

Abstract
Invariant natural killer T (iNKT) cells are a subset of nonconventional T cells recognizing endogenous and/or exogenous glycolipid antigens in the context of CD1d molecules. It remains unclear whether innate stimuli can modify the profile of endogenous lipids recognized by iNKT cells on the surface of antigen-presenting cells (APCs). We report that activation of human APCs by Toll-like receptor ligands (TLR-L) modulates the lipid biosynthetic pathway, resulting in enhanced recognition of CD1d-associated lipids by iNKT cells, as defined by IFN-γ secretion. APC-derived soluble factors further increase CD1d-restricted iNKT cell activation. Finally, using soluble tetrameric iNKT T cell receptors (TCR) as a staining reagent, we demonstrate specific up-regulation of CD1d-bound ligand(s) on TLR-mediated APC maturation. The ability of innate stimuli to modulate the lipid profile of APCs resulting in iNKT cell activation and APC maturation underscores the role of iNKT cells in assisting priming of antigen-specific immune responses.
Keywords: APC, glycosphingolipids, innate immunity, NKT cells
Natural killer T (NKT) cells are a subset of nonconventional T cells recognizing lipid antigens presented by the nonpolymorphic CD1d molecule (1). The majority of human and murine NKT cells, referred to as invariant NKT cells (iNKT), display a highly restricted TCR repertoire, consisting of a semiinvariant TCR α chain (Vα24Jα18 in humans and Vα14Jα18 in mice) and a more variable β chain (Vβ11 in humans and Vβ2, Vβ7, and Vβ8 in mice). iNKT cells play a role in the control of bacterial, parasitic, viral infections, and natural immunosurveillance (2). A number of synthetic glycolipids have been shown to activate iNKT cells. Although the best-characterized agonist to date is α-galactosylceramide (α-GalCer) (3), it has recently been shown that iNKT cells also recognize bacterial glycosphingolipids (GSLs), glycolipids, and cellular phospholipids (4–9).
CD1d molecules have a restricted pattern of expression limited to APCs, including dendritic cells (DCs), B cells, macrophages, cortical thymocytes, and some epithelial cells (2). Professional APCs, such as DCs, are essential players in eliciting adaptive immune responses. DCs reside in peripheral tissues and can be activated by inflammatory stimuli, direct recognition of microbial components via pattern recognition receptors [such as Toll-like receptors (TLR)] or T cell-derived signals (10). Activation of iNKT cells via exogenous CD1d ligands, such as α-GalCer, induces the maturation of DCs and B cells, resulting in powerful adjuvant activity, leading to enhanced CD4, CD8, and antibody responses to coinjected protein antigens (11, 12). In this setting, iNKT cell mediated recognition of bacterial lipids may serve as a “surrogate pattern recognition receptor” for pathogens lacking TLR-ligands (5–7). For some pathogens, however, recognition of exogenous microbial antigens is not required, and iNKT cells are activated after amplification of weak self-reactivity by IL-12 released from DCs upon TLR signaling (6, 13). A TCR-independent mode of bacterial recognition has also been recently reported (14).
Recognition of endogenous antigens is essential for iNKT cell development and selection by double positive cortical thymocytes (1). The identity of the selecting lipid ligand(s) remains unknown. Evidence from a β-glucosylceramide synthase mutant cell line unable to stimulate Vα14 iNKT cells has suggested that the natural ligand(s) is a GSL (15). More recently, it has been proposed that the natural selecting ligand presented by cortical thymocytes and LPS matured DCs is the neutral GSL isoglobotrihexosylceramide (iGb3) (6, 8). However, there is no biochemical evidence for the presence of iGb3 in mouse or human thymus or DCs (16), and iGb3 synthase deficient mice have normal number and subsets of iNKT cells (17). Further studies are therefore required to identify other lipid species responsible for iNKT thymic selection and/or peripheral expansion.
Because it has been shown that iNKT cells can rapidly license DCs for T cell priming via CD40L signaling (11, 12), it is important to assess whether TLR-dependent DC maturation also results in the up-regulation of natural lipid ligands that can be directly recognized by iNKT cells via CD1d. To address this question, we set up an experimental system to study the relative contribution of soluble factors versus self-lipid antigens in eliciting CD1d-restricted human iNKT cells activation after stimulation of APCs with a panel of TLR ligands.
Results
DC Activation with TLR Ligands Enhances NKT Cell Basal Autoreactivity.
Immature human DCs were incubated with a panel of TLR ligands at different concentrations, and DC activation was studied in the presence or absence of iNKT cells. Stimulation through TLR-2 (Pam3Cys), TLR-3 [polyinosinic:polycytidylic acid (poly I:C)], TLR-4 (LPS), TLR-5 (flagellin), and TLR-8 (ssRNA-40 and R-848) led to different degrees of DC activation, as evidenced by secretion of IL-12 (Fig. 1A). In most cases, the presence of iNKT cells resulted in enhanced DC activation. In addition, coculture of TLR-L matured DCs and iNKT cells resulted in iNKT cell activation, as shown by IFN-γ secretion (Fig. 1B) and up-regulation of activation markers (data not shown). Subsequent experiments were performed with LPS and R-848, prototype ligands of a cell surface (TLR-4) and an endosomal (TLR-8) receptor and of two different signaling pathways. Both TLR-4 and TLR-8 signal through the adaptor molecule MyD88, whereas TLR-4 also utilizes the TRIF adaptor molecule (18). Of note, R-848 also signals through the endosomal receptor TLR-7, however, human monocyte-derived DCs only express TLR-8 (19).
Fig. 1.

DC maturation by TLR-L enhances iNKT cells basal recognition. (A and B) Human DCs were left untreated (Imm) or matured with a panel of TLR-L at the indicated concentrations in the presence or absence of the iNKT line MS mix. The means of triplicate values and standard deviation of IL-12 p40 (A) and IFN-γ (B) of one ELISA experiment representative of three are shown. IL-12 p70 was detected only in samples stimulated with R-848 (data not shown). Soluble factors, like type I IFN, may influence the overall secretion of IL-12 by DC, hence the differences between different TLR-L. (C and D) Activation of different subsets of NKT cells by TLR-L-matured DC. Four iNKT cell lines and a noninvariant Vα24-negative NKT cell line, were incubated with DCs and R-848 or LPS. The means of triplicate values and standard deviation of IFN-γ (C) and IL-12 p40 (D) of one ELISA experiment representative of three are shown. DCs from different donors were used in A and B and C and D, hence the variability in the absolute amounts of IFN-γ released by iNKT cells.
Recognition of R-848, and to a certain extent of LPS-matured DCs, was not restricted to any iNKT cell subset but could be observed with a panel of different iNKT lines and clones (CD4+, CD8+, and DN), including a Vα24-negative Vβ11+ NKT line (Fig. 1 C and D). In all experiments, iNKT cell activation by TLR-L stimulated DCs resulted in IFN-γ but not IL-4 secretion (data not shown).
Requirements for iNKT Cell Activation by TLR-L Matured DCs.
To investigate the relative contribution of DC metabolic processes and soluble factors released during DC maturation, we compared iNKT cell recognition of live versus fixed DCs (Fig. 2A). DC fixation, before exposure to either LPS or R-848, prevented iNKT cell activation, demonstrating the requirement for an active APC metabolic process. These data also rule out a direct effect of the two TLR ligands on iNKT cells. By contrast, when DCs were fixed after overnight maturation with the TLR-8 ligand R-848, we observed a 30–50% reduction in their iNKT cell stimulatory capacity compared with live DCs.
Fig. 2.

The role of soluble factors and CD1d in TLR-dependent iNKT cell activation. (A) Fixation of DCs reduces iNKT cell activation. Live human DCs (black bars) were either matured with R-848 or LPS in the presence of the iNKT cell line MS mix or matured overnight with R-848, then washed and incubated with iNKT cells (R848 O/N). As a control, DCs were fixed with glutaraldehyde (gray bars) and then incubated with iNKT cells in the presence or absence of the indicated TLR-L. Only live cells secreted IL-12 p40 (SI Fig. 6A). (B) iNKT cells' recognition of R-848-matured DCs is CD1d- and IL-12-dependent. Human DCs were matured with R-848 in the presence of the iNKT cell line MS mix and each of the indicated blocking antibody. The means of triplicate values and standard deviation of IFN-γ of one ELISA experiment representative of three are shown.
To establish whether the iNKT cell stimulatory effect on TLR-L activation was mediated by CD1d-restricted antigen presentation, we demonstrated that, in the presence of anti-CD1d blocking antibodies, iNKT cells recognition of R-848 matured DCs was reduced by 50% (Fig. 2B). The reduction of iNKT cell stimulation after fixation of R-848 matured DCs (Fig. 2A) also suggested a contribution of DC-derived soluble factors. Indeed, we confirmed the role of IL-12 in amplifying iNKT cell autoreactivity (13) by demonstrating that anti-IL-12 antibodies resulted in 50% reduction of DC recognition. The combination of anti-CD1d and anti-IL-12 antibodies abrogated DC recognition (Fig. 2B).
Soluble Factors Amplify iNKT Cell Basal Autoreactivity.
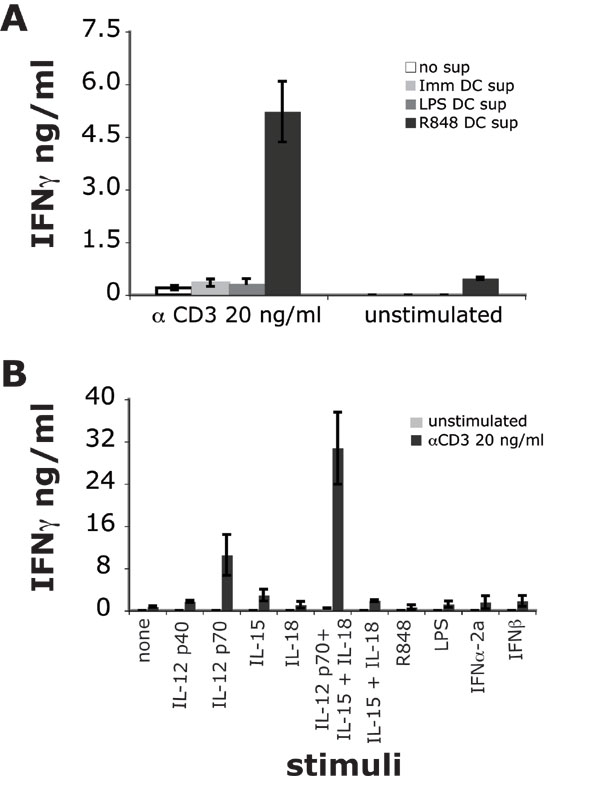
To further investigate the role of soluble factors in mediating iNKT cell activation, we incubated iNKT cells with supernatants from immature or matured DCs, in the presence of suboptimal amounts of plate bound anti-CD3 antibodies, to mimic weak TCR stimulation. Anti-CD3 stimulation alone resulted in minimal iNKT cell activation, which was not enhanced by supernatants from immature or LPS-matured DCs, most likely because of a lack of bioactive IL-12 [supporting information (SI) Fig. 6A and data not shown]. Conversely, supernatants from R-848 matured DCs markedly enhanced iNKT cell activation in the presence of anti-CD3 stimulation (8-fold) and induced minimal activation, even in the absence of TCR triggering.
We excluded direct stimulation of iNKT cells via TLR-7/8 or TLR-4, because neither R-848 nor LPS were capable of enhancing anti-CD3 stimulation (SI Fig. 6B). IL-12 p70 alone or in combination with IL-15 and IL-18 potently enhanced anti-CD3 stimulation (7- and 30-fold, respectively). The combination of IL-12 p70, IL-15, and IL-18 induced IFN-γ secretion (but not IL-4) by iNKT cells even in the absence of suboptimal TCR stimulation (SI Fig. 6B and data not shown). Conversely, IFNα-2a and IFNβ did not enhance anti-CD3 stimulation and had no effect in the absence of TCR triggering.
iNKT Cell Autoreactivity Is Associated with Increased GSL Biosynthesis.
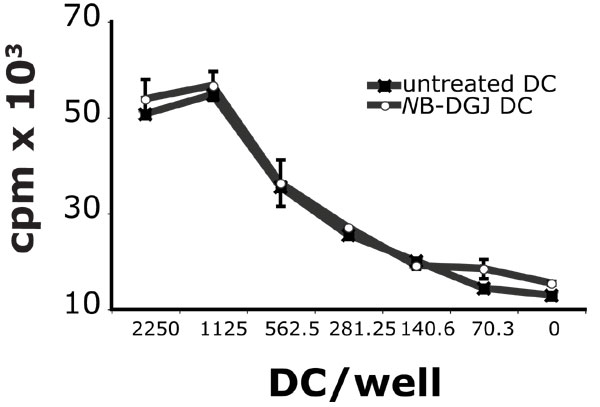
To investigate the possibility that de novo biosynthesis of GSLs is important for iNKT cell activation, we treated human DCs for 4 days with the specific inhibitor of GSL biosynthesis, N-butyldeoxygalactonojirimycin (NB-DGJ) (20). NB-DGJ treatment reduced the stimulation by R-848 and LPS matured DCs by 50% and 33%, respectively (Fig. 3A). The antigen-presenting capacity of the same DCs was unaffected when the exogenous antigen C20:2 [analogue of α-GalCer (21)] was used to stimulate iNKT cells and when DCs were used to stimulate alloreactive T cells (SI Fig. 7), ruling out a nonspecific effect of NB-DGJ. Consistent with TLR-L-dependent up-regulation of GSL antigen(s), by quantitative PCR (Q-PCR) we observed that R-848 matured DCs increase the expression of genes encoding enzymes responsible for the synthesis of GlcCer and LacCer (Fig. 3 B and C). There was a slight reduction in the gene encoding for the enzyme leading to GSLs of the lacto/neolactoseries and a marked increase in genes encoding for enzymes synthesizing GSLs of the globo and ganglio series. We did not detect any amplification of iGb3 synthase, consistent with reports of it being a pseudogene in humans (data not shown and ref. 22). Similar results were obtained by using LPS matured DCs (data not shown).
Fig. 3.

Increased GSL biosynthesis in TLR-L-matured DCs contributes to iNKT cell activation. (A) Inhibition of GSL biosynthesis reduces iNKT cell activation by TLR-L-matured DCs. Monocytes were differentiated to DCs in the presence of NB-DGJ. Untreated and NB-DGJ-treated DCs were incubated with the iNKT line MS mix in the presence or absence of LPS or R-848. As control, untreated and NB-DGJ-treated DCs were pulsed with C20:2. The means of triplicate values and standard deviation of IFN-γ of one ELISA experiment representative of five are shown. (B and C) Q-PCR analysis of enzymes involved in the GSL biosynthetic pathway. (B) Diagram of the GSL biosynthetic pathway. Red arrows indicate genes that have been up-regulated. (C) The expression of the genes involved in the GSL biosynthetic pathway and of CD1d and CD83 is shown in immature and R-848-matured DCs, relative to two housekeeping genes (HKG), β-actin, and ubiquitin C. Data represent mean values of triplicates and standard deviation of one experiment representative of three.
The increased iNKT cells stimulatory capacity of R-848-treated DCs could also be due to increased cell surface expression of CD1d and co-stimulatory molecules. The results of our experiments indicated that CD1d up-regulation does not play a major role. Using three different antibodies in flow cytometry assays, CD1d expression was undetectable on the surface of immature and R-848-matured human DCs (data not shown). In addition, Q-PCR analysis of CD1d mRNA confirmed equal levels of expression of CD1d transcripts between immature and mature DCs (Fig. 3C).
Myelomonocytic Cell Line THP-1 Elicits iNKT Cell Activation.
To further investigate the contribution of CD1d and GSLs to iNKT cell activation, we extended our investigations to the myelomonocytic cell line THP-1. Unlike monocyte-derived DCs, THP-1 cells have low but detectable levels of CD1d expression (data not shown and SI Fig. 8A). Preliminary experiments revealed that THP-1 cells responded only to TLR-4 and TLR-8 ligands, but not to other TLR-L, including the TLR-7 ligand R-837, which was used as negative control. THP-1 cells stimulated with R-848, and two other TLR-8 ligands, CL097 and CL075 (23), induced efficient iNKT cell activation (Fig. 4A and data not shown). Overexpression of CD1d by lentiviral transduction (SI Fig. 8A) enhanced the basal iNKT cell autoreactivity and led to increased response to R-848 and LPS but not to R-837 (Fig. 4A). In all experiments, iNKT cell activation by TLR-L stimulated THP-1 cells resulted in IFN-γ but not IL-4 secretion (data not shown). The iNKT cell autoreactivity to untreated THP-1 CD1d cells was completely blocked by anti-CD1d antibodies (data not shown). Reactivity to R-848-treated THP-1 cells was blocked by a combination of anti-CD1d and anti-IL-12 antibodies, consistent with release of IL-12 upon maturation (data not shown). These results confirmed our previous findings with DCs.
Fig. 4.

THP-1 cells and DCs express ligands recognized by iNKT cells. (A) THP-1 cells transduced with control (mock, gray bars) or with human CD1d lentivirus (black bars) were stimulated with LPS, R-848, or R-837 in the presence of the iNKT line MS mix. (B) Total lipid fractions extracted from immature and R-848 matured DCs were pulsed on immature DCs. Cells were incubated with the iNKT line MS mix in the presence of IL-12 p70. Controls are unpulsed DCs in the presence or absence of IL-12 p70. (C) Total lipid fractions extracted from immature and R-848 matured THP-1 cells were pulsed on immature THP-1 cells. Activation of the iNKT line MS mix was assessed in the presence of IL-12 p70 and isotype control antibody or blocking anti-CD1d. The means of triplicate values and standard deviation of IFN-γ of one ELISA experiment representative of five are shown.
Total APC-Derived Lipid Fractions Sensitize CD1d-Positive Targets.
To further investigate the contribution of endogenous lipids to iNKT cell activation, we isolated the total lipid fraction from immature and R-848-treated DCs and THP-1 cells. DCs and THP-1 cells pulsed with a total lipid extract were capable of activating both iNKT cells and the Vα24-negative line in the presence, but not in the absence, of soluble recombinant bioactive IL-12 (Fig. 4 B and C and data not shown). As a specificity control, iNKT cell activation was abolished in the presence of blocking anti-CD1d antibodies (Fig. 4C). Consistent with the observed autoreactivity against immature DCs and THP-1 cells, lipid fractions isolated from immature DCs and untreated THP-1 cells were only marginally less potent that those isolated from R-848-treated cells at inducing iNKT cell activation. These results also underscore the important role of soluble factors released during maturation in amplifying iNKT cell autoreactivity.
Direct Detection of iNKT Cell Ligand(s) at the Surface of CD1d-Positive APCs.
To confirm the role of CD1d-lipid complexes in eliciting iNKT cell activation, irrespective of any soluble factor released during APCs stimulation with TLR ligands, we probed CD1d-natural ligand complexes at the surface, using tetrameric soluble iNKT T cell receptors (TCR) as staining reagents (24). Soluble iNKT TCRs stained specifically THP-1 CD1d cells but not THP-1 mock transduced cells (Fig. 5A), confirming the ELISA results showing an increased iNKT cell stimulatory capacity of THP-1 CD1d cells. We did not detect staining of cells expressing low levels of surface CD1d (THP-1 and DCs), consistent with the low-affinity interaction between iNKT TCR and CD1d molecules (24, 25). However, activation of THP-1-CD1d cells with R-848 and with LPS, but not with R-837, led to increased staining with the soluble iNKT TCR (Fig. 5A). The specificity of the staining with the soluble TCR was demonstrated by blocking experiments with anti-CD1d antibody, which reduced the tetramer staining to near baseline levels (Fig. 5A). In addition, competition of the natural ligand(s) with excess GM3 ganglioside reduced the intensity of the staining with the soluble iNKT TCR, both on immature and R-848-treated THP-1 cells (Fig. 5B and SI Fig. 8B). No staining of immature or mature THP-1 CD1d cells was observed with an irrelevant soluble TCR (SI Fig. 8C).
Fig. 5.

Direct detection of iNKT cell ligand(s) by soluble iNKT TCR staining. (A) THP-1 CD1d cells were activated for 36 h with LPS, R-848, or R-837. The presence of iNKT cell ligands on the surface of THP-1 CD1d cells was revealed by staining with the soluble iNKT TCR in the presence of isotype control or blocking anti-CD1d antibodies. (B) THP-1 CD1d cells were activated for 24 h with R-848 and incubated with an excess of the ganglioside GM3 overnight or blocking anti-CD1d antibodies before staining with the soluble iNKT TCR. Streptavidin staining is included as negative control. THP-1 mock transduced cells and DCs did not show any detectable staining (data not shown). Numbers in brackets represent the mean fluorescence intensity (MFI) values of the iNKT TCR staining. CD1d MFI remained unchanged with maturation (data not shown). One experiment representative of six is shown.
Discussion
We set up an experimental system to dissect the contribution of APC-derived soluble factors and CD1d/lipid antigens in activating human iNKT cells in response to TLR-L engagement of APCs. We demonstrated that DC maturation by TLR-L induces an active metabolic process, which leads to enhanced recognition by iNKT cells of CD1d-associated GSLs, as shown by experiments using the specific inhibitor of GSL biosynthesis, NB-DGJ (20). Activation of iNKT cells by both DCs and the myelo-monocytic cell line THP-1 resulted in secretion of IFN-γ, but not IL-4, as observed with LPS-matured DCs (13).
Recognition of TLR-L matured DCs was not restricted to specific iNKT cell subsets and was also observed with a Vα24-negative Vβ11+ NKT line. These results are consistent with the similar crystal structure and binding affinities for CD1d-α-GalCer monomers of Vα24 positive and negative NKT TCRs (25). In addition, clones derived from both lines display nearly identical CDR3-α sequences, which have been recently shown to directly interact with CD1d α1-, α2- helixes, and α-GalCer (26).
Activation of iNKT cells by TLR-L matured DCs depended on both the up-regulation of endogenous ligands and on soluble factors, consistent with previous studies (13). Indeed, we observed that iNKT cell recognition of self-lipid antigens was abolished by a combination of anti-CD1d and anti-IL-12 antibodies. Moreover, there was a synergistic effect of IL-12, IL-15, and IL-18 in enhancing iNKT cell responses to suboptimal TCR-mediated stimulation. In addition, these cytokines were capable of activating iNKT cells in the absence of TCR stimulation, suggesting an important role for inducing iNKT homeostatic proliferation as reported for murine iNKT cells in vivo (14, 27).
DC maturation results in increased expression of adhesion and costimulatory molecules, which could also contribute to enhanced iNKT cell activation. Although we and others have observed increased CD1d expression in murine DCs and macrophages, after activation by bacteria, cytokines, and TLR-L (data not shown and refs. 28 and 29), we have not been able to detect increased CD1d expression after activation of monocyte-derived DCs by either FACS or Q-PCR. Our results are in agreement with previous reports demonstrating no change in CD1d expression in human DCs matured by TNF-α or CD40L (30) and a predominant effect of MTB and TLR-2 stimulation on group 1 CD1 molecules (31, 32).
To further confirm the role of GSLs in eliciting iNKT cell activation, we demonstrated that total lipids from APCs were able to elicit iNKT cell activation in the presence of recombinant IL-12, suggesting a low-affinity interaction. We were unable to further characterize the class of lipids responsible for direct activation of iNKT cells by either base treatment (to eliminate all lipids except those based on β-GlcCer and β-GalCer) or ceramide glycanase treatment (to release oligosaccharides from the GlcCer-based GSLs) (33), because of reduced viability of DCs pulsed with base-treated fractions and maturation of DCs pulsed with ceramide glycanase-treated factions (data not shown). Likewise, pulsing of DCs with total lipids separated into neutral and charged fractions resulted in DC maturation (data not shown). However, we detected by Q-PCR an up-regulation of transcripts of genes involved in GSL biosynthesis. Although there is no substitute for biochemically measuring GSLs in tissues and cell lines, these results suggest changes in GSL biosynthesis in agreement with previous reports on the effect of LPS on lipid metabolism and presentation of self-antigens in association with CD1a and CD1b molecules (34, 35). In addition, these results do not exclude that other classes of lipids can be iNKT-activating ligands, because reduction of up to 80% of the cellular GSL content after NB-DGJ treatment does not abrogate iNKT cell recognition.
Over-expression of CD1d molecules on THP-1 cells provided the opportunity to assess whether a soluble tetrameric iNKT TCR could be used to directly bind to CD1d molecules loaded with natural ligands. The results of these experiments unequivocally demonstrated that THP-1 cells stimulated with both LPS and R-848 could be specifically stained by the soluble iNKT TCR at higher levels than unstimulated THP-1 cells. The ability to specifically stain CD1d molecules loaded with natural ligands is of great importance because it validates the iNKT cell-based results and provides a tool to biochemically characterize the identity of natural ligand(s) capable of stimulating/selecting iNKT cells.
Recent studies have reported that TLR signaling coordinates pathogen recognition with antigen uptake and efficient activation of class II-restricted T cell responses (36, 37). We have now shown that TLR-mediated activation of APCs alters lipid metabolism, resulting in iNKT cell activation and enhancing DC maturation. Our data complement results on the effect of TLR agonists on the synthesis of group 1 CD1 molecules and some of their lipid ligands (31, 34). Importantly, these results also suggest that, in addition to directly or indirectly sensing bacteria, iNKT cells can be activated during viral infections, despite the absence of unique glycolipid antigens within viruses. Viral recognition by human iNKT cells can be accomplished upon triggering of the TLR-8 pathway in myeloid cells in addition to the reported cross-talk between myeloid and plasmacytoid dendritic cells upon TLR-9 engagement (38, 39). Because iNKT-dependent DC maturation is an important step in linking innate and adaptive immune responses, understanding the molecular mechanisms and the ligands capable of fine-tuning these interactions is of great importance for harnessing these cells for clinical use in treating and studying infections, autoimmune diseases, and cancer.
Materials and Methods
Medium and Reagents.
The complete medium (CM) used throughout was RPMI medium 1640 (Gibco) supplemented with 10% FCS (Sigma) or 5% human AB serum as described in ref. 40. The following TLR-L were used: poly I:C and LPS from Salmonella abortus equi (5 μg/ml) (Sigma); R-848 (10 μg/ml) (PharmaTech); R-837, ssRNA-40, CL097, CL075, and flagellin (InvivoGen); Pam3Cys4K (EMC Microcollections).
Anti-human CD3 and recombinant human IL-12 were purchased from PharMingen, and recombinant human IL-15 and IL-18 were from Peprotech. IFNα-2a (Roferon-A) was purchased from Roche, and IFNβ-1a (Avonex) was purchased from Biogen.
GM3 was purchased from Matreya LLC (Pleasant Gap, PA). α-GalCer and C20:2 were synthesized as described in ref. 41.
APCs and NKT Cells.
DCs and NKT cells were generated from healthy blood donors as described in refs. 24 and 40. THP-1 cells [American Type Culture Collection (ATCC)] were transduced with a lentiviral vector encoding for human CD1d (M.S., unpublished data).
Stimulation Assays.
APCs were plated at 50,000 cells per well in 96-well plates in CM and incubated with iNKT cells (2–3 × 104 per well, in triplicate) in the presence or absence of different concentrations of TLR-L. The TLR-L were maintained in the cocultures for the duration of the assay. In some experiments, DCs were matured overnight with 5 μg/ml R848, washed and used either live or after fixation. DC fixation was performed and controlled as described in ref. 34. For blocking experiments, APCs were preincubated for 2 h with TLR-L and subsequently for 2 h with 25 μg/ml anti human-CD1d [CD1d42.1, a kind gift from S. Porcelli (Albert Einstein College of Medicine, Bronx, NY)], anti-human IL-12 (PharMingen), or anti TNP-isotype control (ATCC), before the addition of iNKT cells.
APCs and iNKT cell activation was assessed by ELISA (IFN-γ, IL-12 p40, and IL-12 p70, all from PharMingen) on supernatants harvested after 36 h.
Inhibition of GSL Synthesis.
NB-DGJ (Calbiochem) was added at a final concentration of 50 μM during DC differentiation and maintained at this concentration during the iNKT cell stimulation assay. To rule out toxicity of the inhibitors, graded numbers of treated and untreated DCs were irradiated (1,500 rad) and cultured with allogeneic peripheral blood mononuclear cells (250,000 per well). The T cell proliferative response was measured on day 5 by [3H]thymidine (Amersham) incorporation (0.037 MBq per well). The extent of GSL inhibition (≈50%, data not shown) was determined by normal phase HPLC analysis of the GSL content as described in ref. 16.
RNA Extraction and Q-PCR.
DCs were untreated or stimulated for 36 h with 1 μg/ml LPS or 5 μg/ml R-848. RNA was extracted with RNAeasy (Qiagen). cDNA was synthesized by using the High-Capacity cDNA Archive kit (Applied Biosystems). Real-time quantitative PCR was performed in triplicate, using the Corbett Research Rotor Gene RG-3000. Conditions for the PCR, primers, and probes are summarized in SI Table 1. Relative quantitation of gene expression was carried out according to the method described by Pfaffl (42).
GSL Extraction and Treatment.
Total lipid fractions, base treatment, and ceramide glycanase treatment were performed as described in ref. 33. The total fraction was dried under nitrogen, resuspended in FCS at 5 × 105 cell equivalent per microliter, and extensively sonicated. DCs or THP-1 cells were pulsed with 1 μl of each fraction in a final volume of 200 μl, in the presence or absence of recombinant IL-12 p70 (0.05 ng/ml), and used to activate the NKT cells.
iNKT TCR Tetramer Staining.
The generation of soluble TCR heterodimers has been described (24). Immature and TLR-L matured THP-1 and THP-1 CD1d cells were incubated with 0.5–1 μg of iNKT-tetramer or equivalent amounts of streptavidin on ice for 1 h. The irrelevant 1G4-NY-ESO-1 specific soluble TCR (43) was used as negative control at the same concentration. Cells were washed, and samples were analyzed on a Cyan flow cytometer (Dako). Data were processed by using Flowjo software (Treestar). To block the TCR staining, cells were incubated for 30 min on ice with 30 μg/ml of anti-CD1d antibody (CD1d42.1) or isotype control, washed, and stained with the tetramer. To compete the TCR staining, cells were treated with the TLR-L for 24 h, then the ganglioside GM3 was added to the same wells at 2 μg/ml overnight before staining.
Supplementary Material
ACKNOWLEDGMENTS.
This work was supported by Cancer Research United Kingdom Grant C399/A2291 Medical Research Council (to M.S., D.S., and V.C.); an Immunanomap Marie Curie Studentship (to P.P.); a scholarship from the Glycobiology Institute, University of Oxford (to A.O.S.); and a Personal Research Chair from Mr. James Bardick, a former Lister Institute-Jenner Research Fellowship, the Medical Research Council, and the Wellcome Trust (G.S.B.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0710145104/DC1.
References
- 1.Bendelac A, Savage PB, Teyton L. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 2.Kronenberg M. Annu Rev Immunol. 2005;23:877–900. doi: 10.1146/annurev.immunol.23.021704.115742. [DOI] [PubMed] [Google Scholar]
- 3.Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, et al. Science. 1997;278:1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 4.Kinjo Y, Tupin E, Wu D, Fujio M, Garcia-Navarro R, Benhnia MR, Zajonc DM, Ben-Menachem G, Ainge GD, Painter GF, et al. Nat Immunol. 2006;7:978–986. doi: 10.1038/ni1380. [DOI] [PubMed] [Google Scholar]
- 5.Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, Tsuji M, Kawahara K, Wong CH, Kronenberg M. Nature. 2005;434:520–525. doi: 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- 6.Mattner J, Debord KL, Ismail N, Goff RD, Cantu C, III, Zhou D, Saint-Mezard P, Wang V, Gao Y, Yin N, et al. Nature. 2005;434:525–529. doi: 10.1038/nature03408. [DOI] [PubMed] [Google Scholar]
- 7.Sriram V, Du W, Gervay-Hague J, Brutkiewicz RR. Eur J Immunol. 2005;35:1692–1701. doi: 10.1002/eji.200526157. [DOI] [PubMed] [Google Scholar]
- 8.Zhou D, Mattner J, Cantu C, III, Schrantz N, Yin N, Gao Y, Sagiv Y, Hudspeth K, Wu YP, Yamashita T, et al. Science. 2004;306:1786–1789. doi: 10.1126/science.1103440. [DOI] [PubMed] [Google Scholar]
- 9.Gumperz JE, Roy C, Makowska A, Lum D, Sugita M, Podrebarac T, Koezuka Y, Porcelli SA, Cardell S, Brenner MB, et al. Immunity. 2000;12:211–221. doi: 10.1016/s1074-7613(00)80174-0. [DOI] [PubMed] [Google Scholar]
- 10.Steinman RM. Nat Med. 2007;13:1155–1159. doi: 10.1038/nm1643. [DOI] [PubMed] [Google Scholar]
- 11.Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. J Exp Med. 2003;198:267–279. doi: 10.1084/jem.20030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, Schmidt R, Harris AL, Old L, Cerundolo V. J Immunol. 2003;171:5140–5147. doi: 10.4049/jimmunol.171.10.5140. [DOI] [PubMed] [Google Scholar]
- 13.Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB. Nat Immunol. 2003;4:1230–1237. doi: 10.1038/ni1002. [DOI] [PubMed] [Google Scholar]
- 14.Nagarajan NA, Kronenberg M. J Immunol. 2007;178:2706–2713. doi: 10.4049/jimmunol.178.5.2706. [DOI] [PubMed] [Google Scholar]
- 15.Stanic AK, De Silva AD, Park JJ, Sriram V, Ichikawa S, Hirabyashi Y, Hayakawa K, Van Kaer L, Brutkiewicz RR, Joyce S. Proc Natl Acad Sci USA. 2003;100:1849–1854. doi: 10.1073/pnas.0430327100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Speak AO, Salio M, Neville DC, Fontaine J, Priestman DA, Platt N, Heare T, Butters TD, Dwek RA, Trottein F, et al. Proc Natl Acad Sci USA. 2007;104:5971–5976. doi: 10.1073/pnas.0607285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porubsky S, Speak AO, Luckow B, Cerundolo V, Platt FM, Grone HJ. Proc Natl Acad Sci USA. 2007;104:5977–5982. doi: 10.1073/pnas.0611139104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawai T, Akira S. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 19.Jarrossay D, Napolitani G, Colonna M, Sallusto F, Lanzavecchia A. Eur J Immunol. 2001;31:3388–3393. doi: 10.1002/1521-4141(200111)31:11<3388::aid-immu3388>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 20.Platt FM, Neises GR, Karlsson GB, Dwek RA, Butters TD. J Biol Chem. 1994;269:27108–27114. [PubMed] [Google Scholar]
- 21.Yu KO, Im JS, Molano A, Dutronc Y, Illarionov PA, Forestier C, Fujiwara N, Arias I, Miyake S, Yamamura T, et al. Proc Natl Acad Sci USA. 2005;102:3383–3388. doi: 10.1073/pnas.0407488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milland J, Christiansen D, Lazarus BD, Taylor SG, Xing PX, Sandrin MS. J Immunol. 2006;176:2448–2454. doi: 10.4049/jimmunol.176.4.2448. [DOI] [PubMed] [Google Scholar]
- 23.Gorden KB, Gorski KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, Tomai MA, Alkan SS, Vasilakos JP. J Immunol. 2005;174:1259–1268. doi: 10.4049/jimmunol.174.3.1259. [DOI] [PubMed] [Google Scholar]
- 24.McCarthy C, Shepherd D, Fleire S, Stronge VS, Koch M, Illarionov PA, Bossi G, Salio M, Denkberg G, Reddington F, et al. J Exp Med. 2007;204:1131–1144. doi: 10.1084/jem.20062342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gadola SD, Koch M, Marles-Wright J, Lissin NM, Shepherd D, Matulis G, Harlos K, Villiger PM, Stuart DI, Jakobsen BK, et al. J Exp Med. 2006;203:699–710. doi: 10.1084/jem.20052369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borg NA, Wun KS, Kjer-Nielsen L, Wilce MC, Pellicci DG, Koh R, Besra GS, Bharadwaj M, Godfrey DI, McCluskey J, et al. Nature. 2007;448:44–49. doi: 10.1038/nature05907. [DOI] [PubMed] [Google Scholar]
- 27.Matsuda JL, Gapin L, Sidobre S, Kieper WC, Tan JT, Ceredig R, Surh CD, Kronenberg M. Nat Immunol. 2002;3:966–974. doi: 10.1038/ni837. [DOI] [PubMed] [Google Scholar]
- 28.Raghuraman G, Geng Y, Wang CR. J Immunol. 2006;177:7841–7848. doi: 10.4049/jimmunol.177.11.7841. [DOI] [PubMed] [Google Scholar]
- 29.Skold M, Xiong X, Illarionov PA, Besra GS, Behar SM. J Immunol. 2005;175:3584–3593. doi: 10.4049/jimmunol.175.6.3584. [DOI] [PubMed] [Google Scholar]
- 30.Spada FM, Borriello F, Sugita M, Watts GF, Koezuka Y, Porcelli SA. Eur J Immunol. 2000;30:3468–3477. doi: 10.1002/1521-4141(2000012)30:12<3468::AID-IMMU3468>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 31.Roura-Mir C, Wang L, Cheng TY, Matsunaga I, Dascher CC, Peng SL, Fenton MJ, Kirschning C, Moody DB. J Immunol. 2005;175:1758–1766. doi: 10.4049/jimmunol.175.3.1758. [DOI] [PubMed] [Google Scholar]
- 32.Moody DB. Nat Immunol. 2006;7:811–817. doi: 10.1038/ni1368. [DOI] [PubMed] [Google Scholar]
- 33.Neville DC, Coquard V, Priestman DA, te Vruchte DJ, Sillence DJ, Dwek RA, Platt FM, Butters TD. Anal Biochem. 2004;331:275–282. doi: 10.1016/j.ab.2004.03.051. [DOI] [PubMed] [Google Scholar]
- 34.De Libero G, Moran AP, Gober HJ, Rossy E, Shamshiev A, Chelnokova O, Mazorra Z, Vendetti S, Sacchi A, Prendergast MM, et al. Immunity. 2005;22:763–772. doi: 10.1016/j.immuni.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 35.Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. J Lipid Res. 2004;45:1169–1196. doi: 10.1194/jlr.R300019-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Blander JM, Medzhitov R. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 37.West MA, Wallin RP, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, Prescott AR, Watts C. Science. 2004;305:1153–1157. doi: 10.1126/science.1099153. [DOI] [PubMed] [Google Scholar]
- 38.Marschner A, Rothenfusser S, Hornung V, Prell D, Krug A, Kerkmann M, Wellisch D, Poeck H, Greinacher A, Giese T, et al. Eur J Immunol. 2005;35:2347–2357. doi: 10.1002/eji.200425721. [DOI] [PubMed] [Google Scholar]
- 39.Montoya CJ, Jie HB, Al-Harthi L, Mulder C, Patino PJ, Rugeles MT, Krieg AM, Landay AL, Wilson SB. J Immunol. 2006;177:1028–1039. doi: 10.4049/jimmunol.177.2.1028. [DOI] [PubMed] [Google Scholar]
- 40.Salio M, Shepherd D, Dunbar PR, Palmowski M, Murphy K, Wu L, Cerundolo V. J Immunol. 2001;167:1188–1197. doi: 10.4049/jimmunol.167.3.1188. [DOI] [PubMed] [Google Scholar]
- 41.Figueroa-Perez S, Schmidt RR. Carbohydr Res. 2000;328:95–102. doi: 10.1016/s0008-6215(00)00092-6. [DOI] [PubMed] [Google Scholar]
- 42.Pfaffl MW. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen JL, Stewart-Jones G, Bossi G, Lissin NM, Wooldridge L, Choi EM, Held G, Dunbar PR, Esnouf RM, Sami M, et al. J Exp Med. 2005;201:1243–1255. doi: 10.1084/jem.20042323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}