

Abstract
Mutant human presenilins cause early-onset familial Alzheimer's disease and render cells susceptible to apoptosis in cultured cell models. We show that loss of presenilin function in Drosophila melanogaster increases levels of apoptosis in developing tissues. Moreover, overexpression of presenilin causes apoptotic and neurogenic phenotypes resembling those of Presenilin loss-of-function mutants, suggesting that presenilin exerts a dominant negative effect when expressed at high levels. In Drosophila S2 cells, Psn overexpression leads to reduced Notch receptor synthesis affecting levels of the intact ∼300-kD precursor and its ∼120-kD processed COOH-terminal derivatives. Presenilin-induced apoptosis is cell autonomous and can be blocked by constitutive Notch activation, suggesting that the increased cell death is due to a developmental mechanism that eliminates improperly specified cell types. We describe a genetic model in which the apoptotic activities of wild-type and mutant presenilins can be assessed, and we find that Alzheimer's disease-linked mutant presenilins are less effective at inducing apoptosis than wild-type presenilin.
Keywords: presenilin, Alzheimer's disease, apoptosis, Notch signaling, neurodegeneration
Alzheimer's disease is the most common cause of senile dementia in humans, and is characterized neuropathologically by the accumulation of amyloid plaques and selective neuronal loss in certain brain regions (reviewed in Price et al. 1998). Dying neurons display characteristic features of apoptosis, a genetically controlled mechanism for the removal of harmful or superfluous cells in development and in many diseases (reviewed in Jacobson et al. 1997; Raff 1998). Mutations in three genes, amyloid precursor protein (APP)1, presenilin 1 (PS1), and presenilin 2 (PS2), account for most cases of early-onset Alzheimer's disease (reviewed in Haass 1997; Selkoe 1998). APP undergoes complex proteolytic processing that generates the amyloid peptides present in the amyloid plaques seen in Alzheimer's disease brain tissue (reviewed in Haass and Selkoe 1993). The two human presenilin proteins, PS1 and PS2, are closely related membrane proteins with multiple transmembrane domains and a large hydrophilic loop. Human and murine presenilins are widely expressed and localized predominantly in the ER and Golgi compartments (reviewed in Haass 1997). Several studies have shown that Alzheimer's disease-associated mutant variants of presenilin lead to elevated levels of the more neurotoxic 42-amino acid amyloid peptide (Aβ42), relative to the more benign 40-amino acid peptide (Aβ40; reviewed in Haass 1997; Selkoe 1998). In the nematode Caenorhabditis elegans, presenilin proteins have been identified independently as facilitators of Notch/Lin-12 developmental signaling (Levitan and Greenwald 1995; Li and Greenwald 1997).
Numerous studies have examined pro- and antiapoptotic activities of wild-type and mutant PS proteins in mammalian cultured cells or primary neuronal cultures in view of the potential involvement of apoptosis in Alzheimer's disease. Initially, overexpression of an Alzheimer's disease-linked mutant of PS1 in cultured PC12 cells revealed that mutant, but not wild-type, PS1 increases cellular vulnerability to apoptosis induced by trophic factor withdrawal or addition of β-amyloid (Guo et al. 1996, Guo et al. 1997; Weihl et al. 1999). Similarly, overexpression of wild-type PS2 and an Alzheimer's disease-related mutant PS2 form in PC12 cells increases the sensitivity of cells to these apoptotic stimuli, and mutant forms exhibit enhanced apoptotic activity (Deng et al. 1996; Wolozin et al. 1996; Janicki and Monteiro 1997). Presenilin-1 forms a complex with β-catenin that protects neurons from amyloid-β–induced apoptosis, and this complex is destabilized by familial Alzheimer's disease-linked PS1 mutant proteins in human cell lines and brain tissue (Zhang et al. 1998). These observations support the idea that enhanced neuronal cell death in Alzheimer's disease brain tissues may be due to increased apoptotic activity of presenilin caused by the autosomal dominant Alzheimer's disease-linked mutations. Moreover, COOH-terminal fragments of PS1 or PS2 generated either by caspase-mediated proteolysis or by alternative transcriptional initiation have antiapoptotic properties (Vito et al. 1996, Vito et al. 1997; Vézina et al. 1999). However, the different pro- and antiapoptotic effects of various PS proteins or PS proteolytic fragments deduced from various studies are difficult to reconcile, and a recent study suggests that overexpression of wild-type PS1 and an Alzheimer's disease-linked PS1 mutant have no discernible effects on apoptosis in cultured primary cortical neurons (Bursztajn et al. 1998). All studies examining the apoptotic activities of presenilin so far have been performed in cultured cells using overexpression techniques, which may account for the contradictory results obtained (Barinaga 1998). No data exist pertaining to the apoptotic activities of the C. elegans presenilin homologues Sel-12 and Hop-1. It is therefore important to investigate presenilin-mediated apoptosis in the context of a well-defined, multicellular experimental organism, using both traditional genetic methods and overexpression approaches that parallel the mammalian work performed to date.
We therefore analyzed the apoptotic phenotypes of Drosophila Presenilin (Psn) loss-of-function mutants and transgenic flies expressing wild-type Psn or various mutant proteins. Enhanced apoptosis is observed in both Psn loss-of-function mutants and in transgenic fly tissues that express high levels of Psn. Overexpression of wild-type Psn induces apoptosis in imaginal eye discs in a cell autonomous manner, resulting in a roughened external eye phenotype in adult flies. This phenotype is highly sensitive to Notch gene dosage or Notch receptor activation, suggesting that the apoptosis may occur as a developmental response to aberrant cell signaling and cell-fate specification. We also expressed four different Alzheimer's disease-related mutant forms and three other presenilin fragments in the developing eye. In contrast to previous studies demonstrating that Alzheimer's disease-linked substitutions in PS1 or PS2 have enhanced apoptotic activity, all the Alzheimer's disease-linked mutations that we tested have reduced apoptotic activity compared with wild-type, and thus may represent partial loss-of-function mutations. Since elevated levels of apoptosis are also a feature of the Drosophila Psn loss-of-function mutant phenotype, we propose that the proapoptotic activities of mammalian and fly PS proteins may be due to a dominant negative effect, rather than a gain of Psn function. This idea is supported by our finding that overexpression of Psn in S2 cells specifically blocks Notch protein synthesis, resulting in lower levels of immature 300-kD Notch and processed 120-kD COOH-terminal fragments. Transgenic flies expressing presenilin provide a genetic model in which the activity of additional Alzheimer's disease-related PS mutations can be assayed in vivo, and in which other components of presenilin-mediated biological processes may be identified by genetic screens.
Materials and Methods
Expression Constructs and Drosophila Transformation
Wild-type Psn full-length cDNAs (Psn+14 and Psn−14 variants) were cloned into the pUAST vector as EcoRI–XbaI fragments. Four missense mutation constructs were generated by standard PCR-based site-directed mutagenesis. The mutated Psn cDNAs were cloned into pBS-SK and confirmed by sequencing, then inserted into the pUAST vector downstream of the UAS regulatory region. The D-ALG3 and loop truncation constructs were generated by the same strategy after PCR with the following primer pairs: D-ALG3, 5′-CAAGAGTGGTCAGAATTCAAAATGGAACGTGTGGC-3′, 5′-TACTGTAAGACTCTAGATGTGTCCTTG-3′; Loop, 5′-TCTATTTGGGAATTCAAAATGGTCCTTTCGCC-3′, 5′-GGCCACAAAGCTCTAGATTTAGGTCGTCCAGTC-3′. pHS-GV was made from N+-GV3 (Struhl and Adachi 1998) by PCR with the primer pair: 5′-GAAAGCGGTCGCGGCCGCCAAAATGAAGCTTCTGTC-3′, 5′-AACCCCCGATATCTCACCCACCAAAGTCGTC-3′ and inserted into the NotI/StuI sites of pCaSpeR-hs. To produce transgenic flies, each construct was injected into w1118 embryos as described in Spradling and Rubin 1982.
Fly Genetics
Fly culture and crosses were carried out according to standard procedures. To generate Nts1; vg(quadrant enhancer)-lacZ or Nts1; ac-lacZ larvae, homozygous Nts1females collected at the permissive temperature (18°C) were crossed to homozygous vg(quadrant enhancer)-lacZ flies (P{vg-806}; Kim et al. 1996) or ac-lacZ flies (P{0.9ac-lacZ}; Van Doren et al. 1992). Hemizygous Nts1 male progeny from these crosses were mated to homozygous Nts1 females to generate Nts1 mutant larvae with appropriate lacZ markers. These crosses were kept at the nonpermissive temperature (29°C) for different time periods before larvae were analyzed by acridine orange staining and β-galactosidase staining.
Histology
For scanning electron microscopy (SEM), adult flies were dehydrated sequentially in 25, 50, 75, and 100% ethanol for >12 h each, and two 100% ethanol steps before critical point drying with hexamethyldisilazane (Electron Microscopy Sciences, Inc.). The flies were then mounted on stubs, sputter coated with gold, and imaged using a JEOL 6400 scanning electron microscope. Plastic sections were prepared as described in Tomlinson and Ready 1987. Cobalt sulfide staining of pupal retinae was done as described in Wolff and Ready 1991. Acridine orange stainings were done by dissecting eye imaginal discs in Ringer's solution, placing the tissue in 0.2 mg/ml acridine orange in Ringer's solution for 4 min, and mounting the discs in Ringer's solution for immediate fluorescence photomicroscopy (Spreij 1971). β-galactosidase detection was performed as described in Ye et al. 1999. Immunostainings were done as described in Ye and Fortini 1998, using the following primary antibodies: rat anti-ELAV mAb 7E8A10, 1:200 dilution (Robinow and White 1991); mouse anti-Psn Ab L7, 1:100 dilution (Ye and Fortini 1998). Adult wings were removed and mounted in DPX mountant (Electron Microscopy Sciences, Inc.) for photomicroscopy.
TUNEL Assay and Double TUNEL/Immunohistochemistry Assay
TUNEL assays (Gavrieli et al. 1992) were performed using the Oncor S7110 kit. In brief, imaginal discs were fixed with 2% paraformaldehyde for 20 min and washed in PBS-DT (1× PBS, 0.3% Triton X-100, 0.3% deoxycholate) for 20 min on ice. Discs were then washed with PBS four times for 5 min each, and then transferred to equilibration buffer for 5 min, followed by working-strength reaction buffer containing terminal deoxynucleotide transferase. The discs were incubated with the enzyme in a 96-well plate in a humidified chamber at 37°C for 1.5 h. The reaction was stopped by transferring the discs into working-strength stop/wash buffer for 10 min, and discs were then rinsed in three changes of PBS, followed by staining with either working-strength antidigoxigenin–fluorescein solution or antidigoxigenin–AP solution (Boehringer Mannheim Corp.) for 45 min and two more PBS washes for 10 min each. Discs were then immunoreacted with mouse anti-Psn Ab L7, 1:100 dilution, as described above or developed following the S7110 kit instructions.
S2 Cell Studies
Drosophila S2 cell transfections, Western immunoblot analysis, and antibody stainings were performed as described in Fehon et al. 1990 and Ye et al. 1999, using Notch construct pMTNMg and Notch mAbs C17.9C6 and C458.2H (Fehon et al. 1990), Myc mAb 1-9E10.2, and β-tubulin mAb E7 (University of Iowa Developmental Studies Hybridoma Bank).
Results
Psn Loss-of-function Mutations Lead to Increased Apoptosis in Drosophila Tissues
In view of the extensive neuronal cell death seen in Alzheimer's disease, and the increased susceptibility of cultured cells to apoptotic stimuli caused by overexpression of normal and mutant variants of mammalian PS proteins, we sought to determine whether presenilin is associated with apoptosis in a genetically tractable experimental organism. We therefore produced loss-of-function genetic lesions in the Drosophila Presenilin (Psn) gene (Ye et al. 1999) and analyzed patterns of cell death in developing tissues of the mutant animals. Imaginal discs from the Psn mutant larvae are considerably smaller than age-matched wild-type control discs, and Psn mutant eye discs lack almost all differentiating neurons posterior to the morphogenetic furrow (Ye et al. 1999). To determine whether this neuronal cell loss occurs by apoptosis, we examined third-instar Psn mutant imaginal discs using the TUNEL assay for apoptotic nuclear fragments (Gavrieli et al. 1992). A dramatic increase in the number of apoptotic cell bodies is readily detected in the Psn mutant eye discs (Fig. 1A and Fig. B). Similarly high levels of apoptosis are observed in other imaginal tissues, including the wing and leg discs, when presenilin function is eliminated (data not shown). Our data are consistent with a role for presenilin in either the regulation of apoptosis itself, or in developmental patterning events that lead to high levels of apoptosis when they are not executed properly.
Figure 1.
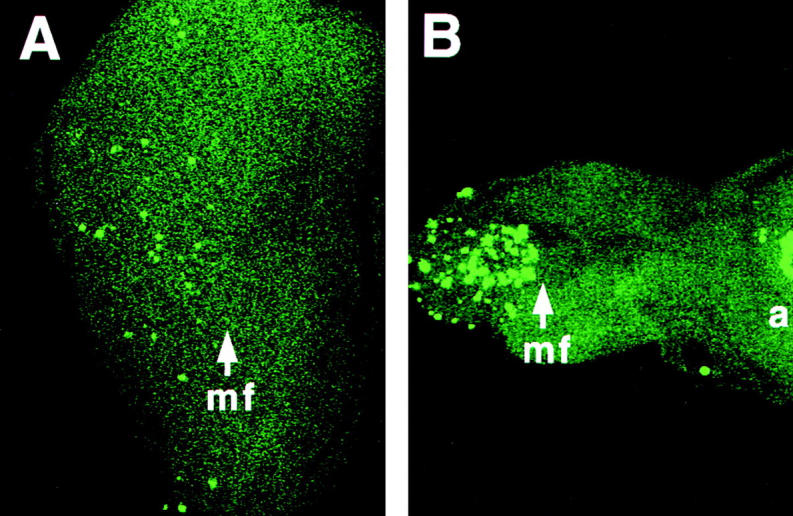
Elevated levels of programmed cell death in Psn loss-of-function mutant tissues. TUNEL labeling reveals apoptotic cell nuclei in imaginal eye discs from third-instar larvae. The genotypes are as follows: A, wild-type; B, homozygous PsnB3 mutant. Removal of Psn gene function results in an increased number of cells undergoing apoptosis in certain disc regions, as visualized by the brightly stained nuclear fragments. Arrows, mf, morphogenetic furrow; a, antennal disc, anterior at right.
Presenilin Overexpression Causes Rough Eye Phenotypes
To investigate further the ability of presenilin proteins to influence apoptotic events in the context of a multicellular organism, we overexpressed Drosophila Psn in transgenic fly tissues and compared the results to analogous experiments that have already been performed exclusively in mammalian cultured cells. The Drosophila Psn gene undergoes alternative splicing to generate two protein isoforms, one of which contains an additional 14 amino acids in the hydrophilic loop domain (Ye and Fortini 1998). We ectopically expressed both the short wild-type isoform (termed Psn−14) and the long wild-type isoform (termed Psn+14), as well as four different Alzheimer's disease-linked missense mutations and three truncated forms of fly presenilin (Fig. 2 A) in third-instar larval eye discs using the GAL4-UAS system (Brand and Perrimon 1993). As driver constructs, we used sev-GAL4, which expresses GAL4 under the control of the sevenless promoter, and GMR-GAL4, in which GAL4 is downstream of a multimerized copy of the binding site for the Glass transcription factor. These regulatory sequences are active in different neuronal subsets of the immature retina and they have been used extensively in studies of cell death in the eye (reviewed in McCall and Steller 1997). The sevenless gene is strongly expressed in the R3, R4, and R7 photoreceptor cell precursors and cone cell precursors, while GMR is more generally expressed in all cells posterior to the morphogenetic furrow (Tomlinson et al. 1987; Ellis et al. 1993). The expression of Psn constructs in eye discs was confirmed by antibody staining, excluding D-ALG3, which does not encode epitopes recognized by available Psn antibodies (Fig. 2, B–D).
Figure 2.
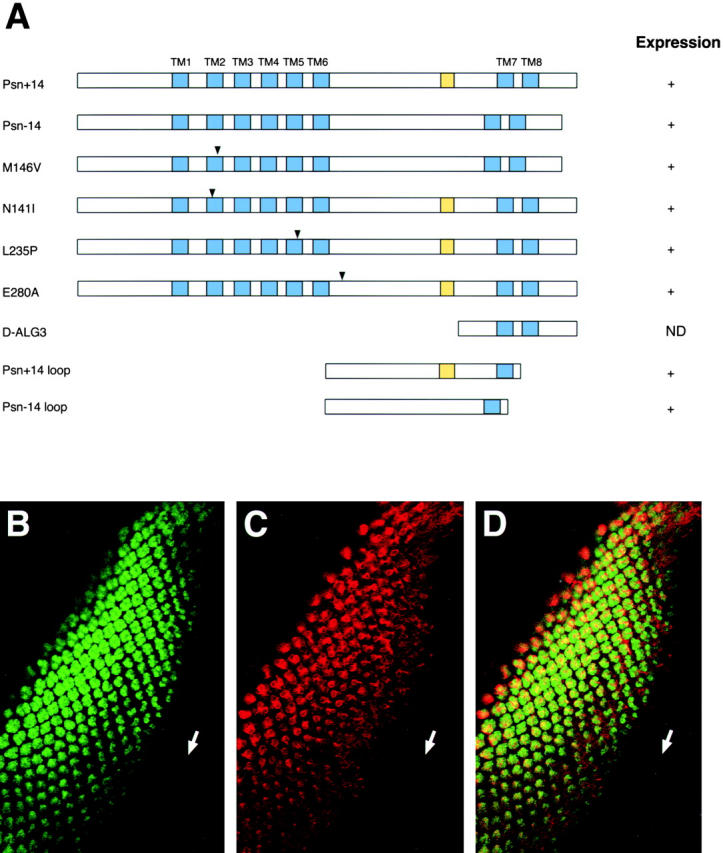
Wild-type and mutant Drosophila presenilins and their expression in transgenic eye imaginal discs. A, Structures of wild-type and mutant forms of Psn encoded by transgenic constructs. The name of each construct is shown at left. Transmembrane domains are shown as blue boxes and the 14-amino acid variable insertion is indicated by the yellow boxes. The positions of different Alzheimer's disease-associated missense mutations are indicated by the arrowheads. Expression of the constructs in imaginal eye discs of transgenic flies is indicated at right, as determined by immunohistochemical staining with anti-Psn polyclonal antibody L7. ND, Not determined. B–D, The expression pattern of GMR-driven Psn protein in transgenic Drosophila. An eye imaginal disc from a third instar larva of genotype GMR-GAL4, UAS-Psn+14, double stained with anti-ELAV antibody (B) that labels a nuclear antigen of all newly born neurons posterior to the morphogenetic furrow (arrow) and with anti-Psn antibody L7 (C) and the merged image of both fluorescence channels (D). The GMR-driven Psn transgene appears to be expressed in all ELAV-positive cells, as expected, and is localized primarily in the cytoplasm. Anterior at lower right in B–D.
Flies bearing one copy of UAS-Psn+14 or UAS-Psn−14, together with either sev-GAL4 or GMR-GAL4, are morphologically indistinguishable from wild-type, as are flies bearing the GAL4 driver constructs alone (Fig. 3A, Fig. E, and Fig. I). At 25°C, two copies of UAS-Psn+14 (termed 2X UAS-Psn+14) with one copy of sev-GAL4 produce a rough eye phenotype, consisting of irregular ommatidial packing, occasional ommatidial fusions, and missing bristles (Fig. 3B and Fig. F). Tangential sections of these eyes revealed that pigment cells are missing while the photoreceptor cell array is largely normal (Fig. 3 J). In general, similar results were obtained with UAS-Psn+14 and UAS-Psn−14, so UAS-Psn+14 was used for subsequent experiments, unless noted otherwise. Two copies of UAS-Psn+14 driven by one copy of GMR-GAL4 result in a stronger rough eye phenotype characterized by fusion of the lens material of adjacent ommatidia and loss of interommatidial bristles, producing a glossy external eye surface (Fig. 3C and Fig. G). A similar glossy eye phenotype is caused by facet alleles of the Notch gene, and is due to a specific defect in primary pigment cell development (Cagan and Ready 1989). Most pigment cells are missing in GMR-GAL4, 2X UAS-Psn+14 retinae, resulting in a nearly complete absence of the pigment cell lattice between photoreceptor cell arrays of different ommatidia (Fig. 3 K). Cobalt staining of retinae from pupae shows that almost all ommatidia have the normal number of cone cells, but display missing primary, secondary, and tertiary pigment cells, consistent with the adult eye phenotype (Fig. 3, M–P). To test whether the two alternatively spliced Psn variants are functionally equivalent or antagonistic, we generated flies bearing two copies of UAS-Psn+14 and one copy of UAS-Psn−14 in addition to GMR-GAL4. The rough eye phenotype caused by two copies of UAS-Psn+14 alone is strongly enhanced by one copy of the short form of Psn. The lens surfaces are completely fused together into a smooth glossy sheet that is almost devoid of interommatidial bristles (Fig. 3D, Fig. H, and Fig. L). These data suggest that the long and short Psn protein isoforms function in a similar manner in this assay.
Figure 3.
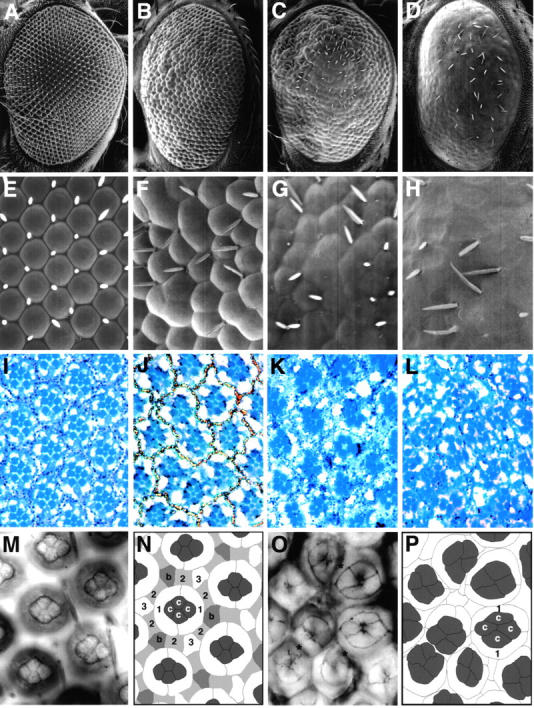
Overexpression of wild-type Psn causes a rough eye phenotype characterized by missing pigment cells. A–L, Scanning electron microscope (SEM) images at low (A–D) and high (E–H) magnification and 1-μm plastic sections (I–L) of adult fly eyes of the following genotypes: A, E, and I, wild-type; B, F, and J, sev-GAL4, 2X UAS-Psn+14; C, G, and K, GMR-GAL4, 2X UAS-Psn+14; D, H, and L, GMR-GAL4, 2X UAS-Psn+14; UAS-Psn−14. The Psn-induced eye phenotype includes irregular corneal lens shapes, ommatidial fusions, and missing bristles. Numbers of photoreceptor cells within each ommatidia are largely unaffected, although the regular trapezoidal arrangement of the photoreceptor cell is partially disrupted. The GMR-GAL4, 2X UAS-Psn+14 phenotype is enhanced by an additional copy of UAS-Psn−14. M–P, Cobalt staining of 60 h pupal eye discs from wild-type (M and N) and GMR-GAL4, 2X UAS-Psn+14 (O and P). N, Schematic tracing of the wild-type pupal eye disc in M, showing that each ommatidium consists of four cone cells (c) surrounded by two primary pigment cells (1), six shared secondary pigment cells (2), three shared tertiary pigment cells (3), and three shared bristle group precursor cells (b). P, Schematic tracing of the GMR-GAL4, 2X UAS-Psn+14 disc in O, revealing missing pigment cells (asterisks in O), but normal cone cell complements. Anterior at left.
Presenilin Overexpression Induces Apoptosis in a Cell Autonomous Manner
Although the sevenless promoter is not active in pigment cells and bristle precursor cells, these two types of cells are the major ones that are affected in our transgenic lines expressing Psn under sev-GAL4 control. One possible explanation for this discrepancy is that Psn overexpression may decrease the number of cells available for eye patterning by affecting cell proliferation or cell death. In this case, cells recruited during the later stages of ommatidial assembly, such as pigment cells and bristle cell groups, would be disproportionately affected because all cell types of the adult eye are recruited from a common pool of uncommitted progenitor cells in the third-instar larval eye disc (Ready et al. 1976).
Given the apoptotic effects of PS1 and PS2 in cultured mammalian cells, we examined programmed cell death in third-instar larval eye discs using acridine orange staining, which specifically labels apoptotic cells, but not necrotic cells, in Drosophila (Abrams et al. 1993). Although a low level of cell death is normally seen posterior to the morphogenetic furrow of the eye disc, a significant increase in apoptosis is consistently observed in discs carrying two copies of UAS-Psn+14 and either the sev-GAL4 or GMR-GAL4 transgene (Fig. 4, data not shown). Since increased levels of apoptosis are also a feature of the Drosophila Psn gene loss-of-function mutant phenotype, our data raise the possibility that the proapoptotic effects of mammalian PS proteins may actually represent a partial loss of Psn function due to protein oligomerization and aggregation when ectopic Psn is expressed at a very high level (Seeger et al. 1997).
Figure 4.

Elevated levels of apoptosis in transgenic eye discs that overexpress wild-type presenilin. Genotypes are as follows: A, sev-GAL4 alone (control); B, sev-GAL4, 2X UAS-Psn+14. Third-instar larval eye discs were stained with acridine orange, a vital dye specific to cells undergoing apoptosis (Abrams et al. 1993). Increased numbers of apoptotic bodies are observed as brightly staining particles when Psn expression is induced by sev-GAL4 in eye disc regions posterior to the morphogenetic furrow (white arrows). Anterior at right.
To determine if Psn-induced apoptosis is cell autonomous, we analyzed third instar eye imaginal discs of genotype sev-GAL4, UAS-Psn+14 with TUNEL labeling in conjunction with Psn antibody staining. The sev promoter is strongly active in only a subset of cells at this stage of eye development, namely the R3, R4, and R7 photoreceptor cell precursors, the cone cell precursors, and one or two so-called mystery cells that are transiently associated with the five-cell ommatidial precluster. The ability of these precursor cells to be identified solely on the basis of their positions in nascent ommatidia enabled us to directly correlate TUNEL-positive apoptotic cell nuclei with the corresponding cell bodies expressing high levels of Psn (Fig. 5). Psn is distributed throughout the cytoplasm, but is excluded from the nucleus (Ye and Fortini 1998). The TUNEL method, however, specifically stains fragmented DNA within nuclei of apoptotic cells and thus labels a different subcellular compartment than the Psn antibody staining. Nevertheless, careful analysis of nearby optical sections using confocal microscopy clearly reveals that both TUNEL and Psn antibodies label numerous identical cells in all the discs analyzed (Fig. 5, C–H), confirming that the Psn-induced cell death is a cell-autonomous process and that only a minority of cells overexpressing Psn undergo apoptosis. We also noticed that the assembly of the photoreceptor cell clusters is largely unperturbed, despite the increase in cell death, as the normal mirror symmetric patterning of R3 and R4 photoreceptor cell precursors along the eye equator can still be visualized by Psn antibody staining (Fig. 5 A). It is apparent that the apoptotic cells are rapidly removed from photoreceptor cell precursor clusters and replaced with neighboring cells that generally are not eliminated by apoptosis.
Figure 5.
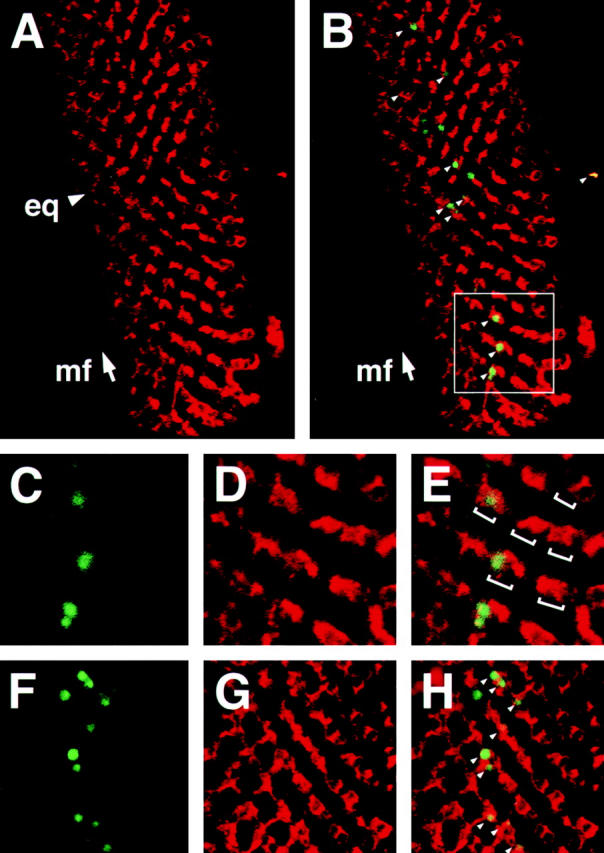
Double TUNEL/Psn antibody labeling using confocal laser microscopy reveals that Psn induces programmed cell death in a cell autonomous manner. A and B, TUNEL labeling (green) in conjunction with Psn antibody staining (red) of a sev-GAL4, 2X UAS-Psn+14 imaginal eye disc reveals apoptotic nuclear fragments (small arrowheads in B) in the developing retina posterior to the morphogenetic furrow (arrow, mf) at a basal focal plane where the sev pattern of Psn transgenic expression is clearly visualized. Overall patterning of photoreceptor cell precursors occur normally, as revealed by the unperturbed mirror symmetrical patterning of R3 and R4 photoreceptor cell precursors along the equator (arrowhead, eq in A). C–E, A disc sector (white square in B) imaged at higher magnification, which clearly shows that TUNEL labeling and Psn antibody staining detect some of the same cells (brackets in E indicate R3/R4 photoreceptor cell precursor pairs of separate ommatidial preclusters). F–H, Double TUNEL/Psn antibody labeling of another sev-GAL4, 2X UAS-Psn+14 eye disc at high magnification, showing that cells undergoing apoptosis express high levels of presenilin (small arrowheads). Anterior at left.
Alzheimer's Disease-linked Mutant Presenilins May Be Partial Loss-of-function Proteins
Although the Alzheimer's disease-linked PS mutations are not complete loss-of-function alleles (Levitan et al. 1996; Davis et al. 1998; Qian et al. 1998), it is not clear if they are partial loss-of-function or gain-of-function mutations. All Alzheimer's disease-linked mutations occur at amino acid residues that are conserved in the Drosophila Psn protein, allowing us to introduce these mutations into the fly protein and assess their apoptotic effects in transgenic animals. Four missense mutations, N141I, M146V, L235P, and E280A were tested in this manner. The N141I mutant of PS2 has been shown to induce more apoptosis in certain cell types, compared with wild-type PS2, and may thus represent a gain-of-function mutation (Deng et al. 1996; Wolozin et al. 1996; Janicki and Monteiro 1997). Both the M146V and E280A mutants of PS1 increase the ratio of Aβ42/Aβ40 by increasing the level of the more neurotoxic and amyloidogenic Aβ42 cleavage product of amyloid precursor protein (reviewed in Selkoe 1998). The L235P mutation was identified in a family with an onset of Alzheimer's disease as early as age 29 (Campion et al. 1996), and may therefore represent a particularly severe mutant form of presenilin. In addition, a truncated Psn protein named D-ALG3, consisting of the most COOH-terminal 100 amino acids, two loop constructs consisting of either the long or short variable hydrophilic loop, and transmembrane domain 7 (TM7) following the loop were also included in our analysis. These constructs were chosen because D-ALG3 resembles truncated mammalian PS proteins that confer resistance to cell death in PC12 cells (Vito et al. 1996, Vito et al. 1997), and the loop may be a cytoplasmic domain required for protein–protein interactions.
The apoptotic activities of mutant presenilins were assessed by the following criteria: their ability to generate a rough eye phenotype in flies bearing two copies of each mutant transgene and GMR-GAL4; their ability to modify the eye phenotype of GMR-GAL4, 2X UAS-Psn+14 flies; and their ability to modify the eye phenotype of flies expressing the Drosophila death-domain protein Reaper under GMR promoter control (Table ). At least five independent transgenic lines were analyzed for each construct. Only the M146V substitution produces a rough eye phenotype in two independent transgenic lines out of five analyzed. The remaining mutations and Psn fragments fail to produce rough eye phenotypes when expressed under GMR-GAL4 control, but all four Alzheimer's disease-associated mutants enhance the rough eye phenotype of GMR-GAL4, 2X UAS-Psn+14 flies, and M146V and N141I also enhance the phenotype of flies bearing GMR-reaper. Although the ALG-3 fragment of PS2 inhibits programmed cell death in PC12 cells, the equivalent segment of Drosophila Psn, D-ALG3, instead weakly enhances the GMR-GAL4, 2X UAS-Psn+14 phenotype, indicating that it may possess weak apoptotic activity. The two loop variant fragments possess no modifying activity in these transgenic assays.
Table 1.
Apoptotic Activities of Alzheimer's-linked Psn Mutants
| GMR-GAL4 driving 2X UAS-construct | GMR-GAL4 driving 1X UAS construct and 2X UAS-Psn+14 | GMR-GAL4 driving 1X UAS construct together with GMR-Rpr | |
|---|---|---|---|
| UAS-construct type | Eye phenotype | Eye phenotype relative to GMR-GAL4, 2X UAS-Psn+14 | Eye phenotype relative to GMR-Rpr |
| Psn+14 | rough eye | enhanced | enhanced |
| Psn−14 | rough eye | enhanced | enhanced |
| M146V | weak rough eye | enhanced | weakly enhanced |
| N141I | wild-type | enhanced | weakly enhanced |
| L235P | wild-type | enhanced | no effect |
| E280A | wild-type | enhanced | no effect |
| D-ALG 3 | wild-type | weakly enhanced | no effect |
| Psn+14 loop | wild-type | no effect | no effect |
| Psn−14 loop | wild-type | no effect | no effect |
Genetic Interactions between Psn and Antiapoptotic Factors
To gain further insight into the mechanism of Psn-induced cell death, we tried to suppress this apoptosis by coexpression of Drosophila cell death inhibitors (DIAP1 or DIAP2) or the baculoviral survival factor p35. DIAP1 and DIAP2 are Drosophila homologues of baculoviral inhibitor of apoptosis proteins (IAPs), and can block programmed cell death induced by proapoptotic factors or mutations (Hay et al. 1995). Baculoviral p35 protein inhibits caspases and thus blocks apoptosis in many species (reviewed in McCall and Steller 1997). In the presence of any of these antiapoptotic proteins, the Psn-induced rough eye phenotype is largely suppressed, as revealed by the more regular external eye surface, the more normal trapezoidal pattern and orientation of R1-6 photoreceptor cells, and a restoration of the pigment cell lattice between ommatidia (Fig. 6). The observation that Psn-induced rough eye phenotypes are suppressed by coexpressing either DIAP1, DIAP2, or p35 confirms that increased cell death is the main cause of the rough eye phenotype. Psn-induced programmed cell death may thus be mediated by the conserved caspase pathway of apoptosis or, alternatively, it may be circumvented by inhibition of this pathway.
Figure 6.
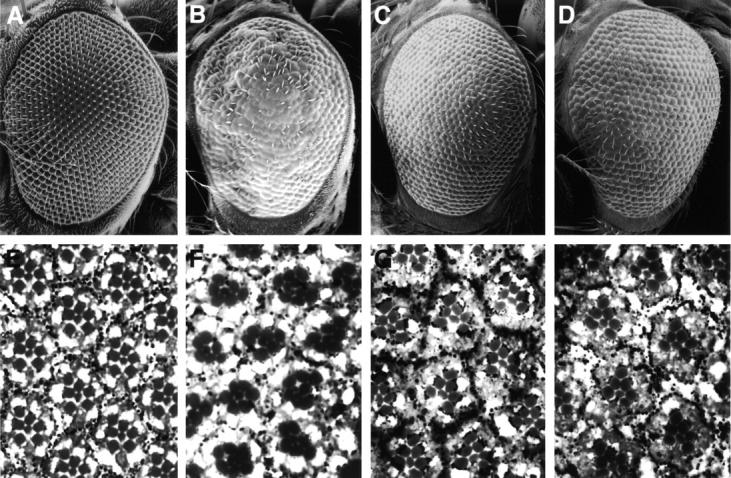
The Psn-induced eye phenotype is suppressed by coexpression of the antiapoptotic factors DIAP and p35. A–D, Adult eyes viewed by SEM. E–H, 1-μm plastic sections of the following genotypes: A and E, wild-type; B and F, GMR-GAL4, 2X UAS-Psn+14; C and G, GMR-GAL4, 2X UAS-Psn+14; GMR-DIAP1; D and H, GMR-GAL4, 2X UAS-Psn+14; GMR-p35. The disrupted ommatidial pattern caused by Psn overexpression is suppressed by coexpressing either DIAP1 or the baculoviral antiapoptosis protein p35, as judged by the relatively normal pigment cell lattice and properly oriented trapezoidal arrays of photoreceptor cells in GMR-GAL4, 2X UAS-Psn+14 transgenic flies also bearing GMR-DIAP1 or GMR-p35. On their own, the GMR-DIAP1 and GMR-p35 transgenes do not produce a noticeable adult eye phenotype, although they suppress a normal round of apoptosis in the pupal retina, leading to a slight excess of pigment cells (Hay et al. 1995). Anterior at left.
Psn-mediated Apoptosis and Notch Signaling
The ability of wild-type and, to a lesser extent, mutant forms of Drosophila Psn to induce low levels of apoptosis similar to that seen in developing imaginal tissues of Psn loss-of-function mutants suggests that the apoptotic effects of Psn may be a secondary consequence of reduced or dominant-negative Psn activity during developmental patterning. In C. elegans and mice, presenilin proteins have been shown to facilitate Notch signaling, and worms or mice lacking presenilin activity display typical Notch or lin-12/glp-1 loss-of-function phenotypes (Levitan and Greenwald 1995; Li and Greenwald 1997; Wong et al. 1997). Similarly, flies lacking functional Psn gene activity exhibit embryonic neurogenic phenotypes and imaginal disc phenotypes that are characteristic of impaired Notch signaling (Struhl and Greenwald 1999; Ye et al. 1999). Moreover, elevated levels of apoptosis have been noted previously in wing imaginal discs of flies having the partial loss-of-function heteroallelic Notch genotype Nts/N55e11; neurA101/+ (Rulifson and Blair 1995). These observations raise the possibility that the apoptotic effects of PS overexpression may be due to a primary interference with Notch signaling, followed by elimination of cells that have not adopted their proper cell fate by a normal corrective mechanism of developmentally controlled apoptosis (reviewed in Bonini and Fortini 1999).
Our in vivo genetic model for Psn-mediated apoptosis allowed us to examine the potential involvement of Notch signaling in the apoptotic response, an important issue that has not been possible to assess in the widely used mammalian cell culture assays for Psn-induced apoptosis. First, we wished to determine if the UAS-Psn constructs that cause apoptosis in the Drosophila eye when driven by GMR-GAL4 are able to produce Notch pathway phenotypes in other tissues when expressed using suitable GAL4 driver constructs. We found that several GAL4 driver lines that are active in the wing and cuticle anlagen are indeed capable of producing adult Notch-like phenotypes in the wing blade and thorax, including wing margin notching, vein thickening, ectopic wing margin bristles, ectopic wing vein campaniform sensilla, ectopic thoracic macrochaetae, and missing thoracic microchaetae (Fig. 7). These phenotypes are consistent with the notion that Psn overexpression leads to dominant-negative effects, since Psn loss-of-function mutants exhibit similar Notch-like phenotypes (Struhl and Greenwald 1999; Ye et al. 1999). To determine if Psn-induced apoptosis might be an indirect effect of reduced Notch activity, we first analyzed apoptosis in imaginal wing discs of the conditional temperature-sensitive Notch mutant Nts1. Increased levels of programmed cell death are spatiotemporally correlated with progressive loss of Notch activity as visualized by reduced wing-pouch–specific expression of the Notch target gene reporter vg(quadrant enhancer)-lacZ and expansion of proneural cell clusters positive for ac-lacZ expression in the presumptive notum region (Van Doren et al. 1992; Kim et al. 1996), suggesting that developmental patterning defects caused by reducing Notch activity directly lead to elimination of affected cells by apoptosis (Fig. 8). Second, we tested whether reduction in the dosage of the wild-type Notch gene or coexpression of constitutively activated Notch is able to suppress or enhance Psn-related apoptotic phenotypes. We found that the rough eye phenotype of GMR-GAL4, 2X UAS-Psn+14 is strongly enhanced in an N54l9 mutant background bearing only one functional copy of the Notch gene (Fig. 9). Apoptosis caused by either Psn overexpression or removal of Psn gene function is also dramatically suppressed by coexpression of constitutively activated Notch in the retina (Fig. 9). These studies show that genetic removal or overexpression of Psn in developing Drosophila tissues is able to induce Notch-like phenotypes, as well as apoptosis, and that when genetic methods are used to compensate for effects on Notch signaling, the levels of apoptosis are dramatically reduced. Our results, together with the observed correlation between impaired Notch signaling and high levels of developmental apoptosis, offer a potential explanation of Psn-mediated apoptosis as a developmental response to a primary failure in cellular patterning events requiring Psn activity for proper Notch synthesis or signaling.
Figure 7.
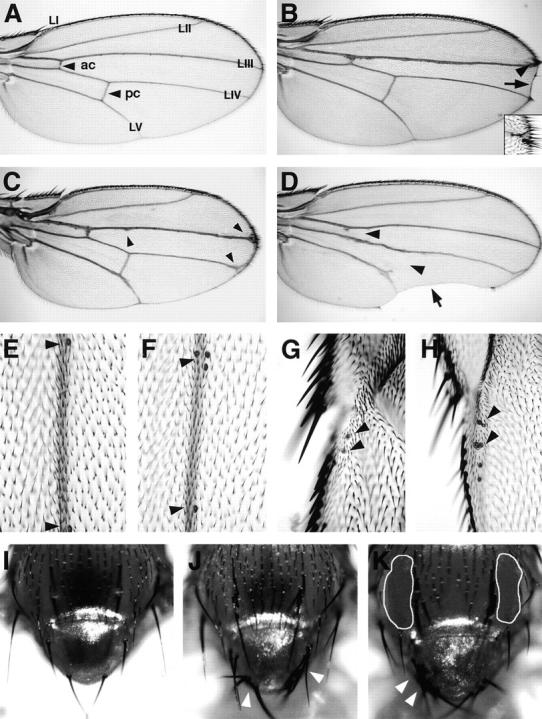
Notch-like phenotypes produced by ectopic expression of Psn in the Drosophila wing and epidermis. A, Wild-type wing blade. LI-LV, Longitudinal wing veins; ac, anterior crossvein; pc, posterior crossvein. B, Distal wing blade notching (arrow) and ectopic margin bristles (arrowhead; inset) in dpp-GAL4; 2X UAS-Psn+14 transgenic flies. C, Thickened vein and Delta-like vein tip phenotypes (arrowheads) in T80-GAL4; 2X UAS-Psn+14 flies. D, Missing crossveins (arrowheads) and posterior wing blade scalloping (arrow) in e16E-GAL4; 2X UAS-Psn+14, in which GAL4 is under engrailed gene control. E–H, Single campaniform sensilla (arrowheads) arise at defined positions along the LIII vein (E) and LI vein (G) in wild-type wings, but multiple sensilla arise along the LIII vein (F) and LI vein (H) in Hc-GAL4; 2X UAS-Psn+14 flies. I, Macrochaetae and microchaetae on the thorax and notum of a wild-type fly. J, Ectopic macrochaetae (arrowheads) on the notum of a C625-GAL4; 2X UAS-Psn+14 fly. K, Ectopic macrochaetae on the notum (arrowheads) and missing microchaetae on the thorax (outlined areas) of a 19A-GAL4; 2X UAS-Psn+14 fly. Both C625-GAL4 and 19A-GAL4 are widely expressed throughout the wing imaginal disc (data not shown). Anterior at top in A–D and I–K; anterior at left in E–H.
Figure 8.
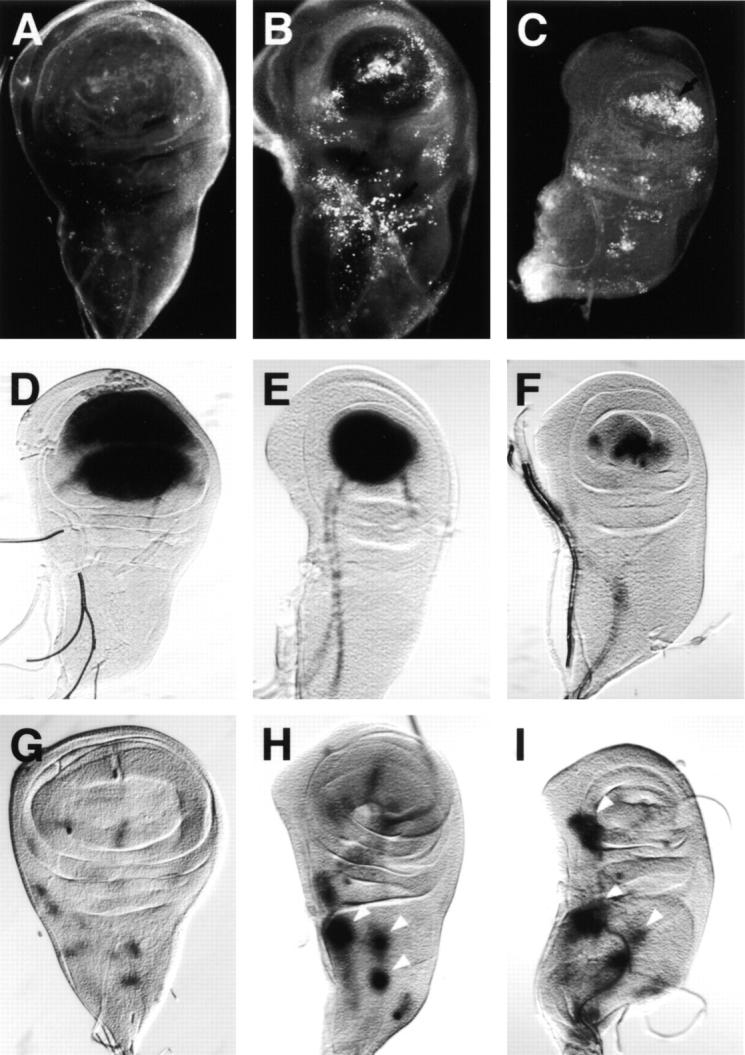
Loss of Notch function is associated with increased levels of apoptosis in imaginal wing discs of the temperature-sensitive, conditional Notch mutant Nts1. Acridine orange staining (A–C) or β-gal detection (D–I) of wing discs from third-instar larvae of genotypes Nts1 (A–C), Nts1; vg(quadrant enhancer)-lacZ (D–F), or Nts1; ac-lacZ (G–I) at either the permissive temperature (A, D, and G), three days after shifting to the nonpermissive temperature (B, E, and H), or five days after the shift (C, F, and I). Loss of Notch signaling in Nts1wing discs can be visualized by the diminished vg(quadrant enhancer)-lacZ expression in the wing pouch (D–F) and the expansion of ac-lacZ expression (G–I, arrowheads). Correlated with these changes, increased levels of apoptosis are first detected mostly in the notum region where the expansion of ac-lacZ expression is first seen (B and H). After the five-day temperature shift, apoptotic cells are mainly limited to the wing pouch zone where vg(quadrant enhancer)-lacZ expression is significantly reduced (C and F). Anterior at left.
Figure 9.
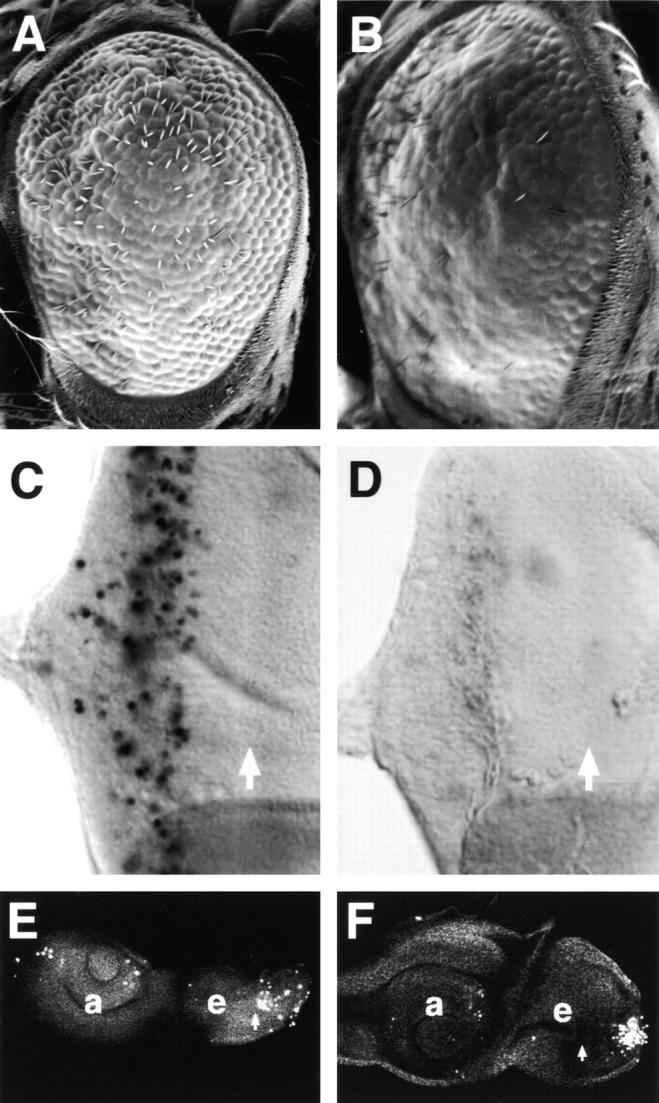
Suppression and enhancement of the Psn-induced apoptotic phenotype by alterations in Notch gene dosage. A and B, SEM images showing that the adult rough eye phenotype caused by increased cell death in GMR-GAL4, 2X UAS-Psn+14 (A) is strongly enhanced by removal of one dose of wild-type Notch in flies of genotype N54l9; GMR-GAL4, 2X UAS-Psn+14 (B). N54l9 is a deletion of Notch (Lindsley and Zimm 1992), which displays no dominant eye phenotype in heterozygotes. C and D, TUNEL assay showing that Psn-mediated programmed cell death seen in GMR-GAL4, 2X UAS-Psn+14 eye discs (C) is strongly reduced by coexpression of a constitutively activated form of Notch, termed sev-Notchact (Fortini et al. 1993; D). E and F, Overexpression of activated Notch using hs-Notch(intra) (Struhl et al. 1993) in homozygous PsnB3 mutant larvae (F) partially rescues the Psn loss-of-function phenotype, producing discs that are larger than tissues from equivalent PsnB3 mutant larvae (E). A, B, E, and F, Anterior at left; C and D, anterior at right.
To further elucidate the molecular mechanism underlying the Psn overexpression phenotypes, we analyzed Notch processing and trafficking in S2 cells cotransfected with Psn and Notch. When expressed before Notch induction, wild-type Psn and the various Psn mutants lead to reduced Notch protein levels, affecting both the full-length and the processed COOH-terminal fragments of Notch (Fig. 10). In agreement with our genetic results, the biochemical effect appears to be more pronounced for wild-type Psn than for the mutant forms. The expression of a nonmembrane-bound control protein, Suppressor of Hairless, is not affected by either the wild-type or the mutant Psn proteins (Fig. 10). The effect on Notch synthesis is unlikely to be due to increased protein degradation, because ectopic expression of the same Psn construct in S2 cells after Notch induction has no detectable effect on Notch protein levels (data not shown). In addition, live cell surface immunostainings reveal that Notch protein is trafficked and inserted into the cell membrane normally in spite of the reduced protein levels caused by Psn overexpression (Fig. 10 C).
Figure 10.
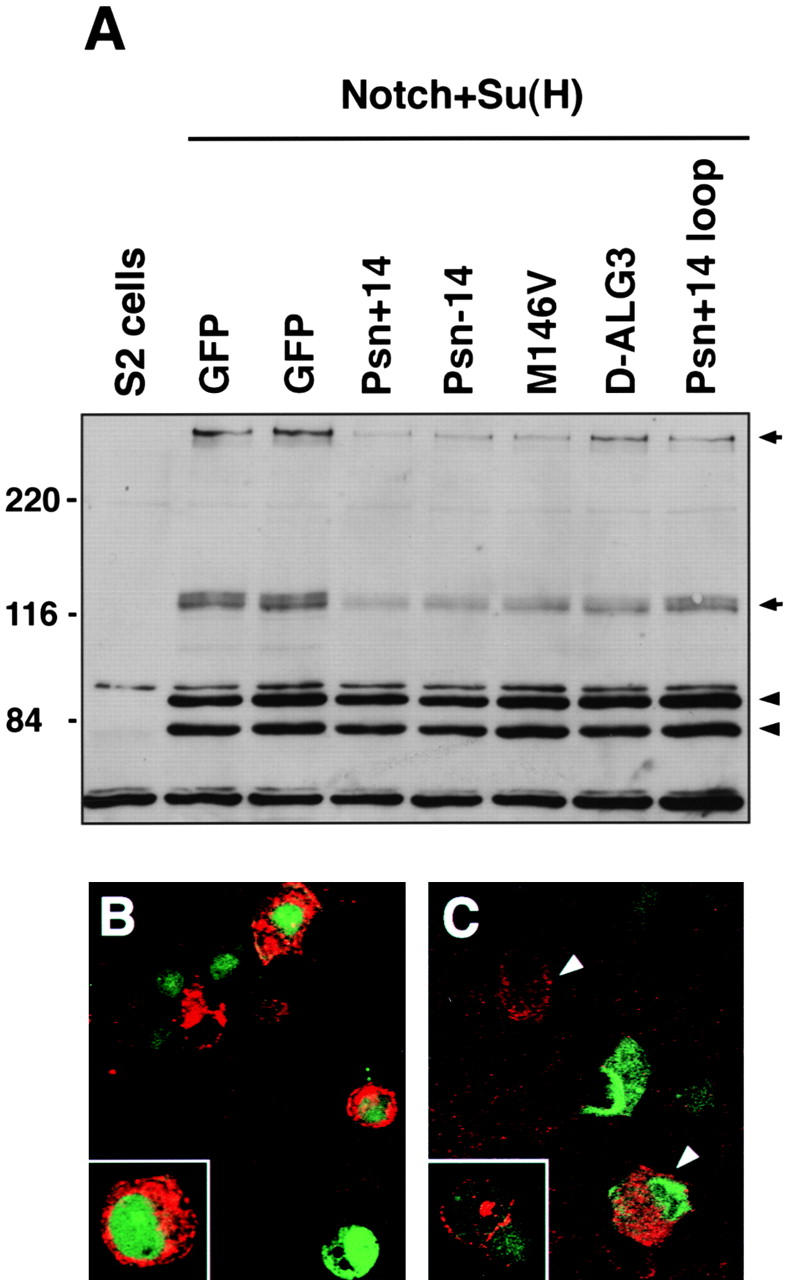
Expression of Psn leads to reduced Notch protein levels in S2 cells. A, S2 cells were transfected with pMTNMg, which expresses full-length Notch; pHS-Su(H)4, which expresses Myc-tagged Su(H); and a pair of overexpression constructs consisting of pHS-GV, which expresses a GAL4-VP16 fusion protein, along with either pUAS-Psn or pUAS-GFP constructs, as labeled above each lane. Cell lysates were analyzed by immunoblotting with a mixture of three antibodies: mAb C17.9C6, which detects both the ∼300-kD full-length and ∼120-kD COOH-terminal Notch fragments (arrows); mAb 9E10, which detects the Myc-tagged Su(H) doublet bands (arrowheads); and mAb E7, which detects β-tubulin (at bottom of gel). Both full-length and COOH-terminal Notch fragment levels are significantly reduced when the Psn constructs are expressed before Notch expression, whereas levels of Su(H) and tubulin are unaffected. B, S2 cells transfected with pMTNMg were fixed and stained with mAb C458.2H, an antibody recognizing the extracellular domain of Notch. In pMTNMg-transfected cells (marked by green GFP cotransfection), Notch accumulates at high levels (red) and shows both vesicular cytoplasmic and cell-surface staining (inset, higher magnification view). When Psn is coexpressed in these cells (C), Notch levels are dramatically reduced (arrowheads). No obvious vesicular staining is detected in these cells, although Notch is still normally localized in the plasma membrane, as shown by cell-surface antibody staining of live, nonpermeabilized S2 cells (inset).
Discussion
The Role of Drosophila Psn in Neuronal Development and Apoptosis
In mice, removal of PS1 results in a severe loss of neural progenitor cells in specific regions of the brain, suggesting that PS1 may normally play a neuroprotective role during neurogenesis (Shen et al. 1997). However, other studies have shown that overexpression of PS1 and PS2 in mammalian cell lines sensitizes the cells to apoptosis induced by trophic factor withdrawal or Aβ peptide, indicating that presenilin may promote cell death triggered by certain stimuli (Guo et al. 1996, Guo et al. 1997; Deng et al. 1996; Wolozin et al. 1996; Janicki and Monteiro 1997; Weihl et al. 1999). In addition, COOH-terminal fragments of PS1 and PS2 are reported to have antiapoptotic activity in similar assays (Vito et al. 1996, Vito et al. 1997; Vézina et al. 1999), and contradictory data exist concerning the proapoptotic effects of wild-type and Alzheimer's disease-associated mutant variants of PS1 (Barinaga 1998; Bursztajn et al. 1998). We therefore sought to analyze the role of presenilin in promoting or preventing apoptosis in a multicellular experimental organism using both classical genetics and a transgenic approach that closely parallels the overexpression strategies used in the mammalian cell culture studies.
Using several loss-of-function lesions in the endogenous Drosophila Presenilin gene, we showed that mutant tissues lacking Psn activity display elevated levels of developmental apoptosis. Imaginal tissues are considerably smaller than age-matched wild-type control tissues, suggesting that apoptosis plays a significant role in reducing cell numbers in the Psn mutants. High levels of apoptosis are correlated with selective loss of photoreceptor neurons in imaginal eye discs of the mutants, consistent with suggestions that mammalian PS proteins may normally function as antiapoptotic factors required to protect neurons from degeneration. However, it is worth noting that apoptotic bodies are also readily detected in other cell types of the Psn mutants, indicating that the antiapoptotic effects are not limited to neuronal tissues in Drosophila. Our loss-of-function genetic analysis is in agreement with a recent study showing that antisense inhibition of PS1 in mammalian cell culture blocks neuronal differentiation and induces apoptosis (Hong et al. 1999).
In our transgenic overexpression model, we find that wild-type Psn is most efficient at inducing apoptosis. Since Psn overexpression causes phenotypes that resemble Psn loss-of-function mutant apoptotic phenotypes, the proapoptotic activity of overexpressed Psn may actually be due to dominant-negative effects. Perhaps cells overexpressing Psn undergo apoptosis not because of a specific regulatory role of Psn in apoptosis, but rather due to a blockage of essential intracellular protein transport pathways by overaccumulation of Psn in the ER and Golgi compartments. Alternatively, overexpression of Psn may interfere with endogenous Psn function, resulting in phenotypes resembling those of Psn loss-of-function mutants. It has been shown previously that both NH2- and COOH-terminal PS1 fragments form oligomers in vitro (Seeger et al. 1997). It is therefore possible that overexpression of wild-type Psn in vivo may promote excess or premature Psn oligomerization, resulting in the formation of nonfunctional complexes that deplete endogenous functional Psn within the cell or disrupt ER/Golgi compartment function.
Apoptosis and Impaired Notch Receptor Synthesis
Our results suggest that the apoptotic effects of presenilin proteins may involve their well-documented role in Notch signaling (Levitan and Greenwald 1995; Li and Greenwald 1997; Wong et al., 1998; Struhl and Greenwald 1999; Ye et al. 1999). In C. elegans, the presenilin homologue Sel-12 is needed for normal cell-surface accumulation of the Lin-12 protein, a member of the Notch receptor family (Levitan and Greenwald 1998), and in Drosophila and mammals, presenilin is required for normal proteolytic processing of Notch during receptor assembly or ligand-induced signaling (De Strooper et al. 1999; Struhl and Greenwald 1999; Ye et al. 1999). We show here that the apoptotic effects of fly Psn are strongly dependent upon Notch gene dosage, and that the effects can be partially suppressed by supplying flies with constitutively activated Notch protein. Moreover, when Psn overexpression is restricted to certain developing structures, we observe typical Notch-like wing and neurogenic phenotypes. Based on these results, we suggest that the apoptotic effects of Psn overexpression may be a secondary consequence of impaired Notch signaling, perhaps due to a dominant-negative effect that interferes with Notch activity, resulting in cells that are developmentally misprogrammed and thus selectively eliminated by a normal corrective mechanism of apoptosis. This interpretation is consistent with the observation that increased amounts of cell death are correlated with reduced Notch signaling (Fig. 9; Rulifson and Blair 1995; Jehn et al. 1999). As discussed above, previous studies have shown that Psn protein self-aggregates under some conditions (Seeger et al. 1997); similar aggregation might interfere with ER/Golgi compartment function in vivo. Our finding that overexpression of Psn in S2 cells leads to reduced Notch protein levels is consistent with this notion and provides a plausible molecular explanation for the Psn overexpression apoptotic and neurogenic phenotypes. This dominant-negative effect on Notch synthesis is distinct from the effect of Psn loss-of-function mutations, which cause overaccumulation of specific ∼120-kD Notch COOH-terminal fragments at the expense of other fragments (Ye et al. 1999).
Alzheimer's Disease-linked Presenilin Mutations Are Partial Loss-of-function Alleles
We have assessed the proapoptotic activity of different Alzheimer's disease-linked mutant variants and found that they possess less apoptotic activity than wild-type presenilin, consistent with the idea that this class of presenilin mutations represents partial loss-of-function mutations in nematodes and transgenic mice (Levitan et al. 1996; Davis et al. 1998; Qian et al. 1998). In contrast to previous studies showing that Alzheimer's disease-associated missense mutations enhance the ability of PS1 and PS2 to induce cell death and that the COOH-terminal ALG-3 fragment blocks the proapoptotic activity of PS2 (Vito et al. 1996, Vito et al. 1997), the corresponding missense mutants of fly Psn induce less cell death than wild-type Psn, and the Drosophila Psn COOH-terminal D-ALG3 fragment appears to induce apoptosis only weakly in vivo. In view of the dominant-negative role that we suggest for overexpressed Psn, the reduced apoptotic activity of these mutant variants may reflect a decreased ability to interfere with endogenous presenilin function or a more rapid clearance of misfolded mutant proteins from the ER/Golgi compartment. Analogous partial loss-of-function effects on Aβ secretion of human mutant presenilins may be relatively weak under physiological conditions, but may contribute to gradual deterioration of brain tissue during the aging process and eventually trigger disease onset when neuronal loss reaches a certain threshold.
Suppression of Psn-induced Apoptosis by Coexpression of DIAP or p35
Using genetic tools developed by others for the analysis of apoptosis in the Drosophila eye (Hay et al. 1995), we have shown that the rough eye phenotypes associated with Psn overexpression are partially suppressed when DIAPs or baculoviral p35 is coexpressed using the same promoter. This result confirms that apoptosis induced by overexpression of Psn is a major cause of the final adult eye phenotype, regardless of its developmental origins. In addition, the Psn effects are most likely to occur at a step upstream of the conserved caspase effector pathway of apoptosis or via a parallel pathway, since Psn-induced cell death is inhibited by p35, which antagonizes caspase activity, and DIAPs, which may block the ability of the Drosophila Reaper protein to activate the caspase effector cascade (Hay et al. 1995; McCall and Steller 1997). It has been previously demonstrated that cells rescued from apoptosis can be physiologically functional, since blocking cell death by inhibiting caspase activity prevents blindness in Drosophila neurodegeneration mutants (Davidson and Steller 1998). In this regard, our results support the notion that preventing neuronal apoptosis may be a useful therapeutic strategy for counteracting the neurodegeneration associated with Alzheimer's disease.
In summary, we demonstrate that both loss of Drosophila Psn activity and overexpression of Psn in the developing fly retina induce programmed cell death in a cell autonomous manner. The apoptotic effects of Psn may result from dominant-negative effects of the overexpressed presenilin, and are likely to be a consequence of interfering with Notch-mediated developmental signaling by impeding Notch synthesis. Apoptotic effects of fly Psn, and perhaps also those caused by mammalian PS proteins, may thus reflect the normal elimination of aberrant cells generated by a primary failure in intercellular communication and cell-fate specification. In addition, we demonstrate that Alzheimer's disease-linked Presenilin mutations may be partial loss-of-function mutations using different genetic criteria. Furthermore, Psn-induced rough eye phenotypes are highly sensitive to transgene dosages and modifying effects of other genes, including evolutionarily conserved components of the caspase-mediated apoptosis pathway. Psn transgenic flies may therefore be a useful genetic tool to dissect Psn function by performing genetic modifier screens for additional factors that interact with presenilin.
Acknowledgments
We thank G.M. Rubin, N.M. Bonini, E. Wilder, and X. Sun for fly stocks, J. Li for making the M146V construct, and N.M. Bonini, T.A. Jongens, and A.B. Jaffe for advice and comments on the manuscript. We also thank L. Peachey and N.M. Bonini for access to their confocal microscope.
This work was funded by National Institutes of Health grant AG14583, the Alzheimer's Association, and a Merck research grant.
Footnotes
1.used in this paper: Aβ40, 40-amino acid amyloid peptide; Aβ42, 42-amino acid amyloid peptide; APP, amyloid precursor protein; DIAP, Drosophila inhibitor of apoptosis protein; PS1, presenilin 1; PS2, presenilin 2
References
- Abrams J.M., White K., Fessler L.I., Steller H. Programmed cell death during Drosophila embryogenesis. Development. 1993;117:29–43. doi: 10.1242/dev.117.1.29. [DOI] [PubMed] [Google Scholar]
- Barinaga M. Is apoptosis key in Alzheimer's disease? Science. 1998;281:1303–1304. doi: 10.1126/science.281.5381.1303. [DOI] [PubMed] [Google Scholar]
- Bonini N.M., Fortini M.E. Surviving Drosophila eye developmentintegrating cell death with differentiation during formation of a neural structure. BioEssays. 1999;In press doi: 10.1002/(SICI)1521-1878(199912)22:1<991::AID-BIES3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Brand A.H., Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Bursztajn S., DeSouza R., McPhie D.L., Berman S.A., Shioi J., Robakis N.K., Neve R.L. Overexpression in neurons of human presenilin-1 or a presenilin-1 familial Alzheimer disease mutant does not enhance apoptosis. J. Neurosci. 1998;18:9790–9799. doi: 10.1523/JNEUROSCI.18-23-09790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagan R.L., Ready D.F. Notch is required for successive cell decisions in the developing Drosophila retina. Genes Dev. 1989;3:1099–1112. doi: 10.1101/gad.3.8.1099. [DOI] [PubMed] [Google Scholar]
- Campion D., Brice A., Dumanchin C., Puel M., Baulac M., De La Sayette V., Hannequin D., Duyckaerts C., Michon A., Martin C. A novel presenilin 1 mutation resulting in familial Alzheimer's disease with an onset age of 29 years. Neuroreport. 1996;7:1582–1584. doi: 10.1097/00001756-199607080-00009. [DOI] [PubMed] [Google Scholar]
- Davidson F.F., Steller H. Blocking apoptosis prevents blindness in Drosophila retinal degeneration mutants. Nature. 1998;391:587–591. doi: 10.1038/35385. [DOI] [PubMed] [Google Scholar]
- Davis J.A., Naruse S., Chen H., Eckman C., Younkin S., Price D.L., Borchelt D.R., Sisodia S.S., Wong P.C. An Alzheimer's disease-linked PS1 variant rescues the developmental abnormalities of PS1-deficient embryos. Neuron. 1998;20:603–609. doi: 10.1016/s0896-6273(00)80998-8. [DOI] [PubMed] [Google Scholar]
- De Strooper B., Annaert W., Cupers P., Saftig P., Craessaerts K., Mumm J.S., Schroeter E.H., Schrijvers V., Wolfe M.S., Ray W.J. A presenilin-1–dependent γ-secretase–like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Deng G., Pike C.J., Cotman C.W. Alzheimer-associated presenilin-2 confers increased sensitivity to apoptosis in PC12 cells. FEBS Lett. 1996;397:50–54. doi: 10.1016/s0014-5793(96)01142-8. [DOI] [PubMed] [Google Scholar]
- Ellis M.C., O'Neill E.M., Rubin G.M. Expression of Drosophila glass protein and evidence for negative regulation of its activity in non-neuronal cells by another DNA-binding protein. Development. 1993;119:855–865. doi: 10.1242/dev.119.3.855. [DOI] [PubMed] [Google Scholar]
- Fehon R.G., Kooh P.J., Rebay I., Regan C.L., Xu T., Muskavitch M.A.T., Artavanis-Tsakonas S. Molecular interactions between the protein products of the neurogenic loci Notch and Delta, two EGF-homologous genes in Drosophila . Cell. 1990;61:523–534. doi: 10.1016/0092-8674(90)90534-l. [DOI] [PubMed] [Google Scholar]
- Fortini M.E., Rebay I., Caron L.A., Artavanis-Tsakonas S. An activated Notch receptor blocks cell-fate commitment in the developing Drosophila eye. Nature. 1993;365:555–557. doi: 10.1038/365555a0. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y., Sherman Y., Ben-Sasson S.A. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q., Furukawa K., Sopher B.L., Pham D.G., Xie J., Robinson N., Martin G.M., Mattson M.P. Alzheimer's PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid β-peptide. Neuroreport. 1996;8:379–383. doi: 10.1097/00001756-199612200-00074. [DOI] [PubMed] [Google Scholar]
- Guo Q., Sopher B.L., Pham D.G., Furukawa K., Robinson N., Martin G.M., Mattson M.P. Alzheimer's presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid β-peptideinvolvement of calcium and oxyradicals. J. Neurosci. 1997;17:4212–4222. doi: 10.1523/JNEUROSCI.17-11-04212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C. Presenilinsgenes for life and death. Neuron. 1997;18:687–690. doi: 10.1016/s0896-6273(00)80309-8. [DOI] [PubMed] [Google Scholar]
- Haass C., Selkoe D.J. Cellular processing of β-amyloid precursor protein and the genesis of amyloid β-peptide. Cell. 1993;75:1039–1042. doi: 10.1016/0092-8674(93)90312-e. [DOI] [PubMed] [Google Scholar]
- Hay B.A., Wassarman D.A., Rubin G.M. Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell. 1995;83:1253–1262. doi: 10.1016/0092-8674(95)90150-7. [DOI] [PubMed] [Google Scholar]
- Hong C.-S., Caromille L., Nomata Y., Mori H., Bredesen D.E., Koo E.H. Contrasting role of presenilin-1 and presenilin-2 in neuronal differentiation in vitro . J. Neurosci. 1999;19:637–643. doi: 10.1523/JNEUROSCI.19-02-00637.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M.D., Weil M., Raff M.C. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Janicki S., Monteiro M.J. Increased apoptosis arising from increased expression of the Alzheimer's disease-associated presenilin-2 mutation (N141I) J. Cell Biol. 1997;139:485–495. doi: 10.1083/jcb.139.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehn B.M., Bielke W., Pear W.S., Osborne B.A. Protective effects of Notch-1 on TCR-induced apoptosis. J. Immunol. 1999;162:635–638. [PubMed] [Google Scholar]
- Kim J., Sebring A., Esch J.J., Kraus M.E., Vorwerk K., Magee J., Carroll S.B. Integration of positional signals and regulation of wing formation and identity by Drosophila vestigial gene. Nature. 1996;382:133–138. doi: 10.1038/382133a0. [DOI] [PubMed] [Google Scholar]
- Levitan D., Greenwald I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer's disease gene. Nature. 1995;377:351–354. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- Levitan D., Greenwald I. Effects of SEL-12 presenilin on LIN-12 localization and function in Caenorhabditis elegans . Development. 1998;125:3599–3606. doi: 10.1242/dev.125.18.3599. [DOI] [PubMed] [Google Scholar]
- Levitan D., Doyle T.G., Brousseau D., Lee M.K., Thinakaran G., Slunt H.H., Sisodia S.S., Greenwald I. Assessment of normal and mutant human presenilin function in Caenorhabditis elegans . Proc. Natl. Acad. Sci. USA. 1996;93:14940–14944. doi: 10.1073/pnas.93.25.14940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Greenwald I. HOP-1, a Caenorhabditis elegans presenilin, appears to be functionally redundant with SEL-12 presenilin and to facilitate LIN-12 and GLP-1 signaling. Proc. Natl. Acad. Sci. USA. 1997;94:12204–12209. doi: 10.1073/pnas.94.22.12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley D.L., Zimm G.G. The Genome of Drosophila melanogaster. Academic Press; San Diego, California: 1992. [Google Scholar]
- McCall K., Steller H. Facing death in the flygenetic analysis of apoptosis in Drosophila . Trends Genet. 1997;13:222–226. doi: 10.1016/S0168-9525(97)01126-8. [DOI] [PubMed] [Google Scholar]
- Price D.L., Sisodia S.S., Borchelt D.R. Genetic neurodegenerative diseasesthe human illness and transgenic models. Science. 1998;282:1079–1083. doi: 10.1126/science.282.5391.1079. [DOI] [PubMed] [Google Scholar]
- Qian S., Jiang P., Guan X.-M., Singh G., Trumbauer M.E., Yu H., Chen H.Y., Van der Ploeg L.H.T., Zheng H. Mutant human presenilin 1 protects presenilin 1 null mouse against embryonic lethality and elevates Aβ1-42/43 expression. Neuron. 1998;20:611–617. doi: 10.1016/s0896-6273(00)80999-x. [DOI] [PubMed] [Google Scholar]
- Raff M. Cell suicide for beginners. Nature. 1998;396:119–122. doi: 10.1038/24055. [DOI] [PubMed] [Google Scholar]
- Ready D.F., Hanson T.E., Benzer S. Development of the Drosophila retina, a neurocrystalline lattice. Dev. Biol. 1976;53:217–240. doi: 10.1016/0012-1606(76)90225-6. [DOI] [PubMed] [Google Scholar]
- Robinow S., White K. Characterization and spatial distribution of the ELAV protein during Drosophila melanogaster development. J. Neurobiol. 1991;22:443–461. doi: 10.1002/neu.480220503. [DOI] [PubMed] [Google Scholar]
- Rulifson E.J., Blair S.S. Notch regulates wingless expression and is not required for reception of the paracrine wingless signal during wing margin neurogenesis in Drosophila . Development. 1995;121:2813–2824. doi: 10.1242/dev.121.9.2813. [DOI] [PubMed] [Google Scholar]
- Seeger M., Nordstedt C., Petanceska S., Kovacs D.M., Gouras G.K., Hahne S., Fraser P., Levesque L., Czernik A.J., George-Hyslop P.S. Evidence for phosphorylation and oligomeric assembly of presenilin 1. Proc. Natl. Acad. Sci. USA. 1997;94:5090–5094. doi: 10.1073/pnas.94.10.5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D.J. The cell biology of β-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- Shen J., Bronson R.T., Chen D.F., Xia W., Selkoe D.J., Tonegawa S. Skeletal and CNS defects in Presenilin-1–deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Spradling A.C., Rubin G.M. Transposition of cloned P elements into Drosophila germ line chromosomes. Science. 1982;218:341–347. doi: 10.1126/science.6289435. [DOI] [PubMed] [Google Scholar]
- Spreij T.E. Cell death during the development of the imaginal discs of Calliphora erythrocephala . Netherlands J. Zool. 1971;21:221–264. [Google Scholar]
- Struhl G., Adachi A. Nuclear access and action of Notch in vivo. Cell. 1998;93:649–660. doi: 10.1016/s0092-8674(00)81193-9. [DOI] [PubMed] [Google Scholar]
- Struhl G., Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila . Nature. 1999;398:522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- Struhl G., Fitzgerald K., Greenwald I. Intrinsic activity of the Lin-12 and Notch intracellular domains in vivo. Cell. 1993;74:331–345. doi: 10.1016/0092-8674(93)90424-o. [DOI] [PubMed] [Google Scholar]
- Tomlinson A., Ready D.F. Cell fate in the Drosophila ommatidium. Dev. Biol. 1987;123:264–275. doi: 10.1016/0012-1606(87)90448-9. [DOI] [PubMed] [Google Scholar]
- Tomlinson A., Bowtell D.D.L., Hafen E., Rubin G.M. Localization of the sevenless protein, a putative receptor for positional information, in the eye imaginal disc of Drosophila . Cell. 1987;51:143–150. doi: 10.1016/0092-8674(87)90019-5. [DOI] [PubMed] [Google Scholar]
- Van Doren M., Powell P.A., Pasternak D., Singson A., Posakony J.W. Spatial regulation of proneural gene activityauto- and cross-activation of achaete is antagonized by extramacrochaetae . Genes Dev. 1992;6:2592–2605. doi: 10.1101/gad.6.12b.2592. [DOI] [PubMed] [Google Scholar]
- Vézina J., Tschopp C., Andersen E., Müller K. Overexpression of a C-terminal fragment of presenilin 1 delays anti-Fas induced apoptosis in Jurkat cells. Neurosci. Lett. 1999;263:65–68. doi: 10.1016/s0304-3940(99)00097-x. [DOI] [PubMed] [Google Scholar]
- Vito P., Lacanà E., D'Adamio L. Interfering with apoptosisCa2+-binding protein ALG-2 and Alzheimer's disease gene ALG-3. Science. 1996;271:521–525. doi: 10.1126/science.271.5248.521. [DOI] [PubMed] [Google Scholar]
- Vito P., Ghayur T., D'Adamio L. Generation of anti-apoptotic presenilin-2 polypeptides by alternative transcription, proteolysis, and caspase-3 cleavage. J. Biol. Chem. 1997;272:28315–28320. doi: 10.1074/jbc.272.45.28315. [DOI] [PubMed] [Google Scholar]
- Weihl C.C., Ghadge G.D., Kennedy S.G., Hay N., Miller R.J., Roos R.P. Mutant presenilin-1 induces apoptosis and downregulates Akt/PKB. J. Neurosci. 1999;19:5360–5369. doi: 10.1523/JNEUROSCI.19-13-05360.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff T., Ready D.F. Cell death in normal and rough eye mutants of Drosophila . Development. 1991;113:825–839. doi: 10.1242/dev.113.3.825. [DOI] [PubMed] [Google Scholar]
- Wolozin B., Iwasaki K., Vito P., Ganjei J.K., Lacanà E., Sunderland T., Zhao B., Kusiak J.W., Wasco W., D'Adamio L. Participation of presenilin 2 in apoptosisenhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- Wong P.C., Zheng H., Chen H., Becher M.W., Sirinathsinghji D.J.S., Trumbauer M.E., Chen H.Y., Price D.L., Van der Ploeg L.H.T., Sisodia S.S. Presenilin 1 is required for Notch1 and Dll1 expression in the paraxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- Ye Y., Fortini M.E. Characterization of Drosophila presenilin and its colocalization with Notch during development. Mech. Dev. 1998;79:199–211. doi: 10.1016/s0925-4773(98)00169-5. [DOI] [PubMed] [Google Scholar]
- Ye Y., Lukinova N., Fortini M.E. Neurogenic phenotypes and altered Notch processing in Drosophila Presenilin mutants. Nature. 1999;398:525–529. doi: 10.1038/19096. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Hartmann H., Do V.M., Abramowski D., Sturchler-Pierrat C., Staufenbiel M., Sommer B., van de Wetering M., Clevers H., Saftig P. Destabilization of β-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998;395:698–702. doi: 10.1038/27208. [DOI] [PubMed] [Google Scholar]