

Abstract
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a heart muscle disorder of unknown cause that is characterised by fibrofatty replacement, primarily of the right ventricular myocardium, which can lead to life-threatening arrhythmias. It is a disease with a very diverse phenotype. In the present article we describe two sisters, each with a different manifestation of this disorder. The first patient died suddenly at the age of 18 during exercise. Her 17-year-old sister did not have any abnormalities at first cardiac consultation, but a few years later she met several diagnostic criteria for ARVC and an internal cardioverter defibrillator was implanted. Genetic analysis identified a mutation in the plakophilin- 2 (PKP2) gene. Cardiac evaluation of a third sister did not reveal any abnormalities and no mutation in the PKP2 gene was found. Thus, ARVC can vary in its clinical presentation, not only between siblings but also in time. This raises difficulties for the physician for diagnosis, treatment and followup. It is important for the physician involved to consider this disease in patients with palpitations and syncope, especially when there is a family history of ARVC or unexplained sudden death. (Neth Heart J 2007;15:348-53.)
Keywords: cardiomyopathy, right ventricle, sudden death, genetics
Arrhythmogenic right ventricular cardiomyopathy (ARVC) was described for the first time by Frank and Fontaine in 1978.1 Histologically, ARVC is characterised by loss of cardiomyocytes and progressive fibrofatty replacement of predominantly the right ventricular myocardium that constitutes a substrate for electrical instability and a focus for ventricular arrhythmias. In the early stages of the disease, in childhood or even in utero, there is presumably only fatty replacement with mild fibrosis and the disease may be better described as arrhythmogenic right ventricular dysplasia (ARVD). At a later stage inflammation may occur with replacement fibrosis. It is likely that environmental factors contribute to the disease at that stage and thus a form of cardiomyopathy may be the preferred terminology. The clinical manifestations can have a very different presentation. The patient may be totally asymptomatic and the condition can be discovered or suspected on a routine electrocardiogram. On the other hand, sudden death may be the first symptom and possibly ARVC counts for up to 20% of cases of sudden death under the age of 30 years.2 Right or left ventricular congestive heart failure can also be the presenting symptom in rare cases.
ARVC is a familial disease in up to 50% of the cases. Therefore, ensuring a proper diagnosis is not only important to the patient but also to the relatives.
The present article will show the various manifestations of the disease and discusses the diagnosis, clinically as well as genetically, and treatment options.
Case report 1
The first patient is a previously healthy 18-year-old woman and competitive athlete in endurance sports, who died suddenly during a race. Autopsy revealed a mildly hypertrophic and dilated left ventricle compatible with the effects of endurance sports activities. There were no coronary artery or valvular abnormalities. The right ventricle was markedly dilated and there was a striking transmural fatty degeneration. Histology showed right ventricular cardiomyopathy. In combination with the sudden cardiac death, arrhythmogenic right ventricular cardiomyopathy was present.
Case report 2
Patient B is a 31-year-old woman and one of the two sisters of the previous patient. She has no medical history and is also a competitive endurance athlete. At the age of 18, after the sudden death of her sister, her cardiac evaluation appeared completely normal. However, 13 years later she was referred again because of dyspnoea on exertion. Careful analysis of the history revealed that this ‘dyspnoea’ was due to the occurrence of paroxysms of palpitations which she does not feel during her endurance races. During that visit she also mentioned that her father had died suddenly three years before, at the age of 65. There were no risk factors for coronary artery disease. On physical examination a healthy, endurance-trained woman was seen. Her blood pressure was 120/70 mmHg and her pulse rate 60 beats/min, regular and equal. An early systolic ejection murmur was heard at the 4th left intercostal space but the rest of the physical examination was normal. This time the electrocardiogram showed a sinus bradycardia and inverted T waves in precordial leads V1 to V3 (figure 1).
Figure 1.
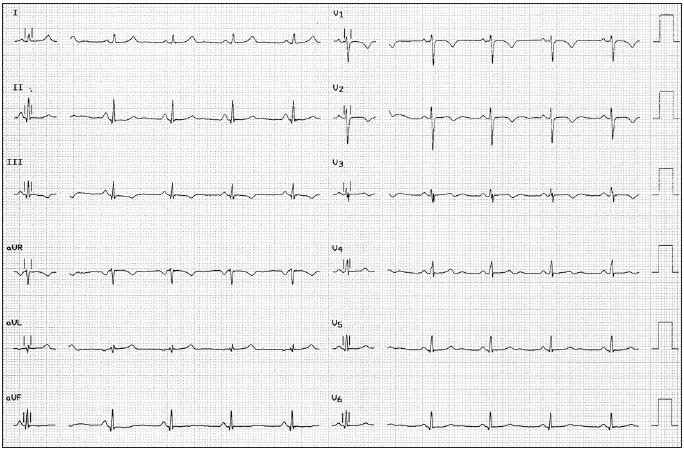
Electrocardiogram from the patient in case report 2, showing a sinus bradycardia with inverted T waves in leads V1 to V3.
Chest X-ray and echocardiogram were normal. During exercise testing, short runs of ventricular complexes were documented on the electrocardiogram, which had a left bundle branch block morphology with superior axis (figure 2). This morphology corresponds with an origin of ectopy situated inferiorly in the right ventricle. Holter monitoring disclosed more than 1000 ventricular complexes per 24 hours. She was advised to avoid peak exercise and a β-blocking agent was prescribed. After that, only solitary ventricular premature complexes were observed on the exercise test.
Figure 2.

Electrocardiogram of the patient in case report 2 showing premature ventricular complexes and ventricular couplets with a left bundle branch block configuration (LBBB) and left axis deviation, during maximal exercise.
Subsequently an MRI showed a diffuse thinning of the right ventricular wall.
After the initiation of the β-blocker she developed symptomatic bradycardia and electrophysiological testing was performed during which ventricular fibrillation could easily be induced. Meanwhile, echodilatation and apical hypokinesia and akinesia. After this an implantable cardioverter defibrillator (ICD) was inserted and she was treated with the β-blocking agent sotalol. No ICD discharges or arrhythmic events have been reported so far.
Four years after the implantation she is leading a normal life, participating in noncompetitive sports regularly and has had two uncomplicated pregnancies.
Genetic analysis
After the recent identification of the plakophilin-2 (PKP2) gene mutation as a causal gene mutation for ARVC, we analysed this patient for the presence of PKP2 mutations. Genomic DNA was isolated from peripheral blood lymphocytes (Gentra Systems, Minneapolis, USA) and the entire PKP2 coding region was screened for mutations. Primer sequences and PCR conditions are available on request.
In patient 2 a Cytosine (C) was replaced by an Adenine (A) at base position 2421 causing a premature termination codon at position 807 (Tyrosine 807X) (figure 3). It is highly likely that this alteration underlies her clinical phenotype. A third sister was also genetically tested and appeared not to have the PKP2 mutation.
Figure 3.

Screening of the PKP2 gene shows a substitution of nucleotide ‘C’ for an ‘A’ at cDNA position 2421 (marked by an arrow) (NCBI ref: NM_004572.2) which causes premature termination of the PKP2 protein at position 807 (Q807X). Left panel shows the nucleotide sequence from a control (wild type).
Discussion
Arrhythmogenic right ventricular cardiomyopathy (ARVC) was initially defined as ‘total or partial replacement of right ventricular muscle by adipose and fibrous tissue associated with arrhythmias of left bundle branch block configuration’.1
The typical anatomical findings are aneurysmatic regions due to myocardial thinning. In these regions the myocardial wall is replaced by fibrofatty tissue with scattered residual myocardial bundles, a perfect substrate for arrhythmias.
Specific sites of involvement include the right ventricular outflow tract, the apex and the infundibular region, which is known as the triangle of dysplasia (figure 4). The fibrofatty tissue replacement can also affect the left ventricular myocardium with a relative sparing of the septum.3,4 These sites form an excellent substrate for re-entry, which can lead to arrhythmias such as premature ventricular complexes, ventricular tachycardia and ventricular fibrillation. Because of the predominant origin in the right ventricle, these arrhythmias have, frequently but exclusively, a left bundle branch block morphology (figure 2).
Figure 4.

The 'triangle of dysplasia' consisting of the right ventricular outflow tract (1), the apex (2) and the infundibulum (3). RA=right atrium, LA=left atrium.
The prevalence of ARVC in the general population is estimated at 0.02 to 0.1%, but this may be underestimated because of diagnostic difficulties and physicians being unaware of this disease. Both sexes are equally affected and there is a peak during the fourth decade. ARVC is an important cause of sudden death in young adults, accounting for approximately 11% of the cases. In athletes, such as the women described in this article, ARVC counts for 22% of cases of sudden deaths.5 Although this suggests that exercise is a risk, sudden death occurs most frequently during sedentary activity.
The definite diagnosis of ARVC is based on the histological demonstration of transmural fibrofatty replacement of right ventricular myocardium at either autopsy or surgery. Myocardial biopsy lacks sufficient sensitivity because, for safety reasons, the biopsy is performed in the septum instead of the ventricular free wall where the typical pathological changes are localised.6
For clinical practice, McKenna et al. defined diagnostic criteria for ARVC. The diagnosis is based on a number of major and minor criteria involving structural, histological, electrocardiographic, arrhythmic and genetic factors.7 To fulfil the appropriate criteria for ARVC, patients have to meet either two major criteria, one major and two minor criteria, or four minor criteria (table 1).
Table 1.
Task force criteria for diagnosis of ARVC (McKenna et al.)7 The diagnosis of ARVC would be fulfilled by the presence of 2 major, 1 major plus 2 minor, or 4 minor criteria from different groups.
| I. Global and/or regional dysfunction and structural alterations |
| Major |
| - Severe dilatation and reduction of right ventricular ejection fraction with no (or only mild) LV impairment |
| Minor |
| - Mild global right ventricular dilatation and/or ejection fraction reduction with normal left ventricle |
| - Mild segmental dilatation |
| - Regional right ventricular hypokinesia |
| II. Tissue characterisation of wall |
| Major |
| - Fibrofatty replacement of myocardium on endomyocardial biopsy |
| III. Repolarisation abnormalities |
| Minor |
| - Inverted T waves in right precordial leads (V2 and V3) in people aged >12 years, in absence of right bundle branch block |
| IV. Depolarisation/conduction abnormalities |
| Major |
| - Epsilon waves or localised prolongation (>110 ms) of the QRS complex in right precordial leads (V1 to V3) |
| Minor |
| - Late potentials on signal-averaged ECG |
| V. Arrhythmias |
| Minor |
| - Left bundle-branch block type ventricular tachycardia (sustained and nonsustained) by ECG, Holter, or exercise testing |
| - Frequent ventricular extrasystoles (>1000/24 hours) on Holter |
| VI. Family history |
| Major |
| - Familial disease confirmed at necropsy or surgery |
| Minor |
| - Family history of premature sudden death (<35 years) due to suspected right ventricular dysplasia. |
| - Familial history (clinical diagnosis based on present criteria) |
The patient in case report 1 died suddenly and the diagnosis was made by autopsy. At that time, patient 2 fulfilled only one major criterion consisting of her sister’s proven ARVC. Hence the diagnosis could not be made. During the second evaluation this second sister also had repolarisation abnormalities on her ECG (minor, figure 1), arrhythmias on exercise test (figure 2) and Holter (minor), and later also localised right ventricular akinesia on echo (major) along with late potentials during electrophysiological testing (minor).
Genetic analysis revealed a mutation in the PKP2 gene which, at present, is not a diagnostic criterion but does confirm the diagnosis.
A third sister also had a major criterion consisting of her sisters death with proven ARVC but otherwise no clinical abnormalities and was screened negative for the PKP2 mutation.
ARVC is found to be familial in 30 to 50% of the cases.8 It is usually inherited in an autosomal dominant manner with reduced and age-related penetrance,8 but autosomal recessive forms have been reported. Several genetic loci have been reported to be associated with the disease and mutations in six genes have been identified: ryanodine receptor, plakoglobin, desmoplakin and recently transforming growth factor-β, plakophilin- 2 and desmoglein.9-16 Patients with mutations in the ryanodine receptor gene, also called ARVC2, are more prone to develop arrhythmias.17 The recognition that ARVC is frequently familial and can cause arrhythmic death leads to the challenge to identify family members who are at risk. Difficulties arise in diagnosing the presymptomatic subjects because identification of minimal structural abnormalities is difficult due to the irregular shape and asymmetrical contractile pattern of the right ventricle. Even on magnetic resonance imaging (MRI) the diagnosis of ARVC cannot be made on the presence of intramyocardial fat or wall thinning alone. A study by Bomma demonstrated a high frequency of misdiagnosis of ARVC on MRI.18
Asymptomatic ARVC patients should have an annual follow-up and currently there is no evidence for medical treatment in this group. However, competitive sports should be avoided. Patients who present with well-tolerated and non-life-threatening ventricular tachycardia can be treated conservatively with antiarrhythmic drugs, guided by Holter monitoring and exercise testing and, in selected cases, by programmed electrical stimulation. The most effective drug used in treating ARVC patients with non-lifethreatening ventricular tachycardia is sotalol with an efficacy rate of 68% followed by amiodarone with an efficacy rate of 15%.19 Radiofrequency ablation is an alternative in this group of patients who are unresponsive or intolerant to antiarrhythmic drugs. Patients who have been resuscitated or who have had a haemodynamically unstable ventricular tachycardia, such as the patient in case report 2, should be treated with an implantable cardioverter-defibrillator.20 Prophylactic ICD implantation will become more important in certain families depending on the aggressiveness of their gene mutation. In these families genetic screening would constitute an important additional tool in diagnosing persons at risk.
Conclusion
The two cases described here illustrate the wide variety in clinical presentation of arrhythmogenic right ventricular cardiomyopathy. The clinical manifestations of ARVC are varied and age-dependent. Therefore, the diagnosis of this illness can be difficult. For every physician, not only cardiologists but also general physicians, internists and sport physicians, it is important to be aware of this disease when a patient presents with syncope or palpitations, especially when there is a family history of sudden death. Because of the major implications and the prevention of sudden death, it is recommended to screen family members of known ARVC patients both clinically and genetically. In the near future, genetic testing will probably provide the gold standard for diagnosis.21
References
- 1.Frank R, Fontaine G. Electrocardiologie de quatre cas de dysplasie ventriculaire droite arythmogene. Arch Mal Coeur Vaiss 1978;71:963-72. [PubMed] [Google Scholar]
- 2.Thiene G, Nava A, Corrado D, Rossi L, Penelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 1998;318:129-33. [DOI] [PubMed] [Google Scholar]
- 3.Richardson P, Mc Kenna WJ, Bristow M, et al. Report of the 1995 WHO/ISFC task force on the definition and classification of the cardiomyopathies. Circulation 1996;93:841. [DOI] [PubMed] [Google Scholar]
- 4.Tabib A, Loire R, Chalabreysse L, et al. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation 2003;108:3000-5. [DOI] [PubMed] [Google Scholar]
- 5.Corrado D, Basso C, Schiavon M, et al. Screening of hypertrophic cardiomyopathy in young athletes. N Engl J Med 1998; 339:364. [DOI] [PubMed] [Google Scholar]
- 6.Angelini A, Basso C, Nava A, et al. Endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy. Am Heart J 1996;132:203. [DOI] [PubMed] [Google Scholar]
- 7.McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Diseases of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the international Society and Federation of Cardiology. Br Heart J 1994;71:215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamid M, Norman M, Quraishi A, et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol 2002;40:1445-50. [DOI] [PubMed] [Google Scholar]
- 9.Tiso N, Stephan DA, Nava A, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 2001;10:189-94. [DOI] [PubMed] [Google Scholar]
- 10.McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular dysplasia with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000;355:2119-24. [DOI] [PubMed] [Google Scholar]
- 11.Rampazzo A, Nava A, Malacrida S, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 2002;71:1200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coonar AS, Protonotarios N, Tsatsopoulou A, et al. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation 1998;97:2049-58. [DOI] [PubMed] [Google Scholar]
- 13.Beffagna G, Occhi G, Nava A, et al. Regulatory mutations in transforming growth factor-β3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res 2005;65:366-73. [DOI] [PubMed] [Google Scholar]
- 14.Gerull B, Heuser A, Wichter T, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet 2004;36:1162-4. [DOI] [PubMed] [Google Scholar]
- 15.van Tintelen JP, Entius MM, Bhuiyan ZA, et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2006; 113:1650-8. [DOI] [PubMed] [Google Scholar]
- 16.Pilichou K, Nava A, Basso C, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 2006;113:1171-9. [DOI] [PubMed] [Google Scholar]
- 17.Bauce B, Rampazzo A, Basso C, et al. Screening for ryanodine receptor type 2 mutations in families with effort-induced polymorphic ventricular arrhythmias and sudden death: early diagnosis of asymptomatic carriers. J Am Coll Cardiol 2002;40:341-9. [DOI] [PubMed] [Google Scholar]
- 18.Bomma C, Rutberg J, Tandri H, et al. Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Cardiovasc Electrophysiol 2004;15:300-6. [DOI] [PubMed] [Google Scholar]
- 19.Wichter T, Borggrefe M, Haverkamp W, Chen X, Breithardt G. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation 1992;86:29-37. [DOI] [PubMed] [Google Scholar]
- 20.Corrado D, Leoni L, Link MS, et al. Implantable cardioverterdefibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 2003;108:3084-91. [DOI] [PubMed] [Google Scholar]
- 21.Van der Wall EE, Schalij MJ, Van der Laarse A. Genetic factors in arrhythmogenic right ventricular cardiomyopathy: need for a DNA bank! Neth Heart J 2006;14:323-4. [PMC free article] [PubMed] [Google Scholar]