

Abstract
We explored the conformational landscape of the proteinogenic amino acid serine [CH2OH CH(NH2)COOH] in the gas phase. Solid serine was vaporized by laser ablation, expanded in a supersonic jet, and characterized by Fourier transform microwave spectroscopy. In the isolation conditions of the jet there have been discovered up to seven different neutral (non-zwitterionic) structures of serine, which are conclusively identified by the comparison between the experimental values of the rotational and quadrupole coupling constants with those predicted by ab initio calculations. These seven forms can serve as a basis to represent the shape of serine in the gas phase. From the postexpansion abundances we derived the conformational stability trend, which is controlled by the subtle network of intramolecular hydrogen bonds formed between the polar groups in the amino acid backbone and the hydroxy side chain. It is proposed that conformational cooling perturbs the equilibrium conformational distribution; thus, some of the lower-energy forms are “missing” in the supersonic expansion.
CH(NH2)COOH] in the gas phase. Solid serine was vaporized by laser ablation, expanded in a supersonic jet, and characterized by Fourier transform microwave spectroscopy. In the isolation conditions of the jet there have been discovered up to seven different neutral (non-zwitterionic) structures of serine, which are conclusively identified by the comparison between the experimental values of the rotational and quadrupole coupling constants with those predicted by ab initio calculations. These seven forms can serve as a basis to represent the shape of serine in the gas phase. From the postexpansion abundances we derived the conformational stability trend, which is controlled by the subtle network of intramolecular hydrogen bonds formed between the polar groups in the amino acid backbone and the hydroxy side chain. It is proposed that conformational cooling perturbs the equilibrium conformational distribution; thus, some of the lower-energy forms are “missing” in the supersonic expansion.
Keywords: amino acids, conformations, laser ablation, microwave spectroscopy, supersonic expansion
Amino acids are distinguished by an outstanding conformational flexibility originating from multiple torsional degrees of freedom, which makes folding and functionality of proteins possible (1). Whereas covalent forces determine the molecular skeleton, conformational isomerism is controlled by weaker nonbonded interactions within the molecule, especially hydrogen bonding. Amino acids are also very sensitive to the chemical medium chosen for their study. In traditional studies in the condensed phase, the extensive intermolecular hydrogen bond interactions fix amino acids as doubly charged zwitterions (2, 3), wiping out the conformational variety of these molecules. The intrinsic structural preferences of amino acids can be revealed only when they are studied as free species in the gas phase, where neutral forms (present in polypeptide chains) are the most stable in detriment of ionic or zwitterionic forms. Levy and coworkers (4) were the first to measure highly resolved electronic spectra of amino acids in the gas phase by seeding tryptophan in a supersonic expansion. Since then, different experimental methods have been used to investigate the electronic spectrum of amino acids and a variety of biological molecules in the gas phase (5–7). The electronic spectrum is interpreted with the help of theoretical calculations to identify different conformations with a high degree of confidence. However, the use of electronic spectroscopy is limited to favorable cases that present aromatic chromophores. Only three of the 20 natural amino acids have aromatic side chains that meet this criterion: phenylalanine, tyrosine, and tryptophan (4, 8–14).
Microwave spectroscopy, often considered the most definitive gas-phase structural probe, can distinguish unambiguously between different conformational structures and provide accurate structural information directly comparable to the in vacuo theoretical predictions. Without any chromophore restriction, all amino acids are potentially amenable to pure rotational studies. However, despite significant instrumental advances in the microwave region carried out in the last two decades (15–18), it is still difficult to extract experimental information on the exact nature of amino acids' conformations and their quantitative distributions. Amino acids are solids with high melting points and low vapor pressures, and consequently they are elusive to gas-phase investigations. Therefore, rotational studies of natural amino acids, started in the late 1970s by Suenram and Godfrey (19–25) on glycine and alanine were restrained for a long time by the difficulties to bring these compounds into gas phase because they quickly decompose under the classical heating methods of vaporization. We have recently revitalized this field with an experiment (see Fig. 1) that combines laser ablation with Fourier transform microwave spectroscopy in supersonic jets (LA-MB-FTMW) (26). Amino acids are vaporized by the green line (532 nm) of a pulsed Nd:YAG laser (irradiances ∼108 W·cm−2), entrained in neon, and supersonically expanded into a vacuum chamber, where they are probed by Fourier transform microwave spectroscopy. The cooling afforded by the expansion, which greatly simplifies the spectrum, and the extreme specificity to molecular structure inherent to rotational spectroscopy make it possible to characterize the most abundant conformers in the supersonic expansion. Thus, their properties can be independently studied, providing the finest structural information available in the gas phase. In the last few years, this experimental approach has allowed us to explore the structural behavior of the α-amino acids alanine (27), valine (28), isoleucine (29), and leucine (30); the amino acids proline (31) and 4-hydroxyprolines (R- and S-) (32); the β-amino acid β-alanine (33); and other related α-amino acids (34–36). These studies have shown that in the supersonic expansion the simplest α-amino acids present two dominant conformers stabilized by either a bifurcated NH···O C hydrogen bond with a cis-COOH interaction (type I) or a N···HO hydrogen bond (type II). Recently, we have even characterized the glycine···H2O complex (37), which constitutes the first step of microsolvation of the simplest amino acid.
C hydrogen bond with a cis-COOH interaction (type I) or a N···HO hydrogen bond (type II). Recently, we have even characterized the glycine···H2O complex (37), which constitutes the first step of microsolvation of the simplest amino acid.
Fig. 1.

Sketch of the LA-MB-FTMW spectrometer.
The presence of polar functional groups in the side chains of α-amino acids is expected to dramatically increase the number of low-energy conformers. This is the case of serine [CH2OHCH(NH2)COOH], with a −CH2OH side chain. The hydroxyl group can establish additional intramolecular hydrogen bonds as a proton donor to the amino group or to the carboxyl group or as a proton acceptor through the nonbonding electron pair at its oxygen atom. These additional interactions, which do not occur in other α-amino acids previously studied, may affect the conformational preferences and give rise to a rich conformational space. Indeed, a complete search of serine conformational space, carried out by Gronert et al. (38) by systematic rotations around the five internal rotation axes, produced 51 conformational minima. In this work we have faced the challenge of investigating the multiconformational behavior of the neutral form of the α-amino acid serine using our LA-MB-FTMW technique.
Results and Discussion
The procedure used for conformer searching was as follows. Before starting the experimental study, we extended the previous ab initio calculations (38) to predict rotational constants, nuclear quadrupole coupling parameters, and electric dipole moments, relevant for the interpretation of the rotational spectrum. Geometry optimizations were carried out with the Gaussian suite of programs (39) using second-order Møller–Plesset perturbation theory (MP2) in the frozen-core approximation and Pople's 6–311++G(d,p) basis set. The relative conformational energies were calculated on the MP2 optimized structures using fourth-order Møller–Plesset (MP4) corrections and the same basis set. Because the highest-energy conformational minima are not sufficiently populated in the jet conditions, we focused our attention on the 11 predicted lower-energy conformers (within ≈1,000 cm−1) of Fig. 2. The conformers have been labeled according to their intramolecular hydrogen bonds. Three possible configurations can be considered attending to the hydrogen bonds established between the amino and carboxylic moieties, which we denoted as configuration I (NH···OC), configuration II (N···HO), and configuration III (NH···OH), according to the convention used for smaller aliphatic amino acids. The different orientations of the side chain have been indicated by adding suffixes to the skeleton configuration i.e., a (corresponding to an +sc configuration for the −OH and −NH2 groups viewed along the Cβ–Cα bond, according to International Union of Pure and Applied Chemistry terminology), b (–sc), and c (ap). Additionally, a prime label indicates a down orientation of the −NH2 along with a OH···N intramolecular interaction with the side chain. For configuration III additional α or β notation is used to indicate which H atom of the −NH2 binds to the −OH of the −COOH group (Fig. 2).
Fig. 2.

Predicted lower-energy conformers of serine and relative energies (MP4, see text) with respect to the global minimum in cm−1. The detected conformers are encircled.
Rotational Spectra.
All of the serine structures of Fig. 2 are predicted to be near-prolate asymmetric tops, whose rotational spectra will show the characteristic patterns of the μa-type R-branch transitions. Initial scans were directed to search for the most intense rotational transitions of a particular conformer. 14N nuclear quadrupole hyperfine structure helped us to confirm that the observed lines belonged to one of the serine conformers. The presence of a single 14N nucleus (I = 1) in serine splits each rotational transition into several close hyperfine components by the interaction of the 14N nuclear electric quadrupole moment with the molecular electric field gradient at the nitrogen position (40). A process of successive predictions and experimental measurements discarded or confirmed the initial assignment until a final set of consistent rotational transitions was collected for each conformer. By using this procedure, the detection of the most abundant conformers of serine could be readily accomplished. However, as the experimental work proceeded to the observation of the least abundant species, very careful scans with coherent averaging of thousands of experimental cycles per frequency step were required to observe the rotational transitions. The thorough analysis of the rotational spectra finally led to the assignment of the seven different conformations of serine encircled in Fig. 2.
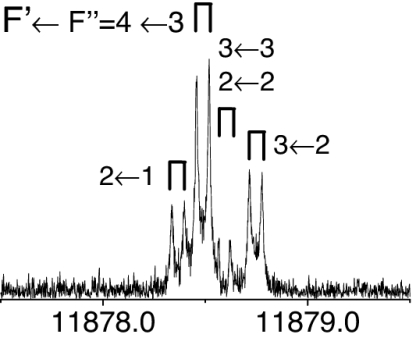
The measured transitions [collected in supporting information (SI) Tables 4–10] corresponded to rotational states with low angular momentum (J < 6) and were analyzed with a Watson's semirigid rotor Hamiltonian [HR(A)] (41) in the asymmetric (A) reduction supplemented with a nuclear quadrupole coupling term (HQ) (40), H = HR(A) + HQ. The Hamiltonian was constructed in the coupled basis set I + J = F and diagonalized. The associated determinable spectroscopic parameters are the rotational and centrifugal distortion constants and the elements of the nuclear quadrupole coupling tensor χαβ (α, β = a, b, c) given in Tables 1–3 and in SI Table 11.
Table 1.
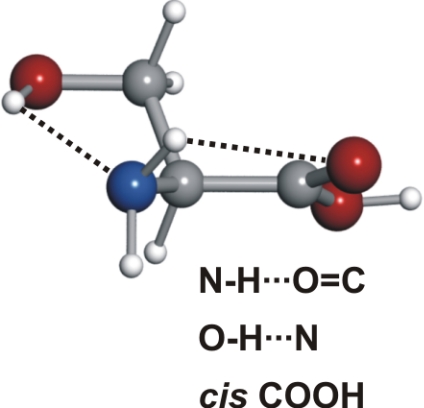
Spectroscopic constants for the observed conformers Ia and IIa of serine, along with their respective 30,3 ← 20,2 rotational transitions
| Constants |
Ia |
IIa |
||
|---|---|---|---|---|
| Exp. | Theor. | Exp. | Theor. | |
| A | 4,479.0320 (12) | 4,481 | 4,517.473 (17) | 4,522 |
| B | 1,830.16170 (25) | 1,825 | 1,846.99360 (30) | 1,850 |
| C | 1,443.79545 (28) | 1,452 | 1,463.79646 (31) | 1,479 |
| χaa | −4.3023 (27) | −4.7 | −0.6066 (55) | −0.4 |
| χbb | 2.82359 (63) | 2.9 | 2.0723 (82) | 2.1 |
| χcc | 1.4788 (46) | 1.7 | −1.466 (30) | −1.6 |
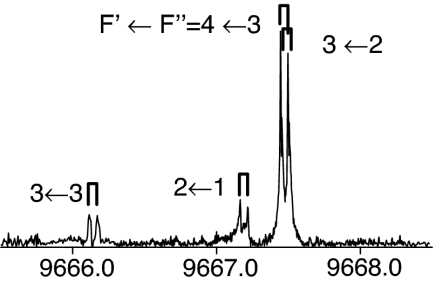 |
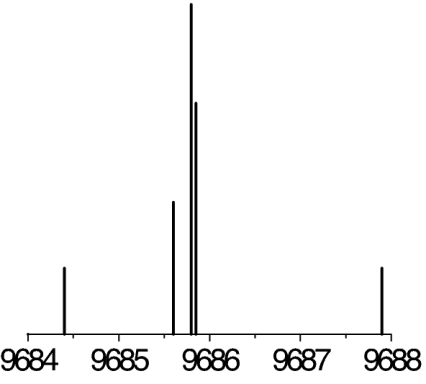 |
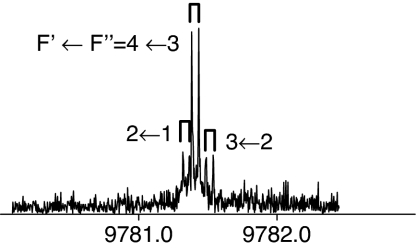 |
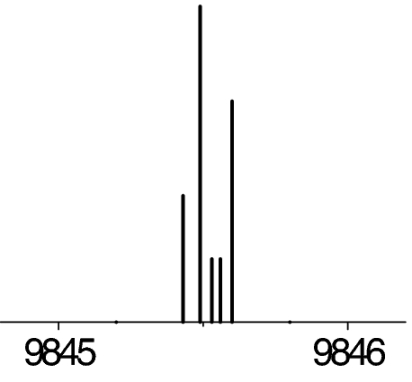 |
|
 |
 |
|||
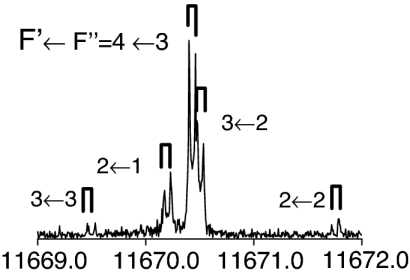
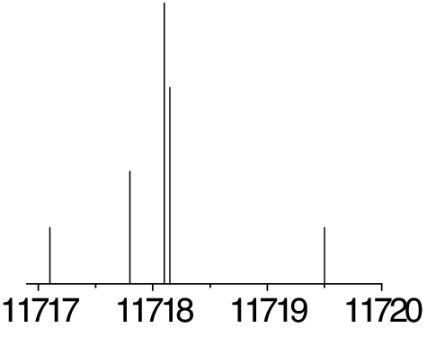
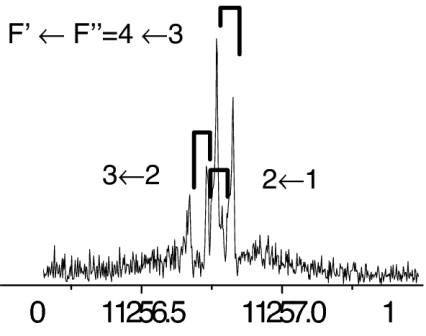
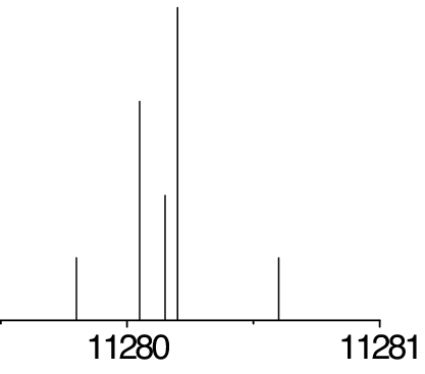
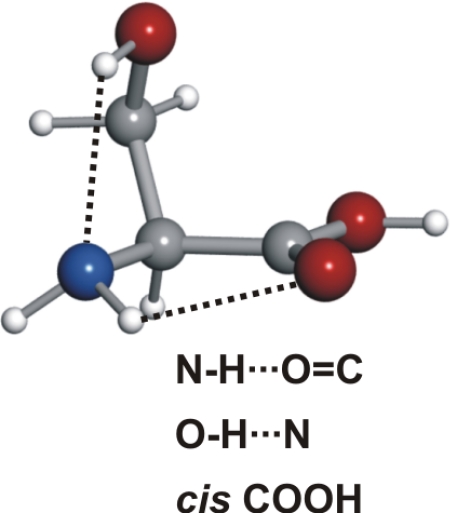
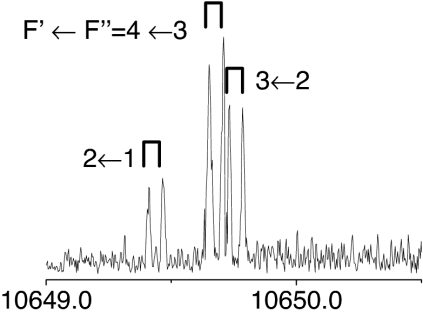
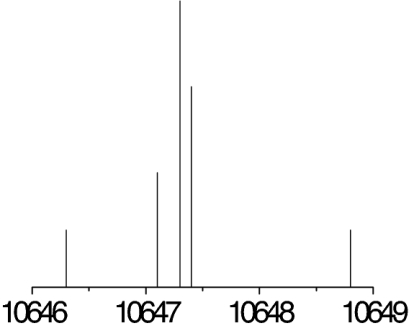
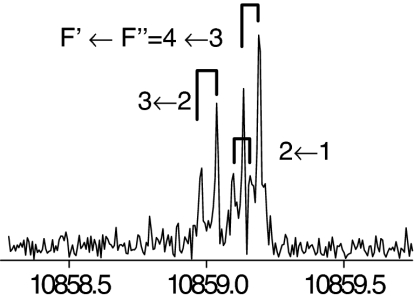
The observed 14N nuclear quadrupole coupling hyperfine structures are compared with those predicted ab initio (sketched below the theoretical values of the spectroscopic constants). The intramolecular hydrogen bonds of each conformer are indicated below each graph. A, B, and C represent the rotational constants; χaa, χbb, and χcc are elements of the 14N nuclear quadrupole coupling tensor. The hyperfine components (F′ ← F″) are labeled by the quantum number F = I + J. Each component appears as a doublet (⊓) due to the Doppler effect. Values are in megahertz, with standard error in parentheses in units of the last digit. Exp., experimental values; Theor., theoretical values.
Table 2.
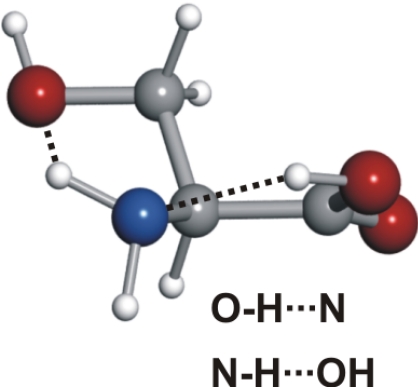
Spectroscopic constants for the observed conformers I′b, IIb, and IIIβb of serine, along with their respective 30,3 ← 20,2 rotational transitions
| Constants |
I′b |
IIb |
IIIβb |
|||
|---|---|---|---|---|---|---|
| Exp. | Theor. | Exp. | Theor. | Exp. | Theor. | |
| A | 3,524.38806 (41) | 3,517 | 3,557.20088 (25) | 3,544 | 3,931.7548 (76) | 3,934 |
| B | 2,307.76826 (70) | 2,339 | 2,380.37208 (40) | 2,396 | 2,242.76701 (70) | 2,249 |
| C | 1,805.20788 (60) | 1,819 | 1,740.92458 (10) | 1,749 | 1,664.53012 (57) | 1,668 |
| χaa | −1.1343 (35) | −0.8 | −3.4616 (19) | −3.7 | −0.6733 (67) | −0.7 |
| χbb | 2.5043 (50) | 2.5 | 2.07974 (93) | 2.2 | −0.456 (16) | −0.5 |
| χcc | −1.3701 (50) | −1.8 | 1.3819 (47) | 1.4 | 1.129 (16) | 1.2 |
 |
 |
 |
 |
 |
 |
|
 |
 |
 |
||||
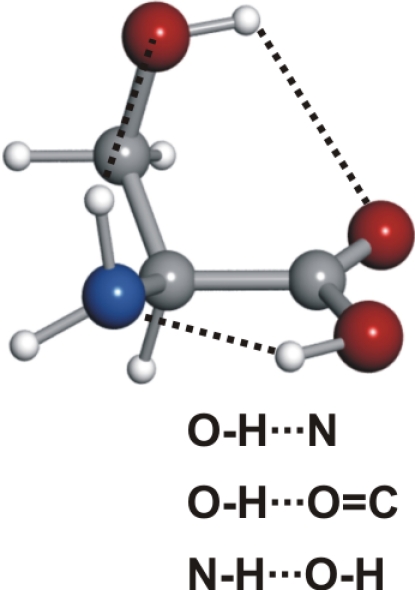
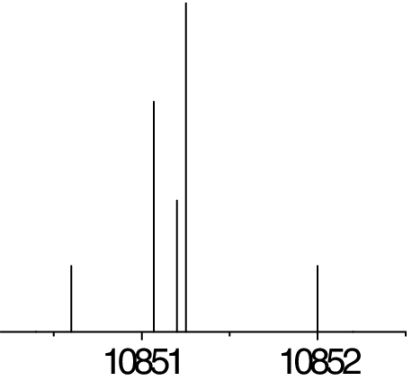
See the legend of Table 1 for details.
Table 3.
Spectroscopic constants for the observed conformers IIc and IIIβc of serine, along with their respective 30,3 ← 20,2 rotational transitions
| Constants |
IIc |
IIIβc |
||
|---|---|---|---|---|
| Exp. | Theor. | Exp. | Theor. | |
| A | 3,638.05784 (38) | 3,656 | 3,510.4015 (35) | 3,519 |
| B | 2,387.89651 (99) | 2,378 | 2,321.90829 (24) | 2,332 |
| C | 1,519.18716 (36) | 1,519 | 1,584.38608 (32) | 1,580 |
| χaa | −3.6527 (57) | −3.9 | −1.0486 (55) | −1.1 |
| χbb | 2.06213 (26) | 2.1 | −0.5637 (53) | −0.7 |
| χcc | 1.5906 (50) | 1.8 | 1.612(21) | 1.8 |
 |
 |
 |
 |
|
 |
 |
|||
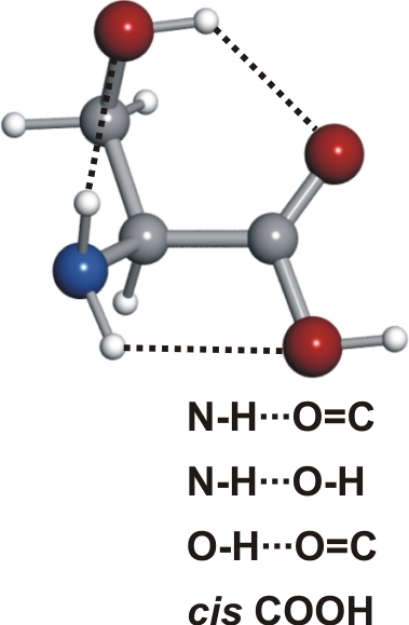
See the legend of Table 1 for details.
Identification of Conformers and Relative Stabilities.
The spectroscopic parameters listed in Tables 1–3 provide unequivocal evidence of the conformers observed. The experimental values of the rotational constants, which depend critically on the masses and geometry of the molecule, agree with those predicted theoretically for conformers Ia, IIb, I′b, IIc, IIIβb, IIIβc, and IIa, supporting the presence of these conformers in our supersonic expansion. For conformers showing only slight geometry changes, i.e., conformers Ia and IIa with the same amino acid backbone but different intramolecular hydrogen bonds, the rotational constants are very similar and they do not allow discrimination. In these cases, conclusive evidence comes from the values of 14N quadrupole coupling constants because they are very sensitive to the orientation of the −NH2 group with respect to the principal inertial axis system and can change dramatically in passing from one conformer to another. The fitted values of the diagonal elements of the 14N quadrupole coupling tensor χaa, χbb, χcc values in Tables 1–3 match nicely with those predicted by ab initio calculations for the above conformers (compare their values for conformers Ia and IIa). All of the above considerations gave the conformer assignments shown in Tables 1–3. The presence of nitrogen in amino acids makes the 14N quadrupole coupling an exceptional tool for conformer identification. The 14N nucleus acts effectively as a structural reporter, informing on the structural arrangement of the amino group through the values of the quadrupole coupling constants.
Our experiment can also provide information on the relative stability of the serine conformers from relative intensity measurements of the rotational transitions. The intensities of the lines from conformer i can be considered proportional to μi·Ni, where μi is the corresponding electric dipole moment component and Ni is the number density of conformer i in the jet on the assumption that cooling in the supersonic jet brings all of the molecules to the lowest vibrational state of each conformer (42). From careful intensity measurements of seven selected μa-type transitions and using the ab initio μa electric dipole moment values, we derived the population ratios (see SI Table 12) of the conformers, which follow the order Ia > IIb > I′b > IIc > IIIβb ≈ IIIβc ≈ IIa, with conformer Ia as the global minimum. This can be taken as indicative of the conformer stability trend, which, on the other hand, is in good correlation with the ordering of the computed relative energies of the conformers (Fig. 2).
Intramolecular Hydrogen Bonding.
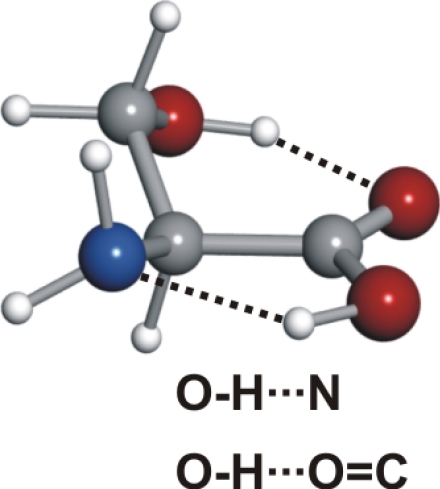
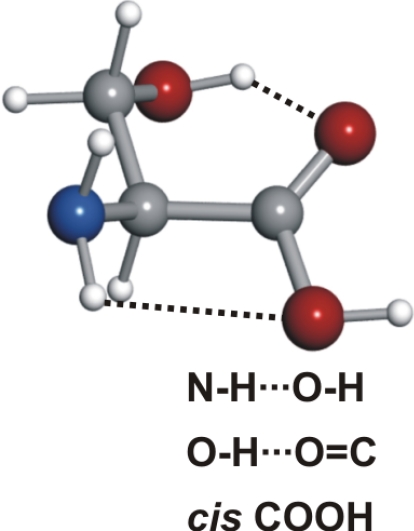
Serine's conformational versatility is likely to be influenced by its ability to reorient the flexible −CH2OH side chain and amino acid group so that they form different intramolecular hydrogen bonds. Conformer Ia exhibits two types of hydrogen bonds, which contribute to an enhanced stability. The amine group shows an interaction with the carbonyl group (NH···OC, type I) analogue to the most stable conformers of the aliphatic amino acids (19–25, 27–30, 34–36). Simultaneously, a second hydrogen bond is established between the hydroxy group (proton donor) and the nonbonding electron pair at the nitrogen atom (CβOH···N). Furthermore, this conformer has the carboxylic group in a cis configuration, which increases its stability (43).
Conformer Ia is followed in abundance by conformer IIb. All serine conformers labeled II possess a trans–COOH, allowing an intramolecular hydrogen bond to be formed between the hydroxyl group of the carboxylic moiety and the nonbonding electron pair at the nitrogen atom (N···HO). The side chain arrangement in conformer IIb allows two additional hydrogen bonds, where the −OH group acts simultaneously as a proton acceptor (through the interaction of the oxygen atom with the closest amino hydrogen) and proton donor (forming a hydrogen bond to the oxygen atom of the carbonyl group). The next conformer in abundance, I′b, shows a NH···OC hydrogen bond, with the −NH2 rotated ≈45° clockwise with respect to conformer Ia to allow the interaction of the electron lone pair of the N atom and the −OH of the side chain.
Conformer IIc, the fourth in abundance, has, besides the N···HO hydrogen bond, the hydroxyl group of the side chain pointing to the domain of one of the nonbonding electron pairs at the carbonyl group. Conformer IIa also presents the N···HO backbone interaction combined with a hydrogen bond between one of the amino hydrogen atoms and the oxygen atom of the side chain. The two other conformers detected, IIIβc and IIIβb (of similar smaller abundances to conformer IIa), show an NH···OH interaction. In conformer IIIβb, one of the amino hydrogen atoms acts as donor to the hydroxyl group in the −COOH and the other interacts with the oxygen atom of the side chain. In conformer IIIβc, the side chain orientation precludes the latter interaction but the terminal hydrogen of the side chain points in the direction of the carbonyl group. It is worth noting that type III conformers are absent from the rotational spectra of the α-amino acids with nonpolar side chains (19–25, 27–30, 34–36).
The stability of some serine conformers can be reinforced as a consequence of hydrogen bonding cooperativity. σ-Bond cooperativity occurs for continuous chains or cycles of hydrogen bonds linking functional groups possessing both donor and acceptor properties (44). For example, in conformer Ia, the three hydrogen bonds adopt a cooperative cyclic arrangement which might contribute to the stability of this form.
Conformational Cooling.
As occurred in other experiments conducted in supersonic expansions, the number of observed conformers does not comprise all thermically accessible forms corresponding to the equilibrium distribution predicted by ab initio calculations. It has been proposed (45) that this is a consequence of collisional relaxation at the onset of the supersonic expansion, which causes higher-energy forms to collapse to the preferred lower-energy ones due to collisions with the carrier gas. The extent of conformational relaxation depends on several factors, most significantly barrier heights between conformers (46–48) and the carrier gas (49–52). Type III conformers are “missing” in the supersonic expansion for all α-amino acids with nonpolar side chains studied up to now (19–25, 27–30, 34–36). This has been attributed to relaxation to configuration I, which is supported by the relatively small III → I interconversion barriers determined for glycine and alanine (19–24, 27).
Contrary to the previously described experimental behavior, type III conformers have been observed in serine. Of the four low-energy type III forms, conformers IIIβb and IIIβc have been detected. Conformer IIIαa, with a predicted relative energy lower than those of other detected conformers but higher than that of conformer Ia (see Fig. 2), is conspicuously absent from the rotational spectrum. Relative stabilities and conformational relaxation processes can be invoked to explain these observations.
Conformers IIIβb and IIIβc are predicted to be more stable than the corresponding Ib and Ic forms. One channel interconversion paths along the −COOH coordinate have been calculated theoretically between type I and III conformers. Fig. 3 shows a low barrier of 140 cm−1 for the interconversion from conformer Ib to conformer IIIβb. The nondetection of conformer Ib can be attributed to relaxation to the most stable form IIIβb, which cannot relax to any other conformer; therefore, it is observed in our experiment. A similar energy profile to that of Fig. 3 has been theoretically calculated between conformers Ic and IIIβc, which explains the observation of conformer IIIβc. In this context, nondetection of conformer Ic can be due to either low abundance in the supersonic expansion or to a I → III relaxation. Conformer Ia is predicted to be more stable than conformer IIIαa. The nondetection of conformer IIIαa can be understood in terms of relaxation to conformer Ia. The interconversion barrier of 220 cm−1 (Fig. 4) is comparable to the III → I interconversion barriers calculated for glycine (364 cm−1) (46) and alanine (274 cm−1) (see figure 2 of ref. 27). The absence of conformer III′αb in the rotational spectrum can be explained by its low abundance in the molecular beam, which causes the intensity of its rotational transitions to be below our detection limit. The changes in the postexpansion abundances induced by these relaxation processes are not expected to significantly alter the proposed stability trend.
Fig. 3.

Interconversion barrier between conformers Ib ⇆ IIIβb calculated at the MP2/6–311++G(d,p) level of theory. This interconversion path also shows that another high-energy conformer (IIIαb, lying at 1,061 cm−1 above the global minimum) can interconvert to Ib or IIIβb.
Fig. 4.

Interconversion barrier between conformers IIIαa ⇆ Ia calculated at the MP2/6–311++G(d,p) level of theory.
Conformers belonging to the a family have the −OH and −COOH groups in an antiperiplanar arrangement; thus, the −OH group can only interact with the amino group forming a OH···N bond that is present in both the Ia and IIIαa conformers. Therefore, the difference in stability between these conformers is entirely attributable to the interaction between the amino and carboxylic groups, and the same conformational preference observed for aliphatic α-amino acids without polar groups in their side chains remains (19–25, 27–30, 34–36). Conformers IIIβb and IIIβc present an NH···OH hydrogen bond plus an OH···OC interaction between the −OH of the side chain and the −COOH group. In the b and c families, the carboxylic group not only interacts with the amino group but also with the polar side chain, and this reverts the stability order, making type III conformers more stable than type I forms.
Conclusions
The detection of seven conformers of neutral serine provides a global picture of the behavior of an α-amino acid with a polar side chain. The hydrogen bonds observed in smaller α-amino acids between the amino and carboxylic groups are supplemented in serine with additional side-chain to main-chain interactions forming a hydrogen bond network that increments the conformational possibilities. The presence of a polar side chain in serine also changes its conformational preferences compared with those of α-amino acids with nonpolar side chains. Type III conformers have been detected in a supersonic expansion. In these cases, the conformational interconversion III → I in α-amino acids with nonpolar side chains is reverted to a I → III relaxation due to an additional interaction between the polar side chain and the carbonyl part of the −COOH group.
Collisional relaxation enhances the notion that the conformational behavior of serine is, to some extent, dynamic. It should be kept in mind that the need of a supersonic expansion for experimental observations perturbs the equilibrium conformational distribution decreasing the number of observable conformers. Despite this, the seven forms observed here can serve as a basis to represent the structure of neutral serine in the gas phase. The data provided can be of use for investigating the nature of serine aggregates, which are known to preferentially occur as octamers (53). These aggregates are believed to be composed of dimeric subunits, but their structure and character are still a matter of controversy.
Serine has been detected as a product of UV irradiation of insterstellar ice analogs (54), which suggests its interstellar origin. Unambiguous identification of serine in space needs precise values of the rotational transitions of its radiofrequency spectrum. The spectroscopic information provided in this work is relevant to check the existence of serine in the interstellar medium, testing the biological theories that propose that life on earth could have an extraterrestrial origin.
The marriage of microwave Fourier transform spectroscopy, pulsed supersonic expansion and laser ablation (LA-MB-FTMW) has been demonstrated to be a valid experimental approach to probe multiple serine conformations in the gas phase. Moving to an environment closer to the biological medium, it remains to be investigated how the observed conformational behavior is altered by docking water molecules to serine; one or several conformers could be selectively stabilized. In the case of glycine···water (37), only conformer I has been observed in the supersonic expansion. Larger amino acids and other solid biomolecules also can be produced in the gas phase by laser ablation and investigated by microwave spectroscopy.
Materials and Methods
The LA-MB-FTMW spectrometer used in this work (26) operates in the frequency range 6–18 GHz. Briefly, cylindrical rods of serine (99%; Aldrich; melting point, 240°C) were held at the exit of a special pulsed nozzle, where they were vaporized with the 532 nm pulses of a Nd:YAG laser and diluted into the expanding stream of neon at 4–6 bar. The supersonic jet fills a Fabry–Pérot microwave resonator where a short fixed-frequency microwave pulse (0.2–0.4 μs; <200 mW) polarizes the species in the jet. The subsequent transient molecular emission (≈400 μs in length) is digitized in the time domain. A Fourier transformation yields the frequency-domain spectrum. The line widths (full-width at half-maximum) of the rotational transitions are <7 kHz. Because the jet expansion is collinear with the resonator axis, all transitions are split into two Doppler components and the rest frequencies are calculated as the arithmetic mean of the frequency doublets. The estimated accuracy of the frequency measurements is better than 3 kHz.
Supplementary Material
ACKNOWLEDGMENTS.
This work was supported by Ministerio de Educación y Ciencia Grant CTQ2006-05981/BQU, and Junta de Castilla y León Grant VA012 C05. M.E.S. was supported by the Ministerio de Educación y Ciencia within the Ramón y Cajal Program and by Fondo Social Europeo Programa Operativo Integrado FEDER-FSE 2000/2006.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0705676104/DC1.
References
- 1.Dill KA, Chan HS. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 2.Kistenmacher TJ, Rand GA, Marsh RE. Acta Crystallogr B. 1974;30:2573–2578. [Google Scholar]
- 3.Görbitz CH, Dalhus B. Acta Crystallogr C. 1996;52:1754–1756. doi: 10.1107/s0108270196002041. [DOI] [PubMed] [Google Scholar]
- 4.Rizzo TR, Park YD, Petaunu LA, Levy DH. J Chem Phys. 1986;84:2534–2541. [Google Scholar]
- 5.Robertson EG, Simons JP. Phys Chem Chem Phys. 2001;3:1–18. [Google Scholar]
- 6.Weinkauf R, Schermann J-P, de Vries MS, Kleinermans K. Eur Phys J D. 2002;20:309–316. [Google Scholar]
- 7.Zwier S. J Phys Chem A. 2001;105:8827–8839. [Google Scholar]
- 8.Snoek LC, Robertson EG, Kroemer RT, Simons JP. Chem Phys Lett. 2000;321:49–56. [Google Scholar]
- 9.Lee KT, Sung J, Lee KJ, Kim SK, Park YD. J Chem Phys. 2002;116:8251–8254. [Google Scholar]
- 10.Lee Y, Jung J, Kim B, Butz P, Snoek LC, Kroemer RT, Simons JP. J Phys Chem A. 2004;108:69–73. [Google Scholar]
- 11.Martinez SJ, Alfano JC, Levy DH. J Mol Spectrosc. 1992;156:421–430. [Google Scholar]
- 12.Grace LI, Cohen R, Dunn TM, Lubman DM, de Vries MS. J Mol Spectrosc. 2002;215:204–219. [Google Scholar]
- 13.Snoek LC, Kroemer RT, Hockridge MR, Simons JP. Phys Chem Chem Phys. 2001;3:1819–1826. [Google Scholar]
- 14.Bakker JM, Aleese LM, Meijer G, von Helden G. Phys Rev Lett. 2004;91:2003003. doi: 10.1103/PhysRevLett.91.203003. [DOI] [PubMed] [Google Scholar]
- 15.Balle T, Flygare W. Rev Sci Instrum. 1981;52:33–45. [Google Scholar]
- 16.Andresen U, Dreizler H, Grabow J-U, Stahl W. Rev Sci Instrum. 1990;61:3694–3699. [Google Scholar]
- 17.Grabow J-U, Stahl W, Dreizler H. Rev Sci Instrum. 1996;67:4072–4084. [Google Scholar]
- 18.Storm V, Dreizler H, Consalvo D, Grabow J-U, Merke I. Rev Sci Instrum. 1996;67:2714–2719. [Google Scholar]
- 19.Brown RD, Godfrey PD, Storey JWV, Bassez MP. J Chem Soc Chem Commun. 1978:547–548. [Google Scholar]
- 20.Suenram RD, Lovas FJ. J Mol Spectrosc. 1978;72:372–382. [Google Scholar]
- 21.Suenram RD, Lovas FJ. J Am Chem Soc. 1980;102:7180–7184. [Google Scholar]
- 22.Lovas FJ, Kawashima Y, Grabow J-U, Suenram RD, Fraser GT, Hirota E. Astrophys J. 1995;455:L201–L204. [Google Scholar]
- 23.Godfrey PD, Brown RD. J Am Chem Soc. 1995;117:2019–2023. [Google Scholar]
- 24.McGlone SJ, Elmes PS, Brown RD, Godfrey PD. J Mol Struct. 1999;486:225–238. [Google Scholar]
- 25.Godfrey PD, Firth S, Hatherley LD, Brown RD, Pierlot AP. J Am Chem Soc. 1993;115:9687–9691. [Google Scholar]
- 26.Lesarri A, Mata S, López JC, Alonso JL. Rev Sci Instrum. 2003;74:4799–4804. [Google Scholar]
- 27.Blanco S, Lesarri A, López JC, Alonso JL. J Am Chem Soc. 2004;126:11675–11683. doi: 10.1021/ja048317c. [DOI] [PubMed] [Google Scholar]
- 28.Lesarri A, Cocinero EJ, López JC, Alonso JL. Angew Chem Int Ed. 2004;43:605–610. doi: 10.1002/anie.200352543. [DOI] [PubMed] [Google Scholar]
- 29.Lesarri A, Sánchez R, Cocinero EJ, López JC, Alonso JL. J Am Chem Soc. 2005;127:12952–12956. doi: 10.1021/ja0528073. [DOI] [PubMed] [Google Scholar]
- 30.Cocinero EJ, Lesarri A, Grabow J-U, López JC, Alonso JL. ChemPhysChem. 2007;8:599–604. doi: 10.1002/cphc.200600730. [DOI] [PubMed] [Google Scholar]
- 31.Lesarri A, Mata S, Cocinero EJ, Blanco S, López JC, Alonso JL. Angew Chem Int Ed. 2002;41:4673–4676. doi: 10.1002/anie.200290012. [DOI] [PubMed] [Google Scholar]
- 32.Lesarri A, Cocinero EJ, López JC, Alonso JL. J Am Chem Soc. 2005;127:2572–2579. doi: 10.1021/ja045955m. [DOI] [PubMed] [Google Scholar]
- 33.Sanz ME, Lesarri A, Peña I, Vaquero V, Cortijo V, López JC, Alonso JL. J Am Chem Soc. 2006;128:3812–3817. doi: 10.1021/ja058194b. [DOI] [PubMed] [Google Scholar]
- 34.Lesarri A, Cocinero EJ, López JC, Alonso JL. ChemPhysChem. 2005;6:1559–1566. doi: 10.1002/cphc.200500103. [DOI] [PubMed] [Google Scholar]
- 35.Cocinero EJ, Villanueva P, Lesarri A, Sanz ME, Blanco S, Mata S, López JC, Alonso JL. Chem Phys Lett. 2007;435:336–341. [Google Scholar]
- 36.Sanz ME, Cortijo V, Caminati W, López JC, Alonso JL. Chem Eur J. 2006;12:2564–2570. doi: 10.1002/chem.200501013. [DOI] [PubMed] [Google Scholar]
- 37.Alonso JL, Cocinero EJ, Lesarri A, Sanz ME, López JC. Angew Chem Int Ed. 2006;45:3471–3474. doi: 10.1002/anie.200600342. [DOI] [PubMed] [Google Scholar]
- 38.Gronert S, O'Hair RAJ. J Am Chem Soc. 1995;117:2071–2081. [Google Scholar]
- 39.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, et al. Gaussian03. Pittsburgh PA: Gaussian; 2003. Version B.04. [Google Scholar]
- 40.Gordy W, Cook RL. Microwave Molecular Spectra. New York: Wiley; 1984. [Google Scholar]
- 41.Watson JKG. In: Vibrational Spectra and Structure. Durig JR, editor. Vol 6. Amsterdan: Elsevier; 1977. pp. 1–89. [Google Scholar]
- 42.Fraser GT, Suenram RD, Lugez CL. J Phys Chem A. 2000;104:1141–1146. [Google Scholar]
- 43.Császár AG, Allen WD, Schaefer HF. J Chem Phys. 1998;108:9751–9764. [Google Scholar]
- 44.Blanco S, López JC, Lesarri A, Alonso JL. J Am Chem Soc. 2004;128:12111–12121. doi: 10.1021/ja0618393. [DOI] [PubMed] [Google Scholar]
- 45.Ruoff RS, Klots TD, Emilson T, Gutowski HS. J Chem Phys. 1990;93:3142–3150. [Google Scholar]
- 46.Godfrey PD, Brown RD, Rodgers FM. J Mol Struct. 1996;376:65–81. [Google Scholar]
- 47.Godfrey PD, Brown RD. J Am Chem Soc. 1998;120:10724–10732. [Google Scholar]
- 48.Florio GM, Christie RA, Jordan KD, Zwier TS. J Am Chem Soc. 2002;124:10236–10247. doi: 10.1021/ja0265916. [DOI] [PubMed] [Google Scholar]
- 49.Antolínez S, López JC, Alonso JL. Angew Chem Int Ed. 1999;38:1772–1774. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1772::AID-ANIE1772>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 50.Sanz ME, López JC, Alonso JL. Chem Eur J. 1999;5:3293–3298. [Google Scholar]
- 51.Sanz M. E., López JC, Alonso JL. Angew Chem Int Ed. 2001;40:935–938. [PubMed] [Google Scholar]
- 52.Antolínez S, López JC, Alonso JL. ChemPhysChem. 2001;2:114–117. doi: 10.1002/1439-7641(20010216)2:2<114::AID-CPHC114>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 53.Nanita SC, Cooks RG. Angew Chem Int Ed. 2006;45:554–569. doi: 10.1002/anie.200501328. [DOI] [PubMed] [Google Scholar]
- 54.Bernstein MP, Dworkin JP, Sandford SA, Cooper GW, Allamandola LJ. Nature. 2002;416:401–403. doi: 10.1038/416401a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}