

Abstract
Determining the mechanisms of flux through protein channels requires a combination of structural data, permeability measurement, and molecular dynamics (MD) simulations. To further clarify the mechanism of flux through aquaporin 1 (AQP1), osmotic pf (cm3/s/pore) and diffusion pd (cm3/s/pore) permeability coefficients per pore of H2O and D2O in AQP1 were calculated using MD simulations. We then compared the simulation results with experimental measurements of the osmotic AQP1 permeabilities of H2O and D2O. In this manner we evaluated the ability of MD simulations to predict actual flux results. For the MD simulations, the force field parameters of the D2O model were reparameterized from the TIP3P water model to reproduce the experimentally observed difference in the bulk self diffusion constants of H2O vs. D2O. Two MD systems (one for each solvent) were constructed, each containing explicit palmitoyl-oleoyl-phosphatidyl-ethanolamine (POPE) phospholipid molecules, solvent, and AQP1. It was found that the calculated value of pf for D2O is ∼15% smaller than for H2O. Bovine AQP1 was reconstituted into palmitoyl-oleoyl-phosphatidylcholine (POPC) liposomes, and it was found that the measured macroscopic osmotic permeability coefficient Pf (cm/s) of D2O is ∼21% lower than for H2O. The combined computational and experimental results suggest that deuterium oxide permeability through AQP1 is similar to that of water. The slightly lower observed osmotic permeability of D2O compared to H2O in AQP1 is most likely due to the lower self diffusion constant of D2O.
INTRODUCTION
Aquaporins belong to a family of membrane proteins that passively transport water, glycerol, and other small molecules (Nielsen et al., 2002). The archetypical water channel AQP1 has been shown to be highly specific for water and excludes protons, ammonia, and sugars such as glycerol (Zeidel et al., 1992, 1994). The specificity of the channel arises from the presence of two highly conserved NPA motifs (Asn-Pro-Ala residues) that form the narrow constriction of the pore region, which acts as a molecular sieve. Crystallographic studies suggest that the pore is narrow at the constriction and lets only water diffuse through it (Murata et al., 2000; Sui et al., 2001). Molecular dynamics (MD) simulation studies suggest the presence of another energy barrier that offers both steric and electrostatic selectivity, named the aromatic (ar/R) constriction, which is formed by the four amino acids Phe-56, His-180, Cys-189, and Arg-195 (rat AQP1) in water-selective aquaporins (de Groot et al., 2003; Chakrabarti et al., 2004; Chen et al., 2006). In aquaglyceroporins this constriction region is wider, as His-180 is usually replaced by Gly, which allows for water and glycerol to pass through as shown in GlpF (Fu et al., 2000).
Although general geometry of aquaporin channels differs only slightly between aquaporin family members, its water permeability may differ by several orders of magnitude. Water is thought to hydrogen bond to arginine in the NPA region (Miloshevsky and Jordan, 2004; Vidossich et al., 2004) and the degree of hydrogen bonding or the energy barrier caused by hydrogen bonding is thought to dictate the rate of water flow through aquaporins. Furthermore, in a study of model channel desformylgramicidin, which shows a 100-fold increase in water permeability compared with gramicidin A, the lowered hydrogen bond–mediated energy barrier is thought to be responsible for large differences observed in water permeability (de Groot et al., 2002).
To determine whether such a finely tuned water channel allows the passage of deuterium oxide (D2O), a close analogue of water, we measured the permeability of D2O in AQP1-reconstituted proteoliposomes. D2O differs from H2O in the following respects: D2O has a lower zero-point vibrational energy leading to a stronger O-D bond and a stronger deuterium bond compared with the H-bond (Scheiner and Cuma, 1996). The effect of the stronger deuterium bond plus the heavier mass of D is believed to result in a self diffusion coefficient of D2O that is 18.6% lower than that of water (Mills, 1973; Grigera, 2001). Since water is thought to interact with specific residues Phe-56 and Agr-195 in the pore via hydrogen bonding (Miloshevsky and Jordan, 2004), it would be interesting to study the passage of D2O, which has a stronger deuterium bond. To test if kinetics of D2O permeability is different from H2O we simulated D2O flux and then measured it, allowing us to determine the predictive power of MD simulations. Both MD simulations and experimental results indicate that D2O and H2O permeate AQP1 at similar rates.
MATERIALS AND METHODS
MD Simulations
Molecular dynamics (MD) simulations were used to calculate the diffusion and osmotic permeability of H2O and D2O through AQP1. The X-ray structure of bovine AQP1 was downloaded from the Protein Data Bank (www.rcsb.org) with pdb code 1J4N (Sui et al., 2001). The tetrameric structure was assembled from the coordinates of the monomer using transformation matrices provided in the pdb file. The tetramer was then embedded in a preequilibrated patch of 177 palmitoyl-oleoyl-phosphatidyl-ethanolamine (POPE) molecules and solvated by 15,079 CHARMM modified TIP3P water molecules (Jorgensen et al., 1983; Neria et al., 1996) (referred to here as TIP3P), so that the total system contained 80,434 atoms. This system was energy minimized and equilibrated for 3 ns at 298 K and 1 atm. To construct the AQP1/D2O system, the TIP3P water model was replaced with a TIP3P-HW model (described below) and equilibrated for 1 ns at 298 K and 1 atm. The system equilibrium was checked by monitoring potential energy and volume over the course of the simulation. The standard deviation of potential energy from the mean was ∼0.13% (∼210 kcal/mol) and for volume was ∼0.15% (∼1150 Å3), which suggests that the system is in equilibrium.
Diffusion permeabilities were calculated for both AQP1/H2O and AQP1/D2O systems from equilibrium MD simulations at constant volume and constant temperature of 298 K from 31 and 26 ns trajectories, respectively. A weak harmonic restraining potential of 0.12 kcal/mol/Å2 was applied in all three dimensions to AQP1 backbone α carbons to prevent the protein form drifting. Diffusion permeability was calculated using the following equation (Zhu et al., 2004):
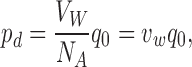 |
(1) |
where pd (cm3/s/pore) is the diffusion permeability coefficient per pore, VW is the molar volume of the solvent, NA is Avogadro's number, q 0 is the unidirectional permeation rate, and vw is the volume of a single solvent molecule. q 0 was calculated by counting the number of H2O or D2O molecules passing from one side of the channel to the other per unit time. The pore region was chosen to be the narrow central section of the channel, 22 Å in length, where water molecules pass in single file or nearly single file fashion (see Fig. 3, below).
Figure 3.

Concentration profile of H2O along z axis (channel axis) at equilibrium conditions (black line) and 100 MPa applied hydrostatic pressure (red line). Note the low concentration region between approximately z = −11 Å and z = +11 Ǻ; this is the “channel pore region” discussed in the text. Left side corresponds to cytoplasmic side.
Osmotic permeabilities were calculated by following the nonequilibrium MD method described by Zhu et al. (2004). A hydrostatic pressure gradient was established across the membrane by applying extra force to a layer of solvent molecules parallel to the membrane interface. Applied pressure MD simulations of different time length were performed at 25, 50, 100, and 200 MPa for both AQP1/H2O and AQP1/D2O systems. A force of 0.013445, 0.02689, 0.05378, and 0.10756 kcal/mol/Å was applied to oxygen atoms of H2O/D2O within an 8-Å solvent layer to generate 25, 50, 100, and 200 MPa pressure, respectively.
To prevent the protein and the lipid bilayer from moving under the influence of this external force, a harmonic restraint of 0.12 kcal/mol/Å2 was applied in all three dimensions to the backbone α carbons of the protein and a harmonic restraint of 0.8 kcal/mol/Å2 to the phosphorus atoms of the POPE molecules. This procedure differs somewhat from the 1D restraining procedure used by Zhu et al. (2004). However, our calculated osmotic permeability for H2O turned out to be similar to Zhu et al. (2004) (see below), implying that small differences in the restraining procedure do not have a major effect on the calculated osmotic permeabilities.
The net flux of H2O/D2O through AQP1 was calculated by averaging the number of molecules that crossed three planes located 5 Å away from each other inside the channel per pore per unit time. Osmotic permeabilities were calculated from the slope of the best (linear least squares) fit line of H2O/D2O flux versus applied hydrostatic pressure according to the following equation (Zhu et al., 2004):
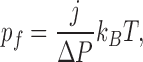 |
(2) |
where pf (cm3/s/pore) is the osmotic permeability coefficient per pore, j (molecules/s) is the net solvent flux through a single water channel, ΔP is the applied hydrostatic pressure, kB is the Boltzmann constant, and T is the absolute temperature.
For our MD simulations we used the CHARMM 27 force field (MacKerell et al., 1998), which was derived to be consistent with the TIP3P model (Neria et al., 1996). For the parameterization of deuterium oxide we started with the TIP3P model as a reference, doubled the mass of the hydrogen atoms, and adjusted the partial charges of both the deuterium and oxygen atoms. The physical motivation for modifying these partial charges is to take into account the quantum effect that liquid D2O is characterized by a stronger deuterium bond than the H bond found in liquid H2O. This quantum effect arises because D2O has a lower zero-point vibrational energy than H2O (Scheiner and Cuma, 1996). It is well known that TIP3P water considerably overestimates (by more than a factor of 2) the self diffusion constant of water (van der Spoel et al., 1998; Mark and Nilsson, 2001). For this reason, we chose as a target for parameterization to reproduce the experimentally measured 18.6% difference in the self diffusion constants of H2O vs. D2O (Mills, 1973) rather than the experimental self diffusion constant of heavy water. A similar reparameterization of the SPC/E model for deuterium oxide has been reported recently in which only the mass and partial charges of the hydrogen atoms were changed in order to reproduce the experimental self diffusion constant, the molar volume and the potential energy of heavy water (Grigera, 2001). For our reparameterization, a simulation box of 3921 TIP3P water molecules was constructed, energy minimized, and equilibrated at 298 K and 1 atm. The mass of hydrogen atoms was set to 2 amu and atomic partial charges were incremented by fractions of a percent. The system was equilibrated for 0.5 ns followed by 1 ns production runs performed at a constant temperature of 298 K and a constant pressure of 1 atm. The self diffusion constant was calculated from the collected coordinate time series using the mean-square-displacement method (Allen and Tildesley, 1987). Adjustment of D2O atomic partial charges along with recalculation of the self diffusion constant was repeated until we generated a diffusion coefficient ∼18% smaller than the value obtained with TIP3P under the same conditions.
All MD simulations were performed using the NAMD package (Kale et al., 1999) version 2.5. For long range electrostatics, the Particle Mesh Ewald method (Essmann et al., 1995) was used as implemented in the NAMD program along with periodic boundary conditions. The MD time step was set to 2 fs and the bonds between each hydrogen atom and its mother atom were fixed via the SHAKEH algorithm (Kale et al., 1999) to the nominal bond length given in the parameter file. MD coordinates of AQP1 simulations were saved every 1 ps and for the parameterization of TIP3P-HW water model every 100 fs. Most simulations were performed on Lemieux supercomputer at the Pittsburgh Supercomputing Center (www.psc.edu). It took ∼4.6 h to run 1 ns of AQP1 simulations on 160 CPUs of Lemieux. All visualizations and analysis of MD trajectories were made with the VMD program (Humphrey et al., 1996).
Reconstitution of AQP1 into Liposomes
Bovine AQP1 was purified from bovine red blood cells using methods similar to that used for purifying human AQP1 (Zeidel et al., 1992). The AQP1 reconstitution procedure was similar to that described earlier (Zeidel et al., 1994). In brief, 6–8 mg of palmitoyl-oleoyl-phosphatidylcholine (POPC) lipids (Avanti Lipids) was bath sonicated for three cycles of 3 min duration at 4 mW setting in 20 mM MOPS, pH 7.4. N-octylglucoside (OG) was added to the sonicated lipids to achieve a final concentration of 1.2% (vol/vol). To this, 50–60 μg of AQP1 in 1.5% OG was added and incubated for 30 min on ice. The mixture was diluted 25-fold into reconstitution buffer (150 mM NaCl, 20 mM MOPS, pH 7.4) containing 10 mM carboxyfluorescein (CF). The proteoliposomes formed were collected by centrifugation at 100,000 g for 1 h. The external CF was removed by two additional centrifugal washes. The final pellet was resuspended in 300 μl of reconstitution buffer and used for permeability studies.
Measurement of Osmotic Permeabilities
Macroscopic osmotic permeabilities (Pf) of H2O and D2O were measured as described earlier (Zeidel et al., 1994). In brief, the proteoliposomes were subjected to a doubling of external osmotic pressure in a stopped-flow fluorimeter (Applied Photophysics, SX.17 MV), and the fluorescence decrease of carboxyfluorescein due to self quenching caused by shrinkage of the vesicle was measured as a function of time. The data were fit using a single exponential function. Pf was calculated by comparing the single-exponential time constants fitted to a family of simulated curves generated using the water permeability equation in which Pf was varied to that obtained experimentally. MathCad was used to numerically integrate the water permeability equation:
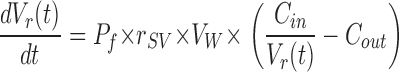 |
(3) |
In Eq. 3, Vr(t) is the relative volume of the vesicle at time t (i.e., volume at time t, divided by the initial volume), Pf (cm/s) is the macroscopic osmotic water permeability coefficient, rSV is the surface area to volume ratio of a vesicle, and Cin and Cout are initial solute concentrations inside and outside the vesicle, respectively. The size of the vesicle was measured by laser light scatter using a DynaPro particle sizer. All other chemicals were obtained from Sigma-Aldrich.
RESULTS AND DISCUSSION
Earlier studies before discovery of aquaporins showed that permeation of D2O across erythrocytes and red cell ghosts was inhibited by mercury (Karan and Macey, 1980; Ye and Verkman, 1989). However, mercurial compounds are nonspecific and do not inhibit the AQP1 channel completely (Zeidel et al., 1992). In this study we have performed a quantitative permeability measurement using purified AQP1, reconstituted into proteoliposomes, which eliminates the background permeability due to other proteins (Roudier et al., 1998; Yang et al., 2001). Fig. 1 shows the time course of relative volume change of liposomes on application of an osmotic gradient, causing the efflux of H2O (Fig. 1 A) or D2O (Fig. 1 B). It was found that the measured osmotic permeability of D2O (1.8 × 10−2 ± 0.42 × 10−2 cm/s) is ∼21% lower than that for H2O (2.3 × 10−2 ± 0.310−2 cm/s). This difference in permeability can be attributed to the higher viscosity of D2O vs. H2O and an 18.6% lower self diffusion coefficient of D2O compared with that of water (Mills, 1973). In red blood cells, before discovery of aquaporins, the decreased permeability of D2O was attributed to higher viscosity of D2O (Karan and Macey, 1980). A similar decrease of conductance of D2O through gramicidin was also attributed to higher viscosity of D2O (Tredgold and Jones, 1979). The background osmotic permeabilities of H2O and D2O in the absence of a water channel through POPC lipid membranes are 0.0032 cm/s and 0.0034 cm/s respectively. To confirm that the observed difference in permeability is not an isotope effect, we measured the osmotic permeability of AQP1 at various osmotic pressure values. Fig. 1 C shows that there is a linear increase in rate of efflux of H2O and D2O with increasing osmotic pressure. Furthermore the efflux of H2O and D2O shows a parallel increase with increasing osmotic pressure, suggesting that there are no additional interactions between the permeant and the channel as flux increases at higher osmotic pressures.
Figure 1.

Osmotic permeability of H2O and D2O. Shown is the time course of volume change of AQP1-containing vesicles and control lipid vesicles on abrupt exposure to a doubling of external osmotic pressure in H2O (A) and D2O (B). C shows the effect of varying osmotic pressure (mOsm/kg) on permeability of H2O and D2O through AQP1.
The force field parameters of our TIP3P-HW (D2O) model are summarized in Table I. Partial atomic charges are 0.96% higher compared with the TIP3P model. This implies a larger dipole moment of 7.904 × 10−30 Cm which is also 0.96% larger than for the TIP3P model. The dimer potential energy in the most favorable configuration is −6.759 kcal/mol, which is 2.4% larger in magnitude than for the TIP3P model. The heat of vaporization for TIP3P-HW is 10.68 kcal/mol, which is 2.9% larger than for the TIP3P model and compares well with the experimentally determined value of 11.11 kcal/mol for D2O (Nemethy and Scheraga, 1964). In our MD simulations, the TIP3P-HW model was characterized by a bulk diffusion coefficient of 4.00 × 10−5 cm2/s, which is 18.7% lower than that of the TIP3P model of water (4.92 × 10−5 cm2/s). This difference is in good agreement with the experimentally measured difference of 18.6% between H2O and D2O (Mills, 1973). The densities of TIP3P and TIP3P-HW models were found to be 1.00 g/cm3 and 1.13 g/cm3, respectively. This is also in good agreement with experimental densities of 0.997 g/cm3 for H2O and 1.104 g/cm3 for D2O (Weast, 1977). These solvent models were subsequently used to calculate the osmotic and diffusion permeabilities of H2O and D2O in AQP1.
TABLE I.
Force Field Parameters of TIP3P-HW Model
|
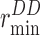
|
ɛOO | ɛDD | qO | qD | bOD | θDOD |
|---|---|---|---|---|---|---|---|
| Å | Å | kcal/mol | kcal/mol | e units | e units | Å | deg |
| 3.5365 | 0.449 | 0.1521 | 0.046 | −0.842 | −0.421 | 0.9572 | 104.52 |
A snapshot of the AQP1/H2O system from the equilibrium MD trajectory is presented in Fig. 2, with only one monomer and channel shown for the sake of clarity. The single file arrangement and rotation of the orientation of the water dipole, which are unique properties of the AQP family (de Groot and Grubmuller, 2001; Tajkhorshid et al., 2002), are correctly reproduced in our simulations. The single file channel occupancy for H2O and D2O are 9.7 and 9.5 molecules, respectively. The concentration profile of H2O across the channel system at equilibrium conditions is compared in Fig. 3 with that obtained at 100 MPa applied hydrostatic pressure. When hydrostatic pressure was applied, the concentration of water became slightly higher on the left side and lower on the right side of the membrane due to the compressibility of water. The main results of our equilibrium MD simulations for H2O and D2O are summarized in Table II. The unidirectional permeation rate (q 0) of H2O was found to be 0.29, which compares well to the value of 0.2 predicted by another AQP study (de Groot and Grubmuller, 2001). This difference can be attributed to the fact that the authors used a different (lower resolution) structure of AQP1 and a different water model. We found that q 0 for D2O is ∼7% higher than for H2O, albeit with relatively large error bars (Table II).
Figure 2.
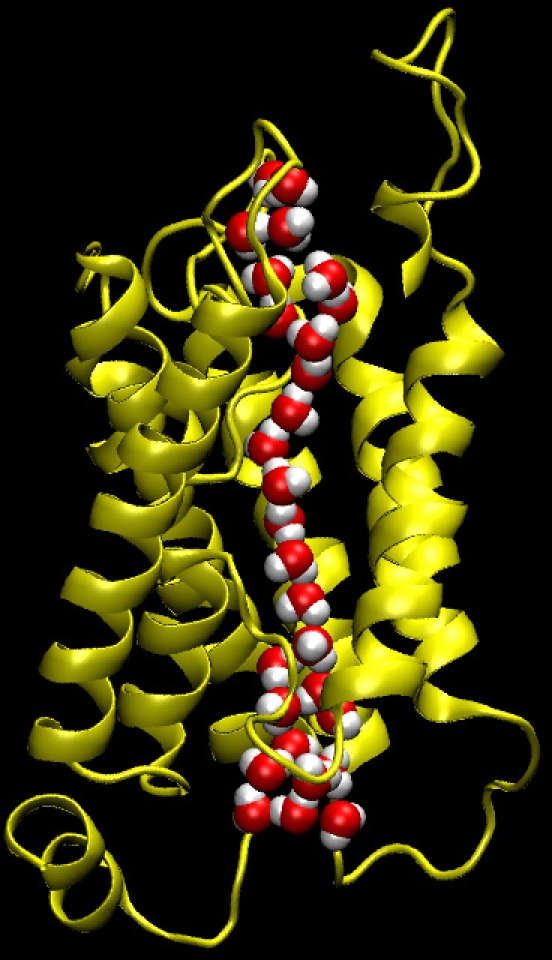
A snapshot from MD simulation of AQP1/H2O system. Only one monomer of the water channel is shown for clarity. Note the single file arrangement of water molecules inside the channel.
TABLE II.
Results of Equilibrium MD Simulations
| Time | No. of permeations | q 0 | pd × 1014 | |
|---|---|---|---|---|
| ns | molecules/ns/pore | cm3/s/pore | ||
| H2O | 31.1 | 71 | 0.29 ± 0.026 | 0.85 ± 0.078 |
| D2O | 26.3 | 65 | 0.31 ± 0.085 | 1.0 ± 0.28 |
Results of our osmotic pressure simulations are reported in Table III and Fig. 4 A for H2O, and in Table IV and Fig. 4 B for D2O. We found that the osmotic permeability of D2O (8.5 × 10−14 cm3/s/pore) is 15% lower than that of H2O (10.0 × 10−14 cm3/s/pore). This difference compares well with the experimentally measured difference of ∼21%. We could not compare the individual values of the osmotic permeabilities because of the experimental uncertainty in the number of the AQP1 channels per proteoliposome. The osmotic permeability of H2O 10.0 × 10−14 cm3/s/pore obtained in our MD simulations is similar to that predicted by another MD study where a value of 7.1 × 10−14 cm3/s/pore was reported (Zhu et al., 2004). In addition it also compares well with the experimentally measured unit osmotic conductance (pf) of 11.7 × 10−14 ± 1.8 × 10−14 cm3/s/pore reported by Zeidel et al. (1994). Since water moves in a single file fashion through the AQP1 channel, the pf/pd ratio should give the number of solute molecules lining the pore (Finkelstein, 1987). The calculated pf/pd ratios for H2O and D2O are 11.8 and 8.4, respectively. The pf/pd ratio of H2O is in good agreement with the value 11.9 predicted in Zhu et al. (2004) and 13.2 measured experimentally (Mathai et al., 1996).
TABLE III.
Results of Applied Pressure MD Simulations for H2O
| Applied pressure | Time | No. of H2O permeations |
Flux | pf × 1014 |
|---|---|---|---|---|
| MPa | ns | H2O/ns/pore | cm3/s/pore | |
| 25 | 8 | 21 | 0.65 | 10.0 ± 0.56 |
| 50 | 26.8 | 107 | 1.0 | |
| 100 | 21.9 | 171 | 1.9 | |
| 200 | 22 | 455 | 5.2 |
Figure 4.

Computed flux versus applied hydrostatic pressure for H2O (A) and D2O (B). Circles (black) represent the calculated (average) data point values, which are shown along with the best-fit line (red). Standard error for the best fit line for H2O is 5.6% and for D2O 5.3%.
TABLE IV.
Results of applied pressure MD simulations for D2O
| Applied pressure | Time | No. of D2O permeations |
Flux | pf × 1014 |
|---|---|---|---|---|
| MPa | ns | D2O/ns/pore | cm3/s/pore | |
| 25 | 7 | 21 | 0.74 | 8.5 ± 0.45 |
| 50 | 22 | 126 | 1.4 | |
| 100 | 34 | 239 | 1.8 | |
| 200 | 20 | 333 | 4.2 |
In conclusion, a new TIP3P-HW model for D2O was developed for simulation of D2O transport through AQP1. This model reproduces the experimental differences observed in the density and the self diffusion coefficient of H2O vs. D2O. Both MD simulations and experimental measurements confirm that D2O and H2O permeate the AQP1 channel. The observed lower permeability of D2O is attributed to the lower self diffusion coefficient and higher viscosity of D2O compared with H2O. Our study showed that MD could accurately predict the decreased permeability of D2O in AQP1 in advance of experimental measurements. This study will be helpful for designing MD simulations to study permeation of solutes through AQP1 that are not easily amenable to experiments such as carbon dioxide, hydrogen sulfide, and oxygen.
Acknowledgments
We would like to thank Zuzana Valnickova, Jan Enghild, Niels Chr. Nielsen, and Astrid C. Sivertsen (University of Aarhus, Aarhus, Denmark) for providing us purified AQP1 for initial studies.
The authors gratefully acknowledge computer time provided by PSC grant MCB030021P. M.L. Zeidel and J.C. Mathai are supported by a grant from the National Institutes of Health, DK 43955. A.B. Mamonov and R.D. Coalson were supported by National Science Foundation grant CHE-0518044 and Army Research Office grant DADD19-02-1-0227.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: AQP1, aquaporin 1; MD, molecular dynamics.
References
- Allen, M.P., and D.J. Tildesley. 1987. Computer Simulation of Liquids. Clarendon Press; Oxford University Press, Oxford Oxfordshire, New York. 408 pp.
- Chakrabarti, N., E. Tajkhorshid, B. Roux, and R. Pomes. 2004. Molecular basis of proton blockage in aquaporins. Structure. 12:65–74. [DOI] [PubMed] [Google Scholar]
- Chen, H., Y. Wu, and G.A. Voth. 2006. Origins of proton transport behavior from selectivity domain mutations of the aquaporin-1 channel. Biophys. J. 90:L73–L75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot, B.L., T. Frigato, V. Helms, and H. Grubmuller. 2003. The mechanism of proton exclusion in the aquaporin-1 water channel. J. Mol. Biol. 333:279–293. [DOI] [PubMed] [Google Scholar]
- de Groot, B.L., and H. Grubmuller. 2001. Water permeation across biological membranes: mechanism and dynamics of aquaporin-1 and GlpF. Science. 294:2353–2357. [DOI] [PubMed] [Google Scholar]
- de Groot, B.L., D.P. Tieleman, P. Pohl, and H. Grubmuller. 2002. Water permeation through gramicidin A: desformylation and the double helix: a molecular dynamics study. Biophys. J. 82:2934–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann, U., L. Perera, M.L. Berkowitz, T. Darden, H. Lee, and L.G. Pedersen. 1995. A smooth Particle Mesh Ewald Method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- Finkelstein, A. 1987. Water Movement through Lipid Bilayers, Pores and Plasma Membranes. John Wiley & Sons, Inc., New York. 228 pp. [DOI] [PubMed]
- Fu, D., A. Libson, L.J. Miercke, C. Weitzman, P. Nollert, J. Krucinski, and R.M. Stroud. 2000. Structure of a glycerol-conducting channel and the basis for its selectivity. Science. 290:481–486. [DOI] [PubMed] [Google Scholar]
- Grigera, J.R. 2001. An effective pair potential for heavy water. J. Chem. Phys. 114:8064–8067. [Google Scholar]
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W., J. Chandrasekhar, and J. Madura. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Kale, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151:283–312. [Google Scholar]
- Karan, D.M., and R.I. Macey. 1980. The permeability of the human red cell to deuterium oxide (heavy water). J. Cell. Physiol. 104:209–214. [DOI] [PubMed] [Google Scholar]
- MacKerell, A.D., D. Bashford, M. Bellott, R.L. Dunbrack, J.D. Evanseck, M.J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, et al. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Physiol. Chem B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- Mark, P., and L. Nilsson. 2001. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Physiol. Chem A. 105:9954–9960. [Google Scholar]
- Mathai, J.C., S. Mori, B.L. Smith, G.M. Preston, N. Mohandas, M. Collins, P.C. van Zijl, M.L. Zeidel, and P. Agre. 1996. Functional analysis of aquaporin-1 deficient red cells. The Colton-null phenotype. J. Biol. Chem. 271:1309–1313. [DOI] [PubMed] [Google Scholar]
- Mills, R. 1973. Self-diffusion in normal and heavy water in the range 1-45. J. Phys. Chem. 77:685–688. [Google Scholar]
- Miloshevsky, G.V., and P.C. Jordan. 2004. Water and ion permeation in bAQP1 and GlpF channels: a kinetic Monte Carlo study. Biophys. J. 87:3690–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata, K., K. Mitsuoka, T. Hirai, T. Walz, P. Agre, J.B. Heymann, A. Engel, and Y. Fujiyoshi. 2000. Structural determinants of water permeation through aquaporin-1. Nature. 407:599–605. [DOI] [PubMed] [Google Scholar]
- Nemethy, G., and H.A. Scheraga. 1964. Structure of water and hydrophobic bonding in proteins. IV. The thermodynamic properties of liquid deuterium oxide. J. Chem. Phys. 41:680–689. [Google Scholar]
- Neria, E., S. Fischer, and M. Karplus. 1996. Simulation of activation free energies in molecular systems. J. Chem. Phys. 105:1902–1921. [Google Scholar]
- Nielsen, S., J. Frokiaer, D. Marples, T.H. Kwon, P. Agre, and M.A. Knepper. 2002. Aquaporins in the kidney: from molecules to medicine. Physiol. Rev. 82:205–244. [DOI] [PubMed] [Google Scholar]
- Roudier, N., J.M. Verbavatz, C. Maurel, P. Ripoche, and F. Tacnet. 1998. Evidence for the presence of aquaporin-3 in human red blood cells. J. Biol. Chem. 273:8407–8412. [DOI] [PubMed] [Google Scholar]
- Scheiner, S., and M. Cuma. 1996. Relative stability of hydrogen and deuterium bonds. J. Am. Chem. Soc. 118:1511–1521. [Google Scholar]
- Sui, H., B.G. Han, J.K. Lee, P. Walian, and B.K. Jap. 2001. Structural basis of water-specific transport through the AQP1 water channel. Nature. 414:872–878. [DOI] [PubMed] [Google Scholar]
- Tajkhorshid, E., P. Nollert, M.O. Jensen, L.J. Miercke, J. O'Connell, R.M. Stroud, and K. Schulten. 2002. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science. 296:525–530. [DOI] [PubMed] [Google Scholar]
- Tredgold, R.H., and R. Jones. 1979. A study of gramicidin using deuterium oxide. Biochim. Biophys. Acta. 550:543–545. [DOI] [PubMed] [Google Scholar]
- van der Spoel, D., P.J. van Maaren, and H.J.C. Berendsen. 1998. A systematic study of water models for molecular simulation: derivation of water models optimized for use with a reaction field. J. Chem. Phys. 108:10220–10230. [Google Scholar]
- Vidossich, P., M. Cascella, and P. Carloni. 2004. Dynamics and energetics of water permeation through the aquaporin channel. Proteins. 55:924–931. [DOI] [PubMed] [Google Scholar]
- Weast, R. 1977. CRC Handbook of Chemistry and Physics. 64th Edition. CRC Press, Cleveland, OH. F-4.
- Yang, B., T. Ma, and A.S. Verkman. 2001. Erythrocyte water permeability and renal function in double knockout mice lacking aquaporin-1 and aquaporin-3. J. Biol. Chem. 276:624–628. [DOI] [PubMed] [Google Scholar]
- Ye, R.G., and A.S. Verkman. 1989. Simultaneous optical measurement of osmotic and diffusional water permeability in cells and liposomes. Biochemistry. 28:824–829. [DOI] [PubMed] [Google Scholar]
- Zeidel, M.L., S.V. Ambudkar, B.L. Smith, and P. Agre. 1992. Reconstitution of functional water channels in liposomes containing purified red cell CHIP28 protein. Biochemistry. 31:7436–7440. [DOI] [PubMed] [Google Scholar]
- Zeidel, M.L., S. Nielsen, B.L. Smith, S.V. Ambudkar, A.B. Maunsbach, and P. Agre. 1994. Ultrastructure, pharmacologic inhibition, and transport selectivity of aquaporin channel-forming integral protein in proteoliposomes. Biochemistry. 33:1606–1615. [DOI] [PubMed] [Google Scholar]
- Zhu, F., E. Tajkhorshid, and K. Schulten. 2004. Theory and simulation of water permeation in aquaporin-1. Biophys. J. 86:50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]