

Abstract
Insulin-like growth factor (IGF) II is overexpressed in many human cancers and is reactivated by, and crucial for viral oncogene (SV40 T antigen, [TAg])–induced tumorigenesis in several tumor models. Using a double transgenic murine hepatic tumor model, we demonstrate that tissue inhibitor of metalloproteinase 1 (TIMP-1) blocks liver hyperplasia during tumor development, despite TAg-mediated reactivation of IGF-II. Because the activity of IGFs is controlled by IGF-binding proteins (IGFBPs), we investigated whether TIMP-1 overexpression altered the IGFBP status in the transgenic liver. Ligand blotting showed that IGFBP-3 protein levels were increased in TIMP-1–overexpressing double transgenic littermates, whereas IGFBP-3 mRNA levels were not different, suggesting that TIMP-1 affects IGFBP-3 at a posttranscriptional level. IGFBP-3 proteolysis assays demonstrated that IGFBP-3 degradation was lower in TIMP-1–overexpressing livers, and zymography showed that matrix metalloproteinases (MMPs) were present in the liver homogenates and were capable of degrading IGFBP-3. As a consequence of reduced IGFBP-3 proteolysis and elevated IGFBP-3 protein levels, dissociable IGF-II levels were significantly lower in TIMP-1–overexpressing animals. This decrease in bioavailable IGF-II ultimately resulted in diminished IGF-I receptor signaling in vivo as evidenced by diminished receptor kinase activity and decreased tyrosine phosphorylation of the IGF-I receptor downstream effectors, insulin receptor substrate 1 (IRS-1), extracellular signal regulatory kinase (Erk)-1, and Erk-2. Together, these results provide evidence that TIMP-1 inhibits liver hyperplasia, an early event in TAg-mediated tumorigenesis, by reducing the activity of the tumor-inducing mitogen, IGF-II. These data implicate the control of MMP-mediated degradation of IGFBPs as a novel therapy for controlling IGF bioavailability in cancer.
Keywords: TIMP-1, insulin-like growth factor II, signal transduction, extracellular proteolysis, tumor suppression
Extracellular matrix (ECM)1 serves as the immediate microenvironment for interactions with the cell surface, besides providing the structural support for all tissues. The ECM is not static. Rather, it is dynamic in nature with a continuous turnover of its protein constituents and growth factor pools. A major determinant of ECM turnover and integrity is the extracellular proteolytic balance between secreted matrix metalloproteinases (MMPs) and their biological inhibitors (TIMPs) (for reviews see Matrisian 1992; Denhardt et al. 1993; Mignatti and Rifkin 1993). The function of extracellular proteolysis extends beyond ECM degradation to the processing of cell surface receptors and ligands and release of protein-bound growth factors (for review see Werb 1997). Therefore, it is conceivable that extracellular proteolytic activity within the cellular microenvironment can directly impact cell proliferation. Despite transgenic studies showing that cellular proliferation is altered by ectopic expression of MMPs or TIMPs (Sympson et al. 1994; D'Armiento et al. 1995; Witty et al. 1995; Martin et al. 1999), the question of whether TIMP/MMP-mediated proteolysis affects growth factor bioavailability in vivo to regulate cell proliferation has not been investigated.
Insulin-like growth factors (IGFs) are important mitogens in many normal and pathological systems. IGFs are essential for normal embryonic and postnatal growth (Murphy et al. 1987; Han et al. 1988), and their overexpression is frequently observed in hyperproliferative states that occur in human cancers of the prostate, breast, and liver (Osborne et al. 1989; D'Errico et al. 1994; Tennant et al. 1996). Moreover, IGF overexpression in cancer is associated with poor patient prognosis (Chan et al. 1998; Hankinson et al. 1998). IGFs are considered survival factors, and depending on cell type and situation, demonstrate either a direct proliferative effect (Schoenle et al. 1985; Osborne et al. 1989) or an antiapoptotic effect (Bozyczko-Coyne et al. 1993; Resnicoff et al. 1995). In accordance with their diverse biological activities, there is a complex regulation of IGF bioactivity. In the extracellular compartment, IGF activity is controlled by six known high affinity IGF-binding proteins (IGFBPs) which bind the IGFs with equal or higher affinity than the type I IGF receptor (IGF-IR; McCusker et al. 1991; Shimisaki and Ling 1991). The molar excess of IGFBPs, along with their higher affinity, leads to effective sequestration of IGFs by IGFBPs, resulting in little or no free IGFs in most biological systems. Nevertheless, proteolytic cleavage of IGFBPs by aspartic, serine, and metalloproteinases (Cohen et al. 1992; Conover et al., 1994; Fowlkes et al. 1994a) has been shown to release IGFs in vitro, because IGFBP fragments demonstrate significantly lower affinities for IGFs than the intact IGFBPs (Gargosky et al. 1992; Lassarre and Binoux 1994; Lassarre et al. 1994). Once released from IGFBPs, both IGF-I and IGF-II exert their mitogenic effects through IGF-IR, a protein tyrosine kinase receptor (Osborne et al. 1989; Rubin and Baserga 1995; Fowlkes 1997). Therefore, the mitogenic activity of IGFs may be regulated at the level of IGF expression, IGFBP sequestration, proteinase-mediated IGFBP proteolysis to release free IGF, or IGF-IR signaling.
We explored whether the TIMP/MMP balance alters cell proliferation by regulating IGF-II bioactivity in vivo by using a double transgenic mouse model that carries the tumor-inducing viral oncoprotein, (SV40 T antigen, [TAg]) and the tumor-suppressing TIMP-1 transgene (Martin et al. 1996). In this model, we have already shown that hepatic TIMP-1 overexpression inhibits TAg-induced hepatocyte hyperplasia and liver tumorigenesis (Martin et al. 1996, Martin et al. 1999). Since TAg transformation is unsuccessful following IGF-IR ablation (Sell et al. 1993), and TAg-induced pancreatic tumorigenesis is substantially reduced by IGF-II ablation (Christofori et al. 1994), the IGF pathway appears to be crucial for TAg oncoprotein transformation. Our double transgenic model presented an ideal opportunity to investigate the potential effects of transgenic TIMP-1 overexpression on IGF bioactivity. Here, we demonstrate that despite IGF-II reactivation during TAg-induced tumorigenesis, transgenic TIMP-1 overexpression attenuates IGF-II bioactivity. This is due to the inhibition of MMP-mediated IGFBP-3 proteolysis leading to elevated IGFBP-3 levels and reduced dissociable IGF-II levels in hepatic tissues. Reduced IGF-II bioactivity in vivo was confirmed by decreased signaling through the IGF-IR signal transduction pathway. To our knowledge, this is the first in vivo example of the modulation of growth factor bioactivity by the regulation of extracellular proteolysis.
Materials and Methods
Transgenic Mice
Transgenic mice expressing the TIMP-1 (Ts+) or TAg (TAg+) transgenes in liver were generated and bred as described previously (Martin et al. 1996). Single transgenics were crossed to generate four categories of littermates designated as wild-type controls (TAg−/Ts−), TIMP-1 controls (Ts+), TAg controls (TAg+), and double transgenic TIMP-1–overexpressing (TAg+/Ts+) mice. Female littermates were killed at specified ages, and the liver tissue was processed and embedded or flash frozen for analyses.
Immunoprecipitation and Western Blotting
Liver tissue was homogenized in lysis buffer (20 mM Tris-HCl, pH 7.4, 1.0% NP-40, 150 mM NaCl, 0.5 M PMSF, 1 mM EDTA, 10 μg/ml pepstatin, 10 μg/ml leupeptin) at 4°C. Samples were centrifuged for 10 min at 16,000 g, the supernatants collected, and the protein content determined by the Bradford assay. Aliquots containing 2.5 mg of protein were adjusted to a volume of 500 μl. TAg was immunoprecipitated by adding 0.5 μg/ml of anti-SV40 large T small t antibody (clone PAb 108; PharMingen) and 50 μl of Gamma Bind Plus Sepharose (Amersham Pharmacia Biotech), and rocking for 2 h at 4°C. Immunoprecipitates were collected by centrifugation in a refrigerated Eppendorf centrifuge and washed three times with NET/gel buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 0.1% NP-40, 1 mM EDTA, 0.25% gelatin, 0.02% sodium azide). The samples were subjected to 10% SDS-PAGE and electroblotted to Hybond N nylon membrane (Amersham Pharmacia Biotech). The membranes were sequentially probed with primary antibodies PAb 108, anti-p53 (clone PAb 421; obtained from Dr. S. Benchimol, Ontario Cancer Institute), and anti-RB (clone G3-245; PharMingen), followed by hybridization with peroxidase-conjugated anti–mouse IgG antibodies and ECL chemiluminescence (Amersham Pharmacia Biotech).
Northern Blotting
Total RNA isolated from individual liver tissue samples (20 μg) was electrophoresed in formaldehyde agarose gels, subjected to Northern blotting and sequential hybridizations with [α-32P]dCTP-labeled and random primed cDNA probes for murine IGF-II (cDNA obtained from Dr. G. Bell, University of Chicago, Chicago, IL), rat IGFBP-3 (cDNA, obtained from Dr. A. Herington, Queensland University of Technology, Brisbane, Australia), 18S ribosomal RNA, and glyceraldehyde 6-phosphate dehydrogenase (GAPDH) were performed. The latter two probes were used to control for equal loading and transfer of samples.
Immunohistochemistry and In Situ Hybridization
Paraffin sections of formalin-fixed liver tissue were generated as described previously (Martin et al. 1999). Clone PAb 108, biotinylated goat anti–mouse IgG, and streptavidin-peroxidase conjugate (Zymed) were used for TAg immunohistochemistry, and peroxidase-conjugated anti–proliferating cell nuclear antigen (PCNA) antibodies (Dako) were used to detect proliferating cells. The detection of bound antibody was carried out using diaminobenzidine (Kirkegaard & Perry), which forms a reddish-brown pigment at sites of peroxidase activity. Digoxygenin-labeled (Boehringer Mannheim) IGF-II riboprobes were synthesized using the rat IGF-II cDNA, which was cloned in Bluescript KS. In situ hybridization was performed as described previously (Martin et al. 1999). Specific signal appears as purple pigment.
Ligand Blotting
Liver samples were homogenized as for Western blotting. Equivalent amounts of protein from each sample (40 μg) were subjected to 10% SDS-PAGE followed by electroblotting to Hybond N. Membranes were blocked by incubating for 1 h in blocking buffer (10% BSA in TBS, 0.01 M Tris-HCl, pH 7.5, 0.15 M NaCl) followed by overnight hybridization with 50,000 cpm/ml 125I–IGF-II (Amersham Pharmacia Biotech) in blocking buffer. Membranes were washed three times for 15 min with TBS, air dried, then subjected to autoradiography.
IGFBP-3 Substrate Zymography
Liver samples were prepared as for Western blotting. Protein from each sample (40 μg) was subjected to SDS-PAGE in a 10% polyacrylamide gel containing 1.0 μg/ml recombinant human IGFBP-3 (kindly provided by Dr. C. Maack, Celtrix Pharmaceuticals, Santa Clara, CA). Following electrophoresis, gels were washed for 30 min with 2.5% Triton X-100. Gels were equilibrated with transfer buffer (50 mM Tris-HCl, pH 8.0, 5 mM CaCl2), then the proteins were subjected to capillary transfer onto PVDF membranes as described previously (Fowlkes et al. 1997). The transfer was performed either in the absence or presence of 10.0 μg/ml recombinant human TIMP-1 (kindly provided by Dr. H. Nagase, University of Kansas, Kansas City, KS). Transfers took place overnight at 37°C. Under these conditions, only proteolytic fragments of IGFBP-3 transfer from the substrate-gel to the membrane. Intact IGFBP-3 (30 kD) impregnated in the polyacrylamide matrix remains in the gel (Fowlkes et al. 1997). Following transfer, the PVDF membrane was probed with polyclonal antibody against IGFBP-3 (Fowlkes et al. 1997).
IGFBP-3 Degradation Assay
Liver tissue was homogenized as for Western blotting, except that NP-40 and proteinase inhibitors were not added. 50 μg of protein from each sample was brought up to a volume of 28 μl with homogenization buffer containing 50 μM CaCl2. 125I–IGFBP-3 (50,000 cpm/ml; Diagnostic Systems Laboratories) was added, and the samples were incubated for 5 h at 37°C, followed by SDS-PAGE, electroblotting, and autoradiography (Davenport et al. 1991).
Affinity Chromatography
Liver protein homogenates were prepared as above. Total levels of IGF-II were quantified according to a published protocol (De Leon and Asmerom 1997). In brief, 10 μl from each sample was dot blotted onto nitrocellulose in triplicate. The membrane was incubated in blocking buffer and then with mAb against IGF-II. The IGF-II concentrations were determined from a standard curve generated with known quantities of purified recombinant IGF-II (kindly provided by Eli Lilly and Co.). To assess the relative levels of dissociable IGF-II, equivalent quantities of total IGF-II from each sample (1.2 μg) were incubated with IGFBP-4–conjugated Affigel overnight at 4°C with gentle rocking. Samples were centrifuged for 10 min at 1,000 g and the pellets washed three times with TBS. The pellets were then incubated with 0.5 M acetic acid for 5 min to elute IGF-II bound to the immobilized IGFBP-4. Supernatants were collected and dried under vacuum. Samples were resuspended in TBS and the relative levels of dissociable IGF-II determined using the IGF-II dot blot assay. To validate that increasing the molar ratio of IGFBP-3 to IGF-II affects IGF-II binding to the IGFBP-4 Affigel affinity media, increasing amounts of recombinant IGFBP-3 were mixed with a constant amount of recombinant IGF-II and the mixtures were subjected to the IGFBP-4 chromatographic separation, as described above.
Receptor Kinase Assays
Frozen liver tissue samples were homogenized in TBS containing 1% Triton X-100, 1 mM PMSF, 100 μM Na3VO4, 1 mM EDTA, 10 μg/ml pepstatin, 10 μg/ml leupeptin. Aliquots from each supernatant containing 5.0 μg of protein were immunoprecipitated as above, using antibodies against the IGF-IR (clone aIR3; Calbiochem). Immunoprecipitates were then incubated with kinase reaction buffer (10 mM Hepes, pH 7.4, 5 mM MnCl2, 5 mM MgCl2, 100 μM Na3VO4, 50 mM NaF) containing 2.0 μCi/μl [γ-32P]ATP either with or without 50 μg Poly (Glu, Tyr) 4:1 (Sigma Chemical Co.) for 30 min at 37°C. Samples were electrophoresed on 10% SDS-PAGE gels, dried onto Whatman 3 MM paper, and autoradiographed.
Phosphorylation Levels
Liver proteins were isolated as for the receptor kinase assays. For insulin receptor substrate (IRS)-1, 2.5 mg of protein from each sample was immunoprecipitated with polyclonal antibodies against rat IRS-1 (Upstate Biotechnology) as described above. The immunoprecipitates were then Western blotted and the membranes probed sequentially with antiphosphotyrosine (clone 4G10; Upstate Biotechnology) and anti–IRS-1 antibodies. For mitogen-activated protein kinase (MAPK), replicate 10% SDS-PAGE gels were run with 40 μg of protein from each sample loaded on each gel. Following electroblotting, membranes were probed with antibodies specific for phospho-MAPK or MAPK (New England Biolabs).
Densitometry and Quantification
Autoradiographs were scanned using a Molecular Dynamics Densitometer. Absorbance was quantified using ImageQuant® software. Statistical significance was determined using t test. For all gel electrophoresis, each lane corresponds to a tissue sample taken from an individual mouse. All samples were obtained from 185-d-old female mice unless indicated otherwise, as we determined previously that TIMP-1 modulation significantly affected TAg-induced preneoplastic proliferation at this age (Martin et al. 1999).
Results
TAg Antigen Molecular Interactions Are Maintained in TIMP-1–overexpressing Mice
We have shown previously that transgenic TIMP-1 expression does not affect TAg oncoprotein levels in double transgenic mice that coexpress TAg and TIMP-1 transgenes (TAg+/Ts+) (Martin et al. 1996). Equivalent TAg protein levels in both TAg+ and TAg+/Ts+ animals are confirmed here in Fig. 1 a (top panel). Since TAg binds to and inactivates the tumor suppressor gene products p53 and Rb to induce hyperplasia (Tan et al. 1986; DeCaprio et al. 1988), we investigated whether TAg interactions with these tumor suppressor proteins are altered by TIMP-1 overexpression. The amount of p53 and Rb that coimmunoprecipitated with TAg protein was examined by Western blotting and was found to be similar between TAg+ and TIMP-1–overexpressing (TAg+/Ts+) livers, as shown in Fig. 1 a (middle and bottom panels). This indicates that TAg interactions with p53 and Rb proteins are intact, and unaffected by the elevation of TIMP-1.
Figure 1.

Reactivation of IGF-II in TAg+ and TAg+/Ts+ livers. (a) Western blots showed equivalent TAg levels in anti-TAg immunoprecipitates from the livers of 185-d-old TAg+ and TAg+/Ts+ littermates. Sequential probing of the blot showed that the levels of p53 and pRB that coimmunoprecipitated with TAg were similar in TAg+ and TAg+/Ts+ livers. (b) Northern blot analysis of IGF-II mRNA levels in the livers of TAg+ and TAg+/Ts+ transgenic littermates. IGF-II mRNA was undetectable in livers of non-TAg expressing littermates, but was reactivated between 153 and 191 d of age in TAg-expressing littermates. (c) IGF-II mRNA levels were the same in 185-d-old TAg+ and TAg+/Ts+ littermates. RNA was isolated from livers from mice of the indicated age and genotype (wt, wild-type; Ts+, TIMP-1 sense transgene–expressing; Ta+, TIMP-1 antisense transgene–expressing; TAg+, TAg transgene–expressing; TAg+/Ts+, double transgenic TIMP-1–overexpressing). 18S ribosomal RNA and GAPDH provided a means of determining RNA loading on the Northern blots.
Cellular Proliferation Is Inhibited by Transgenic TIMP-1 Despite IGF-II Reactivation
IGF-II is a fetal mitogen in rodents, and its transcription is normally repressed in adult tissues by p53 (Zhang et al. 1996). However, focal reactivation of IGF-II and its localization to proliferating cells during TAg-induced tumorigenesis have been reported in two independent transgenic tumor models (Schirmacher et al. 1992; Christofori et al. 1994). Cellular proliferation and tumor development have been shown to be profoundly inhibited in one such model when crossed onto an IGF-II–null background (Christofori et al. 1994), revealing the fundamental importance of IGF-II reactivation in TAg-induced tumorigenesis. IGF-II reactivation has been frequently observed during TAg-induced hepatocarcinogenesis (Casola et al. 1995; Haddad and Held 1997). To investigate whether IGF-II was reactivated in our hepatocellular carcinoma model, we examined its spatiotemporal expression in the livers of experimental and control littermates. IGF-II mRNA was not detected by Northern blot analysis of liver tissue from wild-type mice of all ages, or in the livers of TAg+ mice before 165 d of age. However, from 170 d of age, multiple IGF-II transcripts commonly observed in mouse and human tissues (Daughaday and Rotwein 1989; Casola et al. 1995) were expressed (Fig. 1 b, and data not shown). Fig. 1 c shows that IGF-II mRNA was expressed in liver tissue from TAg+ and TAg+/Ts+ littermates at 185 d of age. Densitometric analysis of the major IGF-II transcript (4.2 kb) confirmed that expression was comparable in TAg+ and TAg+/Ts+ littermates (3.3 ± 1.2 vs. 3.8 ± 0.6, n = 5 per group). The timing of IGF-II reactivation coincided with TAg-induced liver enlargement, and remained unaffected by TIMP-1 overexpression. In contrast to IGF-II mRNA reactivation, IGF-I mRNA expression remained very low and unchanged during all stages of TAg tumorigenesis (data not shown).
The liver-specific C reactive protein promoter, which directs TAg expression (Rüther et al. 1993), resulted in uniform TAg expression in almost all hepatocytes by 185 d of age, as shown by immunohistochemistry (Fig. 2 a). In adjacent sections, in situ hybridization revealed IGF-II mRNA production by most hepatocytes (Fig. 2 b), and extensive hepatocytic hyperplasia was detected by PCNA immunostaining (Fig. 2 c). The liver of a 185-d-old double transgenic TIMP-1–overexpressing (TAg+/Ts+) littermate demonstrated the same ubiquitous expression patterns of both the TAg oncoprotein (Fig. 2 d) and IGF-II mRNA (Fig. 2 e). In contrast, probing the adjacent sections with anti-PCNA antibody revealed far less proliferation in the livers of TAg+/Ts+ mice (Fig. 2 f) compared with TAg+ mice. In our previous study using morphometric analysis, we found this proliferation to be significantly suppressed in TIMP-1 transgenic liver tissue (Martin et al. 1999). Together, the results demonstrate that the effects of TIMP-1 are exerted downstream of IGF-II reactivation, but before cell proliferation.
Figure 2.
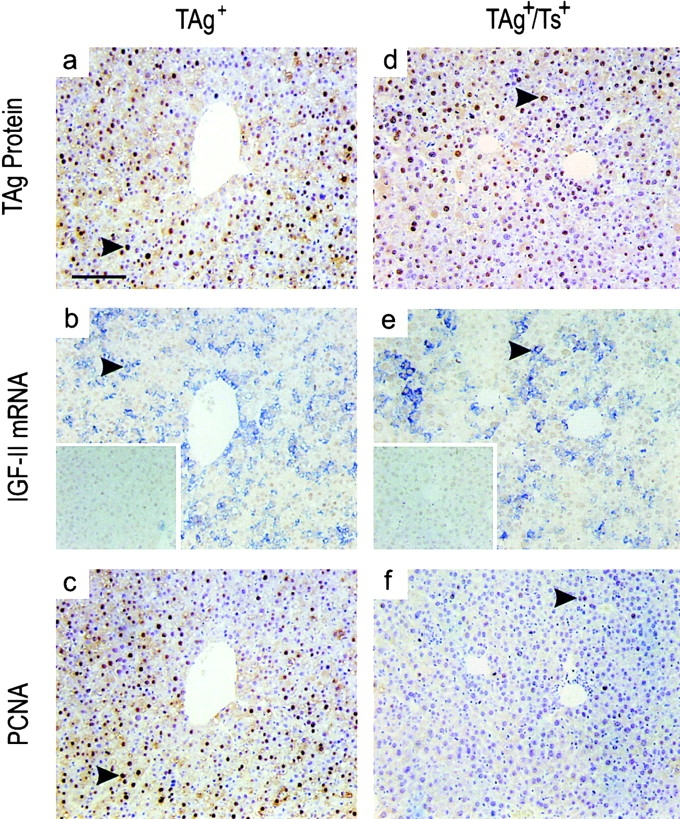
Reduced hepatocyte proliferation in TIMP-1–overexpressing (TAg+/Ts+) livers. The TAg oncoprotein, IGF-II mRNA, and proliferating hepatocytes were localized on adjacent sections of liver from 185-d-old TAg+ and TAg+/Ts+ littermates. (a and d) Immunohistochemical localization showed the same consistent, intense expression of TAg throughout the livers of both TAg+ and TAg+/Ts+ mice. (b and e) Reactivated IGF-II expression also occurred throughout the liver of both TAg+ and TAg+/Ts+ animals, as seen by in situ hybridization. Inset, sense–IGF-II riboprobes. (c and f) In contrast to the equivalent TAg protein and IGFII mRNA levels in TAg+ and TAg+/Ts+ mice, the lower level of PCNA immunostaining in TAg+/Ts+ livers showed that there were fewer proliferating hepatocytes in TIMP-1–overexpressing mice. The arrowheads indicate specific signal in each panel. Bar, 100 μm.
For further analyses of the effects of TIMP-1 on IGF-II bioactivity, we chose to use liver specimens from 185-d-old mice. This choice was based on our previous pilot study encompassing 20–250 d, in which we found that hepatocyte proliferation was maximal (>65%) in TAg+ mice at 185 d of age, and was inhibited 3.3-fold in TAg+/Ts+ littermates at this age (Martin et al. 1999). The difference in proliferation was most accentuated at this age, and therefore we anticipated that in vivo analysis of molecular factors in the IGF-II signaling pathway would be most clearly resolved at this point in time. The above observations that IGF-II is reactivated in both TAg+ and TAg+/Ts+ livers (Fig. 1 b and Fig. 2b and Fig. e), yet hepatocyte proliferation is only prevalent in TAg+ tissue at 185 d (Fig. 2c and Fig. f), further supported the use of this age group for IGF-II bioactivity studies.
IGFBP-3 Levels Are Elevated Due to Reduced Proteolysis
IGFPBs regulate IGF activity by sequestering free IGFs, thus preventing ligand–receptor interactions. Previous studies from one of our laboratories have demonstrated that MMPs, the primary proteinases inhibited by TIMP-1, can degrade IGFBPs both in vitro and in vivo (Fowlkes et al. 1994a,Fowlkes et al. 1994b; Thrailkill et al. 1995; Fowlkes 1997). Since the amount of high-affinity, intact IGFBP may be regulated in part by MMP-mediated proteolysis, we compared the levels of intact hepatic IGFBPs using 125I–IGF-II Western ligand blotting. Of the hepatic IGFBPs that bound 125I–IGF-II on ligand blots, only IGFBP-3 (42–46-kD doublet) levels were strongly affected by TIMP-1 modulation (Fig. 3 a). The levels of this binding protein, which is also the major serum carrier protein for IGFs (Jones and Clemmons 1995), were increased by more than twofold in the livers of TIMP-1–overexpressing mice (TAg+/Ts+) compared with levels in TAg+ littermates (Fig. 3 d, left panel; P < 0.02). TIMP-1 overexpression did not affect the levels of IGFBP-4 (24–26 kD), although there were notable minor elevations of a doublet of proteins at 28–32 kD that, based on molecular mass, likely represent IGFBP-1, -2, and/or -5 (Fig. 3 a).
Figure 3.
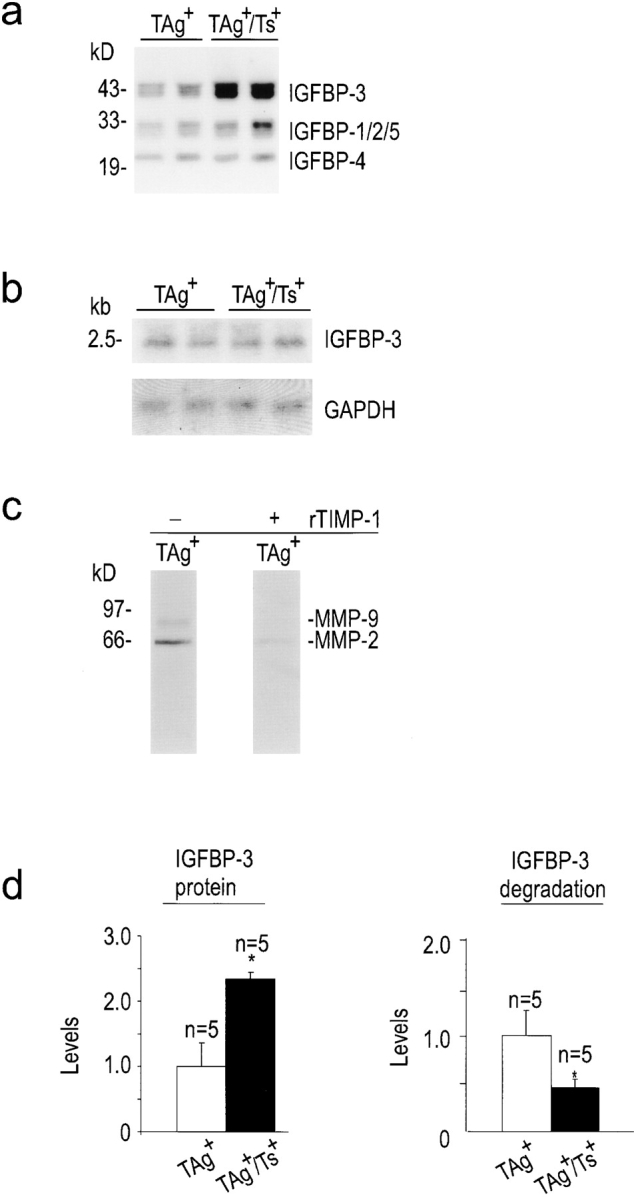
Increased protein levels of IGFBP-3 and reduced IGFBP-3 degradation in the livers of TIMP-1–overexpressing (TAg+/Ts+) mice. (a) Ligand blot showing an increase in hepatic IGFBP-3 in TAg+/Ts+ compared with TAg+ mice. (b) Northern analysis showed that hepatic IGFBP-3 mRNA levels were the same in TAg+ and TAg+/Ts+ littermates. (c) IGFBP-3 substrate zymography showed the presence of proteinases in liver homogenates that were capable of degrading IGFBP-3 and identified them as MMPs, possibly MMP-2 and MMP-9, since they were inhibited by recombinant TIMP-1. (d) Bar graphs of IGFBP-3 protein levels and IGFBP-3 degradation showed that a twofold reduction in IGFBP-3 degradation resulted in a twofold increase in IGFBP-3 levels. The mean absorbance values for positive controls were assigned a value of 1.0. Bars, SD. Asterisk indicates statistical significance in a t test of P < 0.02.
In contrast to IGFBP-3 protein levels, Northern blot analysis showed that TIMP-1 overexpression did not affect the levels of IGFBP-3 mRNA (Fig. 3 b), attributing the differences in IGFBP-3 protein levels to posttranscriptional events. MMP-1, -2, -3, and -9 have been shown to proteolytically cleave IGFBP-2, -3, and -5, a process inhibited by TIMP-1 in vitro (Fowlkes et al. 1994a,Fowlkes et al. 1994b; Thrailkill et al. 1995; Fowlkes 1997). Here, IGFBP-3 substrate zymography (Fowlkes et al. 1997) was used to identify IGFBP-3–degrading proteases in liver homogenates. We found two IGFBP-3–degrading activities with molecular masses of ∼62 and ∼84 kD (Fig. 3 c). Both activities were substantially reduced in the presence of recombinant TIMP-1 (Fig. 3 c), demonstrating that the IGFBP-3–degrading proteinases were MMPs. Next, we determined whether a decreased proteolysis of IGFBP-3 is evident in liver tissue obtained from TIMP-1–overexpressing mice. Liver homogenates were analyzed for their ability to degrade 125I–IGFBP-3 into smaller molecular weight species, as described in Materials and Methods. There was significantly less degradation of 125I–IGFBP-3 by liver homogenates from TIMP-1–overexpressing (TAg+/Ts+) mice compared with TAg+ littermates (Fig. 3 d, right panel; P < 0.02). These data suggest that TAg can induce MMPs that are capable of degrading IGFBP-3, and that coexpression of TIMP-1 can reduce MMP activity, thereby inhibiting IGFBP-3 degradation. Together, these actions allow for a net increase in tissue IGFBP-3 protein levels.
Dissociable IGF-II Levels Are Reduced by Transgenic TIMP-1
We determined whether the increased IGFBP-3 levels in the TIMP-1–overexpressing liver tissue affected the amount of dissociable IGF-II. Fig. 4 a shows that using a dot blot procedure developed to quantify total IGF-II (bound and unbound; De Leon and Asmerom 1997), recombinant IGF-II can be measured linearly over a range of concentrations (0.4–3.2 μg). Next, dissociable IGF-II was measured by IGFBP-4–conjugated Affigel affinity chromatography. Using recombinant IGF-II and recombinant IGFBP-3 in different molar ratios, we were able to confirm that increasing molar ratios of IGFBP-3 reduced the amount of IGF-II that bound to the Affigel (Fig. 4 b). To measure difference in levels of dissociable IGF-II in liver samples, we first quantified total IGF-II using the dot blot assay. Aliquots containing equivalent amounts of total IGF-II were subjected to the affinity chromatography procedure. TIMP-1 overexpression resulted in a sixfold decrease in dissociable IGF-II levels in the livers of TAg+/Ts+ mice compared with their control TAg+ littermates (Fig. 4 c; P < 0.02). This demonstrates that despite an equivalent extent of IGF-II reactivation, the level of dissociable or bioavailable IGF-II is reduced in TIMP-1–overexpressing animals.
Figure 4.
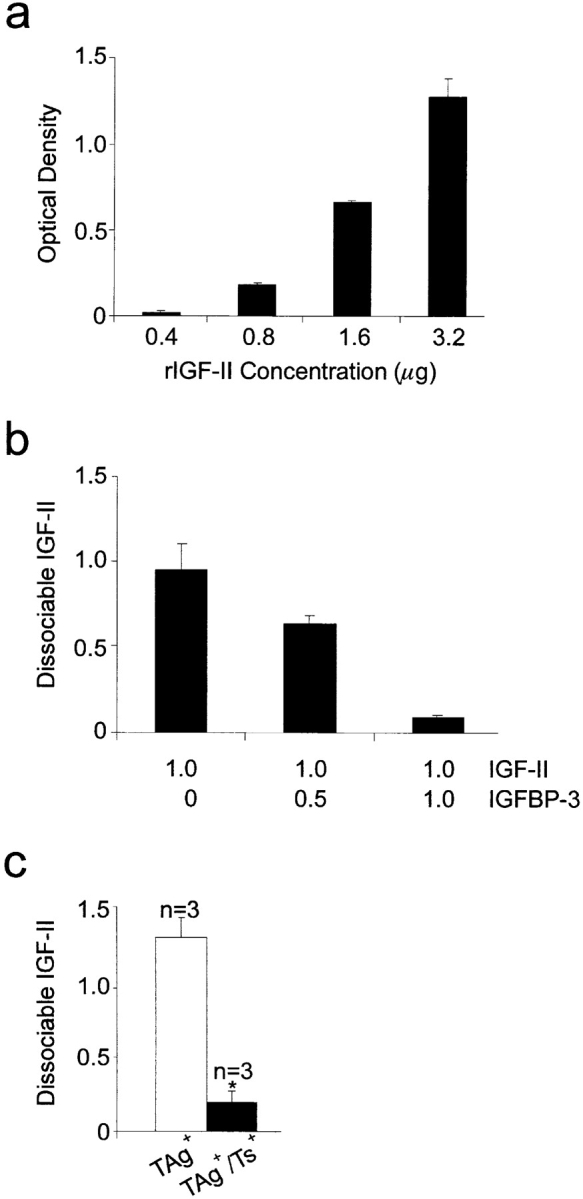
Reduced dissociable IGF-II in the livers of TIMP-1–overexpressing (TAg+/Ts+) mice. (a) Bar graph showing the optical density of dot blotted samples containing known concentrations of recombinant IGF-II (rIGF-II). (b) Bar graph showing the impact of increasing molar ratios of IGFBP-3 to IGF-II on levels of dissociable IGF-II using IGFBP-4 Affigel chromatography. (c) Bar graph showing the relative levels of dissociable IGF-II in liver tissue extracts from TAg+ and TAg+/Ts+ mice. n, number of mice. For all graphs, samples were dot blotted in triplicate. Bars, SD.
Reduced Signaling through the IGF-IR
A reduction in the levels of dissociable or bioavailable IGF-II should result in decreased signaling through the IGF-IR pathway in the livers of TAg+/Ts+ mice. Unlike postnatal IGF-II inactivation that occurs in normal rodent liver, the IGF-IR is expressed at constitutive levels in adult mouse liver, and IGF-II exerts its mitogenic effect through this receptor (Osborne et al. 1989; Rubin and Baserga 1995). Therefore, IGF-IR kinase activity, as well as the phosphorylation status of the downstream signaling effectors, IRS-1 and MAPK, were assessed. IGF-IR immunoprecipitated from TAg+/Ts+ liver tissue exhibited lower autophosphorylation (data not shown) and showed reduced kinase activity on an exogenous substrate (Fig. 5 a), compared with receptor from TAg+ controls. Moreover, tyrosine phosphorylation of IRS-1, which binds to IGF-IR following activation, was also reduced in TIMP-1–overexpressing livers (Fig. 5 b, top panel). Sequential probing of the same blot with an antibody against nonphosphorylated IRS-1 showed that IRS-1 protein levels were not altered in TIMP-1–overexpressing animals (Fig. 5 b, bottom panel). Phosphorylation of the downstream signaling molecules, the MAPKs extracellular signal regulatory kinase (Erk)-1 and Erk-2, was also reduced in TIMP-1–overexpressing liver tissue (Fig. 5 c, top panel), whereas the absolute levels of these proteins were unaffected (Fig. 5 c, bottom panel). These data provide direct evidence that the protein levels of the IGF-IR downstream signaling mediators were not altered, but that signaling from the IGF-IR was attenuated in TIMP-1–overexpressing transgenic tissue.
Figure 5.

Reduced signaling through the IGF-IR in TIMP-1–overexpressing (TAg+/Ts+) livers. (a) Receptor kinase assay showed greatly decreased kinase activity of IGF-IR immunoprecipitated from TAg+/Ts+ compared with TAg+ liver. (b and c) Downstream of the IGF-IR, IRS-1 and Erk-1 and -2 had greatly reduced levels of phosphotyrosine, but equivalent protein levels in double transgenic TAg+/Ts+ livers compared with TAg+ littermates. Quantification of the phosphotyrosine and specific protein signal intensities illustrate the extent of decrease of IRS-1 and Erk-1/2 tyrosine phosphorylation in TAg+/Ts+ livers. Asterisk indicates statistical significance (P < 0.05).
Discussion
Regulation of the IGF-II Pathway by TIMP-1
IGFs are critical growth factors involved in growth, transformation, and tumorigenesis and act through the IGF-IR (for reviews see Jones and Clemmons 1995; Rubin and Baserga 1995, Werner and Le Roith 1997). Studies have shown that cells null for the IGF-IR do not display the normal increase in proliferation in response to growth factors or serum as seen in normal cells, and that all phases of the growth cycle are prolonged (for review see Resnicoff and Baserga 1998). Indeed, in several cell lines, abrogation of the IGF-IR has resulted in enhanced apoptosis. Furthermore, IGF-IR–null cells cannot be transformed by TAg (Sell et al. 1993), activated Ha-ras, a combination of both, or by the overexpression of other growth factor receptors. And finally, a number of studies have shown that ablation of this receptor in tumor cell lines significantly decreases their tumorigenic potential in vivo. Together, these data demonstrate that interruption of the ligand–receptor interaction between IGFs and the IGF-IR effectively disrupts several aspects of the tumorigenic process.
In nature, under homeostatic circumstances, little or no IGFs are present in the free or bioavailable form due to their sequestration by one or more of the six known high-affinity IGFBPs. Because IGFBPs demonstrate equal or higher affinities for IGFs than does the IGF-IR, little or no IGFs are normally available to interact with receptors. Recent studies have begun to elucidate mechanisms by which IGFs can be released from IGFBPs so they may interact with cell-surface receptors and exert their mitogenic and metabolic effects. These studies have demonstrated that a primary phenomenon invoked to release IGFs from IGFBPs is through decreasing the affinities of IGFBPs for IGFs. The best-characterized mechanism involved in decreasing the affinities of IGFBPs is proteolytic degradation (for review see Fowlkes 1997).
Proteolytic cleavage has been demonstrated for at least five IGFBPs, IGFBP-2 to IGFBP-6, and occurs in several physiologic as well as pathologic circumstances (Jones and Clemmons 1995; Fowlkes 1997), yet little is known of the identity of these IGFBP-degrading proteinases in vivo, or the mechanisms that regulate the proteolytic cleavage. In vitro, we and others have provided evidence that production of proteinases, a common feature of transformed cells (Alexander and Werb 1989), can result in degradation of IGFBP–IGF complexes, releasing IGFs to interact with cell surface receptors, thereby triggering proliferation of target cells. We have also shown recently that in vitro, MMPs function as IGFBP-3– and IGFBP-5–degrading proteinases (Fowlkes et al. 1994a,Fowlkes et al. 1994b; Thrailkill et al. 1995). In addition to degrading IGFBPs, extensive data also support the role of MMPs in tumorigenesis, angiogenesis, and metastasis (Alexander and Werb 1989; Flaumenhaft and Rifkin 1991; Liotta et al. 1991; Werb 1997). In the current studies, we now demonstrate the importance of MMP-mediated IGFBP degradation in neoplastic proliferation in vivo, and a means of controlling this degradation.
We have used a double transgenic, TAg-based tumor model to determine directly whether TIMP-1 inhibits IGF bioactivity in vivo to suppress hepatocyte proliferation that leads to tumorigenesis. We selected this model as it provided important features: (a) IGFs are crucial mitogens for TAg-induced transformation, proliferation, and tumorigenesis (Schirmacher et al. 1992; Sell et al. 1993; Christofori et al. 1994; Casola et al. 1995; Haddad and Held 1997); and (b) transgenic TIMP-1 overexpression in this model substantially inhibits TAg-induced proliferation and hepatocellular carcinoma (Martin et al. 1996, Martin et al. 1999), making it possible to measure in vivo differences in molecular factors and IGF-IR signal transduction in transgenic tissue. A systematic analysis has now been undertaken to explore connections that might exist between TAg expression and IGF-II bioactivity on cell proliferation to define the molecular mechanisms behind TIMP-1–mediated tumor suppression in vivo. The events explored herein and the results are summarized in Fig. 6, and are discussed below.
Figure 6.

The molecular mechanism underlying TIMP-1–mediated suppression of hepatocarcinogenesis. The schematic highlights the molecular events that were examined here to uncover the mechanism of TIMP-1–mediated inhibition of hepatocyte proliferation. The primary event in the pathway between TAg-induced IGF-II reactivation and hepatocyte proliferation affected by TIMP-1 overexpression was an increase in IGFBP-3 levels, due to the inhibition of MMP-mediated proteolysis of IGFBP-3 by TIMP-1. The direct result of increased IGFBP-3 levels was decreased dissociable IGF-II levels concomitant with decreased signaling through the IGF-IR and decreased hepatocyte proliferation.
First, an examination of p53 and Rb in these mice showed that TIMP did not interfere with the molecular interactions between TAg and these tumor suppressor proteins. Next, we found that IGF-II expression was indeed reactivated at the onset of cell proliferation, similar to the model of TAg-induced pancreatic tumorigenesis (Christofori et al. 1994), indicating a key mitogenic role for IGF-II in our tumor model. IGF-II reactivation is frequent in TAg-induced liver tumorigenesis (Casola et al. 1995; Haddad and Held 1997), as well as in liver tumor formation induced by oncogene or growth factors (Liu et al. 1997; Harris et al. 1998). Furthermore, our data showing that TIMP-1 significantly inhibited hepatocellular proliferation despite IGF-II reactivation suggested that TIMP-1 might directly act at a posttranscriptional level to modulate IGF-II activity. Our investigations reveal the novel finding that hepatic TIMP-1 overexpression specifically inhibits IGFBP-3 proteolysis, leading to a significant elevation of hepatic IGFBP-3 levels. We have determined that MMPs induced during TAg tumorigenesis (Martin et al. 1999) appear to function in degrading IGFBP-3. Furthermore, we demonstrate that dissociable IGF-II levels are decreased in TIMP-1–overexpressing hepatic tissue. The physiologic consequence of reducing dissociable IGF-II levels (e.g., a reduction in IGF-II bioactivity) was confirmed by demonstrating a significantly reduced signal transduction from the IGF-IR, as measured by reduced IGF-IR kinase activity and tyrosine phosphorylation levels of IRS-1 and MAPK. Thus, despite TAg-induced reactivation of IGF-II in the liver, the transgenic TIMP-1–mediated increase of IGFBP-3 levels blocks TAg-induced hepatocyte proliferation by effectively reducing bioavailable IGF-II levels. Together, our data provide direct evidence that the inhibition of extracellular proteolysis by TIMP-1 attenuates the bioactivity of the tumor-inducing growth factor IGF-II.
Transgenic TIMP or MMP Modulation and Effects on Early Tumor Development
Based on previous studies in transgenic systems, a relationship has begun to emerge between TIMP/MMP expression within a tissue and the tissue's susceptibility to tumor development. We have demonstrated that overexpression of TIMP-1 in the liver inhibits hepatocellular carcinoma (Martin et al. 1996), and its elevation in the skin compromises the ability of transplanted lymphoma cells to grow as a primary tumor (Krüger et al. 1997), whereas a reduction of TIMP-1 in these tissues augments tumor development (Martin et al. 1996; Krüger et al. 1997). Consistent with these observations, the overexpression of MMP-3 (stromelysin-1) in mammary tissue leads to spontaneous mammary tumor development (Sympson et al. 1995) and the transgenic expression of MMP-I (type I collagenase) in skin augments carcinogenesis (D'Armiento et al. 1995). In addition, the ablation of MMP-7 (matrilysin) impairs colorectal tumor development in the min mouse tumor model (Wilson et al. 1997). In many of these studies, ectopic TIMP or MMP expression results in altered cellular proliferation. For example, hepatic TIMP-1 overexpression inhibits hepatocyte hyperplasia (Martin et al. 1999), MMP-3 overexpression leads to mammary epithelial hyperplasia (Sympson et al. 1994; Witty et al. 1995), and MMP-1 overexpression to epidermal hyperproliferation (D'Armiento et al. 1995). Despite the many reports indicating that shifts in the extracellular proteolytic balance have a strong influence on early tumor development and on cell proliferation within the afflicted organ, the molecular mechanisms for these effects have remained elusive. Having previously excluded the effects of TIMP-1 on hepatocyte apoptosis (Martin et al. 1999), our present investigation provides in vivo evidence of a link between the inhibition of extracellular proteolysis, reduced growth factor bioavailability, and reduced cellular proliferation. We have shown previously that this reduction in cellular proliferation precedes suppressed tumor development in this model (Martin et al. 1996).
MMP Substrates of Importance other than ECM during Tumorigenesis
TIMPs have traditionally been considered regulators of cell invasion and motility by virtue of their ability to inhibit MMP-mediated ECM degradation. Now it is recognized that in addition to the effects on ECM structural proteins, extracellular proteolysis has the potential to control the release of growth factors tethered to the ECM (Dallas et al. 1995; Whitelock et al. 1996), the processing of soluble growth factor binding proteins, i.e., IGFBPs (Fowlkes 1997), and the processing of cell surface molecules such as membrane-bound TNF-α, the Notch receptor in Drosophila (Blobel 1997), and the FGF receptor 1 (Levi et al. 1996). Although these and other studies (Kimura et al. 1998) support the overall concept that extracellular proteolysis can broadly influence cell proliferation, behavior, and fate, the observations were made in in vitro systems. To our knowledge, our data are the first in vivo evidence to substantiate the importance of proteolytic degradation of a tethering or binding/carrier protein for a growth factor in the pathological development of cancer, and of its regulation by the natural inhibitor, TIMP-1. Our data demonstrate that the dynamics of IGF-II bioactivity can be altered in vivo by proteolytic modulation, and emphasize that factors other than ECM proteins constitute important physiological targets of MMP-mediated cleavage within the extracellular microenvironment.
Role of IGF-II in Tumorigenesis and Hepatocarcinogenesis
The importance of IGFs in cellular transformation has been demonstrated by findings that cells which do not express IGF-IR cannot be transformed by any of a number of dominant oncogenes (for review see Werner and Le Roith 1997). Indeed, TAg was unable to transform IGF-IR–null fibroblasts, despite the presence of various growth factors present in serum (Sell et al. 1993). Thus, IGF-IR is necessary for TAg-mediated transformation to take place. Consistent with these findings, TAg expression has been shown to reactivate IGF-II gene transcription in vivo. IGF-II reactivation occurs in pancreatic beta cells transgenically expressing TAg (Christofori et al. 1994), and here we show that IGF-II reactivation occurs in hepatocytes expressing TAg transgene coincident with the onset of hepatocyte hyperplasia. Similar to our finding, other investigators have reported that TAg expression in the liver tissue coincides with reactivation of IGF-II gene transcription and this reactivation has been associated with neoplastic transformation (Casola et al. 1995; Haddad and Held 1997). Such reactivation of IGF-II has also been reported during c-myc– and TGF-α–induced hepatocellular carcinomas in transgenic mice (Liu et al. 1997; Harris et al. 1998). The consequence of IGF-II reactivation as it relates to neoplastic transformation has only been addressed recently in TAg models of pancreatic cancer and hepatocellular carcinoma. In the former study, the investigators bred TAg transgenics into an IGF-II–null background (Christofori et al. 1994), whereas in the latter TAg transgenics were bred into an Igf2 (+/−) background (Haddad and Held 1997). In both instances, tumor size and incidence were decreased, strongly suggesting a role for IGF-II in the pathogenesis of these two tumor types. Furthermore, several transgenic lines overexpressing IGF-II demonstrate that IGF-II is associated with tumor formation, including mammary tumors, lymphomas, and hepatocellular carcinomas (Rogler et al. 1994; Bates et al. 1995; for review see Wolf et al. 1998). Although these studies provide compelling support for IGF-II as a causal mitogen in the tumorigenesis evidenced in the current animal model, final resolution of this hypothesis must await studies examining our findings in models that are either null or upregulated for IGF-II. Such studies are currently being pursued in our laboratories.
Significance of Regulated IGF Bioavailability in Clinical Medicine
Both IGF-I and IGF-II are widely implicated in promoting several human cancers, including liver, prostate, and breast cancer, and their expression correlates with poorer prognosis (Osborne et al. 1989; Tennant et al. 1996; Werner and LeRoith 1996; Sohda et al. 1997; Chan et al. 1998; Hankinson et al. 1998). Furthermore, in transgenic models, IGF-I and IGF-II are causally implicated in tumorigenesis (Rogler et al. 1994; Bates et al. 1995; Hadsell et al. 1996; for review see Wolf et al. 1998). These studies point to IGF bioactivity as a target for cancer therapeutics. Our results indicate that TIMP-1–like biomolecules or synthetic MMP inhibitors may be promising candidates for the therapeutic modulation of IGF dosage in novel clinical strategies. Alternatively, strategies to alter specific IGFBP levels or the production of proteinase-resistant IGFBPs (Chernausek et al. 1995; Conover et al. 1995; Imai et al. 1998; Rees et al. 1998) may prove to be effective therapeutic interventions. A distinct feature of all of these approaches will be to target the bioavailability rather than the production of a growth factor.
The results presented here provide compelling evidence for a novel mechanism by which endogenous TIMPs contribute to the cellular microenvironment. We demonstrate that the inhibition of extracellular proteolysis in vivo impairs the activity of a specific growth factor responsible for hyperplasia during TAg-induced tumorigenesis. Because TIMPs are also capable of inhibiting invasion, metastasis, and angiogenesis (Khokha et al. 1989; DeClerck et al. 1992; Khokha 1994; Anand-Apte et al. 1997; Krüger et al. 1997, Krüger et al. 1998; Wang et al. 1997; Martin et al. 1999), all of which are promoted by IGF action (Bae et al. 1998; Dunn et al. 1998), the combined outcome of TIMP-1 elevation may be to suppress multiple stages of tumor development, maintenance, and progression.
Acknowledgments
We thank Dr. P. Waterhouse for constructive comments throughout this work and for critical reading of the manuscript, and Mr. A.T.-V. Ho for tremendous help with the artwork.
This work was supported by funding from the National Cancer Institute of Canada, Medical Research Council of Canada, and Human Frontiers of Science Program to R. Khokha, and partially supported by National Institutes of Health grant DK02776 to J.L. Fowlkes. D.C. Martin was supported in part by an Ontario Graduate Scholarship.
Footnotes
1.used in this paper: ECM, extracellular matrix; Erk, extracellular signal regulatory kinase; GAPDH, glyceraldehyde 6-phosphate dehydrogenase; MMP, matrix metalloproteinase; IGF, insulin-like growth factor; IGFBP, IGF-binding protein; IGF-IR, type I IGF receptor; IRS-1, insulin receptor substrate 1; MAPK, mitogen-activated protein kinase; PCNA, proliferating cell nuclear antigen; TAg, SV40 T antigen; TIMP, tissue inhibitor of metalloproteinase
References
- Alexander C.M., Werb Z. Proteinases and extracellular matrix remodeling. Curr. Opin. Cell Biol. 1989;1:974–982. doi: 10.1016/0955-0674(89)90068-9. [DOI] [PubMed] [Google Scholar]
- Anand-Apte B., Pepper M.S., Voest E., Montesano R., Olsen B., Murphy G., Apte S.S., Zetter B. Inhibition of angiogenesis by tissue inhibitor of metalloproteinase-3. Invest. Opthalmol. Vis. Sci. 1997;38:817–823. [PubMed] [Google Scholar]
- Bae M.H., Lee M.J., Bae S.K., Lee O.H., Lee Y.M., Park B.C., Kim K.W. Insulin-like growth factor II (IGF-II) secreted from HepG2 human hepatocellular carcinoma cells shows angiogenic activity. Cancer Lett. 1998;128:41–46. doi: 10.1016/s0304-3835(98)00044-5. [DOI] [PubMed] [Google Scholar]
- Bates P., Fisher R., Ward L., Richardson L., Hill D.J., Graham C.F. Mammary cancer in transgenic mice expressing insulin-like growth factor II (IGF-II) Br. J. Cancer. 1995;72:1189–1193. doi: 10.1038/bjc.1995.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel C.P. Metalloprotease-disintegrinslinks to cell adhesion and cleavage of TNF alpha and Notch. Cell. 1997;90:589–592. doi: 10.1016/s0092-8674(00)80519-x. [DOI] [PubMed] [Google Scholar]
- Bozyczko-Coyne D., Glickman M.A., Pratner J.E., McKenna B., Conners T., Friedman C. IGF-I supports the survival and/or differentiation of multiple types of central nervous system neurons. Ann. N.Y. Acad. Sci. 1993;692:311–313. doi: 10.1111/j.1749-6632.1993.tb26244.x. [DOI] [PubMed] [Google Scholar]
- Casola S., Ungaro P., Pedone P.V., Lazzaro D., Fattori E., Ciliberto G., Zarrilli R., Bruni C.B., Riccio A. Loss of heterozygosity of imprinted genes in SV40t/T antigen-induced hepatocellular carcinomas. Oncogene. 1995;11:711–721. [PubMed] [Google Scholar]
- Chan J.M., Stampfer M.J., Giovannucci E., Gann P.H., Ma J., Wilkinson P., Hennekens C.H., Pollak M. Plasma insulin-like growth factor-I and prostate cancer riska prospective study. Science. 1998;279:563–566. doi: 10.1126/science.279.5350.563. [DOI] [PubMed] [Google Scholar]
- Chernausek S.D., Smith C.E., Duffin K.L., Busby W.H., Wright G., Clemmons D.R. Proteolytic cleavage of insulin-like growth factor binding protein 4 (IGFBP-4). Localization of cleavage site to non-homologous region of native IGFBP-4. J. Biol. Chem. 1995;270:11377–11382. doi: 10.1074/jbc.270.19.11377. [DOI] [PubMed] [Google Scholar]
- Christofori G., Naik P., Hanahan D. A second signal supplied by insulin-like growth factor II in oncogene-induced tumorigenesis. Nature. 1994;369:414–418. doi: 10.1038/369414a0. [DOI] [PubMed] [Google Scholar]
- Cohen P., Graves H.C., Peehl D.M., Kamarei M., Giudice L.C., Rosenfeld R.G. Prostate-specific antigen (PSA) is an insulin-like growth factor binding protein-3 protease found in seminal plasma. J. Clin. Endocrinol. Metab. 1992;75:1046–1053. doi: 10.1210/jcem.75.4.1383255. [DOI] [PubMed] [Google Scholar]
- Conover C.A., De Leon D.D. Acid-activated insulin-like growth factor-binding protein-3 proteolysis in normal and transformed cells. Role of cathepsin D. J. Biol. Chem. 1994;269:7076–7080. [PubMed] [Google Scholar]
- Conover C.A., Durham S.K., Zapf J., Masiarz F.R., Kiefer M.C. Cleavage analysis of insulin-like growth factor (IGF)-dependent IGF-binding protein-4 proteolysis and expression of protease-resistant IGF-binding protein-4 mutants. J. Biol. Chem. 1995;270:4395–4400. doi: 10.1074/jbc.270.9.4395. [DOI] [PubMed] [Google Scholar]
- Dallas S.L., Miyazono K., Skerry T.M., Mundy G.R., Bonewald L.F. Dual role for the latent transforming growth factor-beta binding protein in storage of latent TGF-beta in the extracellular matrix and as a structural matrix protein. J. Cell Biol. 1995;131:539–549. doi: 10.1083/jcb.131.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Armiento J., DiColandrea T., Dalal S.S., Okada Y., Huang M.T., Conney A.H., Chada K. Collagenase expression in transgenic mouse skin causes hyperkeratosis and acanthosis and increases susceptibility to tumorigenesis. Mol. Cell. Biol. 1995;15:5732–5739. doi: 10.1128/mcb.15.10.5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daughaday W.H., Rotwein P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr. Rev. 1989;10:68–91. doi: 10.1210/edrv-10-1-68. [DOI] [PubMed] [Google Scholar]
- Davenport M.L., Pucilowska J., Clemmons D.R., Lundbland R., Spencer J.A., Underwood L.E. Tissue-specific expression of IGFBP-3 protease activity during rat pregnancy. Endocrinology. 1991;130:2505–2512. doi: 10.1210/endo.130.5.1374007. [DOI] [PubMed] [Google Scholar]
- De Leon D.D., Asmerom Y. Quantification of insulin-like growth factor I (IGF-I) without interference by IGF-1 binding proteins. Endocrinology. 1997;138:2199–2202. doi: 10.1210/endo.138.5.5237. [DOI] [PubMed] [Google Scholar]
- DeCaprio J.A., Ludlow J.W., Frigge J., Shew J.Y., Huang C.M., Lee W.H., Marsilio E., Paucha E., Livingston D.M. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–283. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- DeClerck Y.E., Perez N., Shimada H., Boone T.C., Langley K.E., Taylon S.M. Inhibition of invasion and metastasis in cells transfected with an inhibitor of metalloproteinases. Cancer Res. 1992;52:701–708. [PubMed] [Google Scholar]
- Denhardt D.T., Feng B., Edwards D.R., Cocuzzi E.T., Malyankar U.M. Tissue inhibitor of metalloproteinases (TIMP, aka EPA)structure, control of expression and biological functions. Pharmacol. Ther. 1993;59:329–341. doi: 10.1016/0163-7258(93)90074-n. [DOI] [PubMed] [Google Scholar]
- D'Errico A., Grigioni W.F., Fiorentino M., Baccarini P., Lamas E., De Mitri S., Gozzetti G., Mancini A.M., Brechot C. Expression of insulin-like growth factor II (IGF-II) in human hepatocellular carcinomasan immunohistochemical study. Pathol. Int. 1994;44:131–137. doi: 10.1111/j.1440-1827.1994.tb01697.x. [DOI] [PubMed] [Google Scholar]
- Dunn S.E., Ehrlich M., Sharp N.J., Reiss K., Solomon G., Hawkins R., Baserga R., Barerett J.C. A dominant negative mutant of the insulin-like growth factor-I receptor inhibits the adhesion, invasion, and metastasis of breast cancer. Cancer Res. 1998;58:3353–3361. [PubMed] [Google Scholar]
- Flaumenhaft R., Rifkin D.B. Extracellular matrix regulation of growth factor and protease activity. Curr. Opin. Cell Biol. 1991;3:817–823. doi: 10.1016/0955-0674(91)90055-4. [DOI] [PubMed] [Google Scholar]
- Fowlkes J.L. Insulin-like growth-factor binding protein proteolysisan emerging paradigm in insulin-like growth factor physiology. Trends Endocrinol. Metab. 1997;8:299–306. doi: 10.1016/s1043-2760(97)00112-4. [DOI] [PubMed] [Google Scholar]
- Fowlkes J.L., Enghild J.J., Suzuki K., Nagase H. Matrix metalloproteinases degrade insulin-like growth factor–binding protein-3 in dermal fibroblast cultures J. Biol. Chem. 269 1994. 25742 25746a [PubMed] [Google Scholar]
- Fowlkes J.L., Suzuki K., Nagase H., Thrailkill K.M. Proteolysis of insulin-like growth factor binding protein-3 during rat pregnancya role for matrix metalloproteinases Endocrinology. 135 1994. 2810 2813b [DOI] [PubMed] [Google Scholar]
- Fowlkes J.L., Thrailkill K.M., Serra D.M., Nagase H. Insulin-like growth factor binding protein (IGFBP) substrate zymography. A new tool to identify and characterize IGFBP-degrading proteinases. Endocrine. 1997;7:33–36. doi: 10.1007/BF02778059. [DOI] [PubMed] [Google Scholar]
- Gargosky S.E., Pham H.M., Wilson K.F., Liu F., Giudice L.C., Rosenfeld R.G. Measurement and characterization of insulin-like growth factor binding protein-3 in human biological fluidsdiscrepancies between radioimmunoassay and ligand blotting. Endocrinology. 1992;131:3051–3060. doi: 10.1210/endo.131.6.1280211. [DOI] [PubMed] [Google Scholar]
- Haddad R., Held W.A. Genomic imprinting and IGF2 influence liver tumorigenesis and loss of heterozygosity in SV40 T antigen transgenic mice. Cancer Res. 1997;57:4615–4623. [PubMed] [Google Scholar]
- Hadsell D.L., Greenberg N.M., Fligger J.M., Baumrucker C.R., Rosen J.M. Targeted expression of des (1–3) human insulin-like growth factor I in transgenic mice influences mammary gland development and IGF-binding protein expression. Endocrinology. 1996;137:321–330. doi: 10.1210/endo.137.1.8536631. [DOI] [PubMed] [Google Scholar]
- Han V.K., Lund P.K., Lee D.C., D'Ercole A.J. Expression of somatomedin/insulin-like growth factor messenger ribonucleic acids in the human fetusidentification, characterization, and tissue distribution. J. Clin. Endocrinol. Metab. 1988;66:422–429. doi: 10.1210/jcem-66-2-422. [DOI] [PubMed] [Google Scholar]
- Hankinson S.E., Willett W.E., Colditz G.A., Hunter D.J., Michaud D.S., Deroo B., Rosner B., Speizer F.E., Pollak M. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. 1998;351:1393–1396. doi: 10.1016/S0140-6736(97)10384-1. [DOI] [PubMed] [Google Scholar]
- Harris T.M., Rogler L.E., Rogler C.E. Reactivation of the maternally imprinted IGF2 allele in TGFalpha induced hepatocellular carcinomas in mice. Oncogene. 1998;16:203–209. doi: 10.1038/sj.onc.1201519. [DOI] [PubMed] [Google Scholar]
- Imai Y., Busby W.H., Jr., Smith C.E., Clarke J.B., Garmong A.J., Horwitz G.D., Rees C., Clemmons D.R. Protease-resistant form of insulin-like growth factor-binding protein 5 is an inhibitor of insulin-like growth factor-I actions on porcine smooth muscle cells in culture. J. Clin. Invest. 1998;100:2596–2605. doi: 10.1172/JCI119803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J.I., Clemmons D.R. Insulin-like growth factors and their binding proteinsbiologic actions. Endocr. Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- Khokha R. Suppression of the tumorigenic and metastatic abilities of murine B16-F10 melanoma cells in vivo by the overexpression of tissue inhibitor of metalloproteinase-1. J. Natl. Cancer Inst. 1994;86:299–304. doi: 10.1093/jnci/86.4.299. [DOI] [PubMed] [Google Scholar]
- Khokha R., Waterhouse P., Yagel S., Lala P.K., Overall C., Norton G., Denhardt D. Antisense RNA-induced reduction in murine TIMP levels confers oncogenecity on Swiss 3T3 cells. Science. 1989;244:947–950. doi: 10.1126/science.2465572. [DOI] [PubMed] [Google Scholar]
- Kimura Y., Koga H., Mugita N., Fujita N., Takeshima H., Nishi T., Yamashima T., Saido T.C., Yamasaki T., Moritake K. The involvement of calpain-dependent proteolysis of the tumor suppressor NF2 (merlin) in schwannomas and meningiomas. Nat. Med. 1998;4:915–922. doi: 10.1038/nm0898-915. [DOI] [PubMed] [Google Scholar]
- Krüger A., Fata J.E., Khokha R. Altered tumor growth and metastasis of a T-cell lymphoma in Timp-1 transgenic mice. Blood. 1997;90:1993–2000. [PubMed] [Google Scholar]
- Krüger A., Sanchez-Sweatman O.H., Martin D.C., Orr F.W., Rüther U., Khokha R. Host TIMP-1 overexpression confers resistance to experimental brain metastasis of a fibrosarcoma cell line. Oncogene. 1998;16:2419–2423. doi: 10.1038/sj.onc.1201774. [DOI] [PubMed] [Google Scholar]
- Lassarre C., Binoux M. Insulin-like growth factor binding protein-3 is functionally altered in pregnancy plasma. Endocrinology. 1994;134:1254–1262. doi: 10.1210/endo.134.3.7509737. [DOI] [PubMed] [Google Scholar]
- Lassarre C., Lalou C., Perin L., Binoux M. Protease-induced alteration of insulin-like growth factor binding protein-3 as detected by radioimmunoassay. Agreement with ligand blotting data. Growth Regul. 1994;4:48–55. [PubMed] [Google Scholar]
- Levi E., Fridman R., Miao H.Q., Ma Y.S., Yayon A., Vlodavsky I. Matrix metalloproteinase 2 releases active soluble ectodomain of fibroblast growth factor receptor 1. Proc. Natl. Acad. Sci. USA. 1996;93:7069–7074. doi: 10.1073/pnas.93.14.7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liotta L.A., Steeg P.S., Stetler-Stevenson W.G. Cancer metastasis and angiogenesisan imbalance of positive and negative regulation. Cell. 1991;64:327–336. doi: 10.1016/0092-8674(91)90642-c. [DOI] [PubMed] [Google Scholar]
- Liu P., Terradillos O., Renard C.A., Feldmann G., Buendia M.A., Bernuan D. Hepatocarcinogenesis in woodchuck hepatitis virus/c-myc micesustained cell proliferation and biphasic activation of insulin-like growth factor-II. Hepatology. 1997;25:874–883. doi: 10.1002/hep.510250415. [DOI] [PubMed] [Google Scholar]
- Martin D.C., Rüther U., Sanchez-Sweatman O.H., Orr F.W., Khokha R. Inhibition of SV40 T antigen-induced hepatocellular carcinoma in TIMP-1 transgenic mice. Oncogene. 1996;13:569–576. [PubMed] [Google Scholar]
- Martin D.C., Sanchez-Sweatman O.H., Ho A.T.-V., Inderdeo D.S., Tsao M.-S., Khokha R. Transgenic TIMP-1 inhibits SV40 T antigen-induced hepatocarcinogenesis by impairment of hepatocellular proliferation and tumor angiogenesis. Lab. Invest. 1999;79:225–234. [PubMed] [Google Scholar]
- Matrisian L.M. The matrix-degrading metalloproteinases. Bioessays. 1992;14:455–463. doi: 10.1002/bies.950140705. [DOI] [PubMed] [Google Scholar]
- McCusker R.H., Busby W.H., Dehoff M.H., Camacho-Hubner C., Clemmons D.R. Insulin-like growth factor (IGF) binding to cell monolayers is directly modulated by the addition of IGF-binding proteins. Endocrinology. 1991;129:939–949. doi: 10.1210/endo-129-2-939. [DOI] [PubMed] [Google Scholar]
- Mignatti P., Rifkin D.B. Biology and biochemistry of proteinases in tumor invasion. Physiol. Rev. 1993;73:161–195. doi: 10.1152/physrev.1993.73.1.161. [DOI] [PubMed] [Google Scholar]
- Murphy L.J., Bell G.I., Friesen H.G. Tissue distribution of insulin-like growth factor I and II messenger ribonucleic acid in the adult rat. Endocrinology. 1987;120:1279–1282. doi: 10.1210/endo-120-4-1279. [DOI] [PubMed] [Google Scholar]
- Osborne C.K., Coronado E.B., Kitten L.J., Arteaga C.I., Fuqua S.A., Ramasharma K., Marshall M., Li C.H. Insulin-like growth factor-II (IGF-II)a potential autocrine/paracrine growth factor for human breast cancer acting via the IGF-I receptor. Mol. Endocrinol. 1989;3:1701–1709. doi: 10.1210/mend-3-11-1701. [DOI] [PubMed] [Google Scholar]
- Rees C., Clemmons D.R., Horvitz G.D., Clarke J.B., Busby W.H. A protease resistant form of insulin-like growth factor (IGF) binding protein 4 inhibits IGF-1 actions. Endocrinology. 1998;139:4182–4188. doi: 10.1210/endo.139.10.6266. [DOI] [PubMed] [Google Scholar]
- Resnicoff M., Baserga R. The role of the insulin-like growth factor I receptor in transformation and apoptosis. Ann. N.Y. Acad. Sci. 1998;842:76–81. doi: 10.1111/j.1749-6632.1998.tb09634.x. [DOI] [PubMed] [Google Scholar]
- Resnicoff M., Abraham D., Yutanawiboonchai W., Rotman H.L., Rubin R. The insulin-like growth factor I receptor protects tumor cells from apoptosis in vivo. Cancer Res. 1995;55:2463–2469. [PubMed] [Google Scholar]
- Rogler C.E., Yang D., Rossetti L., Donohoe J., Alt E., Chang C.J., Rosenfeld R., Neely K., Hintz R. Altered body composition and increased frequency of diverse malignancies in insulin-like growth factor-II transgenic mice. J. Biol. Chem. 1994;269:13779–13784. [PubMed] [Google Scholar]
- Rubin R., Baserga R. Insulin-like growth factor I receptor. Its role in cell proliferation, apoptosis and tumorigenesis. Lab. Invest. 1995;73:311–331. [PubMed] [Google Scholar]
- Rüther U., Woodroofe C., Fattori E., Ciliberto G. Inducible formation of liver tumors in transgenic mice. Oncogene. 1993;8:87–93. [PubMed] [Google Scholar]
- Schirmacher P., Held W.A., Yang D., Chisari F.V., Rustum Y., Rogler C.E. Reactivation of insulin-like growth factor II during hepatocarcinogenesis in transgenic mice suggests a role in malignant growth. Cancer Res. 1992;52:2549–2556. [PubMed] [Google Scholar]
- Schoenle E., Zapf J., Hauri C., Steiner T., Froesch E.R. Comparison of in vivo effects of insulin-like growth factors I and II and of growth hormone in hypophysectomized rats. Acta Endocrinol. 1985;108:167–174. doi: 10.1530/acta.0.1080167. [DOI] [PubMed] [Google Scholar]
- Sell C., Rubini M., Rubin R., Liu J.P., Efstratiadis A., Baserga R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type I insulin-like growth factor receptor. Proc. Natl. Acad. Sci. USA. 1993;90:11217–11221. doi: 10.1073/pnas.90.23.11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimisaki S., Ling N. Identification and molecular characterization of insulin-like growth factor binding proteins (IGFBP-1, -2, -3, -4, -5, and -6) Prog. Growth Factor Res. 1991;3:243–266. doi: 10.1016/0955-2235(91)90003-m. [DOI] [PubMed] [Google Scholar]
- Sohda T., Oka Y., Iwata K., Gunn J., Kamimura S., Shijo H., Okumura M., Yun K. Co-localization of insulin-like growth factor II and the proliferation marker MIB1 in hepatocellular carcinoma cells. J. Clin. Pathol. 1997;50:135–137. doi: 10.1136/jcp.50.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sympson C.J., Talhouk R.S., Alexander C.M., Chin J.R., Clift S.M., Bissell M.J., Werb Z. Targeted expression of stromelysin-1 in mammary gland provides evidence for a role of proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J. Cell Biol. 1994;125:681–693. doi: 10.1083/jcb.125.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sympson C.J., Bissell M.J., Werb Z. Mammary gland tumor formation in transgenic mice overexpressing stromelysin-1. Semin. Cancer Biol. 1995;6:159–163. doi: 10.1006/scbi.1995.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T.H., Wallis J., Levine A.J. Identification of the p53 protein domain involved in formation of the simian virus 40 large T-antigen-p53 protein complex. J. Virol. 1986;59:574–583. doi: 10.1128/jvi.59.3.574-583.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant M.K., Thrasher J.B., Twomey P.A., Drivdahl R.H., Brinbaum R.S., Plymate S.R. Protein and messenger ribonucleic acid (mRNA) for the type I insulin-like growth factor (IGF) receptor is decreased and IGF-II mRNA is increased in human prostate carcinoma compared to benign prostate epithelium. J. Clin. Endocrinol. Metab. 1996;81:3774–3782. doi: 10.1210/jcem.81.10.8855837. [DOI] [PubMed] [Google Scholar]
- Thrailkill K.M., Quarles L.D., Nagase H., Suzuki K., Serra D.M., Fowlkes J.L. Characterization of insulin-like growth factor-binding protein 5-degrading proteases produced throughout murine osteoblast differentiation. Endocrinology. 1995;136:3527–3533. doi: 10.1210/endo.136.8.7543045. [DOI] [PubMed] [Google Scholar]
- Wang M., Liu Y.E., Greene J., Sheng S., Fuchs A., Rosen E.M., Shi Y.E. Inhibition of tumor growth and metastasis of human breast cancer cells transfected with tissue inhibitor of metalloproteinase-4. Oncogene. 1997;14:2767–2774. doi: 10.1038/sj.onc.1201245. [DOI] [PubMed] [Google Scholar]
- Werb Z. ECM and cell surface proteolysisregulating cellular ecology. Cell. 1997;91:439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- Werner H., LeRoith D. The role of insulin-like growth factor system in human cancer. Adv. Cancer Res. 1996;68:183–223. doi: 10.1016/s0065-230x(08)60354-1. [DOI] [PubMed] [Google Scholar]
- Werner H., Le Roith D. The insulin-like growth factor-I receptor signaling pathways are important for tumorigenesis and inhibition of apoptosis. Crit. Rev. Oncog. 1997;8:71–92. doi: 10.1615/critrevoncog.v8.i1.40. [DOI] [PubMed] [Google Scholar]
- Whitelock J.M., Murdoch A.D., Iozzo R.V., Underwood P.A. The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. J. Biol. Chem. 1996;271:10079–10086. doi: 10.1074/jbc.271.17.10079. [DOI] [PubMed] [Google Scholar]
- Wilson C.L., Heppner K.J., Laboski P.A., Hogan B.L., Matrisian M.L. Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc. Natl. Acad. Sci. USA. 1997;94:1402–1407. doi: 10.1073/pnas.94.4.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witty J.P., Wright J.H., Matrisian L.M. Matrix metalloproteinases are expressed during ductal alveolar mammary morphogenesis, and misregulation of stromelysin-1 in transgenic mice induces unscheduled mammary alveolar development. Mol. Biol. Cell. 1995;6:1287–1303. doi: 10.1091/mbc.6.10.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E., Hoeflich A., Lahm H. What is the function of IGF-II in postnatal life? Answers from transgenic mouse models. Growth Horm. IGF Res. 1998;8:185–193. doi: 10.1016/s1096-6374(98)80110-x. [DOI] [PubMed] [Google Scholar]
- Zhang L., Kashanchi F., Zhan Q., Zhan S., Brady J.N., Fornace A.J., Seth P., Helman L.J. Regulation of insulin-like growth factor II P3 promoter by p53a potential mechanism for tumorigenesis. Cancer Res. 1996;56:1367–1373. [PubMed] [Google Scholar]