

Abstract
We have studied the mechanism of A-769662, a new activator of AMP-activated protein kinase (AMPK). Unlike other pharmacological activators it directly activates native rat AMPK by mimicking both effects of AMP, i.e. allosteric activation and inhibition of dephosphorylation. It has no effect on the isolated α subunit kinase domain with or without the associated auto-inhibitory domain, on interaction of glycogen with the β subunit glycogen-binding domain, or on binding of AMP to the isolated Bateman domains of the γ subunit. Addition of A-769662 to mouse embryo fibroblasts (MEFs) or primary mouse hepatocytes stimulates phosphorylation of acetyl-CoA carboxylase (ACC), effects that are completely abolished in AMPK-α1−/−α2−/− cells, but not in TAK1−/− MEFs. Phosphorylation of AMPK and ACC in response to A-769662 is also abolished in LKB1−/− mouse muscle. However in HeLa cells, which lack LKB1 but express the alternate upstream kinase CaMKKβ, phosphorylation of AMPK and ACC in response to A-769662 still occurs. These results show that in intact cells the effects of A-769662 are independent of the upstream kinase utilized.
The AMP-activated protein kinase (AMPK) is a regulator of energy balance at both the cellular and whole body levels (1-3). Once activated, it effects a metabolic switch from an anabolic to a catabolic state, both by acutely phosphorylating metabolic enzymes and, in the longer term, by regulating gene expression. AMPK is a heterotrimer composed of a catalytic α subunit and regulatory β and γ subunits. Binding of AMP to the two “Bateman domains” formed by four tandem CBS motifs on the γ subunit (4) triggers increased phosphorylation at Thr-172 on the activation loop of the α subunit, causing >100-fold activation (5). AMP binding was previously thought both to promote phosphorylation (6) and inhibit dephosphorylation (7), although recent results suggest that the effect is exclusively on dephosphorylation (8). AMP binding also causes a further allosteric activation of the phosphorylated kinase by up to 10-fold (5). Phosphorylation of Thr-172 is in most cells catalyzed by the tumor suppressor kinase LKB1 (6,9), which appears to be constitutively active (10,11). In some cells Thr-172 can also be phosphorylated in a Ca2+-mediated process catalyzed by calmodulin-dependent protein kinase kinases such as CaMKKβ (12-14). The protein kinase TGFβ-activated kinase-1 (TAK1) can also activate the Saccharomyces cerevisiae homologue of AMPK (the SNF1 complex) when over-expressed in yeast, as well as phosphorylating Thr-172 on mammalian AMPK in cell-free assays (15). It remains unclear whether this has any physiological relevance in vivo.
Most of the metabolic changes induced by AMPK activation, such as increased glucose uptake into muscle (16), decreased gluconeogenesis in liver (17,18), increased fatty acid oxidation in muscle and liver (16,19), deceased fatty acid synthesis in liver and adipose tissue (20,21) and increased mitochondrial biogenesis (22) would be desirable outcomes for the treatment of type 2 diabetes and the metabolic syndrome. AMPK was therefore proposed (23) to be a promising target for drugs aimed at treatment of these common conditions, which are essentially disorders of energy balance. The first drug shown to activate AMPK in intact cells was 5-aminoimidazole-4-carboxamide riboside (AICAR) (20,21,24). When administered to rodent models of obesity and insulin resistance such as the ob/ob mouse, the fa/fa rat and the high-fat fed rat, AICAR was shown to reverse many of their metabolic abnormalities (25-28). At around the same time, the biguanide metformin was shown to activate AMPK in intact cells and in vivo (29). Metformin is currently the drug of first choice for the treatment of type 2 diabetes, being prescribed to at least 120 million people worldwide. The therapeutic effects of the drug are primarily on the liver, probably because hepatocytes express the organic cation transporter OCT1, resulting in more rapid uptake of the drug into hepatocytes than other cells (30,31). Recent studies involving mice in which AMPK could not be activated in liver due to a tissue-specific knockout of the upstream kinase, LKB1, revealed that the anti-hyperglycemic effects of metformin were abolished, suggesting that the major effect of the drug is to repress gluconeogenesis via activation of liver AMPK (32). It is possible that an AMPK activator that was also effective in organs other than the liver would have additional efficacy.
Although metformin is relatively safe it is not effective in all patients, perhaps due to variability in the efficiency of hepatic uptake by the OCT1 transporter (31). It does not activate AMPK, or affect the phosphorylation or dephosphorylation of the kinase by upstream kinases and phosphatases, in cell-free assays (33). However, metformin and the more potent biguanide, phenformin, have both been reported to be inhibitors of complex I of the respiratory chain (34,35), which suggests that they may activate AMPK indirectly by decreasing cellular ATP and increasing AMP. Significant changes in the cellular AMP:ATP ratio are indeed readily detected after treatment of cultured cells with phenformin (12), and decreases in ATP have recently been reported in primary rodent hepatocytes treated with metformin (36). Intestinal epithelial cells also express transporters of the OCT1 family (Oct1-3 (37)), and inhibition of the respiratory chain in the gut may be responsible for the unpleasant gastrointestinal side effects of biguanides, as well as the more dangerous side effect of lactic acidosis that led to the withdrawal of phenformin. There is evidence that much of the lactic acid produced in animals treated with biguanides is derived from the gut (38).
It therefore seems quite likely that a drug that activated the AMPK system more directly would be more efficacious in the treatment of type 2 diabetes and the metabolic syndrome than the biguanides, while avoiding some of their side effects, which may be related to inhibition of the respiratory chain rather than to activation of AMPK per se. The results of a screen of >700,000 compounds designed to detect AMPK activators were reported recently (39). The thienopyridone A-592017 emerged from the initial screen, and after optimization the more potent activator, A-769662, was developed. When administered to ob/ob mice, A-769662 had many of the effects expected for an AMPK activator, including decreases in plasma glucose and triglyceride, decreases in hepatic triglyceride, decreases in expression of the gluconeogenic enzymes phosphoenolpyruvate carboxykinase and glucose-6-phosphatase and the lipogenic enzyme fatty acid synthase, and even decreases in weight gain (39). Although the utility of the compound as a drug may be limited by its poor oral availability, it does hold considerable promise as an experimental tool for the study of the downstream consequences of AMPK activation. However, the original study provided little information as to the exact mechanism of activation of AMPK by A-769662. We have therefore synthesized A-769662 and have now addressed its mechanism of action in cell-free assays and in intact cells.
Experimental procedures
Material and antibodies
A-769662 was synthesized as described previously (40). STO-609 was from Tocris (Ellisville, Missouri). Protein G– Sepharose and [γ-32P]ATP were from Amersham Bioscience (Little Chalfont, UK), Protease-inhibitor cocktail tablets from Roche (Lewes, UK), precast SDS polyacrylamide gels from Invitrogen (Paisley, UK), phosphocellulose P81 paper from Whatman, and ionomycin and phenformin were from Sigma (Poole, Dorset, UK). Anti pan-AMPKα (anti-AMPK, #2532), phospho-AMPK (anti-pT172, #2535), pan-ACC (anti-ACC, #3661) and phospho-ACC (anti-pACC, #3662) antibodies were from Cell Signaling Technology (New England Biolabs, UK). Anti-TAK1 was from Santa Cruz Biotechnology (#sc-7162). Anti-GST antibodies were from purified as a by-product of production of antibodies against GST-LKB1 (41). Antibodies recognizing GST were removed from the anti-GST-LKB1 antiserum using an immobilized GST column. GST fusions of the rat α1 kinase domain (1-312, wild type and T172D mutant) (42) and human CaMKKβ expressed in bacteria (12) were expressed and purified as described. GST fusions of the rat AMPKα1 kinase domain (T172D mutant) (42) and human CaMKKβ expressed in bacteria (12) were expressed and purified as described. Rat α1 kinase domain (residues 1-310) and rat α1 kinase domain plus the autoinhibitory domain (residues 1-333) were amplified by PCR from a plasmid expressing the full-length α1 subunit (sense oligo: 5′-acctcggaattcgcgagaagcagaagc-3′, 312 antisense oligo: 5′-tggtttctgctcgagaggcagctgagg-3′, 332 antisense oligo: 5′-ggctctcgagattattctcctgttgtc-3′) and inserted into the EcoRI/XhoI sites of the pGEX6P2 vector (GE Healthcare). Positive clones were confirmed by DNA sequencing. Both GST recombinant proteins were expressed and purified as described (4). Recombinant protein phosphatase-2Cα was obtained as described (7).
Animals
All animal studies were approved by the University of Dundee Ethics Committee and performed according to the UK Animals (Scientific Procedures) Act 1986. Muscle-specific LKB1-deficient mice were generated, bred, and genotyped as previously described (43).
Preparations of AMPK and upstream kinases and AMPK assays
Rat liver AMPK (a mixture of the α1 and α2 isoforms with β1 and γ1 (44)) was purified as far as the gel filtration step (45), and rat testis LKB1 complex was purified as described for the rat liver complex (6). AMPK activity was assayed as described previously (46). Immunoprecipitate assays of AMPK in cell lysates were performed as described previously (11).
Preparations and assays of other kinases
The panel of 76 protein kinases was prepared and assayed in the presence and absence of A-769662 as described previously (47).
Creation of a GST-GBD fusion and assays of glycogen binding
The glycogen-binding domain (GBD) of the rat β1 subunit (residues 65–182) was amplified from a pcDNA3-rat β1 plasmid using primers incorporating SalI and EcoR1 restriction sites (sense 5′-ctagaattcacgacctcgaggtgaatgag-3′, antisense 5′-ccaagactggacagctcagatacatcgg-3′). The Sal1/EcoR1 digested product was cloned into the pGEX6P2 vector (GE Healthcare) and positive clones confirmed by DNA sequencing. The GST-GBD fusion was expressed in E. coli (BL21, Invitrogen Life Technologies UK). Cells were cultured in LB ampicillin at 37°C prior to induction of protein expression, at an A600 of 0.6, using 1 mM IPTG (Melford Labs). Cells were cultured for a further 4 hr at 37°C prior to harvesting by centrifugation. The cell pellet was lysed by rapid freezing and subsequent grinding to a fine powder in liquid N2. The cell lysate was then resuspended in a minimal volume of sucrose buffer (0.27 mM sucrose, 50 mM Tris pH 7.5, 1 mM sodium vanadate, 1 mM EDTA, 1 mM EGTA, 10 mM sodium-β-glycerophosphate, 50 mM NaF, 1 mM dithiothreitol (DTT), with one EDTA-free protease inhibitor cocktail tablet (Roche) per 50 ml). The lysate was clarified by centrifugation and applied to a 5ml GSTrap FF column (GE Healthcare) pre-equilibrated with sucrose buffer. Non-specifically bound proteins were removed by extensive washing in wash buffer (50 mM Tris pH 7.5, 200 mM NaCl, 1 mM DTT) and protein eluted in 20 mM reduced glutathione. Fractions containing GST-GBD were identified by protein assay and SDS-PAGE analysis. GST-GBD was dialysed overnight into wash buffer with two buffer changes. To make the glycogen-Sepharose column, CNBr-activated Sepharose 4 Fast Flow (GE Healthcare) was washed with 10 vols of cold 1 mM HCl. Glycogen was then directly coupled by incubation with 1 vol of 50 mg/ml bovine-liver (Type IX) glycogen (Sigma-Aldrich) in 10 mM KH2PO4, pH 8.0, overnight at 4 °C. Excess glycogen was removed by washing the beads with 5 vols of 10 mM KH2PO4 pH 8.0. Unreacted sites on the Sepharose were blocked by incubation with 1 vol of 0.1 M Tris-HCl, pH 8.0, at room temp for 2 hr. The beads were then washed with 6 × 3 vols of 0.1 M Tris-HCl, pH 9.0, 0.5 M NaCl and 6 × 3 vols of 0.1 M Na acetate pH 4.0, 0.5 M NaCl. The beads were finally resuspended and stored in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl. For the binding assay, 50 μl of beads were incubated with 2 μg of protein in a final volume of 150 μl of 50 mM TrisHCl, pH 7.5, 150 mM NaCl at 4°C for 1 hr. The glycogen-Sepharose beads were then pelleted at 13,000 rpm for 30 sec and 10 μl of supernatant retained for analysis. The glycogen-Sepharose beads were then washed with 500 μl of 50 mM Tris-HCl, pH 7.5, 150 mM NaCl prior to resuspension in the original volume of the same buffer. The bead suspension prior to incubation (10 μl), the supernatant post incubation and the resuspended pellet were analyzed on 4-12 % Bis-Tris gels in a MOPS buffer system (Invitrogen).
Proteins were transferred to a nitrocellulose membrane, which was blocked for 1 hr at room temperature in TBS (50 mM Tris-HCl, pH 7.5, 150 mM NaCl) + 5 % non-fat milk powder. The membrane was washed in 4 × 10 ml of TBS. Anti-GST antibody (in 10 mls TBS + 1% milk powder and 0.2 % (v/v) Tween-20) was added and incubated for a further 1 hr at room temp. The membrane was washed 3 × 5 min with TBS + 0.2 % v/v Tween-20. The membrane was then incubated for a further 1 hr with sheep IgG conjugated to IR dye 680 (Molecular Probes). The membrane was finally washed 3 × 5 min with TBS + 0.2 % v/v Tween-20 and 1 × 5 minutes in TBS. The membrane was scanned in the 680 channel of the Odyssey IR imager.
Scintillation proximity assay for AMP binding
Human γ2 (CBS motifs 1-4) was expressed as a GST fusion as described previously (4). GST-γ2 was purified using a 5 ml GST FF column (GE Healthcare) followed by size exclusion chromatography as described previously (48). The protein was incubated with glutathione-coupled scintillation proximity assay (SPA) yttrium silicate beads (GE Healthcare), pre-blocked with 5% gelatin from cold-water fish skin (Sigma). The beads were washed with 50 mM Na Hepes pH 7.4, 200 mM NaCl and resuspended to 10 mg/ml. A 96-well plate was set up with 0.1 mg SPA beads and 120 μM 3H-AMP (GE Healthcare) and made up to 90 μl with buffer. The plate was shaken for 15 min at room temp, and varying concentrations of A-7969662 or AMP were added, to a final volume of 100 μl. The plate was shaken for 15 min, beads allowed to settle and the plate read using a 1450 Microbeta counter (Perkin Elmer).
Cell culture
Mouse embryonic fibroblasts (MEFs) from AMPKα1+/+/α2+/+ and AMPKα1−/−/α2−/− mice were generated as described previously (49). MEFs were cultured in standard Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), 100 units/ml penicillin and 100 mg/ml streptomycin, non-essential amino acids (NEAA) and 1 mM Na pyruvate. HeLa cells were cultured in Minimum Essential Eagle's Medium supplemented with 10% FBS, NEAA and 100 units/ml penicillin and 100 mg/ml streptomycin. MEFs from TAK1+/+ and TAK1−/− mice were generated (50) and cultured as for AMPKα1α2−/− MEFs. Cells cultured in 10 cm dishes in DMEM containing 10% FBS were treated as described in the figure legends, rinsed with PBS and lysed in 500 μl of ice-cold lysis buffer (50 mM Tris-HCl pH 7.5, 1 mM EGTA, 1 mM EDTA, 1 % (w/v) NP-40, 1 mM Na orthovanadate, 10 mM Na β-glycerophosphate, 50 mM NaF, 5 mM Na pyrophosphate, 0.27 M sucrose, 1 mM DTT and complete proteinase inhibitor cocktail (Roche, one tablet per 50 ml). Lysates were centrifuged at 4°C for 15 min at 13,000 rpm and the supernatant collected. Total protein concentration was determined by the Bradford method using bovine serum albumin as standard.
Primary mouse hepatocytes
Primary hepatocytes were prepared from wild type (AMPK-α1+/+, α2+/+) or conditional double knockout (AMPK-α1−/−α2−/−) mice and incubated as described previously (36).
Incubation of isolated mouse skeletal muscle
Mice were fasted overnight and sacrificed by cervical dislocation. Muscles (extensor digitorum longus) were rapidly removed and tendons from both ends were tied with suture and mounted on an incubation apparatus. The muscles were incubated as previously described (11) in Krebs-Ringer bicarbonate (KRB) buffer (117 mM NaCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 24.6 mM NaHCO3, pH 7.4) containing 2 mM Na pyruvate for 1 hr at 37°C in the presence or absence of 100 μM A-769662 (100 mM stock prepared in DMSO) or 2 mM AICAR. DMSO (0.1% final concentration) was added to controls. The buffer was continually gassed directly by bubbling with a mixture of 95% O2/5% CO2. At the end of the incubation, muscles were quickly frozen in liquid N2 and stored at −80°C. Muscles were processed and lysates prepared as described previously (11).
Western blotting (cell culture and muscle studies)
Equal amount of cell lysates (10-30 μg protein) were heated in SDS-PAGE sample buffer, and analyzed by SDS-PAGE and electrotransfer to nitrocellulose membranes. The membranes were blocked for 30 min at room temperature in 50 mM Tris-HCl pH 7.5, 0.15 M NaCl and 0.1% Tween (TBS-T) containing 10% skimmed milk or 5% BSA. The membranes were then incubated with the indicated antibodies in TBS-T containing 5% BSA or 5% skimmed milk for 16 h at 4°C. Detection was performed using horseradish peroxidase conjugated secondary antibodies and enhanced chemiluminescence reagent. Quantification of the bands was performed by digitalizing the ECL films using a Fuji LAS 1000 CCD camera, and analysis of the intensities using the AIDA software.
RESULTS
A-769662 regulates heterotrimeric AMPK but not domains from the α or β subunits
We initially tested the ability of A-769662 to activate the native αβγ complex of AMPK purified from rat liver, which is a mixture of the phosphorylated α1 and α2 catalytic subunit isoforms with β1 and γ1. Both A-769662 and AMP gave a significant stimulation of purified rat liver AMPK, although the former was 50-fold more potent (half-maximal effect (EC50) at 116 ± 25 nM versus 6 ± 3 μM) and also gave a significantly greater stimulation (4.1 ± 0.5 versus 3.1 ± 0.5-fold) than AMP (compare the open circles in Fig. 1A with those in Fig. 1B). The interactions between A-769662 and AMP were complex. In the presence of a saturating concentration of AMP (200 μM), increasing A-769662 up to 5 μM produced a small (1.4-fold) but significant (95% confidence interval 1.2 to 1.6-fold) additional activation (Fig. 1A, filled circles). However, in the presence of a saturating concentration of A-769662 (1 μM) increasing AMP up to 500 μM caused a small (30%) but significant inhibition (Fig. 1B, filled circles).
Fig. 1.
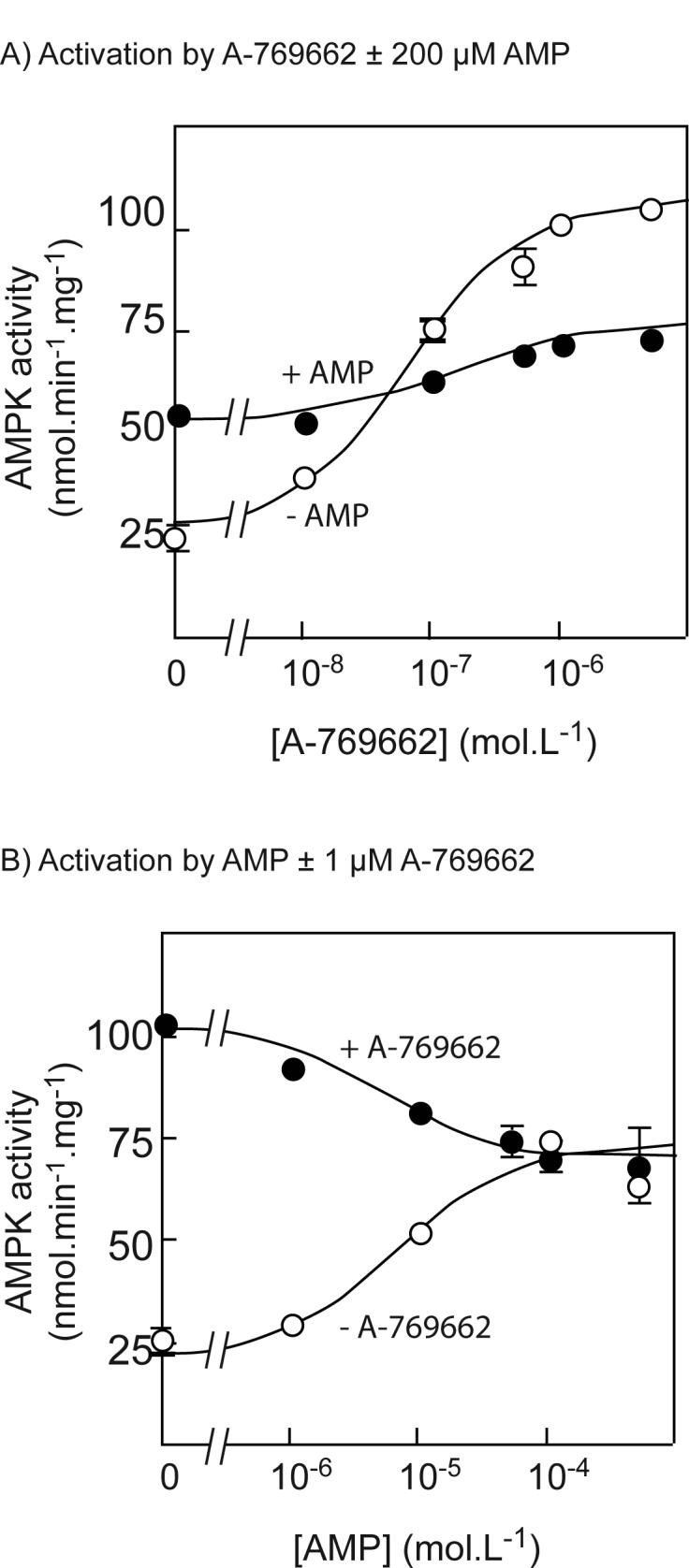
Effects of A-769662 on the native rat liver AMPK complex. (A) Activation of native rat liver AMPK (a mixture of the α1 and α2 isoforms with β1 and γ1) by A-769662 in the presence (filled symbols) and absence (open symbols) of 200 μM AMP. The data were fitted to the equation Activity = Basal + ((((Activation × Basal) − Basal) × [A-769662])/(EC50 + [A-769662])). The circles are the experimental data and the solid line is the theoretical curve obtained using the parameters estimated (in the absence of AMP: Basal = 25 ± 3 nmol.min−1.mg−1; Activation = 4.1 ± 0.5-fold; EC50 = 116 ± 25 nM; in the presence of AMP: Basal = 54 ± 4 nmol.min−1.mg−1; Activation = 1.4 ± 0.1-fold; EC50 = 250 ± 230 nM (all estimates ± standard error of the mean). (B) Activation of rat liver AMPK by AMP in the presence (filled symbols) and absence (open symbols) of 1 μM A-769662. The data were fitted as for (A). The parameters estimated (in the absence of A-769662: Basal = 24 ± 4 nmol.min−1.mg−1; Activation = 3.1 ± 0.5-fold; EC50 = 6 ± 3 μM; in the presence of A-769662: Basal = 102 ± 4 nmol.min−1.mg−1; Activation = 0.69 ± 0.03-fold (i.e. 31% inhibition); EC50 = 5 ± 3 μM (all estimates ± standard error of the mean).
We next tested the ability of A-769662 to regulate the activity and/or ligand binding of various constructs derived from the α or β subunits. The compound had no effect on the activity of a T172D mutant of the α1 kinase domain that had been expressed in bacteria as a glutathione-S-transferase (GST) fusion (data not shown); because of the replacement of Thr-172 by an aspartate residue, this construct is constitutively active and does not require prior phosphorylation (42). Recently it has been reported that a region just C-terminal to the kinase domain (residues 313-335 in human α1) represents an auto-inhibitory domain (AID) (51). To assess whether A-769662 might relieve inhibition by the AID, we expressed the rat α1 kinase domain either with (1-333) or without (1-310) this region. Both were expressed as GST fusions in bacteria, purified on glutathione-Sepharose, and pre-incubated with MgATP in the presence or absence of recombinant CaMKKβ to phosphorylate Thr-172. As expected, the GST-α1 (1-310) construct, which contains the complete kinase domain but lacks the putative AID, was markedly activated by incubation with MgATP in the presence of CaMKKβ (Fig. 2A), and this correlated with phosphorylation of Thr-172 as assessed using an anti-pT172 antibody (Fig. 2B). Neither the dephosphorylated nor the phosphorylated 1-310 construct was activated by A-769662 (Fig. 2A). By contrast, the GST-α1 (1-333) construct that contains the putative AID was not activated by incubation with MgATP in the presence of CaMKKβ even though phosphorylation of Thr-172 still occurred (Fig. 2B). These results confirm the proposal that the region from residues 311-333 in rat α1 (equivalent to 313 to 335 in human) acts as an auto-inhibitory domain (51), and show that the presence of this domain does not prevent phosphorylation of Thr-172. They also show that A-769662 does not activate AMPK by relieving inhibition of the kinase domain by the AID (Fig. 2A).
Fig. 2.
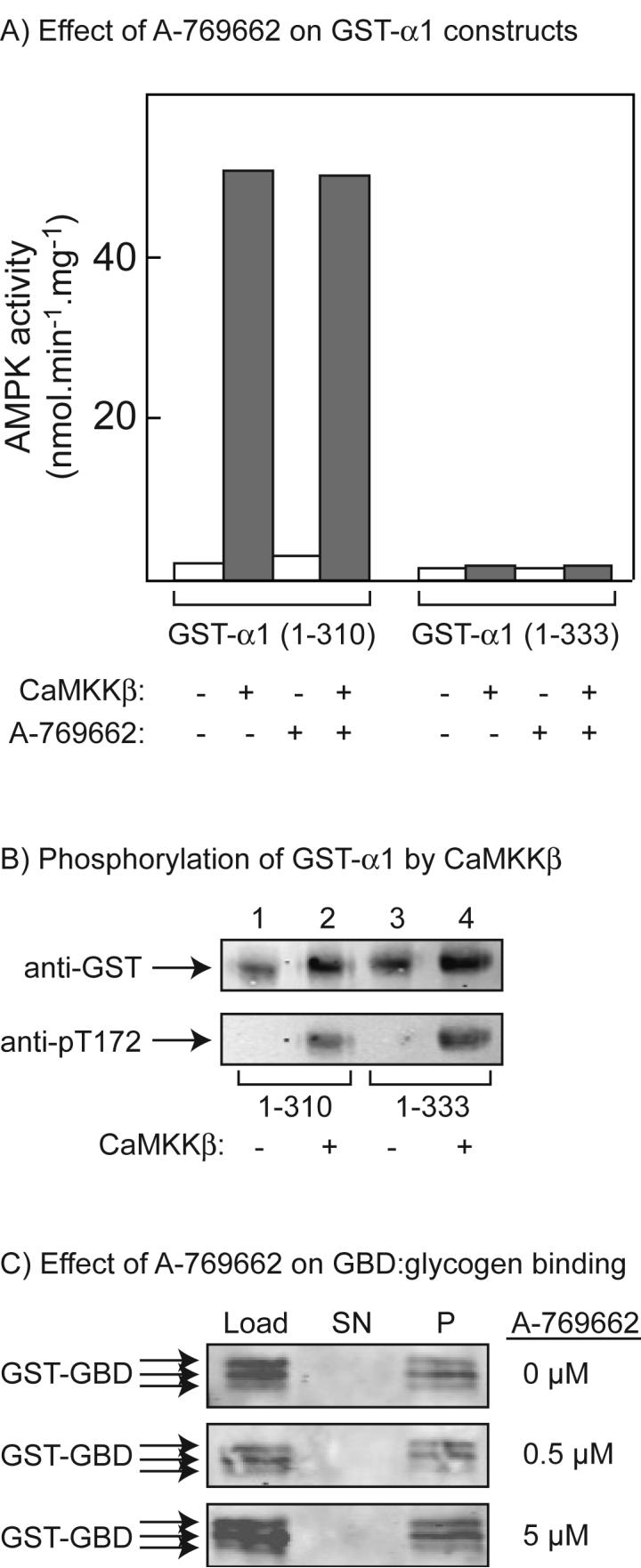
Effects of A-769662 on recombinant constructs derived from the α and β subunits. (A) Effects of phosphorylation of GST fusions of the α1 kinase domain (1-310) or the α1 kinase domain plus the autoinhibitory domain (1-333) by CaMKKβ on AMPK activity, and lack of effect of A-769662. The designated construct was incubated with MgATP with or without CaMKKβ as described under Experimental procedures, and AMPK activity was measured in the presence and absence of 1 μM A-769662. (B) Phosphorylation of GST fusions of the α1 kinase domain or the α1 kinase domain plus the auto-inhibitory domain by CaMKKβ. The designated construct was incubated as for (A), and samples were subject to Western blotting with anti-GST and anti-pT172 antibodies. (C) A GST fusion of the β1 glycogen-binding domain (GST-GBD) was incubated with bovine liver glycogen covalently attached to Sepharose and the concentration of A-769662 indicated on the right for 10 min. The glycogen-Sepharose was removed by centrifugation and the load, the supernatant (SN) and the glycogen-Sepharose pellet (P, resuspended to the original volume with buffer) were analyzed by Western blotting using anti-GST antibody. The two smaller polypeptides detected using this antibody may represent slight degradation of the fusion protein.
The β subunits of AMPK complexes contain a glycogen-binding domain (GBD) that cause the kinase to associate with glycogen particles in intact cells (52,53). Although a crystal structure for this domain has been determined in the presence of a model polysaccharide, β-cyclodextrin (54), its regulatory significance remains unclear. Nevertheless, we developed an assay to test whether A-769662 might cause dissociation of this domain from glycogen. The GBD from β1 (residues 65-182) was expressed in E. coli as a GST fusion and purified on glutathione-Sepharose. When bovine liver glycogen was covalently attached to Sepharose, incubated with the GST-GBD fusion and then centrifuged, all of the fusion protein was recovered in the pellet (P) and none in the supernatant (SN). Note that with this preparation of the GST-GBD fusion, there was a slight degradation of the protein, so that it migrated as a closely spaced triplet. Interaction of the GST-GBD with glycogen-Sepharose was not affected by the presence of 0.5 or 5 μM A-769662 (Fig. 2C). As controls for the specificity of this binding assay, we used GST without the fused GBD, or Sepharose without the attached glycogen, and in both cases all of the GST or GST-GBD fusion appeared in the supernatant (not shown). We also used a double mutant version of the GST-GBD in which Trp-100 and Trp-133 of the GBD were mutated to alanine and glycine respectively (it has been shown that these mutations prevent binding of the GBD to glycogen (53). Once again, all of the GST-GBD fusion appeared in the supernatant (not shown).
A-769662 does not displace AMP from the γ subunit
The interactions between activation by AMP and A-769662 shown in Fig. 1 suggested that the binding sites for these compounds might overlap. To test this, we expressed a GST fusion of the four CBS motifs from human γ2 in bacteria and purified the fusion protein on glutathione-Sepharose (4). We bound the protein to scintillation proximity beads and incubated with [3H]AMP at 120 μM, a concentration that should yield close to maximal binding. We then examined whether unlabelled AMP or A-769662 could displace the labelled AMP. Fig. 3A shows that unlabelled AMP displaced a large amount of the labelled AMP, leaving a small residual radioactivity that represents the background using this method. The dissociation constant for AMP estimated from this curve was 80 ± 15 μM (SEM), which is close to the value of 60 μM estimated previously using a different binding assay (4). By contrast, although high concentrations of A-769662 appeared to cause a small drop in the radioactivity in the scintillation proximity assay, it clearly did not displace [3H]AMP significantly over the concentration range where it caused activation of AMPK (Fig. 3A).
Fig. 3.
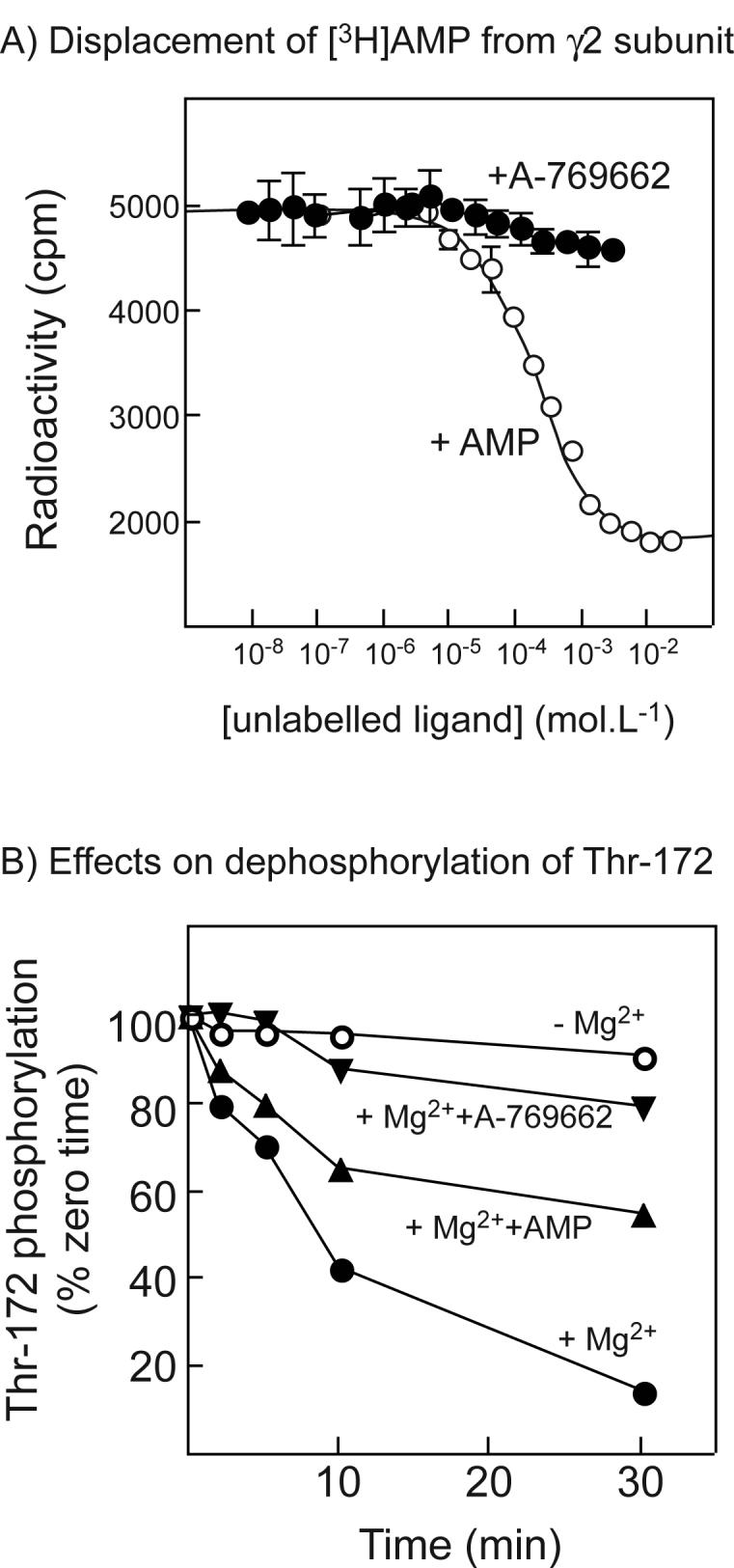
(A) Displacement of [3H]AMP by unlabelled AMP or A-769662 from a GST fusion with the twin Bateman domains of human γ2; (B) Effects of A-769662 on dephosphorylation of AMPK. (A) A GST-γ2 fusion (CBS1-4 (4)) was bound to scintillation proximity beads coated with glutathione, incubated with 120 μM AMP and increasing concentrations of AMP (open circles) or A-769662 (filled circles). Bound radioactivity was determined with a scintillation counter. Data for AMP binding were fitted using GraphPad Prism to the equation Radioactivity = (Maximum.(labelled AMP))/([labelled AMP] + [unlabelled AMP] + KdAMP) + Background. The curve is the theoretical curve generated using the best-fit parameters obtained (Maximum = 5200 cpm; Background = 1800 cpm; KdAMP = 60 ± 15 μM (±SEM)). (B) Purified rat liver AMPK was incubated with protein phosphatase-2Cα (9 ng) without Mg2+ or other addition (open circles), or with 5 mM MgCl2 in the absence (filled circles) or presence of 200 μM AMP (triangles) or 1 μM A-769662 (inverted triangles). Samples were withdrawn at various times and analyzed by Western blotting using anti-pT172 antibody. Intensities of the signal were estimated using the Li-Cor Odyssey infra-red scanner, and results expressed as the percentage of zero time intensity.
Effects of AMP and A-769662 on dephosphorylation by protein phosphatase-2Cα
To test whether A-769662, like AMP (7), inhibited dephosphorylation of Thr-172, AMPK purified from rat liver was incubated with recombinant protein phosphatase-2Cα in the presence and absence of Mg2+, with or without AMP and A-769662 (Fig. 3B). As expected (the protein phosphatase being Mg2+-dependent) the phosphate on Thr-172 was stable in the absence of Mg2+ but was rapidly removed in its presence. As reported previously, 200 μM AMP provided partial protection against dephosphorylation, while we now show that 1 μM A-769662 provided almost complete protection against dephosphorylation.
Effect of A-769662 on other protein kinases
We screened A-769622 at 10 μM and 50 μM in cell-free assays against a panel of 76 protein kinases other than AMPK; data obtained using 10 μM A-769622 is shown in Fig. 4. While one or two kinases appeared to be slightly stimulated at 10 μM, but this may just be experimental variation because none of these were stimulated significantly at 50 μM A-769622 (not shown). The majority of kinases were not significantly affected by 10 μM A-769662, including two kinases that are members of the AMPK-related kinase family (BRSK2 and MARK3). A number of kinases were marginally inhibited, but in only two cases, PIM1 and PIM3, was this by more than 50%. Thus, at a concentration that is ten times the concentration that is saturating for AMPK activation in cell-free assays, the effects of A-769662 on the activities of other protein kinases were negligible or small.
Fig. 4.

Effect of A-769662 (10 μM) on the activities of a panel of 76 protein kinases. Assays were performed in duplicate and the results are shown in rank order as the % activity compared with a control without compound, ± standard deviation. Abbreviations of kinases are as presented elsewhere (47).
Phosphorylation of acetyl-CoA carboxylase in response to A-769662 requires AMPK
To test whether effects of A-769662 in intact cells were dependent on AMPK, we utilized immortalized fibroblasts from wild type and double AMPK knockout (AMPK-α1−/−α2−/−) mouse embryo fibroblasts (MEFs). Fig. 5A shows that expression of the full-length α1 and α2 isoforms of the catalytic subunits of AMPK were readily detectable by Western blotting in the wild type MEFs but were not detectable in the double knockout MEFs, either with isoform-specific antibodies (anti-α1 and -α2) or with a phosphospecific antibody that recognizes both isoforms (anti-pT172). A polypeptide of slightly smaller size was detected by Western blotting, even after prior immunoprecipitation, using anti-α1 antibody in the knockout MEFs but not the wild type MEFs. Since the gene targeting strategy was designed to delete 60 amino acids from the kinase domain that are critical for kinase activity (residues 97-157 (55)), this polypeptide may represent a truncated α1 subunit with this internal deletion. Whatever the identity of this polypeptide, AMPK activity was undetectable in the double knockout MEFs after immunoprecipitation from the extracts (not shown). Pilot experiments revealed that in wild type MEFs, phosphorylation of acetyl-CoA carboxylase (ACC) was maximal after treatment for 15 minutes with 100 μM A-769662 and this increase was sustained for at least 1 hr (not shown). Fig. 5B shows that, in wild type MEFs, there was an increase in the phosphorylation of AMPK at concentrations up to 300 μM A-769662, although the effect was smaller than that seen with 2 mM phenformin. No signal was obtained in the double knockout MEFs (not shown). The phosphorylation of ACC in response to A-769662 in wild type MEFs was saturated at 100 μM, when the level of phosphorylation was essentially identical to that seen in response to 2 mM phenformin. Significantly, the phosphorylation of ACC in response to both A-769662 and phenformin was undetectable in the double knockout MEFs, even after a long exposure (Fig. 5B), and this was not due to a reduced expression of ACC protein. This confirms that the effect of both A-769662 and phenformin on the phosphorylation of ACC in MEFs is completely dependent on AMPK.
Fig. 5.

Effects of A-769662, phenformin and AICAR in wild type and double knockout (AMPK-α1−/−α2−/−) mouse embryonic fibroblasts (MEFs) and primary mouse hepatocytes. (A) Wild type and double knockout MEFs were analyzed by Western blotting with the indicated AMPK antibodies, or with anti-β-actin as a loading control. In one experiment (second panel from top) AMPK-α1 was immunoprecipitated with anti-α1 antibody prior to Western blotting. (B) Wild type or double knockout MEFs were incubated with the indicated concentrations of A-769662 or with 2 mM phenformin for 1 h and lysates analyzed by Western blotting using the indicated phosphospecific or non-phosphospecific antibodies. Results are presented as means ± SEM of duplicates, and are representative of 2 or more experiments. Blots were quantified by densitometry and phosphorylation of ACC is presented in arbitrary units (AU). (C) Primary hepatocytes from wild type and double knockout mice were incubated with the indicated concentrations of A-769662 or 500 μM AICAR for 4 hr and lysates analyzed by Western blotting using the indicated phosphospecific or non-phosphospecific antibodies.
To study this in a more physiological context, we also examined the effect of A-769662 on phosphorylation of AMPK and ACC in primary hepatocytes derived from wild type and double AMPK knockout (AMPK-α1−/−α2−/−) mice (Fig. 5C). The results were similar to those in the MEF cells in that A-769662 at concentrations up to 200 μM, and AICAR at 500 μM, produced a marked phosphorylation of ACC that was abolished in cells from the double knockout mice. A small increase in AMPK phosphorylation in response to A-769662 was also observed, although it was much smaller than the increase in response to AICAR.
Phosphorylation of AMPK and ACC in response to A-769662 requires an upstream kinase but is independent of the upstream kinase utilized
In order to determine whether the phosphorylation and activation of AMPK and the phosphorylation of ACC in intact cells in response to A-769662 requires the upstream kinase LKB1, we examined the effects of A-769662 in muscle tissue isolated from wild type mice or from mice with a muscle-specific deletion of LKB1 (43). Fig. 6A shows an experiment where we incubated isolated wild type (LKB1+/+) extensor digitorum longus muscle ex vivo with various concentrations of A-769662, and compared its effect with those of 2 mM AICAR. This showed that the effect of A-769662 on phosphorylation of Thr-172 on AMPK was smaller than that of AICAR and was most evident at 300 μM. However, the effect on ACC phosphorylation was already evident at 30 μM and was saturated at 100 μM. Fig. 6B shows the effect of 100 μM A-769662 or 2 mM AICAR on LKB1+/+ or LKB1−/− muscles. Both agents stimulated phosphorylation of AMPK and ACC in the wild type muscle, but phosphorylation of both proteins was abolished in muscles lacking LKB1.
Fig. 6.
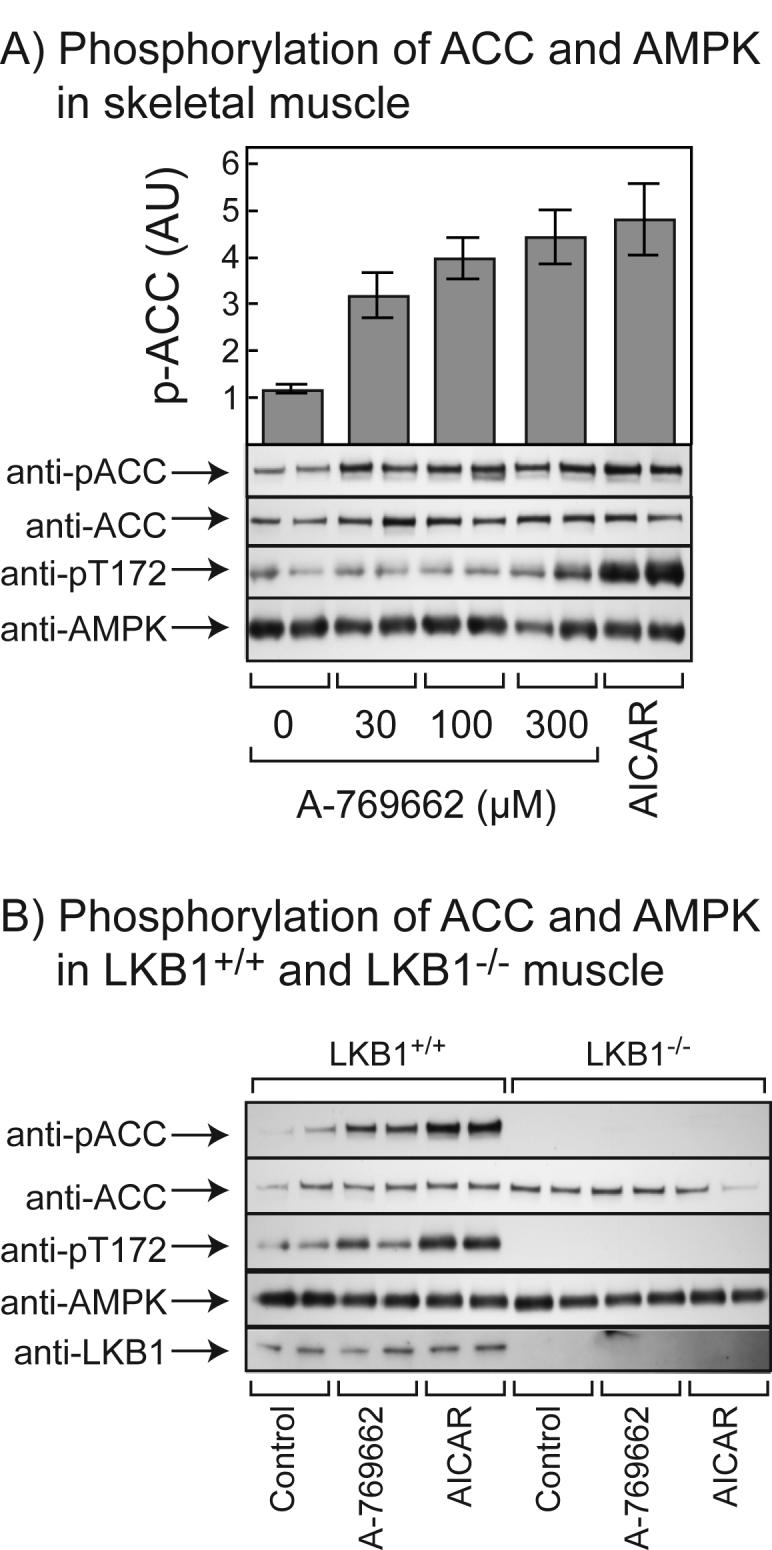
Effects of A-769662 and AICAR on mouse extensor digitorum longus (EDL) muscle. (A) Isolated EDL muscle from LKB1+/+ mice were incubated for 1 hr in the presence or absence of the indicated concentrations of A-769662, or 2 mM AICAR. Extracts were prepared and phosphorylation of ACC and AMPK analyzed by Western blotting. Blots were quantified by densitometry and phosphorylation of ACC is presented in arbitrary units (AU) ± SEM (n = 3-5). (B) As (A) but using 100 μM A-769662 and LKB1+/+ and LKB1−/− muscle.
To determine whether A-769662 would activate AMPK in cells expressing an alternate upstream kinase we studied HeLa cells, which lack expression of LKB1 but do express CaMKKβ (12). Fig. 7A shows that A-769662 caused a small but significant activation and phosphorylation of AMPK in these cells, while ionomycin (which activates AMPK via increased Ca2+ and activation of CaMKKβ (12)) caused a larger effect on both parameters. However, both A-769662 and ionomycin caused a large increase in phosphorylation of ACC. The effect of phenformin was not significant in these cells, as reported previously (6). To test whether the effects of A-769662 and ionomycin were mediated by CaMKKβ, we also performed these experiments in the presence of the CaMKK inhibitor STO-609. This inhibitor reduced the effects of both agents on the phosphorylation and activation of AMPK. It caused a particularly large inhibition of the effect of ionomycin, although inhibition was not complete because some stimulation by ionomycin was still evident. Moreover, a robust phosphorylation of ACC in response to A-769662 was still observed in the presence of STO-609 (Fig. 7A).
Fig. 7.

Effects of A-769662 and other agents in (A) HeLa cells; and (B) TAK1+/+ and TAK1−/− mouse embryo fibroblasts. (A) HeLa cells were incubated with 100 μM A-769662 for 1 hr, 1 μM ionomycin for 5 min, or 2 mM phenformin for 1 hr, with or without prior treatment with STO-609 (10 μg/ml) for 1 hr. Cell lysates were analyzed by immunoprecipitate kinase assays for AMPK activity and by Western blotting using the indicated antibodies. (B) TAK1+/+ and TAK1−/−MEFs were incubated with 100 μM A-769662 for 1 hr and lysates were analyzed by Western blotting using the indicated antibodies. In both (A) and (B), quantification of ACC phosphorylation was performed by densitometry and results are presented in arbitrary units (AU). Results are presented as means ± SEM of duplicates, and are representative of 2 or more experiments.
Phosphorylation of AMPK and ACC in response to A-769662 does not require TAK1
To test whether phosphorylation and activation of AMPK and ACC required the presence of TAK1, we studied TAK1+/+ and TAK1−/− MEFs. Although the effects of A-769662 on AMPK phosphorylation were small in these cells, there were clear increases in the phosphorylation of ACC, and this was identical when the TAK1+/+ and TAK1−/− MEFs were compared (Fig. 7B).
DISCUSSION
The results in this paper provide new information about the mechanism of action of A-769662 and show that the compound is a useful experimental tool to study the downstream consequences of AMPK activation in intact cells and in vivo. A-769662 activated the native αβγ complex of AMPK purified from rat liver extremely potently in cell-free assays, with a half-maximal effect at 120 nM. This is even lower than the EC50 of 800 nM reported for A-769662 by Cool et al (39), although this may be due to differences in the preparation of AMPK used and/or the assay conditions, because our estimated EC50 for the natural activator, AMP, was also lower than that reported by Cool et al (39) (8 versus 56 μM). The ability of A-769662 to directly activate AMPK both in cell-free assays and in intact cells makes it unique among currently known cell-permeable activators. Other activators, such as AICAR, metformin and the thiazolidinediones, do not activate AMPK directly in cell-free assays, and are either pro-drugs that are converted to active components inside the cell (e.g. AICAR, which is converted to the AMP analogue ZMP (21)) or work even more indirectly, e.g. by inhibiting the respiratory chain (metformin) or by triggering release of adiponectin from adipocytes (thiazolidinediones) (3).
Our results also suggest that A-769662 does not act by binding to any of the known ligand-binding sites on the α, β or γ subunits and must utilize a novel binding site. A-769662 had no effect on the activity of the isolated kinase domain from the α1 isoform, either using a T172D mutant that does not require prior phosphorylation (not shown), or after phosphorylation of Thr-172 on the wild type kinase domain by CaMKKβ (Fig. 2A). Neither did A-769662 relieve inhibition of the phosphorylated kinase domain by the auto-inhibitory domain (residues 311-333) previously identified by Pang et al (51) (Fig. 2A). An incidental finding that came out of these experiments was that the presence of the AID did not prevent the phosphorylation of Thr-172 by CaMKKβ (Fig. 2B) or LKB1 (not shown), even though it did completely prevent activation by these upstream kinases. The AID of AMPK-α1 and -α2 aligns with, and show some sequence similarity with, the ubiquitin-associated (UBA) domains in the AMPK-related kinases (56). Indeed, Pang et al (51) have modelled the interaction between the kinase domain and the AID of α1 based on the structure of the kinase and UBA domain of the AMPK-related kinase MARK2. However, the functions of the UBA domain in the AMPK-related kinases and the AID in the α subunits of AMPK appear to be different, because Jaleel et al (56) found that while the UBA domains did not inhibit the AMPK-related kinases, they were required for their phosphorylation by the LKB1 complex. By contrast, we now report that while the AID in the AMPK α subunits is not required for phosphorylation by either CaMKKβ or LKB1, it does completely prevent the activation caused by these phosphorylation events.
A-769662 did not cause dissociation of the glycogen-binding domain from glycogen (Fig. 2C), making it unlikely that the compound binds to the glycogen-binding site on this domain. We also found using a scintillation proximity assay that A-769662 did not displace [3H]AMP from the isolated Bateman domains on the γ2 subunit significantly under conditions where unlabelled AMP clearly did (Fig. 3A). This was a little surprising, because A-769662 mimics not just one but two of the effects of AMP on the AMPK system, i.e. (i) allosteric activation (Fig. 1); and (ii) inhibition of dephosphorylation (Fig. 3B). Our finding that A-769662 did not significantly displace AMP from the isolated Bateman domains suggests that the binding sites for these two ligands must be different, although they may produce a similar change in conformation.
Our studies in intact cells reinforce the idea that A-769662 is a specific and direct activator of AMPK and are also consistent with the idea that it has a dual effect, causing both allosteric activation and inhibition of dephosphorylation. Increased phosphorylation of acetyl-CoA carboxylase (ACC) by A-769662 in mouse embryo fibroblasts and primary mouse hepatocytes was completely dependent on the expression of the two catalytic subunits of AMPK (Fig. 5) showing that, at least when measuring ACC phosphorylation, the compound is completely dependent on AMPK for its effects.
It was noticeable in all of our intact cell studies with A-769662 that, while the effects on AMPK phosphorylation were often quite small, the effects on ACC phosphorylation were generally larger. For example, while phenformin or AICAR had much larger effects than A-769662 on AMPK phosphorylation in MEF cells and primary hepatocytes (Fig. 5), the effects of these agents on ACC phosphorylation were similar. Related observations were made in skeletal muscle (Fig. 6) and in HeLa cells (Fig. 7A). The most likely explanation for these apparent differences is that phosphorylation of ACC is such a sensitive marker of AMPK activation that maximal phosphorylation of ACC occurs when only a small proportion of AMPK has been phosphorylated. The concentrations of AICAR and phenformin chosen for study were designed to give maximal phosphorylation and activation of AMPK in these cells, and are likely to be greater than those required to give maximal ACC phosphorylation. An additional explanation in the case of the effects of the Ca2+ ionophores in HeLa cells (Fig. 7A) is that calcium ions activate phosphorylation by the upstream kinase CaMKKβ (12-14), but do not cause allosteric activation of AMPK. The phosphorylation of ACC in response to A-769662 would reflect a combination of allosteric activation and increased phosphorylation, but the allosteric effect on AMPK is lost during preparation of the extracts, and is not reflected in the kinase assays. By contrast, the effect of increased intracellular Ca2+ is entirely mediated by increased phosphorylation, and the effect on AMPK activity would be fully preserved in the extract. A third potential explanation for these apparent differences is that A-769662 might be able to activate dephosphorylated AMPK, but we could obtain no evidence that this was the case. While the compound did cause some activation of purified AMPK after treatment with protein phosphatase-2Cα (data not shown), the degree of activation of the treated and untreated kinase was the same (4-fold) and A-769662 did not alleviate the large inactivation caused by protein phosphatase treatment. Therefore, the small activation of the protein phosphatase-treated enzyme was most likely due to activation of the small residual amount of phosphorylated kinase left in the preparation.
The results obtained with LKB1−/− muscle (Fig. 6) suggest that an upstream kinase is necessary for the effect of A-769662 on ACC phosphorylation to be observed, presumably because dephosphorylated AMPK is not activated by the compound. However, the effect appears to be independent of the particular upstream kinase being utilized. Thus in HeLa cells, which do not express LKB1 but do express CaMKKβ (6,12), A-769662 still promoted phosphorylation of both AMPK and ACC in a similar manner to the Ca2+ ionophore, ionomycin. Interestingly, while the CaMKK inhibitor STO-609 greatly reduced phosphorylation of ACC in response to ionomycin, a substantial phosphorylation of ACC in response to A-769662 remained (Fig. 7). A likely explanation of these results is that ionomycin acts by increasing Ca2+ and thus activating CaMKKβ and, while this would increase phosphorylation of Thr-172 on AMPK, there would be no concomitant allosteric activation of the kinase. By contrast, A-769662 acts both by inhibiting dephosphorylation of Thr-172 and by causing allosteric activation of AMPK, and even the very low basal activity of CaMKKβ in STO-609-treated cells without ionomycin may be sufficient to observe these effects. This would also explain why the effect of STO-609 on ACC phosphorylation in response to A-769662 was less than its effect on the response to ionomycin.
It was recently reported that the TGFβ-activated kinase-1 (TAK1) could activate the S. cerevisiae homologue of AMPK, the SNF1 complex, when expressed in the yeast, and could also phosphorylate Thr-172 and activate mammalian AMPK in cell-free assays (15). Our findings that phosphorylation of AMPK and ACC by A-769662 was identical in TAK1+/+ and TAK1−/− mouse embryo fibroblasts do not support an important role for TAK1 as an upstream kinase in these cells. However, they do not rule out the possibility that it could act as an upstream kinase for AMPK in other cell types, or in other circumstances.
We would suggest that A-769662 is superior to other AMPK activators, including the nucleoside AICAR and the biguanide drugs, metformin and phenformin, for studies of the downstream actions of AMPK in intact cells and in vivo. AICAR is taken up into cells by adenosine transporters (57) and is converted by adenosine kinase into the mono-phosphorylated nucleotide ZMP, which mimics the effects of AMP on AMPK activation, albeit 50-fold less potently than AMP itself (21). One problem with the use of AICAR is that ZMP has been found to regulate other AMP-sensitive enzymes such as glycogen phosphorylase (58) and fructose-1,6-bisphosphatase (59), which is not the case with A-769662 (39). Another problem is that, while not itself an agonist or an antagonist of adenosine receptors, it does compete with adenosine for re-uptake into cells. In some in vitro systems this can cause effects that are due to increased accumulation of adenosine outside cells, with consequent binding to adenosine receptors (57). Although the biguanide drugs, metformin and phenformin, activate AMPK when incubated with intact cells (29), they have no effect on AMPK or its phosphorylation or dephosphorylation in cell-free assays (33). The mechanism by which they activate AMPK has not been completely elucidated, but it has been reported that their entry into cells is catalyzed by organic cation transporters such as OCT1 (31), and that the drugs then accumulate in mitochondria (driven by the membrane potential across the inner mitochondrial membrane, which would favour uptake of cations) where they inhibit complex I of the respiratory chain (34,35). This may in turn produce an increase in the cellular AMP:ATP ratio that activates AMPK. An increase in the AMP:ATP ratio has indeed been demonstrated in the case of phenformin (12), although it has been more difficult to detect in the case of the less rapidly and potently acting biguanide, metformin, possibly because this was studied in cells that do not express OCT1 (33,60). This is supported by the fact that using primary hepatocytes, which do express OCT1, metformin treatment resulted in a decrease in ATP content (36). If this proposed mechanism of action for the biguanides is correct, they may be no more specific as pharmacological activators of AMPK than are other metabolic poisons such as the mitochondrial ATP synthase inhibitor, oligomycin (33). As a good example of the shortcomings of these widely used AMPK activators, it has recently been shown that AICAR, metformin and oligomycin all inhibit glucose phosphorylation is isolated rodent hepatocytes by inhibiting the translocation of glucokinase from the nucleus to the cytoplasm. However, all three agents were just as effective in this regard in isolated hepatocytes from double knockout (AMPK-α1−/− -α2−/−) mice as in those from wild type controls, showing that these effects do not require AMPK and are probably mediated by ATP depletion (36). Unlike AICAR, metformin or oligomycin, A-769662 is a direct activator of AMPK in cell-free assays and it is certainly the most potent and specific pharmacological activator of AMPK available at present. This drug will be a valuable experimental tool to study the physiological roles of AMPK. A more complete understanding of the mechanism by which it activates AMPK may also facilitate the design of novel AMPK activators that could be used to treat patients with metabolic disorders.
Acknowledgments
We thank Drs. S. Akira and O. Takeuchi for their generous donation of TAK1−/− MEFs. We thank the kinase profiling, protein production and antibody purification teams (especially Jennifer Bain, Hilary McLauchlan and James Hastie) of the Division of Signal Transduction Therapy (DSTT), University of Dundee, for technical help with screening the panel of kinases, and for affinity purification of antibodies. KS and DGH were supported by the companies that fund the DSTT (AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Merck & Co. Inc, Merck KGaA and Pfizer), KS by Diabetes UK and the UK Medical Research Council, DGH by a Programme Grant from the Wellcome Trust, DGH and BV by the EXGENESIS Integrated Project (LSHM-CT-2004-005272) funded by the European Commission, and OG by a Wenner-Gren Fellowship.
The abbreviations used are
- ACC
acetyl-CoA carboxylase
- AICAR
5-aminoimidazole-4-carboxamide riboside
- AID
auto-inhibitory domain
- AMPK
AMP-activated protein kinase
- CaMKK
calmodulin-dependent protein kinase kinase
- CBS
cystathionine β-synthase
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethylsulfoxide
- FBS
fetal bovine serum
- GBD
glycogen-binding domain
- GST
glutathione-S-transferase
- MEFs
mouse embryo fibroblasts
- NEAA
non-essential amino acids
- OCT1
organic cation transporter-1
- PBS
phosphate-buffered saline
- SPA
scintillation proximity assay
- TAK1
TGFβ-activated kinase-1
- TBS
Tris-buffered saline
REFERENCES
- 1.Carling D. Trends Biochem. Sci. 2004;29(1):18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Kahn BB, Alquier T, Carling D, Hardie DG. Cell Metab. 2005;1(1):15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Hardie DG. Annu. Rev. Pharmacol. Toxicol. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- 4.Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG. J. Clin. Invest. 2004;113(2):274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. J. Biol. Chem. 2006;281(43):32207–32216. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- 6.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. J. Biol. 2003;2(4):28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies SP, Helps NR, Cohen PTW, Hardie DG. FEBS Lett. 1995;377:421–425. doi: 10.1016/0014-5793(95)01368-7. [DOI] [PubMed] [Google Scholar]
- 8.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Biochem. J. 2007;403(1):139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. Curr. Biol. 2003;13(22):2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 10.Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG, Alessi DR. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakamoto K, Goransson O, Hardie DG, Alessi DR. Am. J. Physiol. Endocrinol. Metab. 2004;287(2):E310–E317. doi: 10.1152/ajpendo.00074.2004. [DOI] [PubMed] [Google Scholar]
- 12.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Cell Metab. 2005;2(1):9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 13.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Cell Metab. 2005;2(1):21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. J. Biol. Chem. 2005;280(32):29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 15.Momcilovic M, Hong SP, Carlson M. J. Biol. Chem. 2006;281(35):25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- 16.Merrill GM, Kurth E, Hardie DG, Winder WW. Am. J. Physiol. 1997;273(6):E1107–E1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- 17.Lochhead PA, Salt IP, Walker KS, Hardie DG, Sutherland C. Diabetes. 2000;49(6):896–903. doi: 10.2337/diabetes.49.6.896. [DOI] [PubMed] [Google Scholar]
- 18.Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, Takemori H, Montminy M. Nature. 2005;437(7062):1109–1011. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 19.Velasco G, Geelen MJH, Guzman M. Arch. Biochem. Biophys. 1997;337(2):169–175. doi: 10.1006/abbi.1996.9784. [DOI] [PubMed] [Google Scholar]
- 20.Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. FEBS Lett. 1994;353:33–36. doi: 10.1016/0014-5793(94)01006-4. [DOI] [PubMed] [Google Scholar]
- 21.Corton JM, Gillespie JG, Hawley SA, Hardie DG. Eur. J. Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 22.Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. Proc. Natl. Acad. Sci. U S A. 2002;99(25):15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winder WW, Hardie DG. Am. J. Physiol. 1999;277:E1–E10. doi: 10.1152/ajpendo.1999.277.1.E1. [DOI] [PubMed] [Google Scholar]
- 24.Henin N, Vincent MF, Gruber HE, Van den Berghe G. FASEB J. 1995;9(7):541–546. doi: 10.1096/fasebj.9.7.7737463. [DOI] [PubMed] [Google Scholar]
- 25.Song XM, Fiedler M, Galuska D, Ryder JW, Fernström M, Chibalin AV, Wallberg-Henriksson H, Zierath JR. Diabetologia. 2002;45:56–65. doi: 10.1007/s125-002-8245-8. [DOI] [PubMed] [Google Scholar]
- 26.Bergeron R, Previs SF, Cline GW, Perret P, Russell RR, 3rd, Young LH, Shulman GI. Diabetes. 2001;50(5):1076–1082. doi: 10.2337/diabetes.50.5.1076. [DOI] [PubMed] [Google Scholar]
- 27.Iglesias MA, Ye JM, Frangioudakis G, Saha AK, Tomas E, Ruderman NB, Cooney GJ, Kraegen EW. Diabetes. 2002;51(10):2886–2894. doi: 10.2337/diabetes.51.10.2886. [DOI] [PubMed] [Google Scholar]
- 28.Buhl ES, Jessen N, Pold R, Ledet T, Flyvbjerg A, Pedersen SB, Pedersen O, Schmitz O, Lund S. Diabetes. 2002;51(7):2199–2206. doi: 10.2337/diabetes.51.7.2199. [DOI] [PubMed] [Google Scholar]
- 29.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. J. Clin. Invest. 2001;108(8):1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang DS, Jonker JW, Kato Y, Kusuhara H, Schinkel AH, Sugiyama Y. J. Pharmacol. Exp. Ther. 2002;302(2):510–515. doi: 10.1124/jpet.102.034140. [DOI] [PubMed] [Google Scholar]
- 31.Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, Ianculescu AG, Yue L, Lo JC, Burchard EG, Brett CM, Giacomini KM. J. Clin. Invest. 2007;117(5):1422–1431. doi: 10.1172/JCI30558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hawley SA, Gadalla AE, Olsen GS, Hardie DG. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- 34.Owen MR, Doran E, Halestrap AP. Biochem. J. 2000;348:607–614. [PMC free article] [PubMed] [Google Scholar]
- 35.El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. J. Biol. Chem. 2000;275(1):223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 36.Guigas B, Bertrand L, Taleux N, Foretz M, Wiernsperger N, Vertommen D, Andreelli F, Viollet B, Hue L. Diabetes. 2006;55(4):865–874. doi: 10.2337/diabetes.55.04.06.db05-1178. [DOI] [PubMed] [Google Scholar]
- 37.Koepsell H, Lips K, Volk C. Pharm. Res. 2007 doi: 10.1007/s11095-007-9254-z. [DOI] [PubMed] [Google Scholar]
- 38.Bailey CJ, Wilcock C, Day C. Br. J. Pharmacol. 1992;105(4):1009–1013. doi: 10.1111/j.1476-5381.1992.tb09093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, Zhao G, Marsh K, Kym P, Jung P, Camp HS, Frevert E. Cell Metab. 2006;3(6):403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 40.Iyengar RR, Judd AS, Zhao G, Kym PR, Sham HL, Gu Y, Liu G, Zhao H, Clark RE, Frevert EU, Cool BL, Zhang T, Keyes RF, Hansen TM, Xin Z. Thienopyridines as AMPK activators for the treatment of diabetes and obesity. USA: 2005. [Google Scholar]
- 41.Sapkota GP, Boudeau J, Deak M, Kieloch A, Morrice N, Alessi DR. Biochem. J. 2002;362(Pt 2):481–490. doi: 10.1042/0264-6021:3620481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott JW, Norman DG, Hawley SA, Kontogiannis L, Hardie DG. J. Mol. Biol. 2002;317(2):309–323. doi: 10.1006/jmbi.2001.5316. [DOI] [PubMed] [Google Scholar]
- 43.Sakamoto K, McCarthy A, Smith D, Green KA, Hardie DG, Ashworth A, Alessi DR. EMBO J. 2005;24:1810–1820. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woods A, Salt I, Scott J, Hardie DG, Carling D. FEBS Lett. 1996;397:347–351. doi: 10.1016/s0014-5793(96)01209-4. [DOI] [PubMed] [Google Scholar]
- 45.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. J. Biol. Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 46.Hardie DG, Salt IP, Davies SP. Methods Mol. Biol. 2000;99:63–75. doi: 10.1385/1-59259-054-3:63. [DOI] [PubMed] [Google Scholar]
- 47.Bain J, Cummings L, Elliot M, Shpiro N, Hastie J, McLaughlan H, Klevernic I, Arthur S, Alessi D, Cohen P. Biochem. J. 2007 doi: 10.1042/BJ20070797. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scott JW, Ross FA, Liu JK, Hardie DG. EMBO J. 2007;26(3):806–815. doi: 10.1038/sj.emboj.7601542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. Mol. Cell. Biol. 2006;26(14):5336–5347. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S. Nat Immunol. 2005;6(11):1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 51.Pang T, Xiong B, Li JY, Qiu BY, Jin GZ, Shen JK, Li J. J. Biol. Chem. 2007;282(1):495–506. doi: 10.1074/jbc.M605790200. [DOI] [PubMed] [Google Scholar]
- 52.Hudson ER, Pan DA, James J, Lucocq JM, Hawley SA, Green KA, Baba O, Terashima T, Hardie DG. Current Biol. 2003;13:861–866. doi: 10.1016/s0960-9822(03)00249-5. [DOI] [PubMed] [Google Scholar]
- 53.Polekhina G, Gupta A, Michell BJ, van Denderen B, Murthy S, Feil SC, Jennings IG, Campbell DJ, Witters LA, Parker MW, Kemp BE, Stapleton D. Current Biol. 2003;13:867–871. doi: 10.1016/s0960-9822(03)00292-6. [DOI] [PubMed] [Google Scholar]
- 54.Polekhina G, Gupta A, van Denderen BJ, Feil SC, Kemp BE, Stapleton D, Parker MW. Structure (Camb) 2005;13(10):1453–1462. doi: 10.1016/j.str.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 55.Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF. J. Biol. Chem. 2004;279(2):1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 56.Jaleel M, Villa F, Deak M, Toth R, Prescott AR, Van Aalten DM, Alessi DR. Biochem. J. 2006;394(Pt 3):545–555. doi: 10.1042/BJ20051844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gadalla AE, Pearson T, Currie AJ, Dale N, Hawley SA, Randall AD, Hardie DG, Frenguelli BG. J. Neurochem. 2004;88:1272–1282. doi: 10.1046/j.1471-4159.2003.02253.x. [DOI] [PubMed] [Google Scholar]
- 58.Longnus SL, Wambolt RB, Parsons HL, Brownsey RW, Allard MF. Am. J. Physiol. 2003;284(4):R936–R944. doi: 10.1152/ajpregu.00319.2002. [DOI] [PubMed] [Google Scholar]
- 59.Vincent MF, Erion MD, Gruber HE, Van den Berghe G. Diabetologia. 1996;39(10):1148–1155. doi: 10.1007/BF02658500. [DOI] [PubMed] [Google Scholar]
- 60.Fryer LG, Parbu-Patel A, Carling D. J. Biol. Chem. 2002;277(28):25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]