

Abstract
Slow inactivation involves a local rearrangement of the outer mouth of voltage-gated potassium channels, but nothing is known regarding rearrangements in the cavity between the activation gate and the selectivity filter. We now report that the cavity undergoes a conformational change in the slow-inactivated state. This change is manifest as altered accessibility of residues facing the aqueous cavity and as a marked decrease in the affinity of tetraethylammonium for its internal binding site. These findings have implications for global alterations of the channel during slow inactivation and putative coupling between activation and slow-inactivation gates.
INTRODUCTION
What is the conformation of an inactivated channel? There is no crystal structure. However, we have some clues for slow inactivation in K+ channels based on structures of KcsA in low K+ (Morais-Cabral et al., 2001; Zhou et al., 2001b), in the presence of quaternary ammonium blockers (Lenaeus et al., 2005), and of a mutant KcsA that manifests altered inactivation gating (Cordero-Morales et al., 2006). These structural studies have focused mainly on the selectivity filter and outer mouth of the channel, whose local conformational rearrangement is thought to underlie slow inactivation (C-type inactivation) in voltage-gated K+ (Kv) channels (Choi et al., 1991; Yellen et al., 1994; Liu et al., 1996; Harris et al., 1998; Ogielska and Aldrich, 1999). But other parts of the channel could rearrange in the slow-inactivated conformation. For example, the inactivation gate at the selectivity filter communicates with the activation gate at the bundle crossing of S6 segments (Panyi and Deutsch, 2006), and this could implicate a role for the cavity, located between the selectivity filter and the bundle crossing, in the coupling of the two gates. Because the slow-inactivated state is nonconducting, it is difficult to probe electrophysiologically, which precludes determining certain properties of the inactivated state cavity. For example, blocker affinities, though straightforwardly determined for the open channel, are more equivocal for the inactivated channel.
Why is it important to understand the nature of the cavity in the inactivated state? First, the cavity may regulate both permeation (Grabe et al., 2006; Bichet et al., 2006) and gating. Second, therapeutic channel blockers, a major focus in drug design, can target to the cavity of slow-inactivated states (Hanner et al., 2001). Indeed, several drugs bind preferentially to slow-inactivated states of voltage-gated K+, Na+, and Ca2+ channels (McDonough and Bean, 1998; Koo et al., 1999; Martin et al., 2000; Hanner et al., 2001; McNulty and Hanck, 2004). Design of drugs targeted to the inactivated state requires a knowledge of the conformation of this state, including its cavity.
From our previous study of the coupling between the activation and slow-inactivation gates, we observed that a residue in the cavity of Shaker was modified at one-tenth the rate in the slow-inactivated state than in the open state (Panyi and Deutsch, 2006). Is this generally true of the cavity and what does it represent? In the current paper, we probe the cavity of the slow-inactivated Shaker-IR channel. For this purpose, we used cysteine modifying reagents, methanethiosulfonates (Smith et al., 1975; Stauffer and Karlin, 1994) and Cd2+, which have been used to learn about channel structures and their state-dependent conformational changes (Yellen et al., 1994; Yang and Horn, 1995; Larsson et al., 1996; Liu et al., 1996). We report a dramatic change in modification rates in the slow-inactivated state compared with the open state. Moreover, these relative modification rates depend on the location inside the cavity. In addition, we measured the affinity of internally applied TEA, a reversible blocker, for the inactivated state and show it is 20-fold reduced compared with that of the open state. To our knowledge, this is the first report of an affinity for a quaternary ammonium (QA) in the cavity of an inactivated channel. Several mechanisms may account for the altered modification rates and affinities in the slow-inactivated state, however, our results suggest that the cavity of the slow-inactivated Kv channel is conformationally different from that of the open channel.
MATERIALS AND METHODS
Cell Culture
Human embryonic kidney cells transformed with SV40 large T antigen (tsA201) were grown in DMEM-high glucose supplemented with 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin-g, and 100 μg/ml streptomycin (Invitrogen) at 37°C in a 9% CO2 and 95% air humidified atmosphere. Cells were passaged twice per week following a 7-min incubation in Versene containing 0.2g EDTA/L (Invitrogen).
DNA Clones and Site-directed Mutagenesis
Modified Shaker-IR in a GW1-CMV mammalian expression plasmid, under the control of a highly expressing Kozak consensus promoter sequence (Kozak, 1991), was provided by R. Horn (Ding and Horn, 2002). This construct includes a deletion of amino acids 6–46 to remove N-type inactivation, and C301S and C308S point mutations (Holmgren et al., 1996). These two mutations are necessary to exclude possible MTS or Cd2+ modification of endogenous Shaker cysteines and accompanying functional effects. Amino acid substitutions, according to Shaker B numbering, at position 449 (T449A or T449K), 474 (V474C), and 470 (I470C) were introduced using a QuikChange site-directed mutagenesis kit (Stratagene). Mutants were sequenced at the University of Pennsylvania School of Medicine DNA Sequencing Facility using an ABI 3100 16 capillary sequencing apparatus with BigDye Taq FS Terminator V 3.1 chemistry (Applied Biosystems, Inc.). A calcium phosphate transfection kit (Invitrogen) was used to cotransfect CD8 carried in a EBO-pcD vector (Margolskee et al., 1988; Margolskee et al., 1993) with the Shaker-IR mutant construct using 4 or 10 μg of CD8 or Shaker-IR DNA, respectively, per 100-mm dish of tsA201 cells. Transfected cells were replated onto Corning 35-mm polystyrene cell culture dishes pretreated with poly-l-ornithine (Sigma-Aldrich) to improve cell adhesion for excising patches. 12–36 h following transfection, current was recorded from transfected cells, which were identified by decoration with anti-CD8 antibody-coated Dynabeads (Dynal Biotech) as described previously (Margolskee et al., 1993; Jurman et al., 1994).
Electrophysiology
Standard methods were used to record currents in inside-out patches. Data were acquired using a HEKA EPC-9 amplifier, digitized with an ITC-16 analogue-to-digital converter (HEKA Elektronik, GmbH). The filter frequency was set to less than or equal to half the sampling frequency. Pipettes pulled from lead-free 8520 glass (Warner Instruments) were 8–9 MΩ and coated with R-6101 elastomer (Essex Group, Inc.) and fire polished. Adjustments for bath-pipette liquid junction potentials were made before current recording. Typical current amplitudes were 300–600 pA at +50 mV test potential allowing the recording of macroscopic currents. Only those patches with a steady-state current <5% of the peak current were used in these studies. In general, all holding potentials were −100 mV and voltage errors were <3 mV after series resistance compensation. Leakage and capacitance correction, when performed, were designed to avoid opening the channel. All experiments were performed at room temperature (20–24°C). Data were analyzed using Pulse + PulseFit software (V. 8.77 HEKA Elektronik, Dr. Schulze, GmbH). Reported errors are SEM.
Solutions
The standard intracellular (bath) solution contained (in mM) 105 KF, 35 KCl, 10 EGTA, 10 HEPES, titrated to pH 7.36–7.38 with KOH, for a final concentration of 160–165 mM K+ and osmolarity of 285–295 mOsm. For experiments using 50 mM K+ or 150 Cs+, the standard intracellular solution was identical except that it contained 100 NaF or 105CsF plus 35 CsCl, respectively, instead of the corresponding K+ salts and was titrated with NaOH or CsOH, respectively. For experiments using Cd2+ as the modifying reagent, the intracellular solution was identical to the standard K+-based solution except that EGTA was omitted and (in mM) 125 KF, 35 KCl, and 10 HEPES was used. Standard extracellular (pipette) solution was (in mM) 150 NaCl, 2 KCl, 1.5 CaCl2, 1 MgCl2, 10 HEPES, pH 7.38. The osmolarity of the solution was 290 mOsm, and pH was titrated to 7.36–7.38 with NaOH. Ethyltrimethylammonium methanethiosulfonate (MTSET; Toronto Research Corp.) or ethylammonium methanethiosulfonate (MTSEA; Toronto Research Corp.) solution was made fresh in intracellular (bath) solutions from 100 mM stocks in water, which were stored at −80°C. MTS-reagent stock was freshly diluted into the bath solution and loaded immediately in the perfusion apparatus before the start of MTS-reagent application. Fresh MTS-reagent solution was loaded into the perfusion apparatus every 7 min.
Characterization of the Perfusion Kinetics
A Warner Instruments SF-77A Perfusion Fast-Step system with 3-barrel square glass (700 μM internal diameter) was used for rapid solution exchange. Inside-out patches were perfused with standard intracellular solutions at a rate of 0.5 ml/min. The solution exchange is determined by two factors, a delay (d) followed by a rapid exponential decay characterized by the exchange time constant, τe. A description of the test protocol and sample data have been described previously (Panyi and Deutsch, 2006). The medians of d and τe were 33.6 and 3.3 ms, respectively (n = 36). Characteristics of solution exchange were determined for each patch and values were used to calculate the correct cumulative modification time for MTS-reagents and Cd2+.
Cysteine Modification Measurements
Patches were depolarized from a holding potential of −100 to +50 mV for the time required to achieve the desired fraction of inactivation. Pulse protocols were run three to four times in the absence of MTS reagents or Cd2+ to verify the stability of the peak currents. Patches showing >5% variability/rundown in peak current were discarded. MTS-reagent was applied for a given time (indicated for each experiment) during a depolarization to +50 mV. To minimize the effect of hydrolysis on the observed rates of modification with MTSET, we refreshed the MTSET/KF solution every 7 min during the modification reaction. We did not correct for hydrolysis during the modification protocols and thus, any error in k mod, if it exists, will underestimate the absolute rate constants, but will have no effect on the ratio of rate constant for open and inactivated states because the total time of exposure to MTSET was similar. These protocols were repeated for the specified number of times to determine a time course for modification of V474C or I470C. The resulting currents were plotted as normalized peak current versus cumulative modification time, from which a modification rate constant could be extracted. When Cd2+ was used as the modifying reagent, the concentration of Cd2+ ranged from 20 to 100 μM and the protocols were adapted as shown in the figures. Determinations of k on and k off were performed using protocols described in Figs. 5–7 .
Figure 5.

Modification of cysteine 470 by Cd2+. Patches expressing T449K/I470C channels were repeatedly depolarized from a holding potential of −100 to +50 mV using the pulse protocols shown above the corresponding raw current traces. The timing and the duration (L) of the Cd2+ pulse are indicated by the hatched bars. (A) Open-state modification. 10-ms-long depolarizing pulses were applied every 15 s in the presence of 100 μM Cd2+. Perfusion with Cd2+ started 200 ms before each depolarizing pulse and the patch was perfused with KF solution for the remaining time at the interpulse holding potential. After 55 pulses, residual current (R) was 5% of the original peak. For clarity, only every fourth pulse is shown. Peak currents for each pulse were normalized to the peak current of the first pulse, corrected by multiplying by 1.15, and plotted as a function of the cumulative modification time, as described in Fig. 3 A. The data were fit with a single exponential function to give an apparent k on and R, the fraction of residual current. (B) Inactivated-state modification. 1.8-ms-long depolarizing pulses were applied every 41.5 s and perfusion with 20 μM Cd2+ (L = 600 ms) was initiated 1,000 ms after the start of the depolarization. Peak currents for each pulse were normalized to the peak current of the first pulse and plotted as a function of the cumulative modification time. Results represented by different symbols were obtained with L = 400 ms (empty circles), 600 ms (filled circles, data from the experiment shown on the left), and 800 ms (diamonds). Lines are the best fits for single exponential functions, which gave the apparent reaction rates shown. Note that the modification reaction did not reach completion, a significant fraction of channels were not modified by Cd2+ at equilibrium.
Figure 7.

Analytical and experimental determination of the rate constants characterizing Cd2+ block of the inactivated channel. Patches expressing T449K/I470C channels were repeatedly depolarized from a holding potential of −100 to +50 mV and exposed to Cd2+ (20 μM). (A) Cd2+ binding reaction. One representative episode is shown indicating the allocation of three consecutive periods, I, II, and III at +50 mV when the activation gate is open (Panyi and Deutsch, 2006). The durations of periods I and III (no added Cd2+) were 1,000 and 250 ms, respectively. Only Cd2+ dissociation occurs during these periods. Period II (equivalent to L in previous figures) was either 400, 600, or 800 ms. Both Cd2+ association and dissociation occur during period II. This protocol was run repeatedly until equilibrium block was reached. The kinetics of peak current reduction (see Fig. 5 B) was analyzed as described in the text to obtain k on and k off for Cd2+ binding. (B) The protocol in A was run twice only for the period II durations indicated by the filled circles. The ratio of the current recorded after the first exposure to Cd2+ to the control peak was calculated (“first episode” normalized amplitude) and plotted as a function of period II duration. Error bars indicate SEM (n = 4–6). The superimposed solid line is the best fit of the data to a single exponential function. The values of the fitted parameters, apparent k on and R, were used to calculate k on, as described in the text.
Figure 6.

Dissociation of Cd2+ from the inactivated channel. Patches expressing T449K/I470C channels were repeatedly depolarized from a holding potential of −100 to +50 mV and exposed to Cd2+ using pulse protocols shown in Fig. 5 B until equilibrium block was achieved. The patch was subsequently maintained in KF solution and pulsed to +50 mV for variable durations every 41.5 s to measure the washout rate of Cd2+. Fractional recovery (RF) was determined from peak currents (I), the peak currents measured at full recovery from Cd2+ block (Imax), and the peak currents measured at equilibrium block (I0) according to the following equation: RF = (I − I0)/(Imax − I0). RF was plotted as a function of cumulative time spent at +50 mV. A fit of the data to a single exponential function gave the off-rate of Cd2+ from the inactivated state. (A) Independence of k off from Cd2+ loading duration. Equilibrium block was achieved using different durations of the Cd2+ loading pulse (L; see protocol in Fig. 5 B). Application of Cd2+ was initiated at 1,000 ms after the start of the depolarization and the duration of the depolarizing pulse was extended to match L (duration = 1,000 ms + L + 200 ms). (B) Independence of k off from washout pulse duration. Following equilibrium block with Cd2+, washout was monitored using different durations of pulses in the absence of Cd2+. Peak currents were measured during short depolarizations in the absence of Cd2+ (empty circles), following repeated exposure of the inactivated state to 20 μM Cd2+ (filled circles), and after switching to Cd2+-free solution for 1.8- and 6.2-s-long depolarizations to +50 mV (up triangles and down triangles, respectively). Peak current was normalized to the maximum current at the end of the washout period. The best fit of the data to single-exponential functions (superimposed solid lines) gave the k off values plotted in the right panel, along with those obtained for 0.6 and 1.6 s. Error bars indicate SEM.
Blocker-Protection Assays
TEA was used to block modification at 470C in both the open and inactivated states. The main advantage of TEA is that it blocks rapidly and is trapped when the activation gate closes (Fig. 1 A, middle current traces). The duration of exposure to TEA and MTSEA was dictated by the state intended for investigation, as was the concentration of these reagents. The protocol devised to measure open-state protection is shown in Fig. 1 A. It consists of a series of short (12 ms) depolarizations to +50 mV. A step to +50 mV fully activates the current but does not cause detectable inactivation. The first of these depolarizations (leftmost panel) is in KF solution to obtain the maximum current, followed by two consecutive depolarizations in the presence of TEA (second panel, current labeled 1 and 2). Comparison of current 1 and current 2 in TEA shows that the amount of block present at the end of the first pulse in TEA (current 1) was the same as the amount of block present at the beginning of the second pulse (current 2), indicating that TEA remained trapped in the channel when the channel closed at the end of the first pulse. Generally, two pulses were required to reach steady-state block of the open channel by TEA. Subsequent to steady-state block, a third set of depolarizations in the presence of both TEA and MTSEA was used to modify the cysteines (third panel). Each set consisted of four consecutive depolarizations to reduce the number of loading and washout pulses. After MTSEA modification, the TEA was washed out and the current from unmodified channels was recorded (currents 1 and 2, rightmost panel). The slow rising phase of current 1 reflects the unbinding of TEA, which had been locked behind the activation gate in the closed state, from the open channel. Three consecutive pulses were used to ensure complete removal of TEA from the channel and thus obtain the true peak current remaining after MTSEA modification. It is the ratio of this current to that obtained in panel 1 (leftmost panel) that yields the normalized current used in plots such as that shown in Fig. 8 B to calculate kmod. The modification reaction was monitored by sequential cycling through the last three panels shown in Fig. 1 A until all of the channels were modified.
Figure 1.

Pulse sequences for the determination of the blocker protection assay of TEA affinity. Macroscopic K+ currents were recorded in the inside-out patches expressing the Shaker double mutant T449A/I470C. The intracellular (bath) solution was exchanged using a step motor–driven fast perfusion apparatus, as described in Panyi and Deutsch (2006). (A) Blocker protection protocol in the open state. Patches were repeatedly depolarized from a holding potential of −100 to +50 mV for 12 ms every 14 s. The intracellular (bath) solution facing the patch during the current recording is indicated by the bars above the current traces. The sequence started with two pulses in KF solution (1st pulse set), followed by two pulses in KF solution containing 0.5 mM TEA (hatched bar, 2nd pulse set) to reach equilibrium block. This was followed by four pulses in a KF solution containing 0.5 mM TEA and 5 mM MTSEA (crosshatched bar, 3rd pulse set) to modify the cysteines. MTSEA and TEA were washed out completely during three pulses in KF solution (two of the three are shown, 4th pulse set) to give the current remaining after a cumulative MTSEA modification time of 48 ms (4 × 12 ms). The 2nd, 3rd, and 4th sets of pulses were repeatedly cycled until modification was complete. During the interpulse interval (14 s), the intracellular (bath) solution at the holding potential was KF for the 1st and 4th pulse sets and KF + 0.5 mM TEA for the 2nd and 3rd pulse sets. The application of intracellular solutions containing TEA and MTSEA was initiated 400 ms before the test pulse and terminated at the end of the depolarization. (B) Blocker protection protocol in the inactivated state. Patches were repeatedly depolarized from a holding potential of −100 to +50 mV for 1.8 s every 41.5 s. The intracellular (bath) solution facing the patch during the current recording is indicated by the bars above the current traces. The sequence started with three control pulses (left). The patch was depolarized in KF solution for 800 ms to induce full inactivation, and while depolarized the solution was changed to a KF solution containing 5 mM TEA (hatched bar) for 800 ms followed by a 200-ms-long period in KF solution at +50 mV and the repolarization to −100 mV. The patch was bathed in KF solution in between pulses. The second set of identical 1.8-s-long pulses to +50 mV was used to modify cysteines in the presence of TEA (right). The intracellular (bath) solution was KF for the first 800-ms-long segment, then switched to KF + 5 mM TEA solution for 200 ms (hatched bar), followed by a 600-ms-long period in KF + 5 mM TEA + 2 mM MTSEA (crosshatched bar). The intracellular (bath) solution then was switched to KF for 200 ms at +50 mV, which served to wash out TEA and MTSEA, and the patch was then repolarized to −100 mV. This protocol was repeated several times until the modification reaction reached completion. Trace labeled 4 shows the current measured during the 4th repetition of the protocol, the ratio of the peak current to the one measured from trace 1 gives the normalized current shown in Fig. 8 C.
Figure 8.

Affinity of intracellular TEA for the open and inactivated channel. (A) Open-channel block. Patches expressing T449K/I470C channels were repeatedly depolarized from a holding potential of −100 to +50 mV for 12 ms every 14 s. Traces labeled C and W indicate before and after TEA application, respectively. Traces labeled 2 and 3 indicate the second and third pulse recorded after switching to an intracellular (bath) solution containing 1 mM TEA in KF. Traces 2 and 3 superimpose, indicating equilibrium block has occurred. See Fig. 1 A and Materials and methods for additional protocol details. (B) Open-state blocker protection of MTSEA modification of I470C. Patches expressing T449K/I470C channels were exposed to MTSEA (5 mM) in the absence of TEA (empty down triangles) according to the protocol shown in Fig. 4 A. Modification in the presence of 0.5 mM TEA (filled down triangles) was according to the protocol given in Fig. 1 A. (C) Inactivated-state blocker protection. Patches expressing T449K/I470C channels were exposed to MTSEA (2 mM) in the absence of TEA (empty circles) according to the protocol shown in Fig. 4 B. Modification in the presence of 5.0 mM TEA (filled circles) was according to the protocol shown in Fig. 1 B. For both B and C, the peak currents were normalized to the peak of the first pulse, plotted as a function of the cumulative modification time, and the best fit of the data to single exponential functions was used to determine the indicated modification rate constants. (D) Dose–response curves for the interaction of intracellular TEA with open and inactivated channels. The dose–response relationship for the open state (empty up triangles) was obtained from current records shown in A, at TEA concentrations ranging from 0.01 to 20 mM. The fraction of unblocked channels was calculated as I/I0, where I and I0 are the peak currents after equilibrium block and in the absence of the blocker, respectively. The dissociation constant for the open state (Kd,O) was obtained by fitting the data to I/I0 = Kd/(Kd + [TEA]) (solid line). The right ordinate corresponds to the filled symbols, which represent data obtained from blocker protection assays in the open (filled down triangle) and inactivated (filled circle) states. The value of k mod/k mod,max is calculated at each TEA concentration as the ratio of the modification rate constant in the presence of TEA to that in the absence of TEA. The dissociation constant for the inactivated state (Kd,I) was obtained by fitting the k mod/k mod,max = Kd/(Kd + [TEA]) to the data points (solid line). The values of kmod/kmod,max and I/I0 are statistically the same for the interaction of 0.5 mM TEA with the open state (P = 0.36).
The protocol devised to measure inactivated-state protection is shown in Fig. 1 B and entails an initial depolarization to +50 mV for 800 ms in standard internal solution, followed by perfusion with TEA for 800 ms, and finally, perfusion for 200 ms in standard internal solution lacking TEA (left panel). Next, as depicted in the second panel (right panel), the patch was depolarized to +50 mV for 800 ms in standard internal solution, followed by perfusion for 200 ms with TEA alone and then perfusion for 600 ms with a solution that contained both TEA and MTSEA, followed by a return to standard internal solution containing neither reagent for a final 200 ms at +50 mV. The current elicited during this protocol is labeled “1.” A comparison of the peak current in the first panel to that of the second panel (current labeled 1) indicates that the last 200 ms of panel 1 is sufficient to washout TEA completely. Repolarization at −100 mV was sufficient to ensure full recovery of the inactivated, unmodified channels within the interpulse interval of 40 s. The modification reaction was monitored by sequential cycling through the protocol depicted in the second panel of Fig. 1 B until all of the channels were modified. For example, current labeled “4” is the fourth cycle through the protocol. The resulting currents were plotted as normalized peak current versus cumulative modification time, from which a modification rate constant could be extracted.
Analysis of Data
The cumulative modification time for the nth pulse is
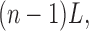 |
(1) |
where L is the duration of the MTS pulse. A single exponential function was fit to the data points, and the modification rate (ρ) was determined. In the simplest case, this rate is proportional to three terms, as follows:
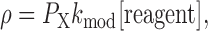 |
(2) |
where P X denotes the probability of being in the modifiable state, k mod is the second-order rate constant, and [reagent] is the MTSET, MTSEA, or Cd2+ concentration. In all cases, k mod was determined while the inactivation gate was in a steady-state closed conformation. Thus no correction was necessary to account for a time-dependent change in the probability of being in the inactivated state, with the activation gate open and the inactivation gate closed, i.e., P OI. If the activation gate was either opening or closing during this time, a correction for the change in P O or P OI (P X in Eq. 2) had to be made. Therefore P X was substituted by
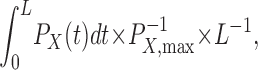 |
(3) |
where P X,max is the maximal occupancy of state X during reagent application and X represents either the O or the OI state. For C→O transitions, the value of Eq. 3 was calculated using P O(t) = P X(t) = (1 − e−t/τ)4 and P O,max = P X,max = 1, where τ is derived from fits of current traces elicited at +50 mV. Although P O,max may be <1, this will affect the estimates of open-state k mod uniformly. For the data shown in Figs. 3 A, 4 A, 5 A, and 9, individual correction factors were derived from the activation kinetics for each patch. Determination of k on for irreversible Cd2+ modification of 474C was made as described above using MTS reagents.
Figure 3.

Modification of cysteine 474 by Cd2+. Patches expressing T449A/V474C channels were repeatedly depolarized from a holding potential of −100 to +50 mV using the pulse protocols shown above the corresponding raw current traces. The timing and the duration (L) of the Cd2+ pulse are indicated by the hatched bars. (A) Open-state modification. 7-ms-long depolarizing pulses were applied every 15 s in the continuous presence of 20 μM Cd2+. After 35 pulses, <1% of the original peak current remained. The peak currents for each pulse were normalized to the peak current of the first pulse, and plotted as a function of the cumulative modification time, as described in the Analysis of Data section of the Materials and methods, Eq. 1, using 0 for both d and τe. The modification rate was corrected by multiplying by 1.27, a factor derived from Eq. 3 using the activation kinetics of the normalized current, P O(t) = (1 − e−t/τ)4 and P O,max = 1 (see Analysis of Data, Materials and methods). (B) Inactivated-state modification. 1.5-s-long depolarizing pulses were applied every 81.5 s and perfusion with 20 μM Cd2+ (L = 400 ms) was initiated 800 ms after the start of the depolarization. The peak currents for each pulse were normalized to the peak current of the first pulse and plotted as a function of the cumulative modification time. (C) Rate constants for 474C modification by Cd2+, MTSET, and MTSEA measured in the open and inactivated states. Data for MTSEA modification were obtained using identical protocols and analysis as described in A and B except the modification rate constants were corrected for the closed-state modification of 474C by MTSEA (1244 ± 94 M−1s−1, n = 2) and the exposure of the patch to MTSEA (40 μM) during the open-state modification assay was limited to 200 ms before the depolarization plus 7 ms during the depolarization. Data for MTSET modification of 474C are from Panyi and Deutsch (2006). Error bars indicate SEM for n = 2–9 independent measurements. (D) Open-to-inactivated state ratios of the rate constants. Ratios were calculated from data shown in C. Error bars for the ratios were calculated using the Gaussian error propagation function.
Cd2+ modification of 470C was reversible, and k off was estimated from washout experiments in which the fraction of recovered current was fit with a 1 − e−t/τ function. To further analyze the kinetics of Cd2+ modification of inactivated channels, we applied a prolonged depolarization to +50 mV and exposed the patch to Cd2+ after a 1-s delay (period I, Fig. 7 A) to ensure a low probability that channels were in the noninactivated open state. Subsequently Cd2+ was applied for a duration of 400, 600, or 800 ms (period II), and was then removed for 250 ms (period III) after which the voltage was returned to the holding potential (see Fig. 7). This pulse/perfusion protocol was repeated every 41.5 s to allow for complete recovery from inactivation between depolarizations. The protocol was repeated until the block reached a steady-state. We assume that the activation gate remained open throughout these prolonged depolarizations (Panyi and Deutsch, 2006).
To obtain estimates of k on and k off for Cd2+ block, we had to consider that Cd2+ could leave its binding site at any time the patch was depolarized and could enter the channel only during period II when the patch was exposed to the blocker. During each of the three periods of the depolarization, the probability of block is expected to change exponentially with rate [Cd2+]k on + k off in the presence of Cd2+, and k off during periods I and III. The predicted equilibrium level of block is [Cd2+]k on/([Cd2+] k on + k off) for period II and zero for periods I and III. Analysis of the development of block is iterative, with the final level of block of any period used as the initial level of block in the subsequent period. The measured levels of block during repeated exposures of Cd2+ were used to estimate the rate constants k on and k off by nonlinear regression using a variable metric algorithm.
RESULTS
We previously demonstrated a decreased rate of modification of a cysteine in the cavity of a slow-inactivated channel compared with a noninactivated, open channel (Panyi and Deutsch, 2006). Several changes in the cavity between these two states could account for these initial observations, namely, a conformational change in the cavity itself, a change in electrostatic potential in the cavity, ion occupancy in intracellular pore sites, and prevention of ion flux through the selectivity filter. To investigate these underlying causes, we first probed the cavity with the cysteine-binding reagent, Cd2+, to confirm our previous conclusion that the rate of MTSET modification of cysteine at position 474 (V474C) was a function of the side chain and its environment, rather than of the specific reagent used. MTS reagents undergo a disulfide exchange to produce a covalently linked MTS-cysteine adduct and a leaving group, methanesulfinate (Karlin and Akabas, 1998). Cd2+ associates noncovalently with cysteines. For these experiments we used a double mutant in the Shaker-IR background (Fig. 2; T449A in blue, V474C in yellow). Residue 449 is located in the outer mouth of the pore and an alanine in this position speeds entry into the slow-inactivated state (Lopez-Barneo et al., 1993; Panyi and Deutsch, 2006; Ray and Deutsch, 2006). Residue 474 faces into the water-filled cavity between the inactivation and activation gates, and accessibility of 474C to hydrophilic modifying reagents reliably tracks the status of the activation gate (Liu et al., 1997; Panyi and Deutsch, 2006). This double mutant was studied in inside-out patches from tsA201 cells and exposed to intracellular Cd2+ to obtain modification rate constants in the open state and the inactivated state. The protocols, designed to capture channels in either the open or the inactivated state, were based on our earlier findings that the channel inactivates with a time constant of ∼130 ms at +50 mV, deactivates with a time constant of 0.6 ms at −120 mV in the noninactivated state and 23 ms in the inactivated state, and recovers with a time constant of 10.6 s at −120 mV in a standard intracellular solution (Panyi and Deutsch, 2006). The closed channel is not modified by Cd2+ on the time scale of our protocols (unpublished data), consistent with previously reported studies on Shaker-IR (Liu et al., 1997). The kinetics of solution changes were determined for each patch (see Materials and methods), and this exchange time was used to calculate the correct cumulative modification time for Cd2+. In general, solution changes were 90% complete within 20 ms (Panyi and Deutsch, 2006).
Figure 2.

The pore region of a Kv channel. A ribbon representation of S5, S6, and the pore helix (red; residues 322–450) of Kv1.2 was made in DS ViewerPro (www.accelrs.com) from Long et al. (2005). Two subunits are shown and residues homologous to Shaker 449 (blue), 474 (yellow), and 470 (cyan) are depicted as space-filing atoms. The location of the activation gate is below residue 474 and the inactivation gate is located at the level of the selectivity filter. K+ ions are shown as green spheres.
Cd2+ Modification of 474C in the Open State
The rate constant for modification of 474C in the open channel was determined by brief (7 ms) depolarizations to +50 mV, repeated every 15 s, in the continuous presence of 20 or 50 μM Cd2+. The 7-ms duration is sufficient to fully activate channels, but is too short to induce significant inactivation (Panyi and Deutsch, 2006). As shown in Fig. 3 A for the open state, modification of V474C renders the channel nonconducting and is manifest as a gradual decrease of the peak currents upon repetitive pulsing in the presence of Cd2+. This is a consequence of the irreversible reaction of Cd2+ with one or more 474C side chains when the activation gate is open. The normalized peak current was plotted as a function of cumulative modification time. The drop in the peak current following the first pulse in Cd2+ (not depicted in figure) represents a rapid, reversible block, that is manifest as a 33 ± 1.8% decrease in the peak (n = 6, using 20 μM Cd2+), and was excluded from the fit. The average k mod of six replicate experiments is 1.8 × 106 ± 0.1 × 106 M−1s−1. These values are consistent with those obtained for V474C in the Shaker-IR/449V open channel (Webster et al., 2004). Modification rates were also determined using 50 μM Cd2+ (unpublished data) to yield a statistically similar k mod for the open state (P = 0.33): 1.7 × 106 ± 0.1 × 106 M−1s−1 (n = 3). Modification of 474C was irreversible, even when the Cd2+-modified channel was treated for 15 episodes at +50 mV with 1 mM 2,3-dimercapto-1-propane sulfonic acid (DMPS), a strong chelator of heavy metals. This is in contrast to the removal of Cd2+ from 474C in a 449V Shaker-IR background using 0.5 mM DMPS (Liu et al., 1997).
Cd2+ Modification of 474C in the Inactivated State
To evaluate the rate of Cd2+ modification of 474C in the inactivated channel, we depolarized a patch to +50 mV for 800 ms to fully inactivate the channels and then perfused with 20 μM Cd2+ for 400 ms (Fig. 3 B), followed by a 300-ms washout period at +50 mV while the activation gate was still open (Panyi and Deutsch, 2006). The patch was repolarized to −100 mV for 80 s and the protocol repeated to give the decaying current traces shown in Fig. 3 B. The decrement of peak current was much slower for the inactivated channel (Fig. 3, compare B with A). In the inactivated channel, there is no detectable reversible block because the open channel is never exposed to Cd2+. In this case, all of the decrease in peak current is due to Cd2+ modification of 474C. The average k mod of five replicate experiments is 1.04 × 105 ± 0.15 × 105 M−1s−1, and is presented in Fig. 3 C along with the MTSET results taken from Panyi and Deutsch (2006).
The ratio of these rate constants for the open state compared with the inactivated state is shown in Fig. 3 D for both MTS and Cd2+ modification. Both reagents reveal a dramatic state dependence of modification; the inactivated state is ∼17-fold slower than the open state. A similar ratio of open-to-inactivated state modification rates was obtained with another MTS reagent, MTSEA (Fig. 3, C and D). MTSEA irreversibly modified 474C to give modification rates of 3.41 × 105 ± 0.49 × 105 M−1s−1 (n = 2) and 0.18 × 105 ± 0.02 × 105 M−1s−1 (n = 3) for the open and inactivated states, respectively. The increased absolute rates of MTSEA versus MTSET in both the open and inactivated states may reflect the smaller size of MTSEA (3.6 Å) compared with MTSET (5.8 Å) or residual uncorrected hydrolysis of MTSET. Fig. 3 D raises the possibility that the activation gate is open only a fraction of the time when the channel is inactivated (i.e., the activation gate flickers) to account for the observed decrease in k mod. This possibility is unlikely, as described below.
Local versus Global Effect on State-dependent Modification
The large decrease in modification rates at position 474 in the cavity could reflect a change localized near the activation gate, particularly at the PVP bend in S6 (del Camino et al., 2000; Long et al., 2005), or it could reflect a more global change in the cavity. To explore this issue, we investigated a site deeper in the cavity (more proximal to the selectivity filter), namely, at position 470. This residue is only 3–4 Å from the T441, the C-terminal residue of the pore helix (βcarbon-to-βcarbon distance, derived from the structure of Kv1.2; Long et al., 2005). A mutation of isoleucine to cysteine at 470, however, inhibits slow inactivation (Holmgren et al., 1997; Olcese et al., 2001). To compensate for this phenotype and ensure reasonable slow inactivation rates, we introduced a second mutation, T449K, which enhances the rate of slow inactivation (Lopez-Barneo et al., 1993; Ray and Deutsch, 2006). This double mutant, 449K/470C Shaker-IR, has inactivation kinetics that are ideal for our experiments, e.g., ∼120 ms. As measured in our normal internal and external solutions (see Materials and methods), this double mutant has activation and inactivation time constants of 0.58 ± 0.03 ms (n =18) and 118.0 ± 3.4 ms (n = 32), respectively, measured at +50 mV. The deactivation kinetics time constants were 0.50 ± 0.02 ms (n = 16) and 1.04 ± 0.07 ms (n = 8), measured at −120 mV and −100 mV, respectively. The inactivated channel recovers completely within 40 s at −100 mV. Residue 470C can be modified by MTSEA but not by MTSET (Liu et al., 1997). Therefore, we used MTSEA in all of our MTS experiments with the 470C mutant. Open-state modification by MTSEA (Fig. 4 A) is slower than MTSET modification of 474C (compare Fig. 3 C and Fig. 4 A), but gives complete modification and excellent fits of the data to yield a k mod for the open state of 3.9 × 103 M−1s−1. The average of five replicate experiments is 3.7 × 103 ± 0.3 × 103 M−1s−1. Modification of the closed state at −100 mV causes only ∼20% modification following 8 s of MTSEA exposure and <5% within the first second of exposure (unpublished data). Thus, closed-state modification does not complicate our analysis of modification of the open and inactivated states because, as shown in Fig. 4 (A and B), modification in these states is complete within 0.3–0.6 s. Modification of the inactivated state is slower than the open state and gives a k mod of 0.59 × 103 M−1s−1. The average of five replicate experiments is 0.66 × 103 ± 0.04 × 103 M−1s−1. The inactivated state modification is approximately sixfold slower than the open state modification. We conclude that a structural change occurs in the cavity of the inactivated conformation of the channel to cause a slower modification of side chains pointing into the cavity and that this change entails more than just a local rearrangement in the immediate vicinity of the activation gate.
Figure 4.

Modification of cysteine 470 by MTSEA. Patches expressing T449K/I470C channels were repeatedly depolarized from a holding potential of −100 to +50 mV using the pulse protocols shown above the corresponding raw current traces. The timing and the duration (L) of the MTSEA pulse are indicated by the hatched bar. (A) Open-state modification. 12-ms-long depolarizing pulses were applied every 12 s in the presence of 2 mM MTSEA. Perfusion with MTSEA started 200 ms before each depolarizing pulse and the patch was perfused with KF solution for the remaining time at the interpulse holding potential. Every fourth pulse is shown. After 35 pulses, <1% of the original peak current remained. The peak currents for each pulse were normalized to the peak current of the first pulse, corrected, and plotted as a function of the cumulative modification time, as described in Fig. 3 A. (B) Inactivated-state modification. 1.8-ms-long depolarizing pulses were applied every 41.5 s and perfusion with 2 mM MTSEA (L = 600 ms) was initiated 1,000 ms after the start of the depolarization. Peak currents for each pulse were normalized to the peak current of the first pulse and plotted as a function of the cumulative modification time.
Cd2+ Modification of 470C
The decreased modification rate for the inactivated state could reflect a structural change in the vicinity of 474 that restricts access to 474C. One possibility is a constricted opening at the bundle crossing. This would predict a higher energy barrier for movement of an MTS reagent or Cd2+ into the cavity, which would manifest as a slower on-rate for the modifier. This scenario also predicts a slower off-rate if the modifier interacts reversibly with a target cysteine residue. Thus, a structural change that only raises the energy barrier for entry into and exit from the cavity would produce a decrease in modification rates of irreversible modifiers such as MTS reagents without a change in the equilibrium dissociation constant, Kd, of reversible modifiers or blockers. A similar prediction applies to the case of a decreased open probability of the activation gate (flicker) for the inactivated versus noninactivated channel. To test this hypothesis, we studied the affinity of Cd2+ at 470C in the 449K mutant, which, as demonstrated below, is a reversible modifier at position 470 in this double mutant (see Figs. 5–7 ; Table I).
TABLE I.
Cd2+ Modification of I470C
| Open Statea | ||
| k on (104 M−1s−1) | 3.36 ± 0.25 (n = 4) | |
| k off (s−1) | 0.18 ± 0.03 (n = 4) | |
| Kd (μM) | 5.47 | |
| Inactivated State | ||
| k on (104 M−1s−1) | 4.8 ± 0.2 (n = 15)b | 5.2d (n = 1) |
| k off (s−1) | 0.083 ± 0.004 (n = 15)b | 0.072 ± 0.002c (n = 16) |
| Kd (μM) | 1.7b | 1.4b c |
| Ratio of O/I | ||
| k on | 0.70 ± 0.06 | 0.65 |
| k off | 2.22 ± 0.36 | 2.53 ± 0.39 |
| Kd | 3.21 | 3.9 |
Determined from τmod = 1/(k on + k off) and R = k off /(k on + k off), as described in the text.
Values were determined from an analytical fit considering binding and unbinding of Cd2+ during periods I–III, as described in the text and Fig. 7 A.
Values were determined from washout experiments as described in text and Fig. 6.
Values were determined from “single period” experiment (Fig. 7 B).
Fig. 5 shows the Cd2+ modification 470C as a decline in current amplitude during successive depolarizations of the appropriate protocols used to study open (Fig. 5 A) and inactivated (Fig. 5 B) channels. For the open state, a plot of normalized current versus cumulative modification time was fit with a single exponential to give a decay constant (apparent k on, which is equal to k on × [Cd2+] + k off) of 4.67 × 104 M−1s−1, the average being 4.21 ± 0.33 M−1s−1 (n = 4). We report this decay as an apparent k on because this is a reversible reaction (notice that there is residual normalized current, R, at steady state) and thus both k on and k off together contribute to the decay observed. We calculate k on from the relationship that the time constant of decay equals (k on + k off)−1 and that the residual normalized current, R, is equal to k off/(k on + k off). The average k on is 3.36 × 104 ± 0.25 × 104 M−1s−1 (n = 4). The modification curves for the inactivated channel (Fig. 5 B, right panel) also revealed a nonzero value of R. Moreover, the steady-state level of modification depends on the duration of exposure to Cd2+ during a single depolarizing pulse. A 400-ms exposure to 20 μM Cd2+ produced 0.24 ± 0.02 (n = 5) fraction of unmodified channels, i.e., R is ∼0.24, significantly different (P = 0.004, ANOVA, all pairwise multiple comparison using the Student-Newman-Keuls method) from 0.18 ± 0.01 (n = 5) and 0.17 ± 0.01 (n = 5) fraction of unmodified channels produced from a 600-ms and 800-ms pulse, respectively. The apparent modification rate for each Cd2+ exposure period was, however, not significantly different (P = 0.74, ANOVA) and was ∼1.2 s−1 (n = 5 in each group).
These observations are consistent with reversible Cd2+ modification at 470C. To demonstrate this more explicitly, we measured both the on- and off-rates for Cd-binding to 470C. First, we measured the off-rates. The off-rate for the open state was calculated from the apparent modification curves similar to the one shown in Fig. 5 A by considering both the steady-state level and the decay of the peak currents, as described above. The average k off is 0.18 ± 0.03 s−1 (n = 4). To determine k off for the inactivated state, a patch was first treated with Cd2+ for 0.4–5.0 s per episode at +50 mV until equilibrium block was reached and then perfused with Cd2+-free bath solution for a washout period of 0.6–6.2 s per episode while the patch was maintained at +50 mV. Cd2+ comes off its binding site when the activation gate is open and no Cd2+ is present in the bath. The total amount of time that the activation gate is open will determine the fraction of Cd2+ washed out. Hence, shorter pulse durations at +50 mV require more episodes to completely wash out Cd2+. The current was recorded during successive washout periods, normalized to the final recovered current, plotted as a function of cumulative pulse duration and fit with a single exponential function to give a value of k off. The average k off is 0.072 ± 0.002 s−1 (n = 16). The k off is independent of both the duration of the loading pulse (Fig. 6 A, P = 0.82, ANOVA) and the duration of the washout pulse (P = 0.91, ANOVA; Fig. 6 B), consistent with the absence of multiple, deeper inactivated states with different affinities for Cd2+. The open-to-inactivated ratio of the off rates is 2.53 ± 0.39.
The on-rates for the inactivated state were more complicated to determine due to the substantial amount of time that the Cd2+-bound channel was exposed to Cd2+-free bath while the activation gate was still open at +50 mV (see Fig. 7 A). During this time, the Cd2+ can unbind and thus, a simple calculation from the decay of the peak currents measured upon consecutive application of Cd2+ to the inactivated state (Fig. 5 B) cannot produce a true k on for the inactivated state. We took two approaches to solve this problem. First, we designed an experimental protocol to permit direct measurement of k on. Second, we derived an analytical solution based on the model depicted in Fig. 7 A. In the first case, the experimental protocol entailed recording the peak current left after only one cycle through periods I, II, and III (see Fig. 7 A) during a depolarization in which Cd2+ solution (20 μM) was present during period II only. We performed this “first episode only” protocol for different period II durations and plotted the decay of the normalized peak current as a function of pulse duration of period II (Fig. 7 B). A single exponential function was fit to the curve to give an apparent rate constant of 1.11 s−1. Because this apparent rate constant is the sum of k on × [Cd2+] + k off, we could use the koff determined from data in Fig. 6, i.e., 0.072 s−1 (see above) to calculate k on. The value of k on is 5.2 × 104 M−1s−1. In addition, we developed an analysis accounting for the progressive modification during repetitive cycles of periods I–III (i.e., yielding traces similar to those in Fig. 5 B (see Materials and methods). This analysis gives estimates of both k on and k off, 4.8 × 104 ± 0.2×104 M−1s−1 and 0.083 ± 004 s−1 (n = 15), respectively. The results of this analysis are shown in Table I, along with estimates of k on and k off from the washout (Fig. 6 A) and “first episode only” experiments (Fig. 7 B). The agreement is excellent (P > 0.1, Mann-Whitney rank sum test) and confirms our assessment of k on and k off for the inactivated state. Both k on and k off are indeed independent of pulse duration (P = 0.4, ANOVA). A comparison of k on values for the open and inactivated states indicates a ratio of 0.65 from the washout and “first episode only” experiments, and a ratio of 2.53 ± 0.39 for the k off values for the open compared with the inactivated states. The decreased ratio for the k on values (0.65) eliminates the possibility that decreased access to cavity sites is due only to increased flicker of the activation gate in the inactivated state. Other mechanisms must be present. Assuming that no change in shape (Hill coefficient) occurs for the binding curve for Cd2+ at position 470, a Kd, i.e., k off/k on, may be approximated from these rate constants for the open and inactivated states, the caveat being that we have only determined these rate constants from a limited number of Cd2+ concentrations. Nonetheless, the open-to-inactivated state ratio of Kd is ∼4, inconsistent with a change in energy barrier alone between the two states. A more precise determination of the Kd, e.g., obtaining a complete binding curve, is technically precluded because of the inordinate number of exposures that would be required to measure modification at lower Cd2+ concentrations. For this reason, we turned to a well-studied reversible class of blockers, quaternary ammoniums (QAs), to compare affinities in the open and inactivated states.
Blocker Affinity of the Inactivated Conformation
The experimental strategy was derived from several considerations. First, we needed to select a blocker with appropriate kinetics relative to the inactivation kinetics of the channel. Second, we needed to engineer a channel with appropriate kinetics. Third, given that block cannot directly be assessed in the inactivated state because there is no current to monitor, we used a blocker protection assay. This assay reveals the extent to which an intracellular blocker, binding inside the cavity, inhibits modification of a cysteine at a nearby cavity site (del Camino et al., 2000). Inhibition, for example, of MTS modification of a cavity cysteine can be evaluated as a function of blocker concentration to determine the equilibrium dissociation constant of intracellular blocker in the inactivated state. At high blocker concentration, the cysteine will be completely protected from MTS modification. At low blocker concentration, the cysteine will be modified. For this purpose, we first investigated 449A/474C because residue 474C was used by Yellen and coworkers to study blocker protection of the open state in a 449V Shaker-IR channel (del Camino et al., 2000). TEA, tetrapropylammonium (TPrA), and tetrabutylammonium (TBA) blocked the open state of 449A/474C, but none were ideal for both the open and inactivated state measurements. TEA and TPrA were fast enough to reach equilibrium block before onset of inactivation, a requirement for the open-state determination, but neither caused dramatic protection of 474C modification in the inactivated state. TBA, although able to protect in the inactivated state, was too slow for use in the open state (unpublished data). The relative potencies on blocker protection are consistent with previous studies indicating that TEA likely binds in the vicinity of residue 441 at the bottom of the pore helix, whereas more hydrophobic QAs bind in the vicinity of residue 469 in S6 (Baukrowitz and Yellen, 1996). Thus, we tested the blocker protection strategy using a different residue in the cavity, 470C, which we reasoned was closer to the putative internal TEA blocking site and therefore would be more sensitive to protection by TEA.
Two features of TEA interaction with the channel make it ideal for determining the Kd of the open state using a blocker protection strategy. First, as shown in Fig. 8 A, onset of block is fast (a marked decrease in current is already manifest by trace 2). Second, the fraction of block at the end of trace 2 is the same as the beginning of trace 3, i.e., equilibrium block is reached quickly. Third, TEA is trapped in the closed state and washes out only upon reopening of the activation gate (see Fig. 1 A, rightmost panel, trace 2 vs. trace 1). Trapping of TEA in closed states of T449V/I470C has been studied previously (Holmgren et al., 1997; Melishchuk and Armstrong, 2001). To determine a Kd for the open state, we first used the traditional method of simply measuring the fraction of residual unblocked current recorded as a function of TEA concentration (e.g., Fig. 8 A). The Kd, determined from these measurements (Fig. 8 D) is 0.12 mM, in excellent agreement with ∼0.16 mM, the value previously reported for Shaker-IR 449V/470C (Holmgren et al., 1997; Melishchuk and Armstrong, 2001).
Our next task was to determine the Kd for TEA in the open state using blocker protection and compare it to the results obtained by simple, traditional open-state block of the current. For open-state measurement using blocker protection, the patch was exposed to 0.5 mM TEA and 5 mM MTSEA continuously (protocol described in Fig. 1, Materials and methods). Under these conditions, there was no significant closed-state modification (unpublished data). The normalized current was plotted as a function of cumulative modification time and fit with a single exponential (Fig. 8 B) to give a k mod of 2.85 × 103 and 1.02 × 103 M−1s−1 in the absence and presence, respectively, of TEA. A comparison of modification rates for 470C by MTSEA in the absence of TEA (Fig. 4 A) and the presence of 0.5 mM TEA will estimate the fraction of open-state block. The ratio of these rates is 0.28 ± 0.02 (n = 9). The fraction of open-state block can also be calculated directly from the results of simple current measurements similar to those shown in Fig. 8 A to give 0 0.25 ± 0.01 (n = 4). The two values for fractional block are virtually identical (P = 0.36), verifying the validity of the blocker protection assay as an accurate means of determining Kd.
Having verified the blocker protection assay for the open state, we could now apply this assay to the inactivated state, as described in Fig. 1 B (Materials and methods). The plot of normalized current versus cumulative modification time yields a k mod of 0.61×103 and 0.17×103 M−1s−1, respectively, in the absence and presence of 5 mM TEA (Fig. 8 C). Repeating these measurements for a range of TEA concentrations (0.5 mM to 20 mM), we obtained a Kd for the inactivated state of 2.0 mM (Fig. 8 D), ∼17-fold lower affinity compared with the open state.
A Test of the Reduced Flux Model
What underlies the altered modification rates and blocker affinities determined for the inactivated state? One possibility (see Discussion for additional possibilities) is that the reduced ion flux through the selectivity filter in the inactivated channel could be responsible. For example, a positively charged modifier will be repelled by cations in the permeation path, e.g., K+ (Crouzy et al., 2001), thus slowing entry of these modifiers into the cavity. In a noninactivated open channel, there are two exit paths for the K+. It can escape via an open activation gate and an open selectivity filter. In the inactivated state, only the former path is available. To test the influence of ion flux through the selectivity filter, we studied open-state MTSET modification by using 50 mM K+ or 160 mM Cs+ in the bath. In both cases, the predicted driving force and single-channel conductance through the pore are reduced (Heginbotham and MacKinnon, 1993; Ray and Deutsch, 2006). If reduced ion flux causes a dramatic decrease in open-to-inactivated ratios for modification rates and affinities, then the open-state k mod should be reduced. As shown in Fig. 9, there is no significant (P = 0.88) difference in k mod values for control (160 mM K+), 50 mM K+, or 160 mM Cs+.
Figure 9.

Effect of reduced ion flux on modification of 474C by MTSET. Patches expressing T449A/V474C channels were repeatedly depolarized from a holding potential of −100 to +50 mV for 7 ms every 15 s in KF solution to monitor the peak currents. Two monitoring pulses were followed by two MTSET (200 μM) pulses in intracellular (bath) solutions containing 160 mM K+, 50 mM K+, or 160 mM Cs+. This sequence of pulses was cycled until complete loss of the current occurred. The normalized current vs. cumulative modification time relationship was constructed and analyzed as described in Figs. 3–5 for open-state modification. The remaining fractions of the current in 50 mM K+ and 160 mM Cs+ solution were normalized to 160 mM K+ to give 0.066 ± 0.004 (n = 8) and 0.011 ± 0.002 (n = 6), respectively.
DISCUSSION
The mechanism of slow inactivation, as well as the structural conformations accompanying entry into and exit from the slow-inactivated state(s), remains relatively unknown. The few structural insights are mostly restricted to the selectivity filter (Lenaeus et al., 2005; Cordero-Morales et al., 2006) and even here, it is unclear which state of the channel is represented. Although we describe slow inactivation as a local rearrangement of the outer mouth of the channel (Choi et al., 1991; Yellen et al., 1994; Liu et al., 1996; Harris et al., 1998), specifically involving residues in the selectivity filter and extracellular turrets of the pore, other regions of the channel could rearrange upon slow inactivation of the channel. We have investigated this possibility for the cavity of Shaker-IR. Cavity sites interact with the N-terminal tethered blocker involved in Shaker fast inactivation (Hoshi et al., 1990; Zhou et al., 2001a) and in KIR channels, cavity-facing side chains govern conduction properties (Bichet et al., 2006; Grabe et al., 2006).
Our investigation of the Shaker cavity suggests its conformation is changed in the slow-inactivated state compared with the open state, and that the affinity of a cavity binding site for TEA is decreased. This bears on two issues. First, an understanding of cavity transformations is key to targeted drug design. The development of drugs that bind preferentially to a slow-inactivated state could offer a distinct advantage, increasing efficacy and minimizing toxicity due to side effects. Second, rearrangements in the cavity may underlie coupling between activation and inactivation gates (Panyi and Deutsch, 2006).
The Experimental Approach
We probed the cavity by evaluating the accessibility of specific pore-facing residues to cysteine-modifying reagents and the affinity of the intracellular TEA binding site. Accessibility was determined with MTS reagents and Cd2+, which each react with ionized cysteine side chains, but by different mechanisms (disulfide exchange versus noncovalent association, respectively). The coordination chemistry of Cd2+ varies from 2 to 8 (Anderson, 1984). Cd2+ is small (Pauling radius = 0.97Å), similar in size to a Na+ but with a higher hydration energy due to its 2+ charge (Marcus, 1997). The MTS reagents, by contrast, are larger (3.6 and 5.8 Å in diameter). The lack of spontaneous dissociation of a cysteine–Cd complex (i.e., relatively high affinity binding) has been interpreted as reflecting Cd2+ bound simultaneously to three or more cysteines (Liu et al., 1997). For Shaker-IR T449A/V474C, we observe irreversible Cd2+ binding in both the open and inactivated states, even after treatment with DMPS, suggesting ≥3 cysteines coordinate with Cd2+. Liu et al. (1997) observed similar irreversibility for Shaker-IR T449V/V474C, but release with DMPS. Sensitivity to DMPS may reflect the different residue at position 449, for unknown reasons.
By contrast, modification of I470C is reversible, suggesting that one Cd2+ binds to two cysteines at position 470 (Liu et al., 1997). This different stoichiometry could be due to (a) steric/orientation differences (perhaps the cavity in the vicinity of 470 is not optimal for four cysteines pointing into the pore at the angles and distances required for tetracoordination), and/or (b) electrostatic repulsion. The ionized cysteines may be too close and repel each other. At position 474, such inhibition might be overcome by small shifts in orientation if this region is relatively flexible, whereas distortion proximal to a more rigid pore (near 470) might be too costly. In general, we do not know the stoichiometry of the Cd2+ modification reactions (nor the MTS-modification reactions), however, Cd2+ binding to multiple cysteines does dictate several constraints in distance and geometry (Loussouarn et al., 2001; Bruhova and Zhorov, 2005).
In addition to accessibility studies with MTS reagents and Cd2+, we determined the affinity of the cavity for intracellular TEA in the open and inactivated states. This required a novel strategy because the inactivated channel does not conduct. Blocker protection of cysteine modification was the method of choice and has been used to study an open Shaker-IR channel that does not exhibit slow inactivation (T449V; del Camino et al., 2000). TEA, at its binding site, blocks MTS modification at I470C, and physical occlusion of the 470 site (i.e., steric overlap) is the likely cause of this inhibition (del Camino et al., 2000).
TEA binds at a site close to the bottom of the selectivity filter (Yellen et al., 1991) and S6 (Choi et al., 1993; Baukrowitz and Yellen, 1996; Zhou et al., 2001a; Lenaeus et al., 2005). The TEA binding affinity is sensitive to mutations in 441, a residue located at the internal end of the selectivity filter (Yellen et al., 1991; Choi et al., 1993; Baukrowitz and Yellen, 1996). The analogous residue in Kv1.2 (373) is within 3–4 Å of 402 (analogous to 470 in Shaker), but 11 Å away from 406 (analogous to 474 in Shaker). These distances are consistent with the observation that TEA blocks MTS modification completely at 470C but not at 474C (unpublished data; del Camino et al., 2000). The size of the cavity may change depending on the side chain at 470 (Melishchuk and Armstrong, 2001). For example, the smaller side chains alanine and cysteine permit the channel to trap a QA in the cavity with the gate closed (Holmgren et al., 1997; del Camino and Yellen, 2001), and this ability likely derives from the size of the cavity (Melishchuk and Armstrong, 2001).
State-dependent Changes in the Cavity
What accounts for the large change in O/I ratios of accessibility? At least five hypotheses may be considered. The first possibility is that the electrostatic potential in the cavity is altered in the inactivated state, which would affect the probability that a reagent/blocker is located in the vicinity of its binding site. In turn, this would affect the modification rate or blocker affinity. The second possibility is that there is a change in ion occupancy in the open versus inactivated states of the channel. The third hypothesis is that the pKa of the cysteine is state dependent. For example, inactivation might cause a change in electron-withdrawing or -donating ability that propagates along the cysteine side chain (Deutsch and Taylor, 1987; Deutsch and Taylor, 1989). The fourth candidate is that reduced flux in the inactivated state, due to a collapsed selectivity filter, is responsible for the altered modification kinetics or TEA equilibrium because the permeant cation in the pore can only escape via one pathway (vis-à-vis two pathways in the open state), through the open activation gate. Finally, a change in conformation of the cavity in the inactivated state could render the cysteine less accessible or the TEA binding site energetically less favorable.
We favor the last mechanism, namely, a conformational change in the cavity, for the following reasons. Pure electrostatics cannot account for the 17-fold decrease in modification kinetics for the inactivated versus open state at 474. For the case of a monovalent cationic reagent, e.g., MTSET and MTSEA, a change in electrostatic potential is exactly balanced by a change in local pH, which changes the concentration of ionized (reactive) thiol (Elinder et al., 2001). According to the formulation of Elinder et al. (2001), only a change in accessibility will change the rate of MTSET modification, not a change in electrostatics. Moreover, if there were a change in electrostatics, then we would have expected a different (but in the same direction) O/I ratio of modification rates for divalent Cd2+. The almost identical O/I ratios for modifying 474C by MTS reagents and Cd2+ makes it likely that electrostatics alone cannot account for the observed O/I ratios. For 470, the k on O/I ratio for MTSEA and Cd2+ are in opposite directions (O/I is 6 for MTSEA and 0.7 for Cd2+), which is inconsistent with a pure electrostatic mechanism. The second possibility, a change in ion occupancy can be eliminated because, through either steric or electrostatic effects, a change in ion occupancy would have the same directional effects on k mod and k on for MTSEA and Cd2+, respectively. But they don't at 470C. The third hypothesis, a change in pKa in the absence of a change in local electrostatics is highly implausible given that there is no other obvious means of altering the electron donating/withdrawing character of the cysteine side chain. The fourth hypothesis, a reduced flux mechanism, is eliminated by our experiments in 50 mM external K+ or in 160 mM external Cs+. In addition, del Camino and Yellen (2001) mimicked closure of the inactivation gate by Ba2+ blockade (Neyton and Miller, 1988; Jiang and MacKinnon, 2000) and found that Ba2+ occlusion caused no difference in Cd2+ modification rates of the open channel. Our results using 50 K+ and 160 Cs+ are consistent with the conclusions of del Camino and Yellen and suggest factors other than reduced flux are responsible for changes in the O/I ratios. Furthermore, these results bear on the argument that differences in ion occupancy do not account for the altered modification rates/blocker affinities in the inactivated state. Ion occupancy of the cavity depends on both ion entry and exit rates. The latter is likely to be slower for Cs+ compared with K+, yet k mod is unchanged. A final consideration is that a change in the O/I ratios might result from a change in stoichiometry of the modification reaction: only a few cysteines are modified in the open state to decrease current, but more cysteines are required to get the same fractional decrease in the inactivated state. Regardless, this implies a conformational change in the cavity.
Based on these arguments, we conclude that the major factor that differs between open- and inactivated-state cavities is a conformational change in the cavity behind the activation gate and that this underlies a change in accessibility when the channel inactivates. There is precedent for small changes in conformation of the cavity. A single point mutation, I-to-C or I-to-A at 470, can make a Shaker-IR channel that does not trap quaternary ammoniums into one that does (Holmgren et al., 1997; Melishchuk and Armstrong, 2001), suggesting a change in volume of the cavity. We propose that such small conformational changes are also manifest in different gating states. Moreover, conformational changes may propagate into other regions of the channel.
Finally, we would like to speculate that conformational changes in the cavity bear on the mechanism by which the activation and inactivation gates are coupled (Panyi and Deutsch, 2006). If S6 moves as a rigid body and pore opening involves simultaneous changes in the bundle crossing and the upstream region of S6 that contacts the pore helix (Perozo et al., 1999; Yifrach and MacKinnon, 2002), then inactivation can cause changes all along S6, with concomitant changes in cavity conformation, and consequently, on activation gate movement. Complementary evidence has emerged from two recent studies to support this hypothesis (Cuello, L.G., S. Chakrapani, J.F. Cordero-Morales, and E. Perozo. 2007. Biophys. J. 189a; Sadovski, Y., and O. Yifrach. 2007. Biophys. J. 617a). Such a propagated conformational change would alter the accessibility of residues pointing into the cavity.
Acknowledgments
We thank R. Horn, T. Hoshi, and Clay Armstrong for critical reading of the manuscript, and R. Horn for writing the Fortran program, use of his Fortran compiler, and invaluable advice.
This work was supported by National Institutes of Health grants GM 069837 and NS 052665, Hungarian Ministry of Health ETT 068/2006, and Hungarian National Research Fund OTKA K 60740. G. Panyi is a Bolyai Fellow.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: DMPS, 2,3-dimercapto-1-propane sulfonic acid; MTSEA, ethylammonium methanethiosulfonate; MTSET, ethyltrimethylammonium methanethiosulfonate; QA, quaternary ammonium.
References
- Andersen, O. 1984. Chelation of cadmium. Environ. Health Perspect. 54:249–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz, T., and G. Yellen. 1996. Two functionally distinct subsites for the binding of internal blockers to the pore of voltage-activated K+ channels. Proc. Natl. Acad. Sci. USA. 93:13357–13361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichet, D., M. Grabe, Y.N. Jan, and L.Y. Jan. 2006. Electrostatic interactions in the channel cavity as an important determinant of potassium channel selectivity. Proc. Natl. Acad. Sci. USA. 103:14355–14360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhova, I., and B.S. Zhorov. 2005. KvAP-based model of the pore region of Shaker potassium channel is consistent with cadmium- and ligand-binding experiments. Biophys. J. 89:1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K.L., R.W. Aldrich, and G. Yellen. 1991. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc. Natl. Acad. Sci. USA. 88:5092–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K.L., C. Mossman, J. Aube, and G. Yellen. 1993. The internal quaternary ammonium receptor site of Shaker potassium channels. Neuron. 10:533–541. [DOI] [PubMed] [Google Scholar]
- Cordero-Morales, J.F., L.G. Cuello, Y. Zhao, V. Jogini, D.M. Cortes, B. Roux, and E. Perozo. 2006. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 13:311–318. [DOI] [PubMed] [Google Scholar]
- Crouzy, S., S. Berneche, and B. Roux. 2001. Extracellular blockade of K+ channels by TEA: results from molecular dynamics simulations of the KcsA channel. J. Gen. Physiol. 118:207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Camino, D., M. Holmgren, Y. Liu, and G. Yellen. 2000. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 403:321–325. [DOI] [PubMed] [Google Scholar]
- del Camino, D., and G. Yellen. 2001. Tight steric closure at the intracellular activation gate of a voltage-gated K+ channel. Neuron. 32:649–656. [DOI] [PubMed] [Google Scholar]
- Deutsch, C.J., and J.S. Taylor. 1987. Intracellular pH as measured by 19F NMR. Ann. N. Y. Acad. Sci. 508:33–47. [DOI] [PubMed] [Google Scholar]
- Deutsch, C.J., and J.S. Taylor. 1989. New class of 19F pH indicators: fluoroanilines. Biophys. J. 55:799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, S., and R. Horn. 2002. Tail end of the s6 segment: role in permeation in Shaker potassium channels. J. Gen. Physiol. 120:87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinder, F., R. Mannikko, and H.P. Larsson. 2001. S4 charges move close to residues in the pore domain during activation in a K channel. J. Gen. Physiol. 118:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabe, M., D. Bichet, X. Qian, Y.N. Jan, and L.Y. Jan. 2006. K+ channel selectivity depends on kinetic as well as thermodynamic factors. Proc. Natl. Acad. Sci. USA. 103:14361–14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanner, M., B. Green, Y.D. Gao, W.A. Schmalhofer, M. Matyskiela, D.J. Durand, J.P. Felix, A.R. Linde, C. Bordallo, G.J. Kaczorowski, et al. 2001. Binding of correolide to the K(v)1.3 potassium channel: characterization of the binding domain by site-directed mutagenesis. Biochemistry. 40:11687–11697. [DOI] [PubMed] [Google Scholar]
- Harris, R.E., H.P. Larsson, and E.Y. Isacoff. 1998. A permanent ion binding site located between two gates of the Shaker K+ channel. Biophys. J. 74:1808–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heginbotham, L., and R. MacKinnon. 1993. Conduction properties of the cloned Shaker K+ channel. Biophys. J. 65:2089–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren, M., M.E. Jurman, and G. Yellen. 1996. N-type inactivation and the S4-S5 region of the Shaker K+ channel. J. Gen. Physiol. 108:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren, M., P.L. Smith, and G. Yellen. 1997. Trapping of organic blockers by closing of voltage-dependent K+ channels. Evidence for a trap door mechanism of activation gating. J. Gen. Physiol. 109:527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi, T., W.N. Zagotta, and R.W. Aldrich. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:533–538. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., and R. MacKinnon. 2000. The barium site in a potassium channel by x-ray crystallography. J. Gen. Physiol. 115:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurman, M.E., L.M. Boland, Y. Liu, and G. Yellen. 1994. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques. 17:876–881. [PubMed] [Google Scholar]
- Karlin, A., and M.H. Akabas. 1998. Substituted-cysteine accessibility method. Methods Enzymol. 293:123–145. [DOI] [PubMed] [Google Scholar]
- Koo, G.C., J.T. Blake, K. Shah, M.J. Staruch, F. Dumont, D. Wunderler, M. Sanchez, O.B. McManus, A. Sirotina-Meisher, P. Fischer, et al. 1999. Correolide and derivatives are novel immunosuppressants blocking the lymphocyte Kv1.3 potassium channels. Cell. Immunol. 197:99–107. [DOI] [PubMed] [Google Scholar]
- Kozak, M. 1991. Structural features in eukaryotic mRNAs that modulate the initiation of translation. J. Biol. Chem. 266:19867–19870. [PubMed] [Google Scholar]
- Larsson, H.P., O.S. Baker, D.S. Dhillon, and E.Y. Isacoff. 1996. Transmembrane movement of the Shaker K+ channel S4. Neuron. 16:387–397. [DOI] [PubMed] [Google Scholar]
- Lenaeus, M.J., M. Vamvouka, P.J. Focia, and A. Gross. 2005. Structural basis of TEA blockade in a model potassium channel. Nat. Struct. Mol. Biol. 12:454–459. [DOI] [PubMed] [Google Scholar]
- Liu, Y., M. Holmgren, M.E. Jurman, and G. Yellen. 1997. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 19:175–184. [DOI] [PubMed] [Google Scholar]
- Liu, Y., M.E. Jurman, and G. Yellen. 1996. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 16:859–867. [DOI] [PubMed] [Google Scholar]
- Long, S.B., E.B. Campbell, and R. MacKinnon. 2005. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 309:897–903. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo, J., T. Hoshi, S.H. Heinemann, and R.W. Aldrich. 1993. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1:61–71. [PubMed] [Google Scholar]
- Loussouarn, G., L.R. Phillips, R. Masia, T. Rose, and C.G. Nichols. 2001. Flexibility of the Kir6.2 inward rectifier K+ channel pore. Proc. Natl. Acad. Sci. USA. 98:4227–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus, Y. Ion Properties. 1997. Dekker, New York. 272 pp.
- Margolskee, R.F., P. Kavathas, and P. Berg. 1988. Epstein-Barr virus shuttle vector for stable episomal replication of cDNA expression libraries in human cells. Mol. Cell. Biol. 8:2837–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolskee, R.F., B. McHendry-Rinde, and R. Horn. 1993. Panning transfected cells for electrophysiological studies. Biotechniques. 15:906–911. [PubMed] [Google Scholar]
- Martin, R.L., J.H. Lee, L.L. Cribbs, E. Perez-Reyes, and D.A. Hanck. 2000. Mibefradil block of cloned T-type calcium channels. J. Pharmacol. Exp. Ther. 295:302–308. [PubMed] [Google Scholar]
- McDonough, S.I., and B.P. Bean. 1998. Mibefradil inhibition of T-type calcium channels in cerebellar purkinje neurons. Mol. Pharmacol. 54:1080–1087. [DOI] [PubMed] [Google Scholar]
- McNulty, M.M., and D.A. Hanck. 2004. State-dependent mibefradil block of Na+ channels. Mol. Pharmacol. 66:1652–1661. [DOI] [PubMed] [Google Scholar]
- Melishchuk, A., and C.M. Armstrong. 2001. Mechanism underlying slow kinetics of the OFF gating current in Shaker potassium channel. Biophys. J. 80:2167–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais-Cabral, J.H., Y. Zhou, and R. MacKinnon. 2001. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 414:37–42. [DOI] [PubMed] [Google Scholar]
- Neyton, J., and C. Miller. 1988. Potassium blocks barium permeation through a calcium-activated potassium channel. J. Gen. Physiol. 92:549–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogielska, E.M., and R.W. Aldrich. 1999. Functional consequences of a decreased potassium affinity in a potassium channel pore. Ion interactions and C-type inactivation. J. Gen. Physiol. 113:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcese, R., D. Sigg, R. Latorre, F. Bezanilla, and E. Stefani. 2001. A conducting state with properties of a slow inactivated state in a Shaker K+ channel mutant. J. Gen. Physiol. 117:149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyi, G., and C. Deutsch. 2006. Cross talk between activation and slow inactivation gates of Shaker potassium channels. J. Gen. Physiol. 128:547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo, E., D.M. Cortes, and L.G. Cuello. 1999. Structural rearrangements underlying K+-channel activation gating. Science. 285:73–78. [DOI] [PubMed] [Google Scholar]
- Ray, E.C., and C. Deutsch. 2006. A trapped intracellular cation modulates K+ channel recovery from slow inactivation. J. Gen. Physiol. 128:203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, D.J., E.T. Maggio, and G.L. Kenyon. 1975. Simple alkanethiol groups for temporary blocking of sulfhydryl groups of enzymes. Biochemistry. 14:766–771. [DOI] [PubMed] [Google Scholar]
- Stauffer, D.A., and A. Karlin. 1994. Electrostatic potential of the acetylcholine binding sites in the nicotinic receptor probed by reactions of binding-site cysteines with charged methanethiosulfonates. Biochemistry. 33:6840–6849. [DOI] [PubMed] [Google Scholar]
- Webster, S.M., D. del Camino, J.P. Dekker, and G. Yellen. 2004. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature. 428:864–868. [DOI] [PubMed] [Google Scholar]
- Yang, N., and R. Horn. 1995. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 15:213–218. [DOI] [PubMed] [Google Scholar]
- Yellen, G., M.E. Jurman, T. Abramson, and R. MacKinnon. 1991. Mutations affecting internal TEA blockade identify the probable pore-forming region of K+ channels. Science. 251:939–942. [DOI] [PubMed] [Google Scholar]
- Yellen, G., D. Sodickson, T.Y. Chen, and M.E. Jurman. 1994. An engineered cysteine in the external mouth of a K+ channel allows inactivation to be modulated by metal binding. Biophys. J. 66:1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yifrach, O., and R. MacKinnon. 2002. Energetics of pore opening in a voltage-gated K+ channel. Cell. 111:231–239. [DOI] [PubMed] [Google Scholar]
- Zhou, M., J.H. Morais-Cabral, S. Mann, and R. MacKinnon. 2001. a. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., J.H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. b. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]