ratio of HAGluR1 R837E on neurons treated with saline (control), 8-Br-cGMP or 8-Br-cAMP. Bar graph shows mean±SEM; n=20 for control, 17 for 8-Br-cGMP and 16 for 8-Br-cAMP; ***p<0.001.
We next tested the structural requirements for cGKII action. Our mutagenesis study showed that R837 mutation to Ala reduced the interaction with cGKII, while mutation to Glu completely disrupted the interaction (Fig. 1E). In the in vitro S845 phosphorylation of GST-GluR1C by cGKII, the level of S845 phosphorylation of GST-GluR1C R837A was reduced, and phosphorylation of GST-GluR1C R837E was further reduced, when compared to wt (Fig. 5Di and Dii). Thus the phosphorylations of these mutants directly correlated with their degrees of binding to cGKII (Fig. 5Di and Dii, and compare with Fig. 1E). This shows that interaction of cGKII with GluR1 promotes phosphorylation of the receptor.
We next tested directly the role of the cGKII-GluR1 interaction in the increase of GluR1 surface levels (Fig. 5E). Surface/total ratio was reduced approximately 40% for HAGluR1 R837A, relative to wt (ratio of 1±0.1 in wt and 0.59±0.07 in HAGluR1 R837A, p<0.005), and was even more greatly reduced for HAGluR1 R837E (0.35±0.05, p<0.001 compared to wt) (Fig. 5E and F). Thus, disrupting the cGKII-GluR1 interaction affects GluR1 surface accumulation. Moreover, 8-Br-cGMP increased HAGluR1 R837A surface levels only slightly (ratio of 1.29±0.12 for HAGluR1 R837A+8-Br-cGMP and 1±0.1 for untreated HAGluR1 R837A, p>0.05) (Fig. 5G), and strikingly, 8-Br-cGMP did not increase HAGluR1 R837E surface levels (1.07±0.11 for HAGluR1 R837E+8-Br-cGMP and 1.0±0.08 for untreated HAGluR1 R837E, p>0.05) (Fig. 5H), showing that interaction of cGKII with GluR1 is necessary for the cGMP-dependent increase of surface GluR1. Contrary to 8-Br-cGMP, 8-Br-cAMP still increased surface levels of HAGluR1 R837A (1.54±0.11, p<0.001, compared to untreated HAGluR1 R837A) (Fig. 5G) and HAGluR1 R837E (1.42±0.11, p<0.001, compared to untreated HAGluR1 R837E) (Fig. 5H). Thus progressively destabilizing the cGKII-GluR1 interaction results in a progressive decrease of the constitutive levels of surface GluR1, and progressively impairs GluR1 responsiveness to cGMP. Moreover, these results establish that different molecular mechanisms underly GluR1 phosphorylation and surface increase by PKA and cGKII.
cGKII mediates a critical step in GluR1 trafficking and synaptic plasticity
We next examined the pathway downstream from the NMDAR, specifically whether the NMDAR requires activation of cGKII via nNOS and sGC to increase AMPARs in the plasma membrane. During chemLTP, the glycine-induced surface increase of GluR1 was blocked by AP5 (25 μM), by nNOS inhibitor I (10 μM), by the sGC inhibitor ODQ (10 μM), and by KT5823 (2 μM) or Rp-8-pCPT-cGMPS (10 μM) (Fig. 6A). On the other hand, following treatment with the NO donor, NOR-3, which stimulates the pathway downstream from the NMDAR and nNOS, surface GluR1 increased even in the presence of AP5 or nNOS inhibitor I, while ODQ, or KT5823 or Rp-8-pCPT-cGMPS still blocked the increase (Fig. 6B).
Figure 6. Immunocytochemical and electrophysiological experiments on cultures and slices validate a model in which NMDAR transmission activates cGKII via nNOS and sGC activity, to increase AMPARs at the plasma membrane.

(A) Quantitation of surface levels of GluR1 in neurons treated with saline (control), glycine, or with glycine following pretreatment for 30 min with the following inhibitors: AP5, nNOS inhibitor I, ODQ, KT5823, and Rp-8-pCPT-cGMPS. Bar graph shows mean±SEM; n=20 for each group; ***p<0.001.
(B) Quantitation of surface levels of GluR1 in neurons treated with saline (control), NOR-3, or with NOR-3 following pretreatment for 30 min with the following inhibitors: AP5, nNOS inhibitor I, ODQ, KT5823 and Rp-8-pCPT-cGMPS. Bar graph shows mean±SEM; n=20 for each group; ***p<0.001.
(C) and (D) Average changes in frequency (C) and amplitude (D) of mEPSCs following application of vehicle (n=3, open red circles), glycine (n=9, filled red circles), nNOS inhibitor I paired with glycine (n=6, green squares), ODQ paired with glycine (n=5, black squares), KT5823 paired with glycine (n=8, blue squares) and Rp-8-pCPT-cGMPS paired with glycine (n=5, filled blue squares). Application of glycine for 3 min produced a rapid and long-lasting increase in frequency (p<0.001) and amplitude of mEPSCs (p<0.001). Perfusion of nNOS inhibitor I, ODQ, KT5823 and Rp-8-pCPT-cGMPS blocked glycine-induced increase in frequency and amplitude of mEPSCs. The mean frequencies were 3.5±0.8Hz, 8.3±0.9Hz, 3.6±0.9Hz, 4.1±0.7Hz, 4.1±0.5Hz, and 3.7±0.6Hz for vehicle, glycine, nNOS inhibitor I+glycine, ODQ+glycine, KT5823+glycine and Rp-8-pCPT-cGMPS+glycine groups, respectively. The mean amplitudes were 10.5±0.5pA, 14.7±0.8pA, 10.9±0.6pA, 11.1±0.5pA, 11.1±0.7pA, and 10.7±0.6pA for vehicle, glycine, nNOS inhibitor I+glycine, ODQ+glycine, KT5823+glycine, and Rp-8-pCPT-cGMPS+glycine groups, respectively. The thick line represents the duration of glycine application and error bars represent SEM.
(E) Cumulative probability distribution of mEPSC inter-event interval and mEPSC amplitude in vehicle (control), glycine, nNOS inhibitor I+glycine, ODQ+glycine, KT5823+glycine and Rp-8-pCPT-cGMPS+glycine groups.
(F) LTP is strongly reduced by 20 min perfusion with 10 μM nNOS inhibitor I (F(1,10)=24.30, p<0.001 compared to tetanized vehicle treated slices). The horizontal bar indicates the period during which the drug was added to the bath solution. In this and in the other figures the arrows indicate the tetanus application. The insets show representative fEPSP before (continuous line) and after (dotted line) tetanus.
(G) The sGC inhibitor ODQ (10 μM) blocks LTP (F(1,11)=28.43, p<0.001 compared to vehicle+tetanus).
(H) Treatment with the cGK inhibitor KT5823 (2 μM) for 10 min impaired LTP (F(1,11)=58.94, p<0.0001, compared to vehicle+tetanus).
(I) Treatment with a second specific inhibitor for cGKII, Rp-8-pCPT-cGMPS (10 μM), for 10 min, impaired LTP (F(1,10)=47.77, p<0.0001 compared to vehicle+tetanus).
To confirm the glycine-induced surface increase of GluR1 using electrophysiology, we tested the effects of nNOS inhibitor I, ODQ, KT5823 and Rp-8-pCPT-cGMPS on glycine-induced enhancement in frequency and amplitude of mEPSCs in the cultured hippocampal neurons. Consistent with previous results (Lu et al., 2001), application of glycine (200 μM, 3 min) produced an immediate and long-lasting increase in frequency (176.2±19.7% of baseline at 25 min after glycine application, n=9) and amplitude (136.2±13.9% of baseline at 25 min after glycine application, n=9) of mEPSCs (Fig. 6C, D, and E). Perfusion of nNOS inhibitor I (10 μM), ODQ (10 μM), KT5823 (2 μM) or Rp-8-pCPT-cGMPS (10 μM) blocked glycine-induced enhancement of mEPSC frequency [84.9±32.2% (n=6), 81.1±22.3% (n=5), 78±22.2% (n=8), and 86.6±22.4% (n=5) of baseline at 25 min for nNOS inhibitor I+glycine, ODQ+glycine, KT5823+glycine, and Rp-8-pCPT-cGMPS+glycine groups, respectively; F(5,30)=8.6, p<0.001, two-way ANOVA with repeated measures; Fig. 6C and E] and amplitude [89.7±4.1%, 93.6±5.2%, 89.9±6.8%, and 97.6±12.9% of baseline at 25 min for nNOS inhibitor+glycine, ODQ+glycine, KT5823+glycine, and Rp-8-pCPT-cGMPS+glycine groups, respectively; F(5,30)=3.17, p<0.001, two-way ANOVA with repeated measures; Fig. 6D and E]. Perfusion of nNOS inhibitor I, ODQ, KT5823 or Rp-8-pCPT-cGMPS alone did not affect basal mEPSC frequency and amplitude (data not shown). These results support a model in which NMDAR transmission activates cGKII via nNOS and sGC activity, to increase AMPAR at the plasma membrane.
LTP-inducing stimuli drive GluR1 to synapses (Hayashi et al., 2000), and S845 phosphorylation plays a role in the accompanying increase of GluR1 at the plasma membrane (Esteban et al., 2003; Lee et al., 2003; Oh et al., 2006). To validate our findings in the slice preparation, we studied the role of the NO-cGMP-cGKII pathway in LTP at the Schaeffer collateral-CA1 connection using hippocampal slices. We found that LTP was dramatically reduced when hippocampal slices were exposed to nNOS inhibitor I (10 μM) for 20 min prior to tetanization (114.57±17.59% of baseline slope at 120 min after tetanus vs. 228.42±10.95% in vehicle-treated tetanized slices, n=6 slices from 6 mice; F(1,10)=24.30, p<0.001; Fig. 6F). nNOS inhibitor I alone did not affect baseline transmission (103.12±2.86% vs. 106.16±5.22% in vehicle-treated non-tetanized slices, n=4/4, F(1,6)=0.22, p>0.5, data not shown). Likewise, a 10 min perfusion with an inhibitor of sGC, ODQ (10 μM), suppressed LTP (94.88±15.30% vs. 210.91±20.34 in vehicle-treated tetanized slices, n=6/7, F(1,11)=28.43, p<0.001; Fig. 6G). Treatment with ODQ did not modify baseline transmission (97.03 ±1.57% vs. 100.15±2.31% in vehicle-treated non-tetanized slices, n=4/4, F(1,6)=0.97, p>0.1 compared to vehicle alone; data not shown). When added to slices 10 min prior to tetanization, the cGKII inhibitor, KT5823 (2 μM) also blocked LTP (105.46±11.94% vs. 236.46±10.96% in vehicle-treated tetanized slices, n=6/7, F(1,11)=58.94, p<0.0001, Fig. 6H) without affecting baseline transmission (97.03±1.57% vs. 101.24±1.00% for the non-tetanized slices either treated or not treated with the cGKII inhibitor, n=4/4, F(1,6)=1.75, p>0.1, data not shown). Finally, 10 min perfusion of slice prior to tetanization with the second specific cGKII inhibitor, Rp-8-pCPT-cGMPS (10 μM), severely blocked LTP (106.59±6.51% vs. 234.15±10.34% of baseline slope at 120 min after tetanus, n=6/6, F(1,10)=47.77, p<0.0001, Fig. 6I) without affecting baseline transmission (96.71±2.18% vs. 102.91±1.10% for the non-tetanized slices either treated or not treated with the cGKII inhibitor, n=4/4, F(1,6)=2.90, p>0.1, data not shown). These data suggest a contribution of cGKII to LTP under our experimental conditions.
Directly disrupting cGKII-GluR1 interaction affects synaptic plasticity
While two structurally different inhibitors for cGKII specifically blocked cGMP induction of GluR1 phosphorylation and surface increase, it is still possible that these inhibitors could block other unexpected kinases. To disrupt the cGKII-GluR1 interaction in neurons by a non-pharmacological, kinase-specific approach, we expressed cGKII aa 1-416, which contain the cGKII regulatory and cGMP-binding domains. This truncated regulatory domain fragment of cGKII was shown to have dominant negative properties and to block endogenous cGKII by interacting with the wt cGKII catalytic domain, while not affecting the activity of other kinases, including PKA and even the other cGK isoform, cGKI (Taylor et al., 2002). We fused this construct to GFP (GFP-cGKII-i) and expressed it in neurons from a Sindbis virus. As a control that GFP-cGKII-i binds endogenous cGKII when virally expressed in neurons, we showed that an antiserum against GFP coprecipitates endogenous cGKII from neuronal lysates, when GFP-cGKII-i, but not the control, GFP, is expressed, as expected (Fig. 7A). Furthermore, upon expression in hippocampal pyramidal cells, GFP-cGKII-i fills all neurites and goes to spines (Fig. 7B).
Figure 7. GFP-cGKII-i blocks trafficking of GluR1 in hippocampal neurons after application of 8-Br-cGMP but not 8-Br-cAMP.

(A) Interaction of infected GFP-cGKII-i with endogenous cGKII in cultured neurons. Cells expressing GFP or GFP-cGKII-i were lysed and precipitated with an anti-GFP Ab. The IPs and inputs were probed with an anti-cGKII Ab.
(B) Hippocampal cell in culture expressing GFP-cGKII-i and stained with an Ab that recognizes both cGKII-i and endogenous cGKII. Scale bar, 20 μm; lower panel, 2 μm.
(C) Hippocampal neurons expressing control GFP (green) were treated with saline (control), 8-Br-cGMP or 8-Br-cAMP. Neurons were stained live for surface GluR1 (red). Scale bar, 4 μm.
(D) Quantitation of surface/total ratio of HAGluR1 on neurons from experiments as the ones shown in (C). Bar graph shows mean±SEM; n=15 for control, 24 for 8-Br-cGMP and 18 for 8-Br-cAMP; ***p <0.001.
(E) Hippocampal neurons expressing GFP-cGKII-i (green) were treated with saline (control), 8-Br-cGMP or 8-Br-cAMP. Neurons were stained live for surface GluR1 (red). Scale bar, 4 μm.
(F) Quantitation of surface/total ratio of HAGluR1 on neurons from experiments as the ones shown in (E). Bar graph shows mean±SEM; n=15 for control, 24 for 8-Br-cGMP and 18 for 8-Br-cAMP; ***p <0.001.
We then tested the effects of GFP-cGKII-i on GluR1 trafficking. 8-Br-cGMP or 8-Br-cAMP increased the levels of surface GluR1 in hippocampal neurons expressing GFP (1±0.19, untreated cells; 2.2±0.26, 8-Br-cGMP, p<0.001; and 2.5±0.31, 8-Br-cAMP, p<0.001) (Fig. 7C and D), consistent with previous results in uninfected neurons. However, in neurons expressing GFP-cGKII-i, 8-Br-cGMP application did not increase levels of surface GluR1 compared to untreated cells (surface GluR1 1±0.41 in untreated cells and 1±0.17 in cells treated with 8-Br-cGMP, p>0.05), while 8-Br-cAMP still induced an increase in GluR1 surface expression (2.5±0.33, p<0.001, compared to untreated) (Fig. 7E and F), confirming that GFP-cGKII-i specifically blocks cGMP-dependent trafficking.
We next tested the effects of GFP-cGKII-i expression on glycine-induced synaptic plasticity. Application of glycine failed to increase either frequency (73.7±13.5% of baseline at 25 min after glycine application, n=6, F(3,20)=14.1, p<0.001, two-way ANOVA with repeated measures; Fig. 8A and C) or amplitude (89.7±6.8% of baseline at 25 min after glycine application, n=6, F(3,20)=2.6, p<0.01, two-way ANOVA with repeated measures; Fig. 8B and C) of mEPSCs in neurons expressing GFP-cGKII-i. However, application of glycine significantly increased both frequency (217.5±20.8% of baseline at 25 min after glycine application, n=6) and amplitude (133.6±4.8% of baseline at 25 min after glycine application, n=6) of mEPSCs in neurons expressing control GFP protein (Fig. 8A, B and C). These results further confirm the role of cGKII in NMDAR-mediated synaptic plasticity and increase in AMPARs at the plasma membrane.
Figure 8. Expression of GFP-cGKII-i blocks glycine-induced synaptic plasticity in cultured hippocampal neurons and LTP in hippocampal slices.

(A) and (B) Average changes in frequency (A) and amplitude (B) of mEPSCs following application of glycine (n=9, filled red circles), GFP expression paired with glycine (n=6, green squares), GFP-cGKII-i expression paired with glycine (n=6, black squares), and vehicle (control) (n=3, open red circles). Glycine application (3 min) produced a rapid and long-lasting increase in frequency (p<0.001) and amplitude of mEPSCs (p<0.001) in neurons expressing GFP. However, expression of GFP-cGKII-i completely blocked glycine-induced increase in frequency and amplitude of mEPSCs. The mean basal frequencies were 3.9±0.5Hz, 3.9±0.6Hz, 4.1±0.6Hz and 3.5±0.4Hz for control, glycine, GFP+glycine, and GFP-cGKII-i+glycine groups, respectively. The mean basal amplitudes were 9.8±0.8pA, 10.2±0.8pA, 10.8±0.9pA and 9.8±0.7pA for control, glycine, GFP+glycine, and GFP-cGKII-i+glycine groups, respectively. The thick line represents the duration of glycine application and error bars represent SEM.
(C) Cumulative probability distribution of mEPSC inter-event interval and mEPSC amplitude in vehicle (control), glycine, GFP expression+glycine, GFP-cGKII-i expression+glycine groups.
(D) Cannulas were implanted bilaterally into the dorsal hippocampus (schematic representation).
(E) Lysates from uninfected hippocampus and hippocampus infected with GFP-cGKII-i were analyzed by IB using an anti-GFP Ab. IB for GFP confirmed that slices infused with Sindbis virus carrying a construct for GFP-cGKII-i were infected.
(F) Representative examples of hippocampal slices viewed on a confocal microscope (20X objective) 24 hrs after injection of Sindbis virus expressing GFP-cGKII-i or vehicle. GFP is visible in the CA1 cell body area only in slices infected with GFP-cGKII-i (right panel).
(G) Basal synaptic transmission is not impaired in animals injected with GFP-cGKII-i virus compared to slices from animals injected with GFP virus or vehicle (F(2,14)=0.02, p=0.98)
(H) The GFP-cGKII-i virus impaired LTP (F(1,10)=10.15, p=0.01, compared to vehicle) whereas GFP virus did not affect LTP (F(1,10)=0.77, p=0.39, compared to vehicle).
(Ii) Model for the regulation of GluR1 trafficking by cGKII. Based on the lipid raft location of the kinase and the fact that GluR1 surface levels are increased after S845 phosphorylation, the kinase is depicted at the plasma membrane and the receptor intracellularly, before S845 phosphorylation. However, the subcellular locations of the kinase and the receptor at the time of phosphorylation have not yet been established. See Discussion. (Iii) cGKII autoinhibitory domain homology with GluR1 CTD binding site for cGKII. GluR1 S845 and cGKII S126 (red), and homologous residues (blue), are shown.
To confirm the role of cGKII in hippocampal LTP, we infected mice with Sindbis virus expressing constructs for GFP or GFP-cGKII-i. Cannulas were implanted bilaterally into the dorsal hippocampi (Fig. 8D). After 6-8 days, mice were infused with the virus and after 24 hrs they were sacrificed to prepare hippocampal slices. We confirmed by IB for GFP (Fig. 8E) and by microscopy of GFP fluorescence (Fig. 8F) that the replacement protein was expressed in the hippocampal pyramidal cell layer of the slices (Fig. 8F). We did not find a difference in basal synaptic transmission (BST) among mice injected with vehicle, or virus expressing the GFP or GFP-cGKII-i constructs (Fig. 8G). BST was determined by measuring the slope of the field excitatory postsynaptic potential (fEPSPs) at increasing stimulus intensity in the different groups. The slope of the input-output curve at stimulation intensity equal to 35 V was ∼94% of vehicle-injected mice in GFP-infected mice and ∼96% in mice infected with GFP-cGKII-i (vehicle: 1.09±0.07 V/sec., n=6; GFP: 1.03±0.12 V/sec., n=6; GFP-cGKII-i: 1.05±0.18 V/sec., n=5). A two-way ANOVA showed no difference between the three groups [F(2,14)=0.02, p=0.98]. However, in agreement with results obtained using pharmacologic cGKII inhibitors, slices with virus expressing the GFP-cGKII-i construct showed an impairment of LTP when compared to vehicle-treated slices (142.52±17.37 % of baseline slope vs 242.05±18.76, n=6/6; F(1,10)=10.15, p=0.01, Fig. 8H). GFP expression did not affect LTP (266.29±35.02% of baseline slope, n=6, F(1,10)=0.77, p=0.39, Fig. 8H). Taken together, these data demonstrate that the action of cGKII is required for synaptic plasticity in this system.
Discussion
NMDAR stimulation activates nNOS and production of NO, which results in cGMP production (Garthwaite, 1991; Hofmann et al., 2000), and cGKII activation. A major mechanism for expression of NMDAR-dependent LTP involves the synaptic insertion of GluR1 (Hayashi et al., 2000; Malenka and Bear, 2004; Zamanillo et al., 1999). Here we report that following activation by the NMDAR, cGKII binds to GluR1 and phosphorylates S845, leading to an increase of GluR1 in the plasma membrane (Fig. 8I). Notably, a cGKII dominant negative inhibitor peptide blocked the increase of GluR1 surface expression, prevented the increase in amplitude and frequency of mEPSCs after chemLTP and strongly reduced LTP in hippocampal slices. These results demonstrate a novel mechanism in which the NMDAR regulates AMPAR trafficking during LTP via NO and cGKII.
Because NO is produced at postsynaptic sites (Garthwaite and Boulton, 1995) and can diffuse through lipid membranes, initial studies of NO dependent plasticity focused on presynaptic NO function through retrograde mechanisms (Bohme et al., 1991; O'Dell et al., 1991; Schuman and Madison, 1991). Some results were controversial, possibly because different methodologies were employed (Bon and Garthwaite, 2001). Indeed, cGMP derivatives only facilitate LTP maximally if briefly applied when the NMDA receptor is active (Son et al., 1998) and deviating protocols would lead to conflicting results. More recently, the use of new NO donors and NOS antagonists (Bon and Garthwaite, 2003; Puzzo et al., 2005), both in vitro (Bon and Garthwaite, 2003) and in vivo (Feil et al., 2005) has demonstrated a role of the NO cascade in synaptic plasticity. Interestingly, as reported here, both the sGC inhibitor ODQ and the cGK inhibitor KT5823 were found to block LTP (Bon and Garthwaite, 2001; Chien et al., 2003; Lu et al., 1999). Nonetheless, specific molecular mechanisms underlying the effects of NO, in particular in NO control of AMPAR trafficking in LTP, have been wanting. S-nitrosylation of NSF enhances NSF binding to GluR2, and regulates GluR2 surface expression (Huang et al., 2005). Also activation of the NO-cGMP-cGKI pathway increases both GluR1 and synaptophysin puncta and the phosphorylation of VASP in hippocampal neurons (Wang et al., 2005). However, as yet, a specific pathway for NO control of activity-dependent GluR1 trafficking to synapses, an essential component of LTP, has not been reported. The interaction of cGKII with GluR1 reported here, and its consequent effect on GluR1 surface levels, directly link of the actions of NO to LTP via GluR1 trafficking.
Convergence of PKA and cGKII on S845 phosphorylation
We report a physical association of cGKII with GluR1 that enables the kinase to phosphorylate GluR1 at S845. This phosphorylation is required for cGMP-dependent GluR1 surface accumulation, since block of the phosphorylation by the S845A GluR1 mutation blocked the surface increase. Others have shown that phosphorylation of S845 accompanies increases in GluR1 surface levels (Ehlers, 2000) and is necessary for GluR1 synaptic insertion during LTP (Esteban et al., 2003). S845 is dephosphorylated during hippocampal LTD (Lee et al., 2000; Lee et al., 1998), and S845 phosphorylation on its own is sufficient for increase of GluR1 in the extrasynaptic plasma membrane (Oh et al., 2006; Sun et al., 2005). Thus far only PKA phosphorylation of S845 has been considered, perhaps because it was the initial kinase shown to phosphorylate this site (Roche et al., 1996). The present study demonstrates that cGKII activity also phosphorylates S845.
Increases of surface GluR1 following both PKA and cGKII phosphorylation are restricted to extrasynaptic sites (Oh et al., 2006; Sun et al., 2005, and this report), and AMPAR synaptic incorporation requires at least one additional step (Bredt and Nicoll, 2003; Passafaro et al., 2001; Tardin et al., 2003), possibly mediated by S818 phosphorylation (Boehm et al., 2006). Interestingly, although 8-Br-cGMP on its own did not enhance hippocampal synaptic responses, when paired with a weak tetanus that by itself does not enhance responses, 8-Br-cGMP produced an immediate potentiation (Zhuo et al., 1994). This suggests that cGMP can prime the system for potentiation by a weak tetanic stimulation, possibly by increasing the extrasynaptic surface AMPAR population.
Differential regulation of cGKII and PKA
The NMDAR and nNOS mutually interact with PSD-95 (Brenman et al., 1996; Christopherson et al., 1999) and Ca++ fluxes through the NMDAR activate nNOS in this complex to produce NO (Bredt and Snyder, 1989; Garthwaite et al., 1988), which induces sGC to produce cGMP, which activates cGKII. Ca++ fluxes also stimulate Ca++-regulated adenylate cyclases (Wang and Storm, 2003) which produce cAMP, which activates PKA which also phosphorylates S845. PKA binds the A kinase anchoring protein, AKAP79, which in turn binds the PDZ domain scaffolding protein, SAP97, which binds the GluR1 CTD, thus targeting PKA to the GluR1 CTD and facilitating phosphorylation of S845 (Colledge and Scott, 1999).
Unlike the SAP97-AKAP-PKA pathway, the NO-cGMP-cGKII pathway does not rely on a scaffold since the kinase binds the receptor directly. Interestingly, a knockin mouse expressing GluR1 that lacks the last 7 aa of its CTD and does not bind SAP97, exhibited normal hippocampal LTP and GluR1 trafficking (Kim et al., 2005). This is explained if the NO-cGMP-cGKII pathway phosphorylates S845 in this mutant.
GluR1 interacts with cGKII via auxiliary and core contact CTD sequences that flank S845. Interestingly, a CTD contact sequence resembles an AI domain sequence of cGKII, suggesting that to bind the catalytic domain, GluR1 mimics the AI domain (See Fig. 8ii and Supplemental Discussion). Also, this receptor-kinase interaction resembles the well studied CaMKII binding to the NR2B CTD (Lisman and McIntyre, 2001) (See Supplemental Discussion).
Mechanism of cGKII-dependent GluR1 trafficking
In the absence of cGMP, cGKII is inactive (Fig. 8Ii). Following NMDAR stimulation, binding of cGMP to cGKII induces a cGKII conformational change that causes AI domain autophosphorylation, AI domain release from the catalytic domain and elongation of the kinase. The GluR1 CTD binds the newly exposed cGKII catalytic domain, facilitating GluR1 phosphorylation and the increase of surface GluR1. In one model for this increase, S845 phosphorylation promotes GluR1 trafficking to the plasma membrane, perhaps by releasing of GluR1 from a cytosolic retention factor. Alternatively, GluR1 may cycle into and out of the plasma membrane constitutively, and S845 phosphorylation may stabilize the receptor at the neuron surface. With either model, S845 phosphorylation would regulate the size of an extrasynaptic pool from which receptors may be inserted into the synapse during LTP. Such transport may depend on additional GluR1 phosphorylation (Boehm et al., 2006). Because, as we have shown, a highly selective peptide block of cGKII strongly reduces LTP, such an increase in an extrasynaptic receptor pool is likely to be a requirement for the synaptic potentiation associated with LTP. The present work demonstrates that the NMDAR can control the size of such a receptor pool, acting through nNOS, NO and cGMP production and the activation of cGKII.
Experimental Procedures
See Supplemental Experimental Procedures for details of the mammalian recruitment assay, 293T cell culture and GST pulldown, preparation and fractionation of cell extracts, in vitro phosphorylation experiments, GluR1 phosphorylation in cortical cell cultures, and for Abs and expression vectors used.
Yeast two-hybrid screen
The GluR1 CTD cloned into the pSos vector was transformed into yeast strain cdc25Hα (Stratagene) to screen an adult rat hippocampal cDNA library in the pMyr (Stratagene) expression vector, using Cytotrap Vector Kit (Stratagene) according to the manufacturer's instructions. Interaction of GluR1 CTD deletion mutants with cGKII were similarly analyzed.
Neuronal cell culture and immunocytochemistry
Hippocampal primary culture, Sindbis virus preparation, neuronal infection and immunostaining of endogenous and recombinant proteins have been described (Osten et al., 2000). Details are described in the Supplemental Experimental Procedures.
Image analysis and quantitation
Images were acquired using a Nikon PCM 2000 confocal microscope, and analysis was done blind using ImageJ software. Images for all experimental groups were taken using identical acquisition parameters. With images acquired for quantitation, background image intensity was determined and subtracted by thresholding during image acquisition. To analyze surface expression of HAGluR1 and the different HAGluR1 mutants, the ratio of surface signal to total signal was calculated. To analyze surface expression of endogenous GluR1, neuritic fractions located one soma diameter from the soma were selected, and fluorescence was quantified using Image J software. Approximately 5 neuritic fractions from each cell were taken for analysis. GluR1-SV2 colocalization analysis was done using the Colocalization Threshold plugin from Image J software. The Manders' overlap coefficient was used as the indicator of the proportion of GluR1 signal (red channel) coincident with the SV2 signal (green channel) over its total intensity (Manders et al., 1992). Error bars are standard errors of the mean, and significance was determined by the t test.
Electrophysiological experiments
Both patch clamp experiments and slice recording have been previously described (O'Dell et al., 1991; Puzzo et al., 2005). Patch clamp recordings were performed 10–15 days after plating cell cultures, whereas slices were obtained from 3-month-old male mice (C57BL6; The Jackson Laboratory). The details of these techniques are reported in the Supplemental Experimental Procedures.
Slice infection
Mice were anesthetized with 20 mg/kg Avertin and implanted with a 26-gauge guide cannula into the dorsal part of the hippocampi (coordinates: P=2.4mm, L=1.5mm to a depth of 1.3 mm) (Paxinos, 1998). The cannulas were fixed to the skull with acrylic dental cement (Paladur). The infusion was made after 6-8 days through infusion cannulas that were connected to a microsyringe by a polyethylene tube. The entire infusion procedure took ∼1min, and animals were handled gently to minimize stress. Either vehicle or Sindbis virus carrying constructs for GFP or GFP-cGKII-i were injected bilaterally in a volume of 1 μl over 1 min. After injection, the needle was left in place for another minute to allow diffusion.
Supplementary Material
Acknowledgments
We thank S. Restituito, D. Tukey and Bryen Jordan for critical discussion, R. Bourtchouladze for teaching us the infusion technique onto hippocampi and S. Mahajan for provision of vectors. M.M. was supported by NIGMS grant 5 T32 GM 07308. This work was supported by NIH grant MH 067229 to E.B.Z and NS 049442 to O.A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aronheim A, Zandi E, Hennemann H, Elledge SJ, Karin M. Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol Cell Biol. 1997;17:3094–3102. doi: 10.1128/mcb.17.6.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Derkach V, Soderling T. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. J Biol Chem. 1997;272:32727–32730. doi: 10.1074/jbc.272.52.32727. [DOI] [PubMed] [Google Scholar]
- Barry MF, Ziff EB. Receptor trafficking and the plasticity of excitatory synapses. Curr Opin Neurobiol. 2002;12:279–286. doi: 10.1016/s0959-4388(02)00329-x. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Boehm J, Malinow R. AMPA receptor phosphorylation during synaptic plasticity. Biochem Soc Trans. 2005;33:1354–1356. doi: 10.1042/BST0331354. [DOI] [PubMed] [Google Scholar]
- Bohme GA, Bon C, Stutzmann JM, Doble A, Blanchard JC. Possible involvement of nitric oxide in long-term potentiation. Eur J Pharmacol. 1991;199:379–381. doi: 10.1016/0014-2999(91)90505-k. [DOI] [PubMed] [Google Scholar]
- Bon C, Bohme GA, Doble A, Stutzmann JM, Blanchard JC. A Role for Nitric Oxide in Long-term Potentiation. Eur J Neurosci. 1992;4:420–424. doi: 10.1111/j.1460-9568.1992.tb00891.x. [DOI] [PubMed] [Google Scholar]
- Bon CL, Garthwaite J. Exogenous nitric oxide causes potentiation of hippocampal synaptic transmission during low-frequency stimulation via the endogenous nitric oxide-cGMP pathway. Eur J Neurosci. 2001;14:585–594. doi: 10.1046/j.0953-816x.2001.01680.x. [DOI] [PubMed] [Google Scholar]
- Bon CL, Garthwaite J. On the role of nitric oxide in hippocampal long-term potentiation. J Neurosci. 2003;23:1941–1948. doi: 10.1523/JNEUROSCI.23-05-01941.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Snyder SH. Nitric oxide mediates glutamate-linked enhancement of cGMP levels in the cerebellum. Proc Natl Acad Sci U S A. 1989;86:9030–9033. doi: 10.1073/pnas.86.22.9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- Butt E, Eigenthaler M, Genieser HG. (Rp)-8-pCPT-cGMPS, a novel cGMP-dependent protein kinase inhibitor. Eur J Pharmacol. 1994;269:265–268. doi: 10.1016/0922-4106(94)90095-7. [DOI] [PubMed] [Google Scholar]
- Chien WL, Liang KC, Teng CM, Kuo SC, Lee FY, Fu WM. Enhancement of long-term potentiation by a potent nitric oxide-guanylyl cyclase activator, 3-(5-hydroxymethyl-2-furyl)-1-benzyl-indazole. Mol Pharmacol. 2003;63:1322–1328. doi: 10.1124/mol.63.6.1322. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Hillier BJ, Lim WA, Bredt DS. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem. 1999;274:27467–27473. doi: 10.1074/jbc.274.39.27467. [DOI] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Colledge M, Scott JD. AKAPs: from structure to function. Trends Cell Biol. 1999;9:216–221. doi: 10.1016/s0962-8924(99)01558-5. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Feil R, Hofmann F, Kleppisch T. Function of cGMP-dependent protein kinases in the nervous system. Rev Neurosci. 2005;16:23–41. doi: 10.1515/revneuro.2005.16.1.23. [DOI] [PubMed] [Google Scholar]
- Francis SH, Corbin JD. Cyclic nucleotide-dependent protein kinases: intracellular receptors for cAMP and cGMP action. Crit Rev Clin Lab Sci. 1999;36:275–328. doi: 10.1080/10408369991239213. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Glutamate, nitric oxide and cell-cell signalling in the nervous system. Trends Neurosci. 1991;14:60–67. doi: 10.1016/0166-2236(91)90022-m. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Charles SL, Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988;336:385–388. doi: 10.1038/336385a0. [DOI] [PubMed] [Google Scholar]
- Haley JE, Wilcox GL, Chapman PF. The role of nitric oxide in hippocampal long-term potentiation. Neuron. 1992;8:211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Hemmings HC, Jr, Nairn AC, Greengard P. DARPP-32, a dopamine- and adenosine 3′:5′-monophosphate-regulated neuronal phosphoprotein. II. Comparison of the kinetics of phosphorylation of DARPP-32 and phosphatase inhibitor 1. J Biol Chem. 1984;259:14491–14497. [PubMed] [Google Scholar]
- Hering H, Lin CC, Sheng M. Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J Neurosci. 2003;23:3262–3271. doi: 10.1523/JNEUROSCI.23-08-03262.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F, Ammendola A, Schlossmann J. Rising behind NO: cGMP-dependent protein kinases. J Cell Sci. 2000;113(Pt 10):1671–1676. doi: 10.1242/jcs.113.10.1671. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Huang Y, Man HY, Sekine-Aizawa Y, Han Y, Juluri K, Luo H, Cheah J, Lowenstein C, Huganir RL, Snyder SH. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46:533–540. doi: 10.1016/j.neuron.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun. 1987;142:436–440. doi: 10.1016/0006-291x(87)90293-2. [DOI] [PubMed] [Google Scholar]
- Kim CH, Takamiya K, Petralia RS, Sattler R, Yu S, Zhou W, Kalb R, Wenthold R, Huganir R. Persistent hippocampal CA1 LTP in mice lacking the C-terminal PDZ ligand of GluR1. Nat Neurosci. 2005;8:985–987. doi: 10.1038/nn1432. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, et al. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273:19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- Lisman JE, McIntyre CC. Synaptic plasticity: a molecular memory switch. Curr Biol. 2001;11:R788–791. doi: 10.1016/s0960-9822(01)00472-9. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;19:10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem. 1997;272:32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992;103(Pt 3):857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- O'Dell TJ, Hawkins RD, Kandel ER, Arancio O. Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci U S A. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, Fishman MC. Endothelial NOS and the blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science. 1994;265:542–546. doi: 10.1126/science.7518615. [DOI] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Osten P, Khatri L, Perez JL, Kohr G, Giese G, Daly C, Schulz TW, Wensky A, Lee LM, Ziff EB. Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron. 2000;27:313–325. doi: 10.1016/s0896-6273(00)00039-8. [DOI] [PubMed] [Google Scholar]
- Passafaro M, Piech V, Sheng M. Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci. 2001;4:917–926. doi: 10.1038/nn0901-917. [DOI] [PubMed] [Google Scholar]
- Paxinos G. Mouse brain in stereotaxic coordinates. New York: Academic Press; 1998. [Google Scholar]
- Pfeifer A, Ruth P, Dostmann W, Sausbier M, Klatt P, Hofmann F. Structure and function of cGMP-dependent protein kinases. Rev Physiol Biochem Pharmacol. 1999;135:105–149. doi: 10.1007/BFb0033671. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Vitolo O, Trinchese F, Jacob JP, Palmeri A, Arancio O. Amyloid-beta peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. J Neurosci. 2005;25:6887–6897. doi: 10.1523/JNEUROSCI.5291-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Madison DV. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- Shen L, Liang F, Walensky LD, Huganir RL. Regulation of AMPA receptor GluR1 subunit surface expression by a 4. 1N-linked actin cytoskeletal association. J Neurosci. 2000;20:7932–7940. doi: 10.1523/JNEUROSCI.20-21-07932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Lee SH. AMPA receptor trafficking and the control of synaptic transmission. Cell. 2001;105:825–828. doi: 10.1016/s0092-8674(01)00406-8. [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ER. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- Son H, Lu YF, Zhuo M, Arancio O, Kandel ER, Hawkins RD. The specific role of cGMP in hippocampal LTP. Learn Mem. 1998;5:231–245. [PMC free article] [PubMed] [Google Scholar]
- Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25:7342–7351. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardin C, Cognet L, Bats C, Lounis B, Choquet D. Direct imaging of lateral movements of AMPA receptors inside synapses. Embo J. 2003;22:4656–4665. doi: 10.1093/emboj/cdg463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MK, Ahmed R, Begley M, Uhler MD. Autoinhibition and isoform-specific dominant negative inhibition of the type II cGMP-dependent protein kinase. J Biol Chem. 2002;277:37242–37253. doi: 10.1074/jbc.M202060200. [DOI] [PubMed] [Google Scholar]
- Vaandrager AB, Ehlert EM, Jarchau T, Lohmann SM, de Jonge HR. N-terminal myristoylation is required for membrane localization of cGMP-dependent protein kinase type II. J Biol Chem. 1996;271:7025–7029. doi: 10.1074/jbc.271.12.7025. [DOI] [PubMed] [Google Scholar]
- Wall ME, Francis SH, Corbin JD, Grimes K, Richie-Jannetta R, Kotera J, Macdonald BA, Gibson RR, Trewhella J. Mechanisms associated with cGMP binding and activation of cGMP-dependent protein kinase. Proc Natl Acad Sci U S A. 2003;100:2380–2385. doi: 10.1073/pnas.0534892100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Storm DR. Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol Pharmacol. 2003;63:463–468. doi: 10.1124/mol.63.3.463. [DOI] [PubMed] [Google Scholar]
- Wang HG, Lu FM, Jin I, Udo H, Kandel ER, de Vente J, Walter U, Lohmann SM, Hawkins RD, Antonova I. Presynaptic and postsynaptic roles of NO, cGK, and RhoA in long-lasting potentiation and aggregation of synaptic proteins. Neuron. 2005;45:389–403. doi: 10.1016/j.neuron.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Wang X, Robinson PJ. Cyclic GMP-dependent protein kinase and cellular signaling in the nervous system. J Neurochem. 1997;68:443–456. doi: 10.1046/j.1471-4159.1997.68020443.x. [DOI] [PubMed] [Google Scholar]
- Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Koster HJ, Borchardt T, Worley P, et al. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284:1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]
- Zhao J, Trewhella J, Corbin J, Francis S, Mitchell R, Brushia R, Walsh D. Progressive cyclic nucleotide-induced conformational changes in the cGMP-dependent protein kinase studied by small angle X-ray scattering in solution. J Biol Chem. 1997;272:31929–31936. doi: 10.1074/jbc.272.50.31929. [DOI] [PubMed] [Google Scholar]
- Zhuo M, Hu Y, Schultz C, Kandel ER, Hawkins RD. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature. 1994;368:635–639. doi: 10.1038/368635a0. [DOI] [PubMed] [Google Scholar]
- Zhuo M, Small SA, Kandel ER, Hawkins RD. Nitric oxide and carbon monoxide produce activity-dependent long-term synaptic enhancement in hippocampus. Science. 1993;260:1946–1950. doi: 10.1126/science.8100368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.