

Abstract
Purpose
A phase I study was conducted to assess the safety of adoptive immunotherapy using gene-modified autologous T cells for the treatment of metastatic ovarian cancer.
Experimental Design
T cells with reactivity against the ovarian cancer – associated antigen α-folate receptor (FR) were generated by genetic modification of autologous T cells with a chimeric gene incorporating an anti-FR single-chain antibody linked to the signaling domain of the Fc receptor γ chain. Patients were assigned to one of two cohorts in the study. Eight patients in cohort 1received a dose escalation of T cells in combination with high-dose interleukin-2, and six patients in cohort 2 received dual-specific T cells (reactive with both FR and allogeneic cells) followed by immunization with allogeneic peripheral blood mononuclear cells.
Results
Five patients in cohort 1 experienced some grade 3 to 4 treatment-related toxicity that was probably due to interleukin-2 administration, which could be managed using standard measures. Patients in cohort 2 experienced relatively mild side effects with grade 1to 2 symptoms. No reduction in tumor burden was seen in any patient. Tracking 111In-labeled adoptively transferred T cells in cohort 1revealed a lack of specific localization of T cells to tumor except in one patient where some signal was detected in a peritoneal deposit. PCR analysis showed that gene-modified T cells were present in the circulation in large numbers for the first 2 days after transfer, but these quickly declined to be barely detectable 1month later in most patients. An inhibitory factor developed in the serum of three of six patients tested over the period of treatment, which significantly reduced the ability of gene-modified T cells to respond against FR+ tumor cells.
Conclusions
Large numbers of gene-modified tumor-reactive T cells can be safely given to patients, but these cells do not persist in large numbers long term. Future studies need to employ strategies to extend T cell persistence. This report is the first to document the use of genetically redirected T cells for the treatment of ovarian cancer.
There is increasing interest in the use of immunotherapy for the treatment of malignant disease, and some dramatic clinical responses have led to intense activity in this field. In particular, the success of adoptive immunotherapy as a treatment for melanoma has prompted us to extend this therapy to ovarian cancer. Variables important in the application of this therapy have been identified in melanoma patients, including the requirement for tumor antigen-reactive lymphocytes, high-dose interleukin-2 (IL-2; ref. 1), and more recently the benefits of prior lymphoablation (2).
However, endogenous tumor-reactive cells cannot be reproducibly found in ovarian patients. Nevertheless, several tumor-associated antigens have been identified for ovarian tumors, including Her-2 (3), tumor-associated glycoprotein 72 (4), Lewis-Y (5), and α-folate receptor (FR; ref. 6), and monoclonal antibodies exist recognizing these antigens. Recombinant genes encoding chimeric receptors incorporating antibody specificity can be used to genetically modify T cells to endow them with activity against tumor cells (7–17).
We have previously reported the generation of FR-specific T cells by modification of T cells with a gene encoding a cell surface chimeric receptor linking single-chain (scFv) anti-FR to the transmembrane and cytoplasmic domains of the Fc receptor γ chain. The scFv was derived from the MOv18 monoclonal antibody (18), and the chimeric gene is referred to as MOv-γ. We showed that this gene could endow ex vivo transduced T cells with the ability to respond against FR+ tumor cells Clin Cancer Res 2006;12(20) October 15, 2006 in vitro (19). In addition, adoptive transfer of anti-FR mouse T cells could inhibit tumor growth in lung metastases and i.p. models of disease in mice (20).
More recently, we have generated FR-reactive cells from populations of T cells with endogenous specificity for allogeneic antigen. We showed that these T cells could respond to both tumor and allogeneic antigen and referred to these T cells as dual specific (21). The rationale behind the generation of dual-specific T cells was to provide a population of FR-reactive T cells that could expand in vivo in response to allogeneic immunization, which is not possible in response to FR alone. Indeed, adoptive transfer of mouse dual-specific T cells into mice followed by allogeneic immunization resulted in expansion of transferred cells and enhanced inhibition of s.c. tumor growth without the need for administration of IL-2 (21).
Based on these encouraging results in mice, we initiated a two-cohort phase I clinical study in ovarian cancer patients. Cohort 1 patients were treated with adoptive transfer of bulk peripheral blood–derived T cells gene-modified with the anti-FR chimeric receptor in combination with high-dose IL-2. Cohort 2 involved the generation of dual-specific T cells from autologous peripheral blood mononuclear cells (PBMC) and their transfer into patients followed by s.c. immunization with allogeneic PBMCs.
Materials and Methods
Treatment regimen
Patients received adoptive transfer of autologous T cells gene-modified to express a chimeric receptor specific for the tumor-associated antigen FR. The study was divided into two cohorts, with cohort 1 receiving T cells and high-dose IL-2 and cohort 2 receiving dual-specific T cells and s.c. immunization with allogeneic PBMCs but no IL-2.
Eight patients were enrolled in cohort 1, each receiving up to three cycles of treatment, with each cycle consisting of administration of gene-modified T cells and IL-2 (720,000 IU/kg body weight). Approximately 4 weeks elapsed between the start of each cycle. Following activation, transduction, and G418 selection, T cells were expanded in culture, harvested, washed, and resuspended in 100 mL of saline and given to patients by i.v. drip over 20 to 30 minutes. The first five patients received a dose escalation regimen beginning at 3 × 109 transduced T cells. If no grade 3 or 4 toxicity was observed, not easily rectified within 24 hours, the patient was eligible to proceed to the next dose level of 1 × 1010 T cells at the start of the next cycle and subsequently to the highest test dose level of 3 × 1010 to 5 × 1010 cells at the start of the third cycle. IL-2 was given i.v. on the day of T cell transfer and every 12 hours for up to six doses if tolerated.
Six patients were enrolled in cohort 2, each receiving up to two cycles of treatment, with each cycle consisting of adoptive transfer of gene-modified dual-specific T cells followed by immunization with allogeneic PBMCs. Eight to 12 weeks elapsed between the start of each cycle. Following two in vitro allogeneic stimulations and expansion in culture, T cells were given to patients as in cohort 1. Allogeneic immunization consisted of s.c. injection of ~2.0 × 109 to 4.0 × 109 allogeneic PBMCs (viable and nonirradiated) from the same donor used to stimulate T cells during their generation in vitro. Each dose of allogeneic PBMCs was split into four equal parts and injected s.c. into separate sites on the lower extremities in 1 mL saline per site. Immunization was done 1 day after T cell transfer and again 1 week later because multiple allogeneic immunizations had shown better effect in mouse studies (21).
Patient treatment and monitoring procedures were reviewed by the Institutional Review Board of the Center for Cancer Research, National Cancer Institute, and informed consent was obtained from all patients before treatment.
Patient eligibility
Patients had biopsy-proven recurrent, resected recurrent, or residual epithelial FR+ ovarian cancer that failed standard effective therapy, including cisplatin/carboplatin – or paclitaxel-containing regimens. Patients ranged in age from 33 to 60 years and had clinical Eastern Cooperative Oncology Group performance status of 0 or 1. Eligibility criteria required serum creatinine levels ≤1.6 mg/dL and bilirubin <2.0 mg/dL. Blood eligibility criteria included hemoglobin >9.0 g/dL, WBC >3,000/mm3, and platelets >100,000/mm3 and an intact immune system as evidenced by a positive reaction to Candida albicans skin test, mumps skin test, or tetanus toxoid skin test on a standard anergy panel.
Response assessment
Patients received radiologic evaluation by magnetic resonance imaging, computed tomography, or sonography immediately before treatment and at completion of therapy. Disease response was determined by comparison of pretreatment and post-treatment images. In addition, serum CA-125 levels were determined following treatment and compared with pretreatment CA-125 levels.
T cell generation
A detailed description of the generation and characterization of T cells used in cohort 1 of the study has been published previously (22). Briefly, patient PBMCs derived from leukapheresis were stimulated with anti-CD3 (OKT3, Ortho Biotech, Raritan, NJ) and human recombinant IL-2 (600 IU/mL; Chiron, Emeryville, CA). After 3 days of culture, ~5 × 107 to 1 × 108 lymphocytes were taken and transduced with retroviral vector supernatant (Cell Genesys, San Francisco, CA) encoding the chimeric MOv-γ gene and subsequently selected for gene integration by culture in G418.
For the generation of dual-specific T cells used in cohort 2, stimulation of T cells was achieved by coculture of patient PBMCs with irradiated (5,000 cGy) allogeneic donor PBMCs from cryopre-served apheresis product (mixed lymphocyte reaction). The MHC haplotype of allogeneic donors was determined before use, and donors that differed in at least four MHC class I alleles from the patient were used. Culture medium consisted of AimV medium (Invitrogen, Carlsbad, CA) supplemented with 5% human AB− serum (Valley Biomedical, Winchester, VA), penicillin (50 units/mL), streptomycin (50 mg/mL; Bio Whittaker, Walkersville, MD), amphotericin B (Fungizone, 1.25 mg/mL; Biofluids, Rockville, MD), L-glutamine (2 mmol/L; Mediatech, Herndon, VA), and human recombinant IL-2 (Proleukin, 300 IU/mL; Chiron). Mixed lymphocyte reaction consisted of 2 × 106 patient PBMCs and 1 × 107 allogeneic stimulator PBMCs in 2 mL AimV per well in 24-well plates. Between 24 and 48 wells were cultured per patient for 3 days, at which time transduction was done by aspirating 1.5 mL of medium and replacing with 2.0 mL retroviral supernatant containing 300 IU/mL IL-2, 10 mmol/L HEPES, and 8 μg/mL polybrene (Sigma, St. Louis, MO) followed by covering with plastic wrap and centrifugation at 1,000 × g for 1 hour at room temperature. After overnight culture at 37°C/5% CO2, transduction was repeated on the following day, and then medium was replaced after another 24 hours. Cells were then resuspended at 1 × 106/mL in fresh medium containing 0.5 mg/mL G418 (Invitrogen) in 175-cm2 flasks for 5 days before resuspension in media lacking G418.
Cells were expanded to 2 × 109 and then restimulated with allogeneic PBMCs from the same donor to enrich for T cells specific for the donor allogeneic haplotype. Restimulation was done by incubating patient T cells (1 × 106/mL) and stimulator PBMCs (2 × 106/mL) in 3-liter Fenwall culture bags in AimV + additives and IL-2 (no G418). Cell numbers were adjusted to 1 × 106/mL, and IL-2 was added every 2 days, until sufficient numbers for treatment were achieved.
Cell lines, flow cytometry, and IFN-γ secretion assay
Tumor cell lines used in assays of T cell function were the FR+ human ovarian cancer cell line IGROV-1 (23) and FR− melanoma cell lines Mel 526, Mel 624, Mel 888, and Mel 1866 (Surgery Branch, National Cancer Institute, Bethesda, MD). Tumor cells were maintained in RPMI supplemented with 10% FCS (Invitrogen), penicillin (50 units/mL), streptomycin (50 mg/mL), amphotericin B (1.25 mg/mL), and L-glutamine (2 mmol/L). A melanoma-specific T cell line used in some experiments was derived from tumor-infiltrating lymphocytes of a patient at National Cancer Institute and maintained in T cell culture medium described above.
Expression of chimeric MOv-γ receptor by transduced T cells was determined using flow cytometry following staining with phycoerythrin-conjugated Id18.1, a monoclonal antibody specific for the MOv-18 idiotype (24). T cells were stained with phycoerythrin-conjugated mouse IgG1 as a control for nonspecific binding.
Transduced T cells were assessed for their ability to respond against the FR antigen by coculture with IGROV-1 ovarian cancer cells. T cells (1 × 105) were incubated with IGROV-1 cells (1 × 105; or FR-negative control tumor cells) in triplicate wells of 96-well plates. After overnight culture, supernatant was taken and assayed for IFN-γ using ELISA kits according to manufacturer’s instructions (Endogen, Woburn, MA). T cells in cohort 2 of the study were also assessed for their ability to secrete IFN-γ in response to allogeneic stimulator PBMCs freshly thawed from cryopreserved stocks. Anti-human CD3 (OKT3) was also used to stimulate T cells to gauge their maximal capacity to respond to TCR-CD3 engagement. OKT3 was immobilized on 96-well plastic plates at 0.5 μg/well in 100 μL PBS overnight at 4°C.
In some experiments, a 25% proportion of patient serum was included in T cell cocultures to determine possible effects of patient serum on T cell function. In some assays, protein G (Amersham Biosciences, Piscataway, NJ; 20 μL/mL) was added to serum before coculture and incubated for 1 hour at 4°C with gentle rocking to deplete patient serum of immunoglobulin.
Serum FR titer assay
A double-determinant assay was done essentially as described (25) using MOv19, a non–cross-reacting antibody directed against FR (18), as catcher. Briefly, 96-well flat-bottomed maxisorp plates (Nunc, Roskilde, Denmark) were coated with 200 μL of MOv19, at 1 μg/mL in PBS, and incubated overnight at 4°C. Plates were washed and blocked for 1 hour with 200 μL/well of 0.5% bovine serum albumin in PBS; 100 μL of sample was added to wells. A positive control consisted of tissue culture supernatant from IGROV-1 cells. Plates were incubated 2 hours and washed, and 100 μL of biotinylated MOv18 (0.25 μg/mL) were added followed by incubation at room temperature for 2 hours. Plates were washed, and streptavidin-horseradish peroxidase in PBS/0.5% bovine serum albumin was added 100 μL/well and incubated for 0.5 hour. Plates were washed, and 100 μL/well trimethylbenzidine was added and incubated for 5 to 10 minutes, and reaction was stopped with 1 mol/L H2SO4. Plates were read on spectrophotometer 450 nm within 0.5 hour of stopping reaction. Concentrations of FR in patient sera were expressed as dilution until absorbance reached background levels of media alone.
T cell tracking
A fraction of transduced T cell cultures (17–50%, 1.5 × 109 to 7.5 × 109 cells) were radiolabeled with 111In-oxine as described previously (26). Briefly, this involved incubation with 750 μCi 111In-oxine per 1010 cells in 30 to 50 mL PBS for 15 minutes with gentle rocking. Labeled cells were then washed and resuspended in 100 mL of saline containing 5% human serum albumin and 75,000 IU IL-2 for i.v. infusion into patients over a period of 10 to 20 minutes. Gamma camera images were obtained at intervals for up to 5 days where practicable.
T cell persistence: PCR-ELISA
Patient peripheral blood was analyzed for persistence of transduced T cells by detection of the neomycin phosphotransferase (neo) gene using a PCR-ELISA DIG Detection kit (Roche, Basel, Switzerland), as per the manufacturer’s directions using 50 pmol forward neo primer (ATTGAACAAGATGGATTGCACGCAG), 50 pmol reverse neo primer (TCAGAAGAACTCGTCAAGAAGGCG), 0.25 unit Taq DNA polymerase (Promega, Madison, WI) and 50% by volume of patient PBMC lysate. A series of lysed samples of Jurkat-22 neo cells (containing 1 copy of neo per cell) was prepared as standards, consisting of 1% of cells and decreasing in multiples of 2 down to 0.016%. PCR cycling consisted of 96°C for 6 minutes followed by 35 cycles of 95°C for 1 minute, 57°C for 1 minute, and 72°C for 2 minutes. The ELISA was done using 7.5 μL of the PCR product and 15 pmol/mL probe (Biotin-AGCAAGGTGAGATGACAGGAGAT), with hybridization done at 48°C.
Results
Patient characteristics
All patients had been diagnosed with metastatic ovarian cancer. Sites of metastases varied between patients but involved peritoneal disease with lymph node involvement (Table 1). Previous treatments received by patients before enrollment in the study varied but included surgical removal of primary lesion, debulking, and chemotherapy (Table 1). Previous therapies ceased at least 2 weeks before receiving gene-engineered T cells.
Table 1.
Sites of disease and treatment history
| Patient | Prior treatment | Metastatic disease status on enrollment in study |
|---|---|---|
| 1 | Hysterectomy, BSO, debulked, Taxol, Carboplatin, Cisplatin, bone marrow transplant, etoposide | Lower abdominal s.c. mass, two inguinal nodules |
| 2 | THA/BSO, omentectomy, appendectomy, nodectomy, Taxol, Cisplatin, Topotecan, Hexamethylmelamine | Retroperitoneal and left cervical lymph nodes |
| 3 | Radical hysterectomy, debulked, Carboplatin, Cytoxan, Adriamycin, Mitoxantrane, Tamoxifen, etoposide, radiation | Liver and vaginal cuff |
| 4 | TAH/BSO Taxol, Carboplatin, Doxil, Topotecan, Gemzar | Perihepatic lesion, midabdominal s.c. nodule |
| 5 | TAH/BSO, Carboplatin, Cytoxan, Taxol, Topotecan | Perihepatic, ascites, sigmoid mass, omental disease |
| 6 | Debulked, Carboplatin, Taxol, Doxil | Pelvic mass, para-aortic adenopathy |
| 7 | TAH/BSO, debulked, Carboplatin, Taxol | Multiple sites periaortic retroperitoneal adenopathy |
| 8 | TAH/BSO, debulked, Cisplatin, Cytoxan, Carboplatin, Taxol, Topotecan, Doxel | Liver, pericolonic, pelvic lymph node |
| 9 | TAH/BSO, debulked, Carboplatin, Taxol, Cytoxan | Liver and rectal muscle mets |
| 10 | TAH/BSO, debulked, Carboplatin, Taxol, Cisplatin, Taxol, monoclonal vaccine, Tamoxifen | Omentum, peritoneal implants, diaphragm, right supraclavicular nodes, pelvis |
| 11 | TAH/BSO, omentectomy, appendectomy, pancreatic reduction, splenectomy, Cytoxan, Cisplatin, vincristine, Hexalen, etoposide, Taxol, Carboplatin, Adriamycin, Topotecan, Gemzar | Peritoneal implants, left pleural effusion, liver, retroperitoneal nodes |
| 12 | TAH/BSO, omentectomy, Taxol, Carboplatin, bone marrow transplant, Taxane, Doxil, Herceptin, Gemcitabine, Topotecan | Pelvic and mediastinal mets |
| 13 | Ovarian cystectomy, hysterectomy, pelvic lymphadenectomy, Taxotere, Carboplatin, Topotecan, Doxyl, Gemzar | Omentum, mediastinum |
| 14 | TAH/BSO, omentectomy, pelvic and para-aortic lymphadenectomy, Taxol, Cisplatin, external beam radiation, Topotecan, Thalidomide, etoposide, Hexalen | Epigastric intra-abdominal mass, right pelvic mass |
NOTE: Patients had advanced ovarian cancer with metastases to various sites. Before enrolling in the current study, total abdominal hysterectomy and bilateral salpingo-oophorectomy were done, and most patients had undergone debulking surgery (patients 1–8 enrolled in cohort 1 and patients 9–14 in cohort 2).
Abbreviations: TAH, total abdominal hysterectomy; BSO, bilateral salpingo-oophorectomy.
Characterization of gene-modified T cells used in cohort 1
A complete characterization of T cells used in cohort 1 has already been described previously (22), but briefly, T cell cultures were stimulated with anti-CD3 antibody and were shown to expand from 11,000- to 3,000,000-fold. The mean time of culture of T cells from patients in cohort 1 was 47 days (range, 25–56 days). The T cells secreted IFN-γ specifically in response to FR and could lyse FR+ tumor cells. Phenotypically, the bulk lymphocyte population was composed of both CD4+ and CD8+ T cells and was shown to consist of a diverse range of clones able to secrete a variety of cytokines, including IFN-γ, IL-10, granulocyte macrophage colony-stimulating factor, and IL-2, in response to FR. Percentages of CD4+CD8− T cells varied between 1% and 39%, and CD4− CD8+ T cell percentages ranged from 47% to 94%.
Expansion of T cells used in cohort 2
In this cohort of the study, T cells received an initial stimulation with allogeneic stimulator cells and transduction with retroviral vector for a culture period of between 21 and 38 days. During this stimulation period, T cell expansion from patients varied from 12- to 325-fold. To further expand T cell numbers and enrich for allo-specific T cells, a second stimulation with PBMCs from the original PBMC donor was done. This restimulation resulted in a further expansion in T cells of ~50-fold. A representative growth curve for T cells over two stimulations is presented in Fig. 1A. The mean time of T cell culture for cohort 2 was 40.5 days (range, 37–48 days).
Fig. 1.
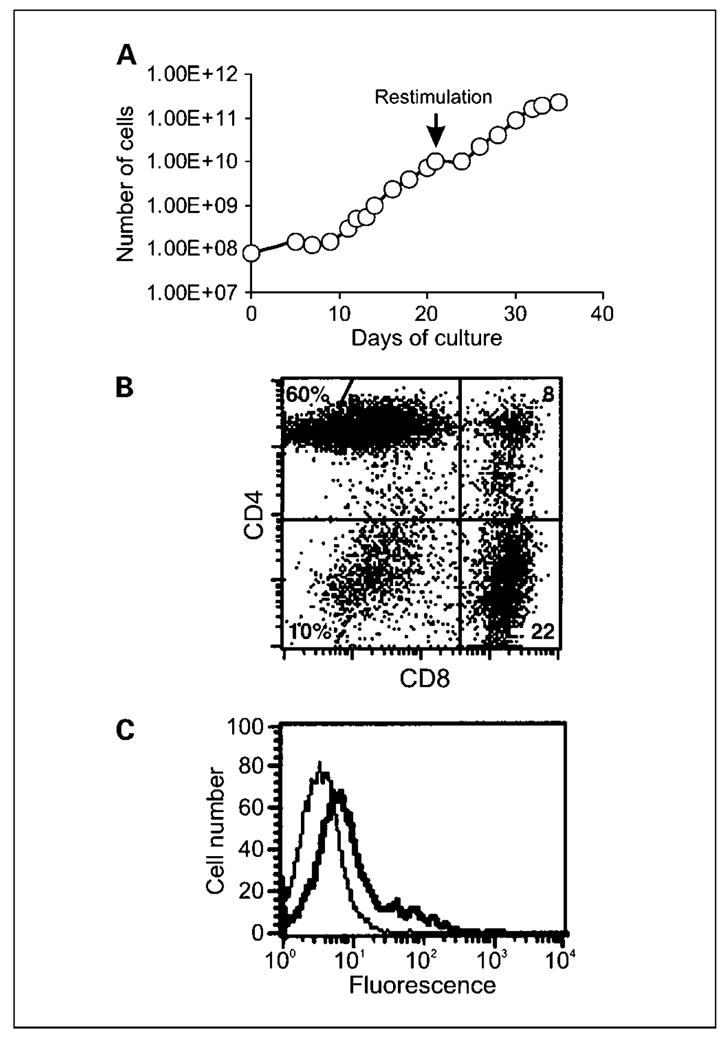
Growth and phenotype of gene-modified T cells. A, following allogeneic stimulation, 8 × 107 Tcells were transduced with retroviral vector encoding the MOv-γ receptor and maintained at 1to 2 × 106/mL in media containing IL-2. Transduced T cells were restimulated with allogeneic PBMCs on day 21, which resulted in further expansion of Tcells. Using this method, large numbers of dual-specificTcells could be generated. T cell expansion depicted is for patient 9. Representative of all six patients in cohort 2.The phenotype of transduced Tcells from cohort 2 of the study was determined with respect to T cell subset markers and chimeric receptor expression using specific antibodies and flow cytometry. B, although the relative proportions of CD4+ and CD8+ T cells varied between patients, the culture was made up predominantly of CD4+ and CD8+ cells as seen in the representative plot. C, expression of the chimeric MOv-γ receptor was evident following staining with anti-idiotype antibody (thick line) compared with isotype control antibody (thin line). Representative of all patients.
To promote high levels of allo stimulation, stimulator PBMCs were HLA typed at MHC class I loci to check for allelic differences to patient HLA type. Patient HLA type was determined to be largely dissimilar to stimulator HLA, with differences in at least four of six alleles.
Phenotype of T cells in cohort 2
T cells are characterized into two major phenotypic subsets, either CD4+ or CD8+, which have different fundamental abilities of helper function or cytotoxic function, respectively. Because this could affect on the overall function of bulk T cell populations and interpretation of clinical results, the relative proportions of CD4+ and CD8+ T cells were determined for each culture. Following two stimulations with allogeneic PBMCs, expression of CD4 and CD8 T cell markers was determined using specific monoclonal antibodies and flow cytometry. Percentages of CD4+CD8− T cells varied between 2% and 82%, and CD4− CD8+ T cell percentages ranged from 13% to 85%. CD4−CD8− cells were present in all cultures but only as a minor population (2–15%; Fig. 1B).
All T cells were transduced, as indicated by G418 resistance, and expressed the chimeric MOv-γ receptor, as determined, in flow cytometry, by an increase in fluorescence staining in presence of Id 18.1, an anti-MOv18 idiotype monoclonal antibody compared with staining in presence of isotype control antibody (Fig. 1C). Although expressed at low level, the chimeric receptor endowed T cells with the ability to respond specifically against FR+ target cells (see next section).
IFN-γ secretion by T cells in response to tumor cells and allogeneic stimulator PBMC
T cell cultures from patients in cohort 1 were shown to secrete IFN-γ specifically in response to FR, and this has been previously reported (22). IFN-γ levels in response to FR+ IGROV-1 cells varied from 1,749 to 28,560 pg/mL. With respect to patients in cohort 2, an important requirement of dual-specific T cells was their ability to respond to both FR and allogeneic stimulator PBMCs that were to be used as immunogen following T cell transfer. IFN-γ secretion following coculture of T cells with tumor or allogeneic PBMC was used as an indicator of T cell response. Although there was some variation between patients in IFN-γ levels in response to FR+ IGROV-1 cells (1,295–9,050 pg/mL; Table 2), secretion was always greater than that in response to FR− melanoma cells (14–54 pg/mL), thereby showing that transduced T cells could respond specifically against FR. The specificity of the response was also supported by the observed lack of IFN-γ secretion upon coculture of nontransduced T cells with IGROV-1 cells (data not shown).
Table 2.
Anti-FR and anti-allo responses of transduced T cells from patients in cohort 1 and 2 of the study
| Cohort 2
|
Cohort 1, median | |||||||
|---|---|---|---|---|---|---|---|---|
| Pt. 9 | Pt. 10 | Pt. 11 | Pt. 12 | Pt. 13 | Pt. 14 | Median | ||
| Media alone | 36 | 60 | 34 | 42 | 0 | 19 | 35 | 153 |
| Melanoma (FR−) | 35 | 54 | 28 | 26 | 14 | 27 | 28 | 139 |
| IGROV-1 (FR+) | 1,375 | 2,960 | 8,010 | 1295 | 1,340 | 9,050 | 2,168 | 6,501 |
| Allogeneic stimulator PBMCs | 2,270 | 1,555 | 2,995 | 5625 | >4,320 | 603 | 2,633 | |
| Autologous PBMCs | 210 | 456 | 36 | 140 | 39 | 298 | 175 | |
| OKT3 | 5,784 | 13,317 | 9,620 | 2890 | >3,760 | >59,000 | 7,702 | 7,457 |
NOTE: T cell reactivity towards the FR tumor antigen and allogeneic stimulator PBMCs was determined by assaying IFN-γ secretion (pg/mL) using ELISA following overnight incubation of Tcells with the targets listed. Plastic-coated anti-CD3 (OKT3) was used as an indicator of maximal T cell response. Transduced T cells from all patients were reactive with FR, and T cells from patients in cohort 2 were reactive with allogeneic PBMCs. Nontransduced Tcells did not respond against IGROV-1, except for patient 14 in whom 548 pg/mL IFN-γ was secreted (data not shown).
Transduced T cells from all patients in cohort 2 also responded against their specific allogeneic stimulator PBMCs, although here again the level of IFN-γ secreted varied between patients (603–5,625 pg/mL; Table 2). No correlation was observed between response against FR and allo PBMCs, with the anti-FR response sometimes greater than, and sometimes less than, the allo response. However, OKT3-induced IFN-γ secretion was always greater than that induced by IGROV-1.
Patient treatment details
Because higher patient response rates have been observed in adoptive immunotherapy of melanoma when high-dose IL-2 is included in the treatment regimen, we included administration of this T cell growth factor in this cohort of the phase I study. Patients without evidence of ischemic heart disease or a history of congestive heart failure or cardiac arrhythmias or other debilitating symptoms received i.v. IL-2 at 720,000 IU/kg every 12 hours up to six doses if adequately tolerated.
The first four patients in cohort 1 received an escalating dose of T cells over three cycles beginning at 3 × 109 before progressing in the next cycle (if tolerated) to 1 × 1010 and ultimately to 3 × 1010 to 5 × 1010. There was ~4 weeks between doses. Having shown no grade 3 or 4 toxicity not easily reversible in the first four patients, the remaining patients in this cohort were eligible to receive doses of T cells at the higher levels.
A variety of symptoms indicative of treatment-related toxicity were observed with grade 1 and 2 fatigue and nausea common. In addition, some patients experienced grade 3 and 4 toxicities often including hypotension and dyspnea (Table 3). Other grade 3 and 4 toxicities less frequently encountered included fatigue, leukopenia, rigors, sinus tachycardia, and diarrhea (Table 3). It is likely that these toxicities were due to the IL-2 component of the treatment regimen because these symptoms have been observed previously in melanoma patients receiving high-dose IL-2.
Table 3.
Summary of treatment regimen and toxicity for patients receiving gene-modified T cells
| Patient | Cycle no. | No. T cells ×10− 9 | No. IL-2 injections | Grades of adverse events | Grade 1 and 2 toxicity events | Grade 3 and 4 toxicity events |
|---|---|---|---|---|---|---|
| 1 | 1 | 3.0 | 6 | 1, 2, 3, 4 | HB, FAT, NAU, PU, PE | LEU, HYP, PCD |
| 2 | 3.0* | 5 | 2, 3 | ED | RIG | |
| 3 | 10.0 | 3 | 2, 3 | ED, LEU, PCD, PU, PE | HB, HYP | |
| 2 | 1 | 3.0† | 5 | 1, 2 | PCD, FAT, NAU, BIL, PU | |
| 2 | 9.0 | 4 | ||||
| 3 | 47.0† | 4 | ||||
| 3 | 1 | 3.0 | 5 | 3 | DIA, FAT | |
| 2 | 10.0 | 3 | 2 | LUO | ||
| 3 | 17.5 | 1 | 3 | HYP, STC | ||
| 4 | 1 | 3.0 | 5 | |||
| 2 | 11.4† | 1 | 3 | DYS | ||
| 3 | 21.9† | 0 | ||||
| 5 | 1 | 3.0 | 3 | 2 | ASC | |
| 6 | 1 | 28.57† | 6 | 2 | FAT, DIA | |
| 2 | 11.0 | 6 | 2 | FAT | ||
| 7 | 1 | 22.0† | 0 | 2, 4 | RIG, VOM | DYS‡ |
| 8 | 1 | 44.0† | 2 | 3 | HYP | |
| 2 | 43.5 | 1 | 2 | DYS | ||
| Cycle no. | No. T cells × 10− 9 | No. allo Immunizations§ | No. allo cells × 10− 9 | Grades of adverse events | Grade 1 and 2 toxicity events | |
|
| ||||||
| 9 | 1 | 46.5 | 2 | 4.55 | 1, 2 | ISR, URT, DYS |
| 2 | 169.0 | 2 | 6.5 | None | ||
| 10 | 1 | 13.17† | 2 | 4.5 | 1, 2 | RIG, ISR, NAU |
| 2 | 50.0 | 2 | 7.0 | None | ||
| 11 | 1 | 4.0 | 2 | 7.8 | 2 | ISR |
| 12 | 1 | 11.7 | 2 | 7.32 | 1 | ISR |
| 13 | 1 | 36.7 | 2 | 6.72 | None | |
| 14 | 1 | 9.0 | 2 | 7.45 | 1, 2 | HYP, RIG, FAT, ISR, NAU, VOM |
Abbreviations: ASC, ascites; HB, hemoglobin; PE, pleural effusion; BIL, bilirubin increased; HYP, hypotension; PU, pulmonary; DIA, diarrhea; LEU, leukopenia; RIG, rigors; DYS, dyspnea; LUO, low urine output; STC, sinus tachycardia; ED, Edema; NAU, nausea; URT, urticaria; FAT, fatigue; PCD, platelet count decreased; VOM, vomiting; ISR, injection site reaction (allogeneic immunization).
No progression to higher dose in this cycle due to grade 4 toxicity in previous cycle.
Some of these cells were labeled with 111In for trafficking.
Off protocol after one cycle due to dyspnea concerns.
Dose divided into two to four injections given on day 1 and 8 following MOv-γ T cells.
A total of six patients were enrolled in cohort 2 of the study, two of whom received two cycles of therapy. Doses of T cells differed between patients, varying between 4.0 × 109 and 169.0 × 109, with the dose given being determined by cell yield from individual cultures after two allogeneic stimulations (Table 3). Numbers of allogeneic PBMCs used for immunization also varied (although not greatly), with doses of 4.5 × 109 to 7.8 × 109 given per cycle. Toxicities associated with cell transfer or allogeneic immunizations were relatively mild, only reaching grade 1 or 2. Skin reaction at the site of allogeneic immunization, consisting of erythema, was the most common side effect, although nausea was also experienced by two patients (Table 3). The low-grade toxicities observed in this cohort that received cells but no IL-2 further supports the likelihood that grade 3 and 4 toxicities observed in patients in cohort 1 were due to IL-2 administration.
Disease status was followed by a computed tomography scan done after each cycle of therapy. In addition, serum CA-125 levels were determined following therapy. No reduction in the size of any tumor was observed, and disease progressed in all patients. In addition, there was no significant reduction in CA-125 levels (data not shown).
Tracking gene-modified T cells
T cell trafficking to tumor is an important requirement for antitumor activity of T cells and has been shown to correlate with response in mice and humans (27). To determine if anti-FR–transduced T cells trafficked to sites of ovarian cancer metastases, a proportion of T cells in cohort 1 were labeled with 111In before adoptive transfer, and imaging was done at intervals for up to 5 days later. Three patients received radiolabeled T cells during a single cycle of therapy, and another two patients received radiolabeled cells during two cycles (Table 3). Radiolabeled T cells accumulated initially in lung and subsequently in liver and spleen (Fig. 2A). Interestingly, T cells persisted longer in the lungs of patients who received T cells that had been subjected to relatively prolonged culture and restimulation in vitro. Restimulation was sometimes done to generate sufficient cell numbers for treatment. Restimulation consisted of a second round of incubation with OKT3, IL-2, and irradiated allogeneic PBMCs (100:1, PBMC/T cells). Relative lung persistence was determined by comparing the ratio of lung/liver and/or spleen 111In signal intensity ranging from 1 to 5 days after transfer. Three of three restimulated T cell transfers persisted longer in lungs compared with four of four nonrestimulated T cell transfers (P2 = 0.03, Fisher’s exact test). A representative image following transfer of 111In-labeled restimulated T cells is depicted in Fig. 2B. Interestingly, increased persistence of restimulated T cells in lungs seemed to be associated with dyspnea (Table 3: patient 4, cycle 2 and patient 7, cycle 1). Reproducible, specific localization of radiolabeled T cells to ovarian tumors was not observed, although some accumulation of T cells was detected in a pelvic mass of patient 4 at 48 hours after cycle 2. (Fig. 2C).
Fig. 2.
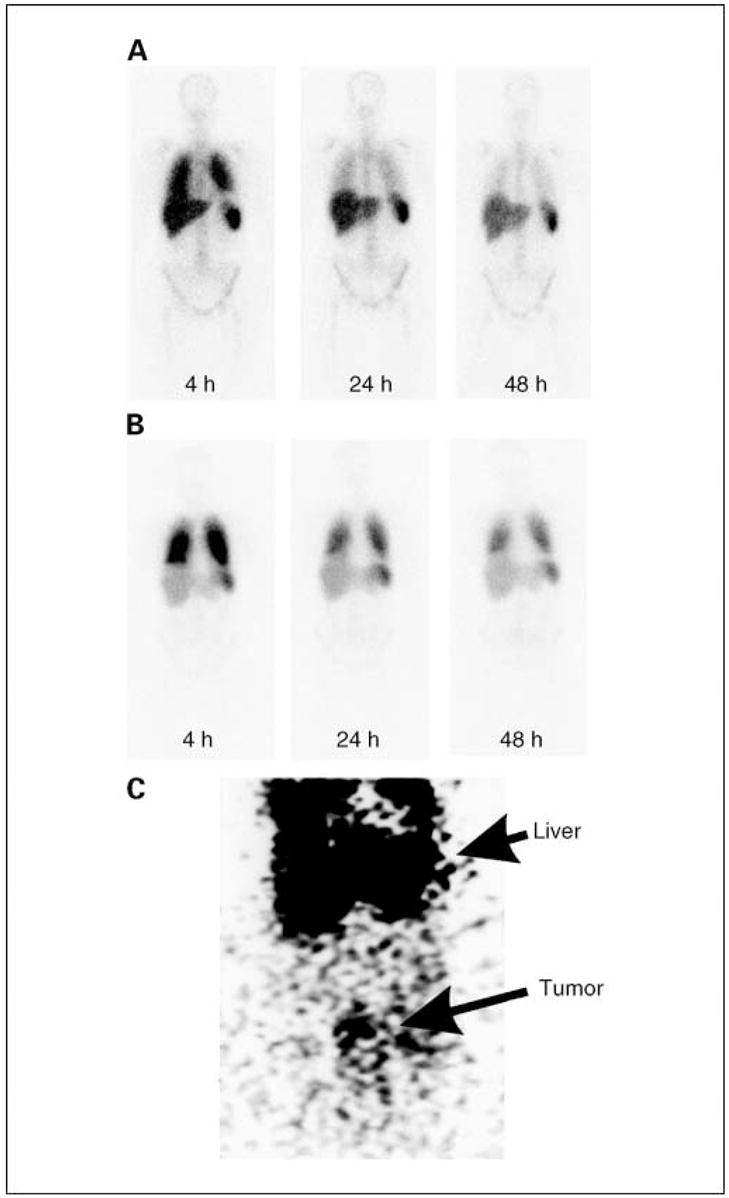
Biodistribution of radiolabeled T cells. Patients received up to 7.5 × 109 111In-labeledTcells, and imaging was done using a gamma camera at intervals followingTcell transfer. A, representative image of four transfers that were done withTcells that received a single stimulation with OKT3. B, representative image of three transfers that were done usingTcells that had received two stimulations with OKT3. Radioisotope signal was detected in lungs, liver, and spleen. Radiolabeled cells were preferentially retained in lungs of patients that received twice stimulated Tcells. C, anteroposterior image of the abdomen of patient 4 at 48 hours after receiving T cells in cycle 2 with evidence of T cell localization to a peritoneal tumor (bottom) in addition to localization to liver (top).
Persistence of gene-modified T cells following adoptive transfer
Expansion and persistence of specific T cells is important in an effective immune response against antigen, and details of this can provide insight into the response against antigen and the regulation of adoptively transferred T cell numbers. We, therefore, determined the persistence of gene-modified anti-FR T cells in the circulation of patients and expressed this as a percentage of total blood leukocytes. Adoptively transferred T cells were detectable in all patients at most times in the days immediately following cell transfer, often making up >1% of circulating lymphocytes (Table 4). However, the percentage of circulating transduced cells quickly declined and was absent or barely detectable after ~3 weeks, with the exception of patient 12, where a low percentage of transduced T cells were detected at 12 months after transfer.
Table 4.
Persistence of gene-modified T cells in blood of patients
| Cycle | Patient 1
|
Patient 2
|
Patient 3
|
Patient 4
|
Patient 5
|
Patient 6
|
Patient 7
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day/h | % | Day/h | % | Day/h | % | Day/h | % | Day/h | % | Day/h | % | Day/h | % | |
| 1 | (1 h) | 0.16 | (1 h) | 0.61 | (1 h) | 0.16 | (1 h) | 3.9 | (1 h) | 2.57 | (1 h) | 2.87 | ||
| (12 h) | 0 | (6 h) | 0 | (6 h) | 0.06 | (6 h) | 0.24 | 1 | 0 | 1 | 0.05 | |||
| 2 | 0 | 1 | 0 | 1 | 0 | (12 h) | 0 | 3 | 0.02 | 5 | 0.12 | |||
| ND | 5 | 0.13 | 3 | 0.22 | 3 | 1.47 | 2 | 0 | 4 | 2.39 | 27 | 0.09 | ||
| 14 | 0 | 6 | 0.5 | 5 | 0.47 | 3 | 0.3 | 5 | 2.97 | |||||
| 21 | 0 | 13 | 0 | 13 | 0 | 5 | 2.74 | |||||||
| 2 | (1 h) | 0.98 | (1 h) | 0 | (1 h) | 0.52 | (1 h) | 1 | ||||||
| 2 | 0 | (6 h) | 0.46 | 1 | 0 | (6 h) | 0.08 | |||||||
| ND | 5 | 0.1 | 1 | 0 | 5 | 4.24 | ND | 1 | 0 | ND | ||||
| 14 | 0 | 13 | 0.26 | 13 | 0.57 | 3 | 0 | |||||||
| 21 | 0.18 | 5 | 0 | |||||||||||
| 13 | 0 | |||||||||||||
| 30 | 0 | |||||||||||||
| 3 | (1 h) | 0 | 2 | 0.14 | (1 h) | 0.48 | (1 h) | 0.89 | ||||||
| (12 h) | 0.04 | 3 | 0.54 | 13 | 0.06 | (6 h) | 0.23 | |||||||
| 1 | 0.27 | 21 | 0.07 | 1 | 0.08 | ND | ND | ND | ||||||
| 2 | 0.8 | 3 | <0.03 | |||||||||||
| 5 | 0.31 | 5 | 0.05 | |||||||||||
| 14 | 0.05 | 13 | 0 | |||||||||||
| 21 | 0 | |||||||||||||
|
Patient 8
|
Patient 9
|
Patient 10
|
Patient 11
|
Patient 12
|
Patient 13
|
Patient 14
|
||||||||
| Day/h | % | Day/h | % | Day/h | % | Day/h | % | Day/h | % | Day/h | % | Day/h | % | |
|
| ||||||||||||||
| 1 | (1 h) | >1 | 1 | 3.72 | 1 | 1.86 | 1 | 6.17 | 1 | 1.52 | 1 | 2.92 | 1 | 2.75 |
| (6 h) | >1 | 2 | 3.66 | 2 | 2.33 | 6 | 0.61 | 2 | 0.78 | 2 | 2.9 | 2 | 2.54 | |
| 1 | >1 | 4 | 0.42 | 12 | 0.23 | 8 | 1.82 | 8 | 0.04 | 8 | 0.02 | 8 | 0.09 | |
| 2 | >1 | 7 | 0.6 | 26 | 0 | 13 | 1.44 | 14 | 0.01 | 9 | 0.06 | 15 | 0.06 | |
| 4 | 0.43 | 12 | 0.44 | 34 | 0 | 27 | 1.44 | 21 | 0.01 | 29 | 0.01 | 21 | 0.09 | |
| 13 | 0.04 | 18 | 0.16 | 55 | 0 | 39 | 1.36 | 26 | 0.06 | 39 | 0 | 28 | 0.07 | |
| 25 | 0 | 48 | 0.01 | 365 | 0.03 | 32 | 0.04 | |||||||
| 34 | 0.04 | 48 | 0.02 | |||||||||||
| 76 | 0 | |||||||||||||
| 2 | (1 h) | >1 | 1 | 2.88 | 1 | 2.03 | ||||||||
| (6 h) | 0.4 | 2 | 0.52 | 7 | 0.06 | |||||||||
| 1 | >1 | 3 | 0.04 | 27 | 0 | ND | ND | ND | ND | |||||
| 3 | 0.82 | 8 | 0 | |||||||||||
| 12 | 0 | 42 | 0 | |||||||||||
| 38 | 0 | |||||||||||||
NOTE: The quantity of neomycin phosphotransferase gene-positive T cells as a percentage of total PBMCs was determined by PCR-ELISA. Although persistence varied between patients, transferred T cells were generally present in large numbers up to day 5 following transfer but were absent or barely detectable by 2 to 3 weeks after transfer.
Abbreviation: ND, not done.
A proportion of patients develop a T cell inhibitory factor in serum
Two particular issues are of potential concern when redirecting T cell activity against an antigen in an non–MHC-restricted manner using a gene encoding a molecule with mouse components. The scFv component of the chimeric receptor was derived from the murine monoclonal antibody MOv18, and human anti-mouse antibody has been detected in a previous clinical study in the serum of patients receiving T cells retargeted with the bispecific F(ab′)2 OC/TR, antiCD3 × anti-FR, in which the anti-FR arm is derived from the MOv18 hybridoma (28). Despite the fact that the chimeric receptor only contained the variable regions of the mouse monoclonal antibody and was present as an integral membrane protein on T cells, it is still possible that a human anti-mouse antibody response occurred that inhibited redirected T cell function by blocking the interaction between chimeric receptor and FR antigen.
In addition, cell surface proteins, such as FR, can be shed from the surface of cells, and high levels of circulating free antigen may impair redirected T cell responses by binding to chimeric receptors, thereby blocking the interaction of T cells with FR on tumor cells. With these two issues in mind, we assessed posttreatment patient serum for the capacity to affect on T cell function against tumor cells in vitro.
Transduced T cells were incubated with FR+ IGROV-1 tumor cells in the presence or absence of serum taken before or after treatment. Following incubation, supernatants were analyzed for IFN-γ as a measure of T cell response against antigen. Sera from three of six patients posttreatment were found to inhibit IFN-γ production by T cells, whereas serum before treatment had no effect on IFN-γ levels compared with assays done in the absence of patient serum (Table 5). This suggested that an inhibitory factor had developed in the a proportion of patients over the course of treatment.
Table 5.
IFN-γ secretion by T cells in the presence of serum from patients
| Cohort 1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| T cells/target | No serum
|
Patient 2 serum
|
Patient 3 serum
|
Patient 4 serum
|
Patient 6 serum
|
|||||
| T cells alone | Anti-id | Pre | Post | Pre | Post | Pre | Post | Pre | Post | |
| MOv-γ T cells vs IGROV-1 | 1,260 | 0 | 1,100 | 0 | 1,000 | 110 | 550 | 780 | 1,200 | 1,460 |
| Melanoma T cells vs Mel624 | 2,615 | 3,615 | 2,900 | 3,440 | 3,080 | 2,600 | 3,635 | 2,910 | 3,665 | 4,080 |
| Cohort 2 | ||||||||||
|
| ||||||||||
| Target cell | Patient 9 T cells as effectors | Patient 10 T cells as effectors | ||||||||
|
|
|
|||||||||
| T cells alone | Anti-id | Pt. 9 pre serum | Pt. 9 post serum | T cells alone | Anti-id | Pt. 10 pre serum | Pt. 10 post serum | |||
|
| ||||||||||
| IGROV-1 | 2,590 | 740 | 3,340 | 390 | 1,170 | 108 | 1,825 | 1,550 | ||
| Allo PBMC | 388 | 3,360 | 828 | 1,250 | 199 | 356 | 291 | 49 | ||
NOTE: Cohort 1: MOv-γ-transduced T cells were incubated with FR+ IGROV-1 tumor cells and IFN-γ secretion (pg/mL) determined by ELISA. Inclusion of anti-idiotype antibody in the coculture inhibited redirected T cell activity as expected. Inclusion of posttreatment serum from two patients was also seen to inhibit IFN-γ secretion (bold underlined values). Inhibition of T cells was specific for FR because no decrease in IFN-γ secretion was observed using posttreatment serum with a melanoma-specific T cell line against its cognate target cell. Cohort 2: Posttreatment serum from one patient (Pt. 9) inhibited anti-FR IFN-γ secretion (bold underlined value). Inhibition was specific for FR because no decrease in reactivity to allogeneic stimulator PBMCs was observed.
To gain insight into the identity of the inhibitory factor we determined the titer of soluble FR in the serum of some patients after treatment (when inhibitory activity was present) and compared it to the level before treatment (when inhibitory activity was absent). The titer, as determined using ELISA, varied between patients with some being low and others high. In addition, FR levels were stable over the observation period in some patients but varied in another patient (Table 6). There was no direct correlation between FR levels and serum inhibitory activity.
Table 6.
Lack of correlation between serum FR titer and inhibitory factor in posttreatment serum
| Patient 2
|
Patient 9
|
Patient 10
|
||||
|---|---|---|---|---|---|---|
| FR titer | IFN-γ response | FR titer | IFN-γ response | FR titer | IFN-γ response | |
| Pretreatment | 3 | 1,100 | 50 | 3,340 | 1,000 | 1,825 |
| Posttreatment | 6 | 0 | 1,650 | 390 | 900 | 1,550 |
NOTE: Serum FR titers were determined using ELISA. FR levels varied between patients, with patient 2 having low amounts and patient 10 high amounts both before and after treatment. Patient 9 experienced an increase in FR levels over the course of treatment. There was no consistent correlation between FR levels and inhibition of IFN-γ response against IGROV-1 tumor cells.
To further investigate the identity of inhibitory activity, posttreatment serum from a patient with inhibitory activity was preincubated with protein G before inclusion in an IFN-γ response assay, reasoning that if inhibition was due to human anti-mouse antibody, then protein G should remove this. Consistent with previous experiments, IFN-γ secretion in response to IGROV-1 tumor cells was decreased by >60% in the presence of posttreatment serum. However, inhibition was totally removed by preclearance of serum using protein G, suggesting that immunoglobulin was responsible for the inhibitory activity (Fig. 3).
Fig. 3.
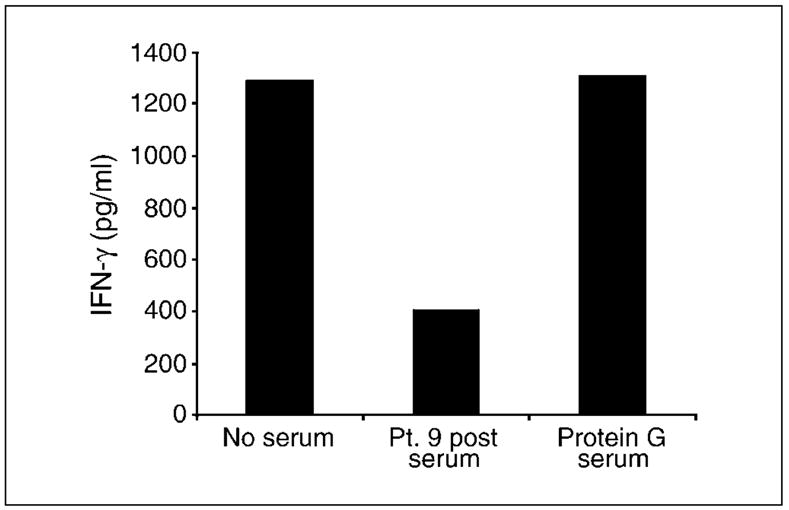
Preincubation of patient serum with protein G removed inhibitory activity. MOv-γ-transduced T cells were incubated overnight with IGROV-1tumor cells, and antitumor activity was determined by the amount of IFN-γ secreted. T cell activity was inhibited in the presence of serum taken from patient 9 afterTcell transfer. This inhibitory activity of posttreatment serum was removed by pretreatment of serum with protein G.
Discussion
The demonstrated effectiveness of adoptive immunotherapy regimens for melanoma prompted us to investigate the use of T cells against ovarian cancer. Due to the difficulty in isolating endogenous ovarian-reactive T cells from patients, we employed a genetic modification strategy to produce T cells reactive with the ovarian cancer-associated antigen FR.
FR-specific T cells could be produced consistently from all 14 patients in the study, and these T cells could be expanded to large numbers in vitro before adoptive transfer. However, restimulation of T cells was necessary for three cultures in cohort 1 and all cultures in cohort 2 to achieve large enough numbers for treatment. A phenotypic analysis of T cell cultures in cohort 2 revealed that CD4+/CD8+ ratios varied widely between patients. The reason for this was not clear but may be due to differing stimulatory capacities of MHC class I or II allogeneic HLA alleles from various donors.
A total of 14 patients in two cohorts of the study were treated with adoptively transferred FR-specific T cells, involving 26 infusions in all. In general, the treatment was well tolerated, although there were some grade 3 and 4 toxicities observed in cohort 1 of the study that were most likely due to the use of high-dose IL-2. Patients in cohort 2 received T cells without IL-2 and experienced relatively mild side effects. Therefore, acceptable safety was shown in answer to the central question of this phase I study.
However, there was no evidence of antitumor responses in any patient as assessed by computed tomography scan or serum CA-125 levels. Poor trafficking of T cells to tumor, as shown by the observed lack of tumor localization of radiolabeled cells, may be at least partially responsible for the absence of patient responses.
The reason for poor trafficking of T cells to tumor is not clear but has been observed in previous studies using adoptively transferred T cells against melanoma and could be due to the large size and/or “sticky” phenotype acquired by T cells in culture that results in their preferential retainment in lung, liver, and spleen. However, it should be noted that T cells could only be tracked for a relatively short time and with low resolution using 111In, and future studies using molecular imaging may enable longer-term studies of trafficking (29). In this study, T cells were delivered i.v., which results in initial passage through the lungs, where many cells lodge at least temporarily. An alternate delivery route, such as i.p. delivery, may reduce nonspecific retention of T cells in lung, leading to enhanced trafficking to tumor (30).
An alternate reason for the lack of patient responses may lie in the low persistence of transferred T cells in patients. Indeed, although transduced T cell numbers were initially high in the circulation in the days immediately following transfer, they quickly decreased by day 5 and did not persist or were barely detectable beyond 3 weeks. The reason for the poor persistence was not clear but could reflect an inability of the cultured T cells to adapt to in vivo conditions that requires expression of specific molecules important in homeostasis, trafficking, and growth factor responsiveness. Long-term culture has been previously shown to be detrimental to in vivo T cell function (31). Alternately, long-term culture may have produced “exhausted” cells proceeding toward an apoptotic program. Enhanced antitumor function may be possible using shorter-term cultured T cells in future studies of this form of therapy.
Cohort 2 of the study, involving dual-specific T cells, was initiated to try and induce expansion and increase persistence of adoptively transferred T cells as had previously been shown in a mouse cancer model using adoptive immunotherapy (21). However, despite the observed ability of transduced T cells to respond to both FR tumor antigen and allogeneic immunogen, no clear expansion or increase in persistence of transduced T cells was seen in patients in cohort 2 over those in cohort 1, although some persistence was observed in a single patient out to 1 year after treatment, albeit at very low levels (0.03% of PBMCs).
The failure of allogeneic immunization to affect T cell persistence may have been due to an insufficient dose of allogeneic PBMCs used as immunogen. Indeed, in the mouse model described previously, optimal expansion of adoptively transferred dual-specific T cells required s.c. immunization with 5 × 107 allogeneic splenocytes. Extrapolation of this to humans may require immunization with in excess of 1 × 1011 allogeneic cells, whereas doses of allogeneic cells between 4.5 × 109 and 7.5 × 109 were used in this study.
The current study has shown safety of s.c. administration of large numbers of allogeneic cells, and it would be of interest to try increasing doses of allogeneic PBMCs, or use alternate allogeneic stimulators, such as dendritic cells, that would be expected to possess enhanced stimulatory capacity that may enhance T cell persistence. It may also be worth considering nonmyeloablative conditioning before adoptive transfer of tumor-reactive T cells because this has been shown to result in increased persistence of tumor-specific T cells in the circulation of patients undergoing adoptive immunotherapy for melanoma (2). In addition, incorporating costimulatory domains into the transgene format may enhance T cell proliferation and persistence, as has been observed in previous studies in vitro (7, 32).
Another contributing factor to the lack of patient responses may be the development of T cell inhibitory activity in serum following treatment. This is a novel observation with respect to the use of adoptively transferred T cells, although serum inhibition of CD3-ζ expression in Jurkat cells has been shown from nonresponding ovarian patients receiving IL-2 (33). In our study, dramatic reduction in T cell responses to FR+ tumor cells was observed when posttreatment serum from some patients was included in in vitro assays. The identity of the inhibitory factor is unclear at present, although neutralization of inhibitory activity by protein G suggests that it could be a human anti-mouse antibody response to the scFv component of the chimeric receptor. However, it is unlikely that this factor alone was responsible for the lack of antitumor responses because inhibitory activity was found in serum of only 50% of patients tested. Nevertheless, it may be worthwhile using a humanized single-chain antibody, derived from the original MOv18 murine monoclonal, or an entire human scFv with the same binding specificity, raised by epitope imprinting as previously described (34) in future chimeric receptor design to reduce potential immunogenic epitopes.
In summary, this study is the first description of the use of adoptive transfer of gene-modified tumor-reactive T cells for the treatment of ovarian cancer. The use of gene-modified cells was shown to be safe in this setting. Although no patient responses were observed using this treatment regimen, important insight was gained into T cell trafficking, persistence, and function in vivo. Future clinical studies involving protocol modifications based on these insights may lead to improved treatment options for patients.
Acknowledgments
We thank My Do for technical assistance and Arnold Mixon and Shawn Farid for flow cytometric assays.
Grant support: The National Health and Medical Research Council (M. Kershaw), National Breast Cancer Foundation of Australia (M. Kershaw), Bob Parker Memorial Fund (J. Westwood), and Peter MacCallum Cancer Centre Foundation (J.Westwood).
References
- 1.Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233:1318–21. doi: 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- 2.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verri E, Guglielmini P, Puntoni M, et al. HER2/neu oncoprotein overexpression in epithelial ovarian cancer: evaluation of its prevalence and prognostic significance. Clinical study Oncology. 2005;68:154–61. doi: 10.1159/000086958. [DOI] [PubMed] [Google Scholar]
- 4.Thor A, Ohuchi N, Szpak CA, Johnston WW, Schlom J. Distribution of oncofetal antigen tumor-associated glycoprotein-72 defined by monoclonal antibody B723. Cancer Res. 1986;46:3118–24. [PubMed] [Google Scholar]
- 5.Yin BW, Finstad CL, Kitamura K, et al. Serological and immunochemical analysis of Lewis y (Ley) blood group antigen expression in epithelial ovarian cancer. Int J Cancer. 1996;65:406–12. doi: 10.1002/(SICI)1097-0215(19960208)65:4<406::AID-IJC2>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 6.Coney LR, Tomassetti A, Carayannopoulos L, et al. Cloning of a tumor-associated antigen: MOv18 and MOv19 antibodies recognize a folate-binding protein. Cancer Res. 1991;51:6125–32. [PubMed] [Google Scholar]
- 7.Haynes NM, Trapani JA, Teng MW, et al. Single-chain antigen recognition receptors that costimulate potent rejection of established experimental tumors. Blood. 2002;100:3155–63. doi: 10.1182/blood-2002-04-1041. [DOI] [PubMed] [Google Scholar]
- 8.Pinthus JH, Waks T, Malina V, et al. Adoptive immuno-therapy of prostate cancer bone lesions using redirected effector lymphocytes. J Clin Invest. 2004;114:1774–81. doi: 10.1172/JCI22284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel SD, Ge Y, Moskalenko M, McArthur JG. Anti-Tumor CC49-zeta CD4 T cells possess both cytolytic and helper functions. J Immunother. 2000;23:661–8. doi: 10.1097/00002371-200011000-00007. [DOI] [PubMed] [Google Scholar]
- 10.Gade TP, Hassen W, Santos E, et al. Targeted elimination of prostate cancer by genetically directed human T lymphocytes. Cancer Res. 2005;65:9080–8. doi: 10.1158/0008-5472.CAN-05-0436. [DOI] [PubMed] [Google Scholar]
- 11.Haynes NM, Trapani JA, Teng MW, et al. Rejection of syngeneic colon carcinoma by CTLs expressing single-chain antibody receptors codelivering CD28 costimulation. J Immunol. 2002;169:5780–6. doi: 10.4049/jimmunol.169.10.5780. [DOI] [PubMed] [Google Scholar]
- 12.Ho WY, Blattman JN, Dossett ML, Yee C, Greenberg PD. Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell. 2003;3:431–7. doi: 10.1016/s1535-6108(03)00113-2. [DOI] [PubMed] [Google Scholar]
- 13.Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64:9160–6. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- 14.Kershaw MH, Teng MW, Smyth MJ, Darcy PK. Supernatural T cells: genetic modification of T cells for cancer therapy. Nat Rev Immunol. 2005;5:928–40. doi: 10.1038/nri1729. [DOI] [PubMed] [Google Scholar]
- 15.Ma Q, Safar M, Holmes E, Wang Y, Boynton AL, Junghans RP. Anti-prostate specific membrane antigen designer T cells for prostate cancer therapy. Prostate. 2004;61:12–25. doi: 10.1002/pros.20073. [DOI] [PubMed] [Google Scholar]
- 16.Mansoor W, Gilham DE, Thistlethwaite FC, Hawkins RE. Engineering T cells for cancer therapy. Br J Cancer. 2005;93:1085–91. doi: 10.1038/sj.bjc.6602839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossig C, Brenner MK. Genetic modification of T lymphocytes for adoptive immunotherapy. Mol Ther. 2004;10:5–18. doi: 10.1016/j.ymthe.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 18.Miotti S, Canevari S, Menard S, et al. Characterization of human ovarian carcinoma-associated antigens defined by novel monoclonal antibodies with tumor-restricted specificity. Int J Cancer. 1987;39:297–303. doi: 10.1002/ijc.2910390306. [DOI] [PubMed] [Google Scholar]
- 19.Hwu P, Shafer GE, Treisman J, et al. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor γ chain. J Exp Med. 1993;178:361–6. doi: 10.1084/jem.178.1.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwu P, Yang JC, Cowherd R, et al. In vivo antitumor activity of Tcells redirected with chimeric antibody/T-cell receptor genes. Cancer Res. 1995;55:3369–73. [PubMed] [Google Scholar]
- 21.Kershaw MH, Westwood JA, Hwu P. Dual-specific T cells combine proliferation and antitumor activity. Nat Biotechnol. 2002;20:1221–27. doi: 10.1038/nbt756. [DOI] [PubMed] [Google Scholar]
- 22.Parker LL, Do MT, Westwood JA, et al. Expansion and characterization of T cells transduced with a chimeric receptor against ovarian cancer. Hum Gene Ther. 2000;11:2377–87. doi: 10.1089/104303400750038480. [DOI] [PubMed] [Google Scholar]
- 23.Benard J, Da Silva J, De Blois MC, et al. Characterization of a human ovarian adenocarcinoma line, IGROV1, in tissue culture and in nude mice. Cancer Res. 1985;45:4970–9. [PubMed] [Google Scholar]
- 24.Mantovani L, Menard S, Mezzanzanica D, Miotti S, Pupa SM, Colnaghi MI. Evaluation of the immunore-active fraction of an anti-tumour monoclonal antibody. Br J Cancer Suppl. 1990;10:15–7. [PMC free article] [PubMed] [Google Scholar]
- 25.Mantovani LT, Miotti S, Menard S, et al. Folatebinding protein distribution innormal tissues andbiological fluids from ovarian carcinoma patients as detected by the monoclonal antibodies MOv18 and MOv19. Eur J Cancer. 1994;30A:363–9. doi: 10.1016/0959-8049(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 26.Griffith KD, Read EJ, Carrasquillo JA, et al. In vivo distribution of adoptively transferred indium-111-labeled tumor infiltrating lymphocytes and peripheral blood lymphocytes in patients with metastatic melanoma. J Natl Cancer Inst. 1989;81:1709–17. doi: 10.1093/jnci/81.22.1709. [DOI] [PubMed] [Google Scholar]
- 27.Pockaj BA, Sherry RM, Wei JP, et al. Localization of 111Indium-labeled tumor infiltrating lymphocytes to tumor in patients receiving adoptive immunotherapy. Augmentation with cyclophosphamide and correlation with response. Cancer. 1994;73:1731–7. doi: 10.1002/1097-0142(19940315)73:6<1731::aid-cncr2820730630>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 28.Miotti S, Negri DR, Valota O, et al. Level of anti-mouse-antibody response induced by bi-specific monoclonal antibody OC/TR in ovarian-carcinoma patients is associated with longer survival. Int J Cancer. 1999;84:62–8. doi: 10.1002/(sici)1097-0215(19990219)84:1<62::aid-ijc12>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 29.Dubey P, Su H, Adonai N, et al. Quantitative imaging of the T cell antitumor response by positron-emission tomography. Proc Natl Acad Sci U S A. 2003;100:1232–7. doi: 10.1073/pnas.0337418100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Freedman RS, Platsoucas CD. Immunotherapy for peritoneal ovarian carcinoma metastasis using ex vivo expanded tumor infiltrating lymphocytes. Cancer Treat Res. 1996;82:115–46. doi: 10.1007/978-1-4613-1247-5_8. [DOI] [PubMed] [Google Scholar]
- 31.Gattinoni L, Klebanoff CA, Palmer DC, et al. Acquisition of full effect nor function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ Tcells. JClin Invest. 2005;115:1616–26. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol. 2002;20:70–5. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 33.Taylor DD, Edwards RP, Case CR, Gercel-Taylor C. Modulation of CD3-zeta as a marker of clinical response to IL-2 therapy in ovarian cancer patients. Gynecol Oncol. 2004;94:54–60. doi: 10.1016/j.ygyno.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 34.Figini M, Obici L, Mezzanzanica D, et al. Panning phage antibody libraries on cells: isolation of human Fab fragments against ovarian carcinoma using guided selection. Cancer Res. 1998;58:991–6. [PubMed] [Google Scholar]