

Abstract
Ionic liquids (ILs) offer new possibilities for epoxide hydrolase (EH) catalyzed resolution of epoxides and for synthesis of chiral 1,2-diols. Soluble EHs from cress and mouse (csEH and msEH) and microsomal EH from rat (rmEH) were tested in several ILs. For all the enzymes tested, higher enantioselectivities were obtained in [bmim][N(Tf)2] and [bmim][PF6]. The optimized amount of water for EH activity in these ILs was established. Classical problems arising from low solubility of epoxides in water or from the high tendency of the oxirane ring to undergo chemical hydrolysis were avoided using these new media.
Introduction
Due to their chemical versatility, enantiopure epoxides and vicinal diols are extensively employed as useful building blocks for the synthesis of bioactive compounds in the pharmaceutical and agrochemical industries.1 An emerging approach involves the use of cofactor-independent epoxide hydrolases, which allow the preparation of these compounds under mild conditions.2–4 These enzymes have long been known in mammalian systems, and stereochemical studies have shown that the reaction catalyzed by EHs generally proceeds with a high product and/or substrate enantioselectivity.5,6 However, the interest of the synthetic chemist towards the application of these biocatalysts has only recently increased, since it has been shown that (i) these enzymes are ubiquitous in nature (they have been detected in plants, fungi, microorganisms) and therefore they are easily accessible;7 (ii) EHs may be cloned, modified and expressed in cells or microorganisms;8 (iii) racemic epoxides can be resolved on a preparative scale.9 This latter aspect is surely important from a synthetic point of view, particularly for industrial applications; however, only a few examples have been reported using two-phase reactors.9 Generally, a bio-catalytic approach allows only the transformation of small amounts of substrate per litre of solution, due to the low solubility and/or stability of many organic compounds in the aqueous buffer solutions used for the reactions. In order to overcome these drawbacks, non-conventional media such as organic co-solvents, and organic–aqueous two-phase systems have been used in several biocatalyzed reactions.9 However, many common solvents are toxic, and activity and selectivity of the enzymes are generally reduced in non-aqueous media, limiting their use.10 The use of organic solvents might be particularly important in the case of epoxides, which easily undergo spontaneous hydrolysis in the presence of water. This process not only depletes the substrate (reducing the overall yield) but it also affects the enantiomeric purity of the product. Chemical hydrolysis gives generally the same diol arising from enzymatic reaction, but in a racemic mixture. There are exceptions where chemical hydrolysis leads to extensive rearrangement and/or polymerization of the substrate. In contrast to other enzymatic systems, however, only a few examples of the use of epoxide hydrolases in organic solvents or in two phase systems have been reported, most likely due to the instability of these enzymes in non-natural media.11 Recently, we showed that soluble epoxide hydrolases are able to perform resolution processes in a very efficient and enantioselective manner in some ionic liquids (ILs),12 although organic solvents (e.g. methyl tert-butyl ether and tert-butanol) completely deactivate the same enzymes. The term ionic liquids identifies a large class of organic salts, liquid at or near room temperature, which have emerged recently as alternative green media for biotransformations, using both whole cell systems and isolated enzymes.13 The use of ionic liquids has shown many advantages with respect to conventional organic solvents, such as a better enzyme stability, substrate and/or product selectivity and suppression of unwanted side reactions.14 Moreover, as solvents for chemical reactions, ILs exhibit excellent physico-chemical properties, in particular they are able to dissolve polar and non-polar organic, inorganic and polymeric materials; they have a high thermal stability and they lack significant vapor pressure.15 Finally, all the physico-chemical properties of ILs can be modified by altering the cation or anion, and in principle the best IL may be designed for each specific reaction system.
As part of our research program, we became interested in ionic liquids as alternative solvents for biotransformations using epoxide hydrolases (EHs) as catalyst. Here, we report our more recent results on the hydrolysis of racemic and meso epoxides (some of these substrates easily undergo spontaneous hydrolysis in buffer solution) catalyzed by different EHs in several ionic liquids.
Experimental
The 1H and 13C NMR spectra were obtained in CDCl3 with a Bruker AC 200 instrument using TMS as the internal reference. GC analyses were performed using a Carlo Erba HRGC 5300 instrument. HPLC analyses were performed on a Waters 600E instrument equipped with a Varian Prostar 325 detector.
Chemicals and enzymes
Racemic 3,3-dimethyl-1,2-butene oxide (1), cyclopentene oxide (3) and cyclohexene oxide (5) were purchased from Aldrich. 4-tert-Butylstyrene oxide (7), α-methylstyrene oxide (9) and dihydronaphthalene oxide (13) were prepared from the corresponding olefins by oxidation with m-chloroperbenzoic acid–KF complex, as previously reported.16 4-Chlorostyrene oxide (11) was prepared by oxidation with m-chloroperbenzoic acid in dichloromethane. The corresponding diols were synthesized by acid catalyzed hydrolysis (0.1 N HClO4) from the corresponding epoxides. [bmim][BF4], [emim][EtSO4] (ECOENG 212) and [mmim][Me2PO4] (ECOENG 1111P) were purchased from Solvent Innovation (GMBH). [bmim][PF6], [bmim][Tf2N], [bmim][BOB] and [bmpyrr][Tf2N] were prepared following reported procedures.17 Attention was paid to the elimination of bases and Cl− ions which may be present in the solvents as impurities. The purity of imidazolium salts was always checked by measuring the absorption spectra between 240 and 400 nm. Purified [bmim]+ salts (containing Cl− < 0.1 ppm) have practically no absorption band in the 250–300 nm region.18 After drying (2 h at 80 °C under vacuum), the amount of water in the ILs was determined by the Karl–Fisher technique using an apparatus composed of a stand titrator and a coulometer. The water content of dried ILs ranged from 150 to 350 ppm. The water activity (aw) was measured with an aw measuring instrument from DELTA Instrument (Trieste, Italy). Measurements were carried out by placing the sensor into the open end of 5 mL glasses vials at 37 °C, until constant readings were obtained. All samples were previously equilibrated for 24 h.
The recombinant soluble and microsomal epoxide hydrolase enzymes of mouse, cress and rat were prepared and purified as previously described.7d,19 Recombinant cDNA of each enzyme was cloned into baculovirus expression system. Trichoplusia ni high five cell cultures were transfected with prepared recombinant baculovirus in order to express the desired enzyme and subsequently were purified from cell lysate using affinity chromatography.
Enantioselectivity assays
Analysis conditions
The enantiomeric composition of residual epoxides 7, 9 and 13 was determined by 1H NMR after addition of the resolving agent, Europium tri[3-hepta-fluoropropylhydroxymethyle)-(+)camphorate] (Aldrich), whereas the enantiomeric composition of all the other epoxides were determined by GC on a chiral 30 m Chiradex G-TA (ASTEC) column (helium flow 50 KPa, with evaporator and detector set at 200 °C) at the following temperatures: 1, 45 °C; 11, 100 °C. The enantiomeric composition of diols 8 and 10 were determined by 1H NMR after addition of the resolving agent, Europium tri[3-heptafluoropropylhydroxymethyl)-(+)camphorate] (Aldrich), whereas that of diol 12 by HPLC on a Daicel Chiralcel OD-H column, using hexane-2-propanol (98 : 2) as solvent (solvent flow, 0.5 mL min−1). The enantiomeric excesses of all the other diols were determined by GC on a chiral 30 m Chiradex G-TA (ASTEC) column (helium flow 50 KPa, with evaporator and detector set at 200 °C) at the following conditions: diol 2, 80 °C for 10 min, at a rate of 4 °C min−1, 120 °C for 10 min; diol 4 (as trifluoroacetyl derivative), 100 °C; diol 6 (as trifluoroacetyl derivative), 85 °C; diol 14 (after transformation into the corresponding 1,2-dimethyl ether), 170 °C. The absolute configurations of the excess enantiomers were determined on the basis of their optical rotation values. The enantiomeric ratios (ES and EP) were calculated using the conversion rate (c) and either the substrate e.e. (ee(S)) or the product e.e. (ee(P)) using the following equations.20
Incubation procedures with mouse and cress sEH and rat mEH. Selection of ionic liquids
Epoxide 1 (150 μmol) and epoxide hydrolase (cress sEH 3.1 mg; mouse sEH 2,8 mg; rat mEH 19 mg) were mixed with ionic liquid (2 mL) or buffer solution (Tris-HCl, pH = 7.4) and the resulting mixture was stirred at 37 °C. After 3 h, the reaction mixture was first extracted with hexane (5 times with 4 mL portion). The combined hexane phases were analyzed by GC on a chiral column, after addition of cyclopentanone as an internal standard, to determine the conversion and the e.e. of the remaining substrate. The ionic liquid was then extracted with ethyl ether (5 times with 4 mL portion) and the combined ethereal phases, containing the formed diol 2, were analyzed by GC on the same chiral column. Control experiments carried out without enzyme or using a deactivated preparation showed that the spontaneous hydrolysis did not contribute to diol formation under the conditions reported here. Finally, after extraction of the remaining epoxide and resulting diol, the ionic liquids were filtered, washed and dried under vacuum at 80 °C for 2 h and reused. The water content of the dried ILs, determined by Karl–Fisher titration ranged from 150–350 ppm. When purified enzyme was used the amount of water was around 1% while in the case of crude extracts it was around 10%.
Incubation of epoxides 3 and 5 with rat mEH in [bmim][PF6]
Epoxide 3 or 5 (250 μmol) and rat mEH (4 × 19 mg: each portion added every 12 h) were added to [bmim][PF6] (2 mL), and the resulting mixture was stirred at 37 °C for 48 h until TLC analysis (hexane–ethyl acetate, 80 : 20) demonstrated the disappearance of epoxides 3 or 5. The resulting trans-diol 4 or 6 was extracted with ethyl ether (5 times with 4 mL portion) and analyzed by GC after conversion to the corresponding bis-trifluoroacetyl derivatives.
Incubation of epoxides 7, 9 and 13
Aryl substituted epoxides 7 or 9 or 13 (400–600 μmol) were incubated at 37 °C in [bmim][PF6] (4 mL) using microsomal and soluble epoxide hydrolase (cress sEH 10 mg; mouse sEH 9.6 mg; rat mEH 36 mg) as biocatalyst. The reactions, stopped at prefixed times (1–3 h) by filtration of the enzyme, were first extracted with hexane (5 times with 4 mL portion). The combined hexane phases were analyzed by GC, HPLC or NMR, as reported above, to determine the conversion and the e.e. of the unreacted substrate. The ionic liquid was then extracted with ethyl ether (5 times with 4 mL portion) and the combined ethereal phases, containing the formed diols, were analyzed by GC, HPLC or NMR. Blank experiments carried out without enzyme or using a deactivated preparation showed that the spontaneous hydrolysis did not contribute to diol formation under the incubation conditions.
Result and discussion
Seven different ionic liquids [bmim][PF6], [bmim][Tf2N], [bmim][BF4], [bmpyr][N(Tf2)], [bmim][Bob], [emim][EtSO4], [mmim][Me2PO4] (where emim = 1-ethyl-3-methylimidazolium, bmim = 1-butyl-3-methylimidazolium, mmim = 1,3-dimethylimidazolium, bmpyr = butylmethyl pyrrolidinium, PF6 = hexafluorophosphate, Tf2N = bis(trifluoromethylsulfonyl)imide, BF4 = tetrafluoroborate, EtSO4 = ethylsulfate, Me2PO4 = dimethylphosphate, [BOB] = bis[oxalate(2-)]borate]) were used as solvents to investigate the influence of these media on epoxide hydrolase activity to hydrolyze racemic 3,3-dimethyl-1,2-butene oxide (1) (Scheme 1), under water controlled conditions (around 10% when crude enzyme extracts were used; around 1% when the purified enzyme was used).
Scheme 1.

EH catalyzed hydrolysis of racemic 1.
The efficiency of each conversion was evaluated on the basis of the conversion and the enantiomeric excess of the remaining epoxide 1 and resulting diol 2, after 3 h. Both of these parameters were evaluated by GC, using a chiral column. The data reported in Table 1 show that three of the investigated ILs ([bmim][BOB], [bmim][EtSO4] and [mmim][Me2PO4]) are not suitable for this kind of reaction. Epoxide 1 probably reacts with the anion of these ionic liquids to give one or more addition products, which have not been isolated. The enzymatic hydrolysis is able to compete with this process only in the case of [mmim][Me2PO4]; in this solvent, we detected small amounts of pure (R)-2. In the other ionic liquids, having [BF4]− [PF6]− or [Tf2N]− as anion, the biocatalyzed reactions were dependent on the structure of the cation and anion, and on enzyme source. Higher conversions and enantioselectivities were generally found in [bmpyr][N(Tf)2] and [bmim][PF6], two hydrophobic ILs. It is worth noting that, in all examined ILs (in which it was possible to isolate the diol 2) the reaction proceeded with a complete substrate enantioselectivity to give a pure diol of R configuration and an enantio-enriched epoxide of S configuration (before 50% conversion), in agreement with the enzyme’s behavior previously observed in buffer solution.21
Table 1.
Hydrolysis of 3,3-dimethyl-1,2-butene oxide (1) catalyzed by crude cress, mouse soluble and rat microsomal EH (csEH, msEH, rmEH) or purified csEH in ionic liquidsa
| Solvent | Conv. (%)b | Residue (S)-1 e.e. (%) | ES | Formed (R)-2 e.e. (%) | EP | |
|---|---|---|---|---|---|---|
| [bmim][PF6] | csEH | 24 | 30 | 52c | >98 | >100 |
| [bmim][Tf2N] | 35 | 54 | >100 | >98 | >100 | |
| [bmim][BF4] | 16 | 18 | >100 | >98 | >100 | |
| [bmpyr][Tf2N] | 25 | 35 | >100 | >98 | >100 | |
| [bmim][BOB] | —d | |||||
| [mmim][Me2PO4] | —e | |||||
| [emim][EtSO4] | —f | |||||
| [bmim][PF6] | msEH | 25 | 34 | >100 | >98 | >100 |
| [bmim][Tf2N] | 36 | 55 | >100 | >98 | >100 | |
| [bmim][BF4] | 10 | 11 | >100 | >98 | >100 | |
| [bmpyr][Tf2N] | 39 | 63 | >100 | >98 | >100 | |
| [bmim][BOB] | —d | |||||
| [mmim][Me2PO4] | —e | |||||
| [emim][EtSO4] | —f | |||||
| [bmim][PF6] | Purified csEH | 8 | n.d.g | >98 | >100 | |
| [bmim][Tf2N] | 13 | n.d. | >98 | >100 | ||
| [bmim][BF4] | 5 | n.d. | >98 | >100 | ||
| Tris-HCl | rmEH | 20 | 24 | >100 | >98 | >100 |
| [bmim][PF6] | 10 | 10 | 21c | >98 | >100 | |
| [bmim][Tf2N] | 10 | 11 | >100 | >98 | >100 | |
| [bmpyr][Tf2N] | 20 | 24 | >100 | >98 | >100 | |
| [bmim][BOB] | —d | |||||
| [mmim][Me2PO4] | —e | |||||
| [emim][EtSO4] | —f |
In typical analytical experiments, the enzymatic reactions were performed with a solution containing substrate (0.15 mmol) and enzyme (cress sEH 3.1 mg; mouse sEH 2.8 mg; rat mEH 19 mg) in solvent (2 mL).
Conversions were calculated on the basis of the unreacted epoxide and formed diols. The values matched within 5%.
Considering the error on conversion, the value may be >100.
Neither unreacted epoxide, nor the resulting diol were recovered from this IL.
Conversions calculated on the basis of the residue epoxide were higher than 80%. Only traces of formed diols were however detected.
Conversions calculated on the basis of the residue epoxide were higher than 80%. No diol was however detected.
n.d.: not determined.
The water content of the reaction medium generally plays an important role in enzymatic catalysis, since it affects the flexibility of the proteins, a factor determining enzyme activity and selectivity.22 In the case of EH catalyzed hydrolysis of epoxides, water is also a reactant. In neat ionic liquid, no activity was observed at all when lyophilized EH from Aspergillus niger was used, underlying the need for reactive water. To investigate the influence of water, we selected two ionic liquids from our first experiment, [bmim][PF6] and [bmim][N(Tf)2]. Using purified cress soluble epoxide hydrolase as catalyst and trans-β-methylstyrene oxide as substrate (trans-β-methylstyrene oxide is a typical substrate for this enzyme) we determined the enzymatic activity in the presence of different amounts of water (1, 5, 10, 15, 25 and 50%), starting from a homogeneous solution (below 5%), going to a biphasic system (above 25%) and passing through a microemulsion, (around 10–15%) as evidenced by the opalescent aspect of the mixture.
As shown in Fig. 1, in both ILs the enzyme activity varies with the water content; the higher activity being found around 10–15%. The apparent activity decrease observed at the higher water percentages, may be attributed to the biphasic system characterizing these latter conditions. Because water activity aw is considered a more precise parameter to compare dependence of the enzyme’s activity on the amount of water present in the incubation medium,23 we determined aw values for the employed mixtures ILs–wet csEH, corresponding to a IL : water ratio around 90 : 10 v/v.
Fig. 1.

The effect of water on the csEH catalyzed hydrolysis of β-methylstyrene oxide in IL–water mixtures. Black, [bmim][PF6]–water mixtures. Grey,[bmim][Tf2N]–water mixtures.
The aw values for the examined mixtures csEH-[bmim][PF6], csEH-[bmim][Tf2N], csEH-[bmim][BF4] and csEH-[bmpyr][Tf2N] were 0.80, 0.82, 0.84 and 0.84, respectively. These values and conditions are very similar to those found to provide mandelate racemase and penicillin G amidase activity in ILs.24
Considering the promising results and optimized conditions obtained for the hydrolysis of epoxide 1, we then examined the behavior of other substrates in these reaction conditions. In particular, we investigated the enantioselective production of diols from simple meso-epoxides, such as cyclopentene oxide (3) and cyclohexene oxide (5) by mEH (Scheme 2) and from aryl substituted epoxides (Scheme 3).
Scheme 2.
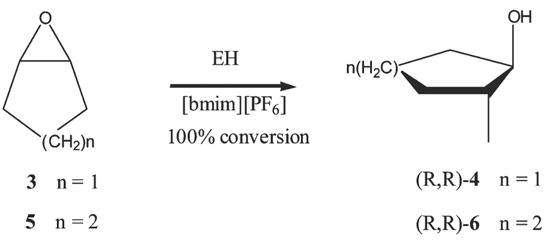
EH catalyzed hydrolysis of meso-epoxides.
Scheme 3.
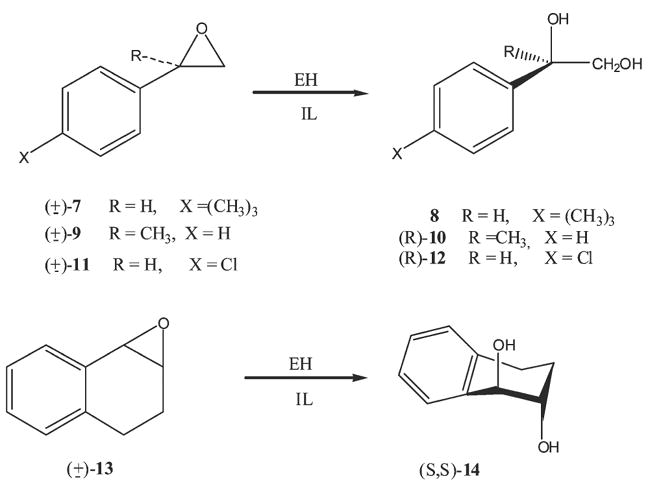
EH catalyzed hydrolysis of racemic aryl substituted epoxides.
Examples of desymmetrization of meso-epoxides are scarce.25 Epoxides 3 and 5 (0.25 mmol) and mEH (4 × 19 mg) were suspended in 5 μl of phosphate buffer; each portion (added at intervals of 12 h) were incubated in 2 mL of [bmim][PF6] at 37 °C for 48 h, until no epoxide was detected. Although on the basis of the data reported in Table 1 the best IL seems to be [bmpyr][Tf2N], in this screening it was replaced with [bmim][PF6], since this latter IL is less expensive and the products were more easily recovered. Pure trans-(R,R)-diols 4 and 6 (e.e. ≥ 98) were isolated in quantitative yield. It is interesting to note that, the e.e. of diols 4 and 6 obtained from the mEH catalyzed hydrolysis of epoxides 3 and 5 in [bmim][PF6] are significantly higher than those previously reported for the reaction carried out in buffer Tris-HCl solution (4, 90%; 6; 76%).25a In the aqueous medium, at least for epoxide 5, an e.e. around 90% was obtained working under highly dilute conditions.26 Control experiments had shown that in buffer solution at pH 7.4, epoxide 5 is partially converted to its vicinal diol 6 via an oxirane ring opening reaction with water, and the contribution of this spontaneous reaction, which proceeds without enantioselectivity, increases with the substrate concentration. The high e.e. found in [bmim][PF6], working at relatively high substrate concentrations (0.12 M) and in the presence of ca. 12% of water, highlights the ability of the IL to reduce the non-enzymatic hydrolysis. In ILs, water-sensitive catalysts and chemical reactions are less affected by the presence of water, compared with the situation in organic solvents, because water dispersed throughout the IL cannot act like bulk water.15g
To investigate in more detail the possibility of the hydrolysis of water sensitive epoxides in ionic liquids we performed the incubations of three aryl substituted oxiranes, 4-tert-butylstyrene oxide (7), α-methylstyrene oxide (9), and 1,2-dihydronaphthalene oxide (13) in [bmim][PF6] (in the presence of ca. 10% of water) using soluble and microsomal epoxide hydrolases (Scheme 3).
Results are reported in Table 2. As expected, product and substrate enantioselectivity of all three aryl substituted epoxides depended on the enzyme source, being generally low with the murine soluble enzyme and relatively high with the cress soluble EH and rat microsomal EH.
Table 2.
Hydrolysis of 4-tert-butylstyrene oxide (7), α-methylstyrene oxide (9), dihydronaphthalene oxide (13) catalyzed by csEH, msEH, rmEH in [bmim][PF6] (in the presence of ca. 10% of water) at 37 °Ca
| Substrate | Conv (%)b | Residue epoxide e.e (%, abs. conf.) | ES | Diol | Formed diol e.e. (%, abs. conf.) | EP | |
|---|---|---|---|---|---|---|---|
| 7 | csEH | 29 | 10 | 1.8 | 8 | 20, (R) | 1.6 |
| msEH | 25 | 10 | 2.0 | 20, (R) | 1.6 | ||
| rmEH | 50 | 20 | 1.8 | 22, (R) | 1.9 | ||
| 9 | csEH | 35 | 20,(S) | 1.1 | 10 | 62, (R) | 5.9 |
| msEH | 30 | 16,(S) | 2.5 | 30, (R) | 2.0 | ||
| rmEH | 33 | 10,(S) | 1.7 | 20, (R) | 1.6 | ||
| 13 | csEH | 10 | 2 | 1.5 | 14 | 80, (1S,2S) | 9.8 |
| msEH | <1 | 0 | |||||
| rmEH | 20 | 16, (1S,2R) | 5.3 | 63, (1S,2S) | 5.1 |
In typical analytic experiments, the enzymatic reactions were performed with a solution containing substrate (0.4–0.6 mmol), enzyme (cress sEH 10 mg; mouse sEH 9.6 mg; rat mEH 36 mg) in solvent (4 mL).
Conversions were calculated on the basis of the unreacted epoxide and formed diols. The values matched within 5%.
In aqueous buffer solution, the product enantioselectivity characterizing the mammalian mEH catalyzed hydrolysis of terminal racemic epoxides is considered to be the outcome of the two selective processes: (i) a substrate enantioselectivity, arising from a different affinity of the two epoxide enantiomers for the enzyme active site; associated with (ii) a high regioselectivity. The enzymatic attack is highly preferential (or even absolute) at the less substituted carbon. In the case of terminal epoxides, therefore, hydrolysis involves retention of configuration at the stereogenic centre and the e.e. of the product is a function of the substrate enantioselectivity and of the conversion. The e.e. of diols 8 and 10, although low, correlate with the e.e. of the corresponding epoxides and with the percent of conversion; the reaction occurs with high regioselectivity but with moderate substrate enantioselctivity.27 The regioselectivity of mEH, a feature probably arising from the mechanism of hydrolysis (nucleophilic attack of an aspartate carboxylic anion on an enzymatically activated epoxide, but without the formation of a true carbonium ion),28 seems to not be affected by the IL.
At variance, the rmEH hydrolysis of 1,2-dihydronaphthalene oxide (13) proceeds with a significant substrate enantioselectivity, resulting in the corresponding trans-diol of (1S,2S) configuration, by formal water attack at the benzylic carbon of the (1R,2S)-enantiomer. In agreement with the proposed topology of the active site, the enantiomer preferentially hydrolyzed is that bearing the phenyl ring on the right back side, when the oxirane ring is oriented with the oxygen upward, as reported in Scheme 4.
Scheme 4.
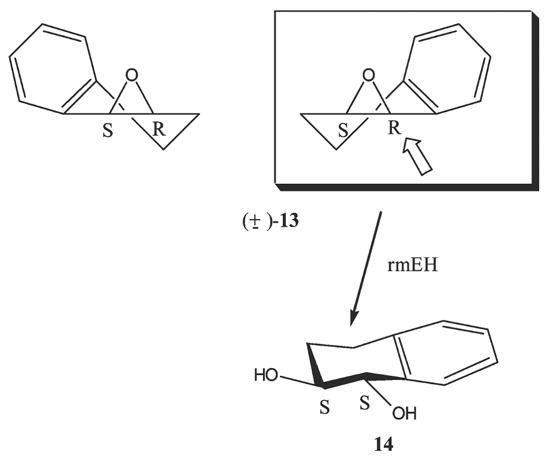
Preferential pathway in the mRH catalyzed hydrolysis of racemic 13.
In aqueous buffer solution, it is more difficult to generally rationalize the enantioselectivity of the sEH catalyzed processes. These enzymes show the potential to convert both substrate enantiomers through two processes having different regioselectivity.6,29 As a consequence, the product enantioselectivity is not only a function of the substrate enantioselectivity and the conversion, but also of the ratio of retention to inversion for each enantiomer. Moreover, many sEH enzymes were found to be regioselective at the benzylic oxirane carbon, supporting the proposed mechanism in which one or more tyrosine residues in the site active of the enzyme act as general acid catalysts in the alkylation half reaction.6 The moderate e.e. characterizing diol 8 and epoxide 7 evidences the lack of enantioselectivity of both the msEH and csEH, analogous to the behavior observed with mEH (Table 2).
On the other hand, the significant e.e. of diol 10, obtained from csEH hydrolysis of 9, is reasonably consistent with a regioselective attack at the terminal carbon. The tertiary benzylic position should not be reactive towards the nucleophilic attack due to the steric hindrance. Related to the hydrolysis of this substrate, it is also worth noting that the high enantioselectivity found in the csEH catalyzed hydrolysis of α-methylstyrene oxide (9) shows that ILs can be efficiently used to perform the hydrolysis of water sensitive epoxides at high substrate concentration (0.1–0.2 M) without the competition of the non-enzymatic process, which generally occurs without any stereoselectivity. Moreover, α-methylstyrene is prone to dimer formation under acidic conditions.
Finally, a comment concerning the sEH catalyzed hydrolysis of epoxide 13 is necessary. Whereas the mammalian enzyme (msEH) hydrolyses epoxide 13 at a negligible rate, csEH more efficiently catalyzes the oxirane ring opening. Moreover, the high product enantioselectivity is associated with a completely non-selective hydrolysis of the two enantiomers of racemic epoxide 13. In analogy with the hydrolysis of meso epoxides, the oxirane ring opening of both enantiomers occurs selectively at the same configured carbon (in this case the (R) carbon) to give the corresponding (S,S) diol with a very high enantiomeric excess. The csEH catalyzed hydrolysis of this substrate is an enantioconvergent process, arising from the different regioselectivity characterizing the oxirane ring opening of each enantiomer (Scheme 5). The fact that EP is much larger than ES (Table 2) illustrates the opposite regioselectivity of csEH for both enantiomers of 13.
Scheme 5.

Hydrolysis pathway for the cress sEH catalyzed hydrolysis of racemic 13.
Ultimately, to verify the general applicability of ILs in EH catalyzed reactions we performed the hydrolysis of 4-chlorostyrene oxide (11, 0.6–0.7 mmol) using a commercial EH from Aspergillus niger (2.5 mg) in “wet” [bmim][PF6] (6 mL), after addition of 5, 10 or 15% of water. The higher conversion (around 25%) has been obtained in the presence of 15% of water. The percentage of hydrolysis was comparable to that characterizing the reaction in Tris-HCl buffer solution (28%). Enantiomerically pure (R) diol 12 was obtained both in buffer solution and in [bmim][PF6]. In [bmim][PF6] the epoxide 11 was completely soluble and even higher concentrations may be used, supporting a synthetic application of the enzymatic hydrolysis.
Conclusions
In summary, we have shown that ionic liquids not bearing nucleophilic anions can be used effectively for the enzymatic hydrolysis of epoxides catalyzed by EHs. Soluble EHs from cress and mouse, and microsomal EH from rat are able to catalyzed the hydrolysis of racemic and meso epoxides in several ILs in the presence of water (around 10%). The optimized amount of water has been established. Problems arising from low solubility of epoxides in water or from the tendency of oxirane rings to undergo chemical hydrolysis have been avoided using these new media, resulting in more enantioselective reactions.
Acknowledgments
This was supported by grants from MIUR, NIEHS (# R37-ES02710), NIEHS Center (# P30-ESO5707), and NIH/NIEHS Superfund Basic Research program (# P42-ES04699).
References
- 1.Savle PS, Lamoreaux MJ, Berry JF, Gandour RD. Tetrahedron: Asymmetry. 1998;9:1843–1846. [Google Scholar]
- 2.Steinreiber A, Faber K. Curr Opin Biotechnol. 2001;12:552–558. doi: 10.1016/s0958-1669(01)00262-2. [DOI] [PubMed] [Google Scholar]
- 3.Archelas A, Furstoss R. Curr Opin Chem Biol. 2001;5:112–119. doi: 10.1016/s1367-5931(00)00179-4. [DOI] [PubMed] [Google Scholar]
- 4.Archer VJ. Tetrahedron. 1997;53:15617–15662. [Google Scholar]
- 5.Chiappe C, Cordoni G, Lo Moro G, Palese CD. Tetrahedron: Asymmetry. 1998;9:341–350. [Google Scholar]
- 6.Williamson KC, Morisseau C, Maxwell JE, Hammock BD. Tetrahedron: Asymmetry. 2000;11:4451–4462. [Google Scholar]
- 7.(a) Kroutil W, Mischitz M, Faber K. J Chem Soc Perkin Trans. 1997;1:3629–3636. [Google Scholar]; (b) Moussou P, Archelas A, Baratti R, Furstoss R. J Org Chem. 1998;63:3532–3537. [Google Scholar]; (c) Weijers CAGM, Botes AL, Van Dick MS, De Bont M. Tetrahedron: Asymmetry. 1998;9:467–473. [Google Scholar]; (d) Morisseau C, Beetham JK, Pinot F, Debernard S, Mewman JW, Hammock BD. Arch Biochem Biophys. 2000;387:321–332. doi: 10.1006/abbi.2000.1810. [DOI] [PubMed] [Google Scholar]
- 8.(a) Stapleton A, Beetham JK, Pinot F, Garbarino JE, Rockhold DR, Friedman M, Hammock BD, Belknap WR. Plant J. 1994;6:251–258. doi: 10.1046/j.1365-313x.1994.6020251.x. [DOI] [PubMed] [Google Scholar]; (b) Zhao L, Mathur EJ, Weiner D, Richardson T, Milan A, Burk MJ, Han B, Short JM. 6979733B2. US Pat. 2005
- 9.(a) Morisseau C, Nellaiah H, Archelas A, Furstoss R, Baratti JC. Enzyme Microb Technol. 1997;20:446–452. [Google Scholar]; (b) Manoj KM, Archelas A, Baratti J, Furstoss R. Tetrahedron. 2001;57:695–701. [Google Scholar]; (c) Monfort N, Archelas A, Furstoss R. Tetrahedron. 2004;60:601–605. [Google Scholar]
- 10.(a) Koshinen AMP, Klibanov AM. Enzymatic Reactions in Organic Media. Blackie Academic & Professional; Glasgow, United Kingdom: 1996. [Google Scholar]; (b) Faber K. Biotransformation in Organic Chemistry. 3. Springer; Berlin, Germany: 1997. [Google Scholar]; (c) Klibanov AM. Trends Biotechnol. 1997;15:97–101. doi: 10.1016/S0167-7799(97)01013-5. [DOI] [PubMed] [Google Scholar]
- 11.(a) Choi WJ, Lee EY, Yoon SJ, Yang ST, Choi CY. J Biosci Bioeng. 1999;88:339–341. doi: 10.1016/s1389-1723(00)80022-5. [DOI] [PubMed] [Google Scholar]; (b) Cleij M, Archelas A, Furstoss R. Tetrahedron: Asymmetry. 1998;9:1839–1842. [Google Scholar]; (c) Gong PF, Xu JH. Enzyme Microb Technol. 2005;36:252–257. [Google Scholar]
- 12.Chiappe C, Leandri E, Lucchesi S, Pieraccini D, Hammock BD, Morisseau C. J Mol Catal B: Enzym. 2004;27:243–248. [Google Scholar]
- 13.(a) Cull SG, Holbrey JD, Vargas-More V, Seddon KR, Ley G. J Biotechnol Bioeng. 2000;69:227. [PubMed] [Google Scholar]; (b) Lau RM, van Rantwijk F, Seddon KR, Sheldon RA. Org Lett. 2000;2:4189–4191. doi: 10.1021/ol006732d. [DOI] [PubMed] [Google Scholar]; Erbeldinger M, Mesiano AJ, Russell A. J Biotechnol Prog. 2000;16:1131. doi: 10.1021/bp000094g. [DOI] [PubMed] [Google Scholar]; (c) Itoh T, Akasaki E, Kudo K, Shirakami S. Chem Lett. 2001:262. [Google Scholar]; (d) Park S, Kazlauskas RJ. J Org Chem. 2001;66:8395–8401. doi: 10.1021/jo015761e. [DOI] [PubMed] [Google Scholar]; (e) Schöfer SH, Kaftzik N, Wassersheid P, Kragl U. Chem Commun. 2001:425–426. [Google Scholar]; (f) Kim KW, Song B, Kim MJ. Org Lett. 2001;3:1507–1509. doi: 10.1021/ol015824f. [DOI] [PubMed] [Google Scholar]; (g) Nara SJ, Harjani JR, Salunkhe MM. Tetrahedron Lett. 2002;43:2979–2982. [Google Scholar]; (h) Lozano P, De Diego T, Carrié D, Vaultier M, Iborra JL. J Mol Catal B: Enzym. 2003;21:9–13. [Google Scholar]; (i) Kaftzik N, Wassersheid P, Kragl U. Org Process Res Dev. 2002;6:553–557. [Google Scholar]; (j) Howarth J, James P, Dai J. Tetrahedron Lett. 2001;42:7517–7519. [Google Scholar]; (k) Howarth J, James P, Day J. Tetrahedron. 2001;42:7517. [Google Scholar]; (l) Kitazume T, Jiang Z, Kasai K, Mihara Y, Suzuki MJ. Fluorine Chem. 2003;121:205. [Google Scholar]; (m) Gaisberger RP, Fechter MH, Griengl H. Tetrahedron: Asymmetry. 2004;15:2959. [Google Scholar]; (n) Matsumoto M, Mochizuki K, Fukunishi K, Kondo K. Sep Purif Technol. 2004;40:97. [Google Scholar]; (o) Matsuda T, Yamagishi Y, Koguchi S, Iwai N, Kitazume T. Tetrahedron Lett. 2006;47:4619. [Google Scholar]
- 14.(a) Kragl U, Eckstein M, Kaftzik N. Curr Opin Biotechnol. 2002;13:565–571. doi: 10.1016/s0958-1669(02)00353-1. [DOI] [PubMed] [Google Scholar]; (b) van Rantwijk F, Lau RM, Sheldon RA. Trends Biotechnol. 2003;21:131–138. doi: 10.1016/S0167-7799(03)00008-8. [DOI] [PubMed] [Google Scholar]; (c) Park S, Kazlauskas RJ. Curr Opin Biotechnol. 2003;14:432–437. doi: 10.1016/s0958-1669(03)00100-9. [DOI] [PubMed] [Google Scholar]; (d) Itoh T, Akasaki E, Nishimura Y. Chem Lett. 2002:154. [Google Scholar]; (e) Itoh T, Nishimura Y, Ouchi N, Hayase S. J Mol Catal B: Enzym. 2003;26:41. [Google Scholar]; (f) Itoh T, Han S, Matsushita Y, Hayase S. Green Chem. 2004;6:437. [Google Scholar]; (g) Chiappe C, Neri L, Pieraccini D. Tetrahedron Lett. 2006;47:5089. [Google Scholar]
- 15.(a) Holbrey JD, Seddon KR. Clean Prod Process. 1999;1:223–236. [Google Scholar]; (b) Earle MJ, Seddon KR. Pure Appl Chem. 2000;72:1391–1398. [Google Scholar]; (c) Welton T. Chem Rev. 1999;99:2071–2083. doi: 10.1021/cr980032t. [DOI] [PubMed] [Google Scholar]; (d) Wasserscheid P, Keim W. Angew Chem, Int Ed. 2000;39:3773–3789. doi: 10.1002/1521-3773(20001103)39:21<3772::aid-anie3772>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]; (e) Sheldon RA. Chem Commun. 2001:2399–2407. doi: 10.1039/b107270f. [DOI] [PubMed] [Google Scholar]; (f) Olivier-Bourbigou H, Magna L. J Mol Catal A: Chem. 2002;182:419–437. [Google Scholar]; (g) Dupont J, de Souza RF, Suarez PAZ. Chem Rev. 2002;102:3667–3692. doi: 10.1021/cr010338r. [DOI] [PubMed] [Google Scholar]
- 16.Bellucci G, Catelani G, Chiappe C, D’Andrea F. Tetrahedron Lett. 1994;35:8433–8436. [Google Scholar]
- 17.(a) Fuller J, Carlin RT, De Long HC, Haworth D. J Chem Soc, Chem Commun. 1994:299–300. [Google Scholar]; (b) Cammarata L, Kazarian SG, Salter PA, Welton T. Phys Chem Chem Phys. 2001:5192–5200. [Google Scholar]; (c) Chiappe C, Pieraccini D, Saullo P. J Org Chem. 2003;68:6710–6715. doi: 10.1021/jo026838h. [DOI] [PubMed] [Google Scholar]; (d) Jork C, Kristen C, Pieraccini D, Stark A, Chiappe C, Beste YA, Arlt W. J Chem Thermodyn. 2005;37:537–558. [Google Scholar]
- 18.Billard I, Moutiers G, El Azzi A, Gaillard C, Mariet C, Lützenkinchen J. Inorg Chem. 2003;42:1726–1733. doi: 10.1021/ic0260318. [DOI] [PubMed] [Google Scholar]
- 19.Grant DF, Stroms DH, Hammock BD. J Biol Chem. 1993;268:17628–17633. [PubMed] [Google Scholar]
- 20.Chen CS, Fujimoto Y, Girdaukas G, Sih CJ. J Am Chem Soc. 1982;104:7294–7299. [Google Scholar]
- 21.(a) Bellucci G, Chiappe C, Conti L, Marioni F, Pierini G. J Org Chem. 1989;54:5978–5983. [Google Scholar]; (b) Bellucci G, Chiappe C, Marioni F, Benetti M. J Chem Soc, Perkin Trans 1. 1991:361–362. [Google Scholar]
- 22.Clark S. Philos Trans R Soc London, Ser B. 2004;359:1299–1307. doi: 10.1098/rstb.2004.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valivety RH, Halling PJ, Macrae AR. Biochem Biophys Acta. 1992;1118:218–222. doi: 10.1016/0167-4838(92)90278-l. [DOI] [PubMed] [Google Scholar]
- 24.(a) Kaftzik N, Kroutil W, Faber K, Kragl U. J Mol Catal A: Chem. 2004;214:107–112. [Google Scholar]; (b) Basso A, Cantone S, Linda P, Ebert C. Green Chem. 2005;7:671–676. [Google Scholar]
- 25.(a) Bellucci G, Capitani I, Chiappe C, Marioni F. J Chem Soc, Chem Commun. 1989:1170–1171. [Google Scholar]; (b) Zhao L, Han B, Huang Z, Miller M, Huang H, Malashock DS, Zhu Z, Milan A, Robertson DE, Weiner DP, Burk MJ. J Am Chem Soc. 2004;126:11156–11157. doi: 10.1021/ja0466210. [DOI] [PubMed] [Google Scholar]
- 26.Bellucci G, Chiappe C, Marioni F. J Chem Soc, Perkin Trans 1. 1989:2369–2373. [Google Scholar]
- 27.Similar enantiomeric ratios can be calculated from the remaining substrate (ES) or formed product (EP).
- 28.(a) Morisseau C, Hammock BD. Annu Rev Pharmacol Toxicol. 2005;45:311–333. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]; (b) Hopmann KH, Himo F. Chem–Eur J. 2006;12:6898. doi: 10.1002/chem.200501519. [DOI] [PubMed] [Google Scholar]
- 29.Bellucci G, Chiappe C, Cordoni A, Marioni F. Tetrahedron Lett. 1994;35:4219–4222. [Google Scholar]