

Abstract
A significant improvement in molecular hydrogen uptake properties is revealed by our ab initio calculations for Li-decorated metal–organic framework 5. We have found that two Li atoms are strongly adsorbed on the surfaces of the six-carbon rings, one on each side, carrying a charge of +0.9e per Li atom. Each Li can cluster three H2 molecules around itself with a binding energy of 12 kJ (mol H2)−1. Furthermore, we show from ab initio molecular dynamics simulations with a hydrogen loading of 18 H2 per formula unit that a hydrogen uptake of 2.9 wt % at 200 K and 2.0 wt % at 300 K is achievable. To our knowledge, this is the highest hydrogen storage capacity reported for metal–organic framework 5 under such thermodynamic conditions.
Keywords: first-principles calculations, molecular adsorption, molecular dynamics, porous materials
Metal–organic frameworks (MOFs) form a class of nanoporous materials with high surface area that are capable of binding gas molecules in a nondissociative manner (1–5). Consequently, MOFs are promising candidates for use in hydrogen storage media. In MOF systems, the hydrogen sorption processes display good reversibility and fast kinetics; however, the weak dispersive interactions that hold H2 molecules require low operation temperatures and/or high pressures to guarantee a significant storage capacity, e.g., MOF-5 reaches an H2 uptake of only 1.3 wt % at 78 K and 1 bar (6). Neither the thermodynamics nor the storage capacity meets the requirements established for onboard applications (7). Therefore, a great deal of effort is being focused on devising ways to strengthen hydrogen adsorption interactions and maximize volumetric and gravimetric surface area densities. The achievement of the latter is being pursued through the following approaches: (i) topological engineering of pore shape (8), (ii) insertion of other adsorbate surfaces inside the pores (9), (iii) synthesis of light-metal MOFs (10, 11), and (iv) entanglement of frameworks (framework catenation) (12, 13). To achieve stronger H2–surface interactions, which is the more challenging problem, most studies have turned to investigating the introduction of electron-donating ligands to the organic linkers and also to the synthesis of so-called “open metal sites” (14). In fact, it is well accepted that to significantly enhance the H2 affinity in these frameworks, the binding mechanism should include other contributions, such as electrostatic and/or orbital interactions, rather than being purely dispersive, as described in ref. 15.
An alternative approach for strong, nondissociative H2 binding comes from the possibility of adsorbing hydrogen molecules on light, nontransition metal ions such as Li+, Na+, Mg2+, and Al3+ (15, 16). These bare metal ions are capable of clustering several H2 molecules, with binding energies in the range of 12–340 kJ (mol H2)−1. For the alkali metals (Li+ and Na+), H2 is bound through electrostatic charge–quadrupole and charge-induced dipole interactions. For Al3+ and Mg2+, which display higher H2 binding energies, other contributions are obtained from orbital interactions. Despite such good binding affinity, these bare metal ions are difficult to realize in real systems. The ions tend to form complexes in which only part of the charge is available for interaction with hydrogen molecules. One promising approach is to have alkali metals adsorbed on a surface displaying high electron affinity. In this case, a charge is transferred from the metal to the surface, producing the desired ionic state. This charge transfer has previously been shown to occur when alkali metals are adsorbed on C60 (17) and carbon nanotubes (18, 19). However, in these systems the ions are not sufficiently isolated from each other, resulting in a lowering of the charge state; furthermore, practical application remains a challenging task.
Here we present an alternative means for obtaining the desired ionic state of Li atoms and the consequent strong nondissociative H2 binding. The scheme is to let Li atoms be adsorbed on the organic linkers of MOFs. In particular, we have studied the adsorption of Li on the 1,4-benzenedicarboxylate (BDC) linker in MOF-5. MOF-5 is made up of BDC linkers joining Zn4O clusters to form a cubic periodic framework, which has the formula unit Zn4O(BDC)3. By means of first-principles calculations, we show that two Li atoms are strongly adsorbed on the surfaces of the BDC six-carbon rings, one on each side, carrying a charge of +0.9e per Li. Each Li is found to be able to cluster three H2 molecules around itself with a binding energy of 12 kJ (mol H2)−1, which is more than two times larger than for H2 adsorption on pure BDC [binding energy ≈5 kJ (mol H2)−1 (20)]. Furthermore, by employing ab initio molecular dynamics (21), we show that a hydrogen uptake of 2.9 wt % at 200 K and 2.0 wt % at 300 K can be achieved. To our knowledge, this is the largest hydrogen storage capacity yet reported for MOF-5 under such thermodynamic conditions.
Results and Discussion
The structure of pure MOF-5 was fully optimized without imposing any symmetry constraints. The resulting structure is displayed in Fig. 1A. The calculated lattice constant a = 26.06 Å, as well as the Zn–Zn bond length of 3.21 Å, C–C average distance in the carbon ring of 1.40 Å, and O–C–O bond angle of 125.6° compare very well with the corresponding experimental values of 25.83 Å, 3.17 Å, 1.39 Å, and 126.4°, respectively (1). The adsorption of Li atoms on the BDC (forming BDC–Li complexes) was investigated by fully optimizing the structures, with Li atoms initially placed close to the center of the six-atom carbon ring (C6H4 units). The geometry modification induced on the BDC unit as a result of Li-binding can be analyzed by comparing the C–C bond lengths given in Table 1(C1, C2, and C3 are defined in Fig. 2D). As can be seen, Li induces a stretching of the aromatic ring with a shortening of C1–C2 and C2–C3 distances and an increase in C3–C3 distance, indicating that a charge transfer takes place from Li atoms to the MOF-5 framework. In fact, Bader analysis (22) of the self-consistent charge density revealed that Li atoms carry a charge of +0.9e/Li. Thus, the difference in the electronegativity between Li and BDC is large enough to produce the desired ionic state of Li atoms, which is beneficial in raising the H2 binding affinity, as we show below. It should also be noted that in this framework, the organic linkers are isolated from each other and, consequently, so are the Li atoms. This is an important feature that guarantees that the entire on-site Li charge will be available to interact with hydrogen molecules.
Fig. 1.

MOF-5 is made up of 1,4-benzenedicarboxylate molecules joining Zn4O clusters to form a cubic periodic porous framework. (A) Pure MOF-5. (B) Li-decorated MOF-5. (C) MOF-5 with Li cluster. C, black; Zn, blue; O, red; Li, green. All H atoms have been omitted for clarity.
Table 1.
Some of the relevant atomic distances (Å) in the 0 K relaxed structures before and after the adsorption of Li and H2
| System | C1–C2 | C2–C3 | C3–C3 | C6CoM–Li | Li–H2CoM | Li–H2CoM | Li–H2CoM | H–H |
|---|---|---|---|---|---|---|---|---|
| MOF | 1.484 | 1.406 | 1.391 | |||||
| MOF:Lia | 1.422 | 1.476 | 1.380 | 1.677 | ||||
| MOF:Lia:1H2 | 1.419 | 1.471 | 1.381 | 1.737 | 2.078 | 0.757 | ||
| MOF:Lia:2H2 | 1.419 | 1.470 | 1.380 | 1.773 | 2.111 | 2.152 | 0.757 | |
| MOF:Lia:3H2 | 1.421 | 1.467 | 1.380 | 1.806 | 2.179 | 2.224 | 2.316 | 0.756 |
The numbering of the carbon atoms is defined in Fig. 2D. Columns 6–8 show the distance from Li to the center of mass of the first, second, and third hydrogen molecules, respectively.
Fig. 2.

Hydrogen adsoprtion on Li-decorated MOF-5. (A–C) Optimized geometries of adsorbed hydrogen molecules on BDC:2Li in the framework of MOF-5 with one H2 per Li (A), two H2 per Li (B), and three H2 per Li (C). C, black; Li, green; H, gray. The rest of the framework has been omitted for clarity. (D) Schematic drawing of the C atoms in the BDC unit.
To estimate the strength of the BDC–Li interaction, we also compared the total energy of two possible competitive configurations: Li adsorbed on BDC (Fig. 1B) and Li forming a cluster (Li12) inside the pores (Fig. 1C). We optimized a cluster containing 12 Li atoms because there are 12 BDC faces pointing toward the center of one pore. The difference in total energies is given by
where ETa (MOF:Lia) and ETc (MOF:Lic) are the total energies of the unit cell containing the adsorbed Li atoms and the one containing the Li12 cluster inside the pore, respectively. The calculated value of ΔETLi = −0.25 eV per Li atom shows that Li adsorbed on the BDC rings is the more stable configuration.
To investigate the hydrogen uptake, we optimized structures with H2 molecules placed in the vicinity of the Li atoms. In Fig. 2 A–C, we display the optimized geometries as the hydrogen uptake increases from two to six molecules around the BDC:2Li complexes. Actually, Fig. 2C shows the equilibrium configuration at saturation, i.e., no more than three hydrogen molecules could be adsorbed around each Li ion. The adsorption energies were calculated as
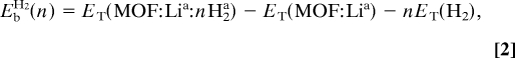 |
where ET(MOF:Lia:nH2a), ET(MOF:Lia), and ET(H2) are the total energies of MOF-5 containing the adsorbed Li-nH2 clusters, of MOF-5 containing the adsorbed Li atoms, and of H2 in the gas phase, respectively. We found binding energies of 18 kJ (mol H2)−1 for n = 1, 16 kJ (mol H2)−1 for n = 2, and 12 kJ (mol H2)−1 for n = 3. It is well known that H2 binds to Li+ through charge–quadrupole and charge-induced dipole interactions (15). Such electrostatic interactions are consistent with the small change in the H–H bond length, as seen in Table 1 (our calculated value for the H–H bond distance in the free molecule is 0.750 Å), and also with the fact that both H2 and Li maintain their original charge state. The decrease in the binding energy as a function of cluster size is a consequence of repulsive H2–H2 interaction. It should also be noted that the optimized geometries of Li–nH2 clusters are in good agreement with previous studies in which H2 molecules were found to bind mainly in a side-on configuration (15, 16). The interaction energy for hydrogen uptake in pure MOF-5 lies in the interval 4–7 kJ (mol H2)−1 (20). In particular, the energy of the dispersive interaction between the six-atom carbon ring (which forms the center of BDC) and one H2 molecule placed on its surface is found to be ≈5 kJ (mol H2)−1 (20). Thus, the calculated adsorption energy of 12–18 kJ (mol H2)−1 appears to be the highest value among the results available in the literature. This stronger interaction is due solely to the introduced electrostatic contribution of Li. Moreover, the hydrogen adsorption interaction around the center of the BDC unit, which was found to play a secondary role among the identified adsorption sites (23), is shown to be significantly strengthened. To estimate the storage capacity of this system, let us assume that all Li sites are saturated, i.e., three H2 are adsorbed around every Li atom, and that no other sites are populated. Because each formula unit (f.u.) can contain six Li atoms, Zn4O(BDC)3Li6, 18 H2 molecules would be adsorbed, corresponding to a hydrogen uptake of 4.3 wt %. Thus, a significant amount of adsorbed hydrogen is strongly bound. At lower temperatures, where the sites around Zn4O are populated as well, the gravimetric density may be even higher. An estimate of the H content considering saturated Li–3H2 clusters and the 5 H2/f.u. (1.3 wt % in pure MOF-5 at 77 K and 1 atm) experimentally found to be adsorbed around the Zn4O clusters (6, 23) yields an impressive H content of 5.4 wt %.
To investigate the temperature effect on the hydrogen uptake in Li-decorated MOF-5, we performed ab initio molecular dynamics simulations with two concentrations, 5 and 18 H2 molecules per formula unit, at 20, 50, 100, 200, and 300 K. The molecules were initially distributed in a uniform grid inside the pore, with their orientation and initial velocities assigned randomly. Control over temperature was achieved by rescaling velocities. Employing a time step of 1 fs throughout, the system was equilibrated until the dynamical equilibrium was reached (≈3,000 steps). After this, the simulations were allowed to run for 10,000 steps (10 ps) at each temperature.
To assess the capability of Li ions to stay attached to the BDC unit, we calculated the pair distribution functions (PDFs) for C6CoM–Li pairings, where C6CoM is the center of mass of the six-atom carbon ring. As seen from Fig. 3 A and B, the Li stay bound to the BDC units for the entire temperature range. To investigate the population of different adsorption sites, we also calculated the PDFs for Zn–H2CoM and Li–H2CoM pairings, where H2CoM is the center of mass of the H2 molecule. The results are displayed in Fig. 3 C–F. As can be observed for both hydrogen loadings (5 and 18 H2/f.u.) used in this simulation, the first Li–H2CoM PDF peak is situated at ≈2.0 Å, which is consistent with the equilibrium distances between Li and H2CoM as obtained from static geometry optimization (see Table 1). For the concentration of 5 H2/f.u., the first Zn–H2CoM PDF peak appears at ≈4.7 Å, which corresponds to the pairing between Zn and those molecules bound to the Li site. However, as the loading is increased from 5 to 18 H2/f.u., a new Zn–H2CoM PDF peak appears at ≈3.8 Å, showing that sites other than those around Li become populated as well. The population of these sites is also illustrated by the Li–H2CoM PDF peak at ≈4.3 Å in Fig. 3D. In fact, we found that only two molecules bind to each Li atom, corresponding to a gravimetric density of 2.9 wt %, which is an uptake below the saturation level (4.3 wt %) found from 0 K calculations. This result could be due to dynamical effects preventing the third molecule from being adsorbed, to the need for higher concentrations of hydrogen molecules that would increase the population probability of the adsorption sites, or to the need for even longer simulation times. From the simulations, we observe that Li stays coordinated by two H2 molecules up to at least 200 K.
Fig. 3.

Pair distribution functions from ab initio molecular dynamics simulations. (A and B) PDF of C6CoM–Li pairs with 5 H2 per formula unit (A) and 18 H2 per formula unit (B). (C and D) PDF of Li–H2CoM pairs with 5 H2 per formula unit (C) and 18 H2 per formula unit (D). (E and F) PDF of Zn–H2CoM pairs with 5 H2 per formula unit (E) and 18 H2 per formula unit (F).
For an approximate estimation of the H2 binding energy required to achieve chemical equilibrium, ΔH = TΔS, ΔS is usually approximated by the hydrogen gas entropy. Thus, the required Li–H2 binding energy for a release temperature of T = 300 K at 1 bar would be TSH2 = 39.3 kJ (mol H2)−1 (24). However, the integration of the first peak from the PDF at 300 K corresponds to a hydrogen atom content of 2.0 wt %, even though the calculated binding energies are in the range of 12–18 kJ (mol H2)−1, showing that here this simple model fails. It should be noted that although the amplitude of the PDF peak decreases with temperature because of broadening, it is the integration of the PDFs, and not the amplitude, that determines the amount of molecules in a given distance interval. A hydrogen uptake of 2.9 wt % at 200 K and 2.0 wt % at 300 K is the highest reported uptake under such thermodynamic conditions in MOF-5. Other adsorption sites in the MOF may also be activated to significantly improve storage capacity by raising the pressure, as has been shown for pure MOFs (6). We hope that our results will motivate further experimental investigations.
Conclusions
We have shown that the adsorption of Li atoms on BDC units of MOF-5 improves the hydrogen storage functional properties of these systems. Each organic linker is able to bind two Li atoms through an ionic interaction in which the desired charge state of +0.9e/Li was obtained. The formation of Li clusters inside the pores was shown to be unlikely because the cluster is not energetically favored. Each Li can bind up to three H2 molecules around itself with a binding energy of 12 kJ (mol H2)−1, which is much higher than that of H2 adsorption on pure BDC [≈5 kJ (mol H2)]. If the saturation level of all Li sites is reached, the storage capacity amounts to 4.3 wt % of hydrogen. However, from ab initio molecular dynamics simulations in a cell containing 18 H2/f.u., it was found that Li is coordinated with only two hydrogen molecules, corresponding to a hydrogen uptake of 2.9 wt %. With those hydrogen molecules staying bound to Li up to at least 200 K, and with approximately two-thirds staying bound even up to 300 K, this system displays the best hydrogen storage properties of MOF-5 found to date. Moreover, we expect that higher storage capacity and higher operation temperatures may be achieved by increasing the gas pressure, as has been done for the intrinsic frameworks (6). It may be worthwhile to investigate the adsorption of guest atoms (other than Li) on the framework in a way that would further strengthen electrostatic and orbital interactions in the hydrogen binding mechanism. Thus, we have shown here a route to improving the hydrogen affinity, and consequently the thermodynamics, of the hydrogen sorption reactions in MOFs.
Methods
The calculations of total energy were carried out within the framework of generalized gradient approximation (25) to density functional theory (26, 27) by using the projector augmented wave (PAW) method (28), as implemented in the Vienna ab initio Simulation Package (VASP) (21). The PAW potentials with the valence states 1s2s for Li, 2s2p for C, 2s2p for O, d10p2 for Zn, and 1s for H were used. All results reported here have been successfully tested for convergence with respect to cutoff energy. The pure MOF-5 was tested for convergence with respect to the number of k-points, but because we are dealing with a large system (at least 106 atoms per primitive cell), the Γ-point alone was sufficient for sampling the Brillouin zone. Ionic positions and cell parameters were relaxed with respect to minimum forces and stress by using conjugate-gradient algorithms. The ab initio molecular dynamics simulations were performed with the VASP code (21).
ACKNOWLEDGMENTS.
We thank the Futura Foundation, the Swedish Research Council, the European Science Foundation (EuroMinScI), and the Swedish Foundation for International Cooperation in Research and Higher Education for financial support.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Eddaoudi M, Kim J, Rosi N, Vodak D, Wachter J, O'Keeffe M, Yaghi OM. Science. 2002;295:469–472. doi: 10.1126/science.1067208. [DOI] [PubMed] [Google Scholar]
- 2.Rosi NL, Eckert J, Eddaoudi M, Vodak DT, Kim J, O'Keeffe M, Yaghi OM. Science. 2003;300:1127–1129. doi: 10.1126/science.1083440. [DOI] [PubMed] [Google Scholar]
- 3.Rowsell JLC, Spencer EC, Eckert J, Howard JAK, Yaghi OM. Science. 2005;309:1350–1354. doi: 10.1126/science.1113247. [DOI] [PubMed] [Google Scholar]
- 4.Li H, Eddaoudi M, O'Keeffe M, Yaghi OM. Nature. 1999;402:276–279. [Google Scholar]
- 5.Mattesini M, Soler JM, Ynduráin F. Phys Rev B. 2006;73 doi: 10.1103/PhysRevLett.96.079701. 094111. [DOI] [PubMed] [Google Scholar]
- 6.Rowsell JLC, Millward AR, Park KS, Yaghi OM. J Am Chem Soc. 2004;126:5556–5667. doi: 10.1021/ja049408c. [DOI] [PubMed] [Google Scholar]
- 7.Schlapbach L, Züttel A. Nature. 2001;414:353–358. doi: 10.1038/35104634. [DOI] [PubMed] [Google Scholar]
- 8.Rowsell JLC, Yaghi OM. Angew Chem Int Ed. 2005;44:4670–4679. doi: 10.1002/anie.200462786. [DOI] [PubMed] [Google Scholar]
- 9.Chae HK, Siberio-Pérez DY, Kim J, Go Y, Eddaoudi M, Matzger AJ, O'Keeffe M, Yaghi OM. Nature. 2004;427:523–527. doi: 10.1038/nature02311. [DOI] [PubMed] [Google Scholar]
- 10.Férey G, Latroche M, Serre C, Millange F, Loiseau T, Percheron-Guégan A. Chem Commun. 2003:2976–2977. doi: 10.1039/b308903g. [DOI] [PubMed] [Google Scholar]
- 11.Côté AP, Shimizu GKH. Chem Eur J. 2003;9:5361–5370. doi: 10.1002/chem.200305102. [DOI] [PubMed] [Google Scholar]
- 12.Batten SR, Robson R. Angew Chem Int Ed. 1998;37:1460–1494. doi: 10.1002/(SICI)1521-3773(19980619)37:11<1460::AID-ANIE1460>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 13.Chen B, Eddaoudi M, Hyde ST, O'Keeffe M, Yaghi OM. Science. 2001;291:1021–1023. doi: 10.1126/science.1056598. [DOI] [PubMed] [Google Scholar]
- 14.Chen B, Eddaoudi M, Reineke TM, Kampf JW, O'Keeffe M, Yaghi OM. J Am Chem Soc. 2000;122:11559–11560. [Google Scholar]
- 15.Lochan RC, Head-Gordon M. Phys Chem Chem Phys. 2006;8:1357–1370. doi: 10.1039/b515409j. [DOI] [PubMed] [Google Scholar]
- 16.Barbatti M, Jalbert G, Nascimento MAC. J Phys Chem A. 2002;106:551–555. [Google Scholar]
- 17.Sun Q, Jena P, Wang Q, Marquez M. J Am Chem Soc. 2006;128:9741–9745. doi: 10.1021/ja058330c. [DOI] [PubMed] [Google Scholar]
- 18.Chen P, Wu X, Lin J, Tan KL. Science. 1999;285:91–93. doi: 10.1126/science.285.5424.91. [DOI] [PubMed] [Google Scholar]
- 19.Froudakis GE. Nano Lett. 2001;1:531–533. [Google Scholar]
- 20.Sagara T, Klassen J, Ganz E. J Chem Phys. 2001;121:12543–12547. doi: 10.1063/1.1809608. [DOI] [PubMed] [Google Scholar]
- 21.Kresse G, Furthmüller J. Phys Rev B. 1996;54:11169–11186. doi: 10.1103/physrevb.54.11169. [DOI] [PubMed] [Google Scholar]
- 22.Henkelman G, Arnaldsson A, Jónsson H. Comput Mater Sci. 2006;36:354–360. [Google Scholar]
- 23.Yildirim T, Ciraci S. Phys Rev Lett. 2005;94:175501. doi: 10.1103/PhysRevLett.94.175501. [DOI] [PubMed] [Google Scholar]
- 24.Stull D, Prophet H, editors. JANAF Thermochemical Tables. 2nd Ed. Washington: US Govt Printing Office; 1971. Natl Std Ref Data Ser 37. [Google Scholar]
- 25.Perdew JP, Wang Y. Phys Rev B. 1992;45:13244–13249. doi: 10.1103/physrevb.45.13244. [DOI] [PubMed] [Google Scholar]
- 26.Hohenberg P, Kohn W. Phys Rev B. 1964;136:864–871. [Google Scholar]
- 27.Kohn W, Sham LJ. Phys Rev A. 1965;140:1133–1138. [Google Scholar]
- 28.Blöchl PE. Phys Rev B. 1994;50:17953–17979. doi: 10.1103/physrevb.50.17953. [DOI] [PubMed] [Google Scholar]