

Abstract
Phosphatidylinositol 3-kinase (PI 3-kinase) is a lipid kinase which has been implicated in mitogenesis, protein trafficking, inhibition of apoptosis, and integrin and actin functions. Here we show using a green fluorescent protein–tagged p85 subunit that phosphatidylinositol 3-kinase is distributed throughout the cytoplasm and is localized to focal adhesion complexes in resting NIH-3T3, A431, and MCF-7 cells. Ligand stimulation of an epidermal growth factor receptor/c-erbB-3 chimera expressed in these cells results in a redistribution of p85 to the cell membrane which is independent of the catalytic activity of the enzyme and the integrity of the actin cytoskeleton. The movement is, however, dependent on the phosphorylation status of the erbB-3 chimera. Using rhodamine-labeled epidermal growth factor we show that the phosphatidylinositol 3-kinase and the receptors colocalize in discrete patches on the cell surface. Low concentrations of ligand cause patching only at the periphery of the cells, whereas at high concentrations patches were seen over the whole cell surface. Using green fluorescent protein–tagged fragments of p85 we show that binding to the receptor requires the NH2-terminal part of the protein as well as its SH2 domains.
Keywords: phosphatidylinositol 3-kinase, green fluorescent protein, erbB-3, focal complexes, tyrosine kinase
Phosphatidylinositol 3-kinase (PI 3-kinase)1 was first identified as a lipid kinase activity associated with the middle T antigen in polyoma virus (SV40) transformed cells (Cantley et al. 1991). The enzyme phosphorylates inositol phospholipids at the D-3 position of the inositol ring. Phosphatidylinositol 3-phosphate (PI-3-P) is constitutively synthesized, whereas the other products of PI 3-kinase, PI-3,4-P2 and PI-3,4,5-P3, are rapidly produced after growth factor stimulation (Auger et al. 1989), and function directly as second messengers. PI 3-kinase is known to be involved in a number of biological processes, such as mitogenesis, protein trafficking, inhibition of apoptosis, and the regulation of actin and integrin functions (Carpenter and Cantley 1996). The enzyme is now known to associate with several growth factor receptors, including the erbB-3 receptor (Prigent and Gullick 1994), platelet-derived growth factor receptor (Klippel et al. 1992), hepatocyte growth factor receptor (Ponzetto et al. 1993), NGF receptor (Ohmichi et al. 1992), and insulin receptor via insulin receptor substrate 1 (IRS-1) (Backer et al. 1992). It also binds to nonreceptor tyrosine kinases such as members of the src kinase family, and to the crk and abl oncogene products (Cantley et al. 1991). PI 3-kinase is a heterodimer consisting of an 85-kD regulatory subunit (p85) and a 110-kD catalytic subunit (p110) (Otsu et al. 1991). The p85 subunit contains several domains that mediate protein–protein interactions. It has one NH2-terminal SH3 domain which binds to proline-rich sequences and two SH2 domains which bind to the specific phosphotyrosine-containing motif pYXXM. It also contains a region that is homologous to the COOH-terminal portion of the break cluster region gene product, which is flanked on either side by proline-rich sequences (Kapeller and Cantley 1994). The p110 catalytic subunit is similar to the Saccharomyces cerevisiae protein, VPS34, which is essential for protein sorting (Herman and Emr 1990). VPS34 only uses phosphatidylinositol as a substrate to produce phosphatidylinositol 3-phosphate (PI-3-P) (Stack and Emr 1994), suggesting that the constitutive production of this phospholipid in mammalian cells might be involved in protein trafficking. There are four known isoforms of p110, termed α, β, δ, and γ (Chantry et al. 1997; Ho et al. 1997). The NH2-terminal region of p110α, β, and δ binds to p85 in the sequence that separates the SH2 domains, whereas p110γ does not bind to p85 but its activity is stimulated by G proteins (Stoyanov et al. 1995).
The downstream signaling events stimulated by the action of PI 3-kinase are poorly understood. Known targets of PI 3-kinase's lipid products are protein kinase B (PKB), also known as Akt and phosphoinositide-dependent protein kinase; phosphoinositide-dependent kinase 1 (PDK-1), which is involved in the inhibition of apoptosis (Hemmings 1997; Anderson et al. 1998); certain isoforms of PKC (Nakanishi et al. 1993; Toker et al. 1994); and p70S6 kinase (Weng et al. 1995). PI 3-kinase has also been described as an upstream regulator of Rac and Rho in mediating focal adhesion complexes, and stress fiber and lamellipodia formation (Reif et al. 1996), and may also be upstream of Ras signaling (Jhun et al. 1994; Hu et al. 1995).
Although PI 3-kinase is thought to be involved in the signaling pathways of a number of growth factor receptors (Kapeller and Cantley 1994), immunoprecipitation experiments have shown that only a small fraction of the cytoplasmic pool of p85 is redistributed to the membrane after ligand stimulation (Soler et al. 1994). Receptor signaling might therefore result in the relocation of p85 to other locations, reflecting its diverse role in cellular functioning. To investigate the distribution and movement of p85 in live cells after ligand stimulation we have tagged the NH2 terminus of human p85α with green fluorescent protein (GFP).
Materials and Methods
All chemicals were obtained from Sigma Chemical Co. unless otherwise stated. The antibodies U1, U13, and U14 were obtained from Ivan Gout (Ludwig Institute, London, United Kingdom). The antibody to the COOH terminus of p85 was obtained from Transduction Laboratories, and the antibody to p110 was obtained from Autogen Bioclear. The Sto x22 antibody to clathrin was a kind gift of Julian Downward (Imperial Cancer Research Fund, London, United Kingdom).
Cloning and Expression of GFP Fusion Proteins
p85 DNA was obtained from a human cDNA clone by PCR and then fused to the COOH terminus of MUT2 GFP cDNA (gift of Stanley Falkow, Stanford University, Stanford, CA; Cormack et al. 1996). GFP-p85 contained all the 724 amino acids of the human p85α, GFP-2SH2 contained amino acids 321–724, GFP-CSH2 contained amino acids 615–724, and GFP-NSH2 contained amino acids 321–474. The chimeric cDNAs were cloned into the CMV promoter-driven plasmid pcDNA3.1/Zeo (Invitrogen), and microinjected at 0.1 ng/nl into the nuclei of C18 cells which had been serum starved (DMEM supplemented with 0.5% FCS) for 18 h.
Cell Culture and Transient Transfections
C18 (NIH-3T3 based), Cos-7, A431, and MCF-7 cells were cultured in DMEM supplemented with 10% FCS. For transfection, 5 × 106 Cos-7 cells were electroporated (0.3 kV, 250 μF, 0.4 mm cuvette) with 10 μg of either the GFP-p85, GFP-2SH2, or pcDNA3.1/Zeo as a control. After 48 h, the cells were lysed at 4°C in Triton buffer (50 mM Tris, pH 7.4, 5 mM EGTA, 1% Triton X-100, 150 mM sodium chloride, 25 mM benzamidine, 10 μg/ml leupeptin, 0.2 mM sodium orthovanadate, 50 mM sodium fluoride, 1 mM PMSF) for immunoprecipitation.
Protein Immunoprecipitations and Immunoblotting
The fusion proteins were in vitro transcribed and translated using a TNT-coupled reticulocyte lysate system (Promega) with [35S]methionine labeling. The protein sizes were deduced using SDS-gel electrophoresis (10% gel) and autoradiography. A 50-μl TNT reaction was sufficient for six immunoprecipitations. The reaction was diluted in 2.5 ml of PBS, and 400 μl of the protein mix was immunoprecipitated for 2 h with antibodies bound to protein A–Sepharose. The immunocomplexes were washed four times with RIPA buffer (PBS with 1% Triton X-100, 0.1% SDS) and once with PBS/0.2% Triton X-100 before being eluted with SDS-PAGE buffer at high temperature.
GFP-p85 and GFP-2SH2 proteins from transiently transfected Cos-7 cells were immunoprecipitated using an antibody to GFP (gift of Dave Shima, Imperial Cancer Research Fund, London) to distinguish them from the endogenous p85. Proteins were separated on a 10% SDS-polyacrylamide gel and subjected to Western blotting with an antibody to p85 (1:250; Transduction Laboratories). The filter was then stripped for 1 h at 60°C with 2% SDS, 100 mM mercaptoethanol in 62.5 mM Tris, pH 6.8. This was then blocked and reprobed with an antibody to the p110 catalytic subunit (1 μg/ml; Autogen Bioclear). Detection was by incubation with horseradish peroxidase–conjugated secondary antibody and visualization was by enhanced chemiluminescence (Amersham).
C18 cells in 9-cm dishes (80% confluent) were stimulated with 5 × 10−7 M EGF for 2.5 min to bring about phosphorylation of the erbB-3 chimera. The cells were lysed in Triton X-100 buffer at 4°C. The chimera was precipitated for 2 h with 5 μg of EGF receptor 1 (EGFR1) which reacts with the extracellular domain of EGFR and 2.5 mg of protein A–Sepharose. Immunocomplexes were washed and analyzed as above. The phosphorylation status of the chimera was assessed using an antibody specific for phosphotyrosine (PY20, 1:1,000; Transduction Laboratories).
Fluorescence Imaging and Immunofluorescence
Microscopy was performed using a Zeiss Axiovert 135 microscope using a 63× objective lens with fluorescein and Cy3 filters. The images were captured with a CCD camera and processed using IPLab and Photoshop 4.0 software. Fluorescence intensity was normalized to the first time point in the films. The temperature was maintained throughout at 37°C using an environmental chamber.
For immunofluorescence, cells were grown on glass coverslips. They were fixed at 4°C for 15 min with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 in PBS, and incubated with 1:50 antivinculin antibody (Sigma Chemical Co.). Antibody detection was by reaction with Cy3-conjugated goat anti–mouse IgG for 20 min at room temperature. Localization of clathrin was performed essentially as for vinculin using the anticlathrin mouse mAb Sto x22 at 1:100. Immunostaining for the chimera was performed using the EGFR1 mAb as described by Gullick et al. 1985.
Results
Construction of GFP-p85 Fusion Proteins
A number of constructs were made containing different portions of p85 fused to GFP: the full length p85 protein (GFP-p85), the two SH2 domains (GFP-2SH2), the NH2-terminal SH2 domain (GFP-NSH2), and COOH-terminal SH2 domain (GFP-CSH2). The constructs were in vitro transcribed and translated in the presence of [35S]methionine, and the resulting proteins were immunoprecipitated with antibodies to GFP and p85 to check the integrity of the encoded proteins (Fig. 1). GFP-p85 was immunoprecipitated with an antibody to GFP, the inter–NSH2-SH3 domain (U1), the SH3 domain (U13), and the COOH-terminal SH2 domain (Transduction Laboratories) (Fig. 1 A, lanes 1–4, respectively). GFP-2SH2 was immunoprecipitated with antibodies to GFP and the NH2- (U14) and COOH-terminal SH2 domains (Transduction Laboratories) (Fig. 1 B, lanes 1–3, respectively). GFP-CSH2 and GFP-NSH2 were immunoprecipitated with anti-GFP and COOH- and NH2-terminal SH2 domain antibodies, respectively (Fig. 1 B, lanes 5 and 6; Fig. 1 A, lanes 6 and 7). GFP-p85 was able to bind weakly to protein A–Sepharose in the absence of antibody (Fig. 1 A, lane 5), possibly due to exposure of hydrophobic sequences in the unfolded protein. The COOH-terminal SH2 antibody immunoprecipitated GFP-p85 and GFP-2SH2 well but GFP-CSH2 very weakly, which suggests that the antibody recognizes the larger protein with a higher affinity.
Figure 1.

Characterization of the GFP-p85 fusion constructs. The cDNA constructs were in vitro translated using [35S]methionine. GFP-p85 was immunoprecipitated with an antibody to GFP, the inter–NSH2-SH3 domain (U1), the SH3 domain (U13), the COOH-terminal SH2 domain (Transduction Laboratories), and with protein A–Sepharose alone (A, lanes 1–5, respectively). GFP-2SH2 was immunoprecipitated with antibodies to GFP, the NH2 (U14) and COOH termini, (Transduction Laboratories), and protein A–Sepharose alone (B, lanes 1–4, respectively). GFP-CSH2 and GFP-NSH2 were immunoprecipitated with anti-GFP, COOH- and NH2-terminal antibodies, respectively, and with a protein A–Sepharose control (B, lanes 5–7; A, lanes 6–8). To assess whether GFP-p85 bound to the p110 catalytic subunit, Cos-7 cells were transiently transfected with the GFP-p85, GFP-2SH2, and pcDNA3.1/Zeo constructs (Mock, C and D). The cells were lysed and immunoprecipitated with an antibody to GFP, transferred to nitrocellulose, probed with an antibody to p85 (C), and stripped and reprobed with an antibody to the p110 catalytic subunit of PI 3-kinase (D).
p85 binds the p110 catalytic subunit between its two SH2 domains to form the holoenzyme. To determine whether the GFP-tagged proteins engineered to encode this region retained their ability to bind p110, the full length and the 2SH2 constructs were transiently transfected into Cos-7 cells. The expressed proteins were immunoprecipitated with an antibody to GFP to distinguish them from the endogenous p85 and were then analyzed by SDS-PAGE and immunoblotting. The proteins were blotted with an antibody against p85 (Fig. 1 C, lanes 2 and 3) and then reprobed with an antibody to p110 (Fig. 1 D, lanes 2 and 3). In both the full length and 2SH2 transfected cells, but not the mock transfected cells (Fig. 1C and Fig. D, lane 1), the p110 subunit was coimmunoprecipitated, indicating that the addition of GFP to their NH2 termini did not interfere with their ability to form heterodimers.
Expression and Visualization of GFP Fusion Constructs in Live Cells
The behavior of the GFP-p85 proteins in live cells was assessed by digital fluorescence microscopy. A stable NIH-3T3 cell line (C18) was used which expressed low levels of endogenous murine EGFRs (<2,000/cell) and a high level of a chimera of the extracellular domain of human EGFR fused to the cytoplasmic domain of human c-erbB-3 (130,000/cell) (Prigent and Gullick 1994). Activation with EGF has been shown previously to recruit p85 to the chimera (Prigent and Gullick 1994; Soltoff et al. 1994), probably as a result of transphosphorylation by endogenous EGFRs because the erbB-3 receptor is thought to be kinase defective (Sierke et al. 1997). Intranuclear microinjection of the C18 cells with the GFP-p85 construct resulted in visible expression as early as 2 h after injection. The protein was distributed throughout the cytoplasm and was excluded from the nucleus (Fig. 2 A). The GFP-tagged 2SH2, CSH2, and the NSH2 domains were distributed throughout the cytoplasm but were also present to a lesser extent in the nucleus (Fig. 2B, Fig. C, and Fig. D, respectively).
Figure 2.

Localization of GFP-p85 and its subfragments in serum-starved cells, not exposed to an activating ligand. The constructs were microinjected into the nuclei of the cells, and the distribution of the proteins was visualized using fluorescence microscopy after 2–5 h. The typical appearance of C18 cells expressing GFP-p85 (A), GFP-2SH2 (B), GFP-CSH2 (C), and GFP-NSH2 (D). (E) The typical appearance of unstimulated A431 cells and (F) MCF-7 cells expressing the GFP-p85 protein.
The GFP-p85 and GFP-2SH2 proteins, but not the smaller constructs, were also localized at discrete points at the periphery of the cell (Fig. 3 A), which were identified as focal complexes by colocalization experiments with an antibody to vinculin (Fig. 3B and Fig. C), demonstrating that both of the SH2 domains have to be present for this localization to occur. This peripheral localization of p85 was also apparent in the epithelial cell lines, A431 and MCF-7, after microinjection with GFP-p85 (Fig. 2E and Fig. F, respectively).
Figure 3.

Colocalization of GFP-p85 with vinculin at focal adhesion complexes. Cells were stimulated with EGF for 5 min then fixed in 4% paraformaldehyde. A GFP image was taken of the cell (A). Vinculin was detected using a mouse antivinculin antibody and a Cy3-labeled rabbit anti–mouse secondary antibody (B). (C) Merge of the two images showing colocalization.
EGF Stimulation of GFP-p85–expressing Cells
Addition of 500 nM EGF to C18 cells expressing the GFP-p85 protein resulted in recruitment of p85 to the cell surface where it formed patches (Fig. 4 and Quicktime movie at http://www.lif.icnet.uk/axp/rbl/index.html). The EGFR/c-erbB-3 chimera was also visualized after 500 nM EGF addition using an anti-EGFR antibody. Fig. 5A and Fig. B, shows that the receptors cluster to form the same patching pattern. To confirm that p85 is localizing to the receptor and not to another cellular site, rhodamine EGF was used to mark the external part of the receptor. Incubation of injected cells with 50 nM rhodamine EGF for 5 min resulted in patching indistinguishable from unlabeled ligand. Paired rhodamine/GFP images of the cells were then taken. A similar pattern was obtained for the rhodamine and GFP images, demonstrating that the majority of p85 relocalizes to the receptor (Fig. 5, C–E). Expression of the GFP-p85 protein in mock-transfected NIH-3T3 cells also resulted in focal complex localization in unstimulated and stimulated cells, but no patching was seen after EGF addition, indicating that p85 was only recruited to the erbB-3 chimera and not to endogenous EGFR homodimers (data not shown).
Figure 4.

Relocalization of GFP-p85 in response to ligand activation in C18 cells. Distribution of GFP-p85 every 20 s after 500 nM EGF addition (for Quicktime movie, see http://www.lif.icnet.uk/axp/rbl/index.html).
Figure 5.
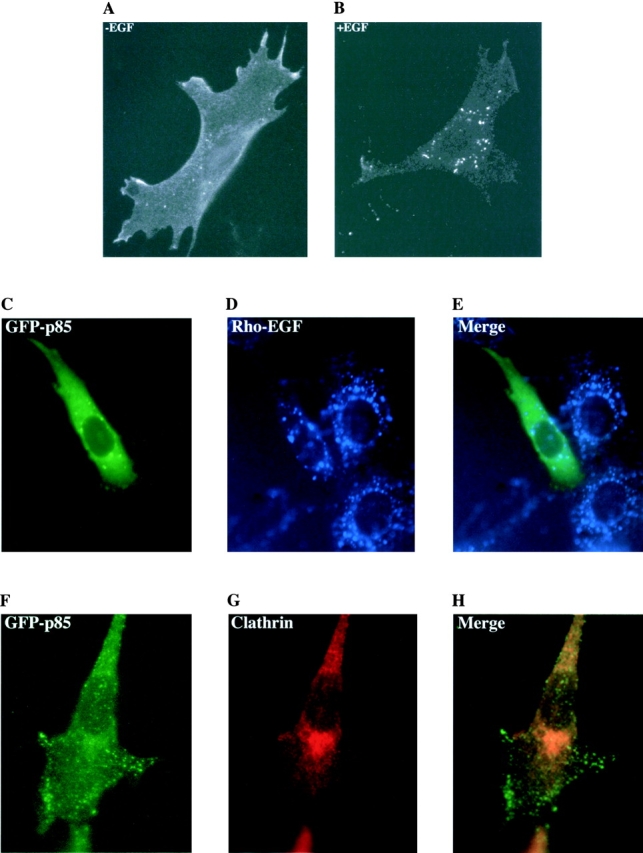
Characterization of the EGF-induced patches. C18 cells were incubated for 5 min with 500 nM EGF and then fixed and stained with an anti-EGFR antibody and a FITC-conjugated secondary antibody. The position of the receptor is therefore visible before (A) and after (B) EGF addition. C18 cells expressing GFP-p85 were incubated with 50 nM rhodamine-labeled EGF for 5 min to again mark the position of the receptor. A GFP (C) and rhodamine image (D) was taken of the same cell. This was then merged to show the colocalization of the two images (E). C18 cells expressing GFP-p85 were stimulated with 500 nM EGF for 5 min, fixed and stained with anti-clathrin antibodies. (F) GFP image, (G) clathrin image, and (H) merged image showing no colocalization.
Recruitment to the receptor was first visible after 30 s and was observed up to 1 h after EGF addition. During this time no visible internalization occurred. Cells were costained with anticlathrin antibodies to demonstrate the lack of coated vesicles or early endosomes associated with the patches (Fig. 5, F–H). There was also no change over 1 h after ligand stimulation in the localization of the tagged PI 3-kinase at the focal complex sites.
EGF addition to A431 cells expressing the GFP-p85 protein also resulted in translocation to the membrane and patching (Fig. 6 A). In A431 cells, the EGFR is normally present at the base of microvilli. Immediately after 500 nM EGF addition, the surface of the cells and the microvilli undergo ruffling, hence GFP-p85 recruitment to the receptor at the base of the microvilli is visible as a wave pattern. After 10 min, when the ruffling has subsided, the familiar patching pattern was observed. The patches are roughly the same size as those formed in C18 cells, which is to be expected as they have similar levels of erbB-3 present (Soltoff et al. 1994). Although internalization was not generally apparent in this cell line, certain individual cells showed signs of internalization after 1 h of EGF incubation (Fig. 6 B).
Figure 6.

Relocalization of GFP-p85 in response to ligand activation in A431 cells. (A) A431 cells were treated with 500 nM EGF and pictures were taken of the same cells over a period of 1 h. (B) Another cell from the same plate of cells.
Visualization of the clustering of the p85–receptor complex also enabled us to identify changes in cluster formation in response to ligand concentration. At 500 and 50 nM EGF, clustering occurs in patches over the complete surface of the C18 cells within 2–3 min. At 5 nM EGF, however, the clustering occurred reproducibly at the cells' periphery (Fig. 7, and see also the Quicktime movie at http://www.lif.icnet.uk/axp/rbl/index.html). At the lower EGF concentration of 0.5 nM and 50 pM no patches were observed (Fig. 8).
Figure 7.

Relocalization of GFP-p85 in response to a low ligand concentration in C18 cells. Distribution of GFP-p85 every 20 s after the addition of 5 nM EGF (for Quicktime movie, see http://www.lif icnet.uk/axp/rbl/index.html).
Figure 8.

Effect of ligand concentration on GFP-p85 distribution. The distribution of GFP-p85 in C18 cells 5 min after the addition of 50 nM EGF (A), 5 nM EGF (B), 0.5 nM EGF (C), and 50 pM EGF (D).
Characterization of the Requirements for Translocation of p85
The mechanism by which p85 translocates to the membrane after activation is not known. Relocation of p85 could be by diffusion or could be regulated by its lipid products (Janmey 1995) or by interaction with the actin cytoskeleton (Fincham et al. 1996). PI 3-kinase activity has been suggested to be involved in actin cytoskeleton–dependent translocation of src to the cell membrane because of its affinity for v-src and also because of its role in protein trafficking (Brown et al. 1995; Joly et al. 1995). To address whether p85 redistribution is controlled in a similar manner, cells expressing GFP-p85 were incubated with cytochalasin D to disrupt the actin cytoskeleton (Fig. 9 D). Although cytochalasin D is not thought to depolymerize all cellular actin, phalloidin staining of the treated cells showed that most was disrupted and the cell shape was visibly distorted. Despite this treatment, p85 was still able to translocate to the membrane after EGF addition (Fig. 9 B). Treatment of GFP-p85–expressing cells with Ly294002, an inhibitor of PI 3-kinase activity, also did not affect p85 movement (Fig. 9 F). These data suggest that p85 translocation to the membrane is not dependent on the catalytic activity of the enzyme or the integrity of the actin cytoskeleton. However, relocalization was inhibited by the EGFR tyrosine kinase inhibitor AG1478 (Fig. 10 A) at a concentration (1 μM) sufficient to completely inhibit receptor tyrosine phosphorylation (Fig. 10 B), demonstrating that this activity is required. Thus, although SH2 binding is required it does not appear to be sufficient for relocalization since EGF addition to cells expressing either the two SH2 domains or the NH2- or COOH-terminal SH2 domain alone did not result in patching (data not shown).
Figure 9.
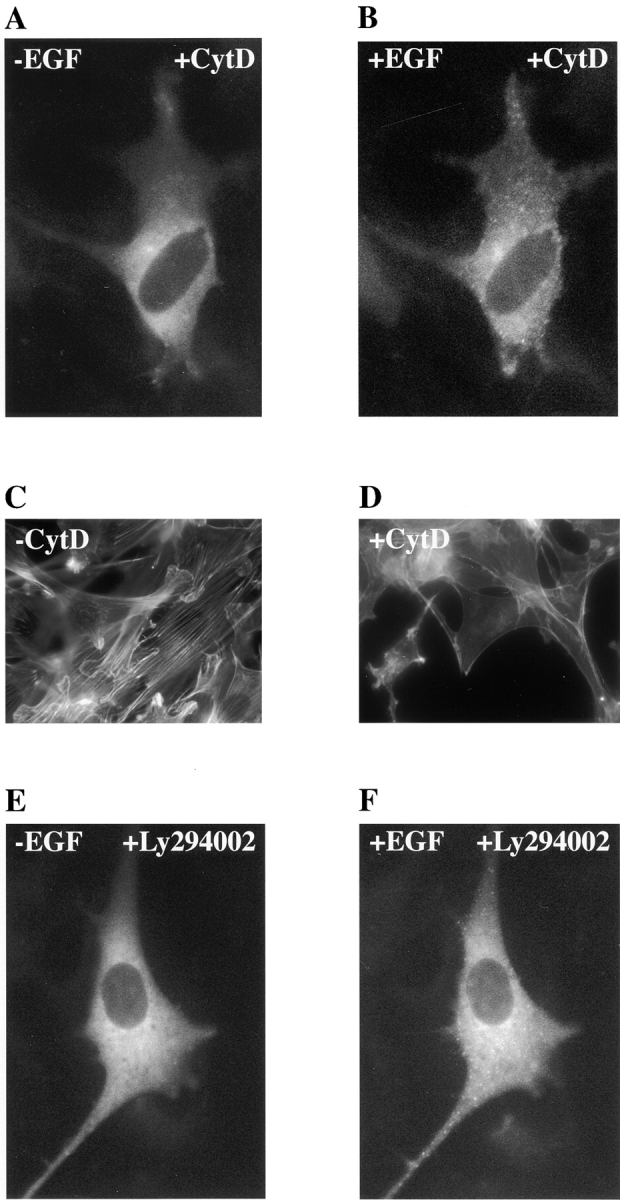
The effect of specific inhibitors on GFP-p85 relocation in response to 500 nM EGF. Cells were treated for 1 h with 0.1 μg/ml cytochalasin D, and the GFP-p85 was visualized before (A) and after (B) a 5-min incubation with 500 nM EGF. The appearance of the actin cytoskeleton was visualized with Cy3-labeled phalloidin in untreated serum-starved C18 cells (C) and in serum-starved cells treated with 0.1 μg/ml cytochalasin D for 1 h (D). The PI 3-kinase inhibitor Ly294002 was added to C18 cells at 10 μM for 15 min, and the GFP-p85 was visualized before ligand addition (E) and 5 min after the addition of 500 nM EGF (F).
Figure 10.

Inhibition of EGF-induced chimera autophosphorylation and GFP-p85 translocation by tyrphostin AG-1478. (A) C18 cells were treated for 30 min with 1 μM AG-1478, and the GFP-p85 was visualized before and 5 min after the addition of 500 nM EGF. (B) The effect of AG-1478 on chimera phosphorylation was assessed by incubating cells with varying concentrations of inhibitor followed by cell stimulation with 500 nM EGF for 2.5 min. The chimera was immunoprecipitated with an mAb to the extracellular domain, transferred to nitrocellulose, and Western blotted with an antibody against phosphotyrosine (PY20).
Discussion
Direct visualization of p85 within live cells results in positional information about the signaling molecule that cannot be easily obtained by traditional biochemical methods. We have observed a focal complex localization for PI 3-kinase in resting cells, which is dependent on the binding of the two SH2 domains of p85. Focal adhesions not only act as structural links between the extracellular matrix and the actin cytoskeleton but also transduce biochemical signals from the extracellular matrix (Burridge et al. 1988). Clustering of cell surface integrins is associated with the tyrosine phosphorylation of a number of focal adhesion proteins. In fibroblasts these include focal adhesion kinase, tensin, p130cas, and paxillin (Burridge et al. 1992; Bockholt and Burridge 1993; Clark and Brugge 1995; Vuori et al. 1996). These proteins can then interact with signaling molecules which contain SH2 domains, such as src kinases (Vuori et al. 1996), csk, and adapter proteins, such as crk and Grb2 (Bergman et al. 1995; Nakamoto et al. 1996; Schlaepfer et al. 1994). p85 association with focal adhesion kinases via its SH2 domains in vitro has been described previously (Chen and Guan 1994). p120cbl, an adapter protein, has also been reported to bind PI 3-kinase in macrophages and in PC12 cells. In PC12 cells, p120cbl was thought to link PI 3-kinase to the EGFR independently of the presence of erbB-3 (Soltoff and Cantley 1996). However, we did not see any evidence of EGFR/PI 3-kinase linkage in NIH-3T3 cells without chimera after EGF stimulation (data not shown), although this may be due to very low levels of EGFR expression. In macrophages, p120cbl has been reported to link PI 3-kinase to the integrin signaling pathway (Ojaniemi et al. 1997). Both focal adhesion kinase and p120cbl are potential p85 binding sites because they both have at least two pYXXM motifs (Soltoff et al. 1994; Chen and Guan 1994). Association of PI 3-kinase with other focal adhesion proteins such as src kinase has been described, but these interactions are mediated via SH3 domains and cannot be responsible for the binding seen during these experiments (Liu et al. 1993; Pleiman et al. 1994).
It is interesting that PI 3-kinase is only associated with focal complexes at the periphery of the cells, and not to focal adhesions underneath the cell. Focal complexes share many constituents with focal adhesions but they are morphologically distinct (Hall 1998). PI 3-kinase at these complexes may be involved in the signal pathways that are responsible for membrane ruffling and cell migration, especially since it is known to be involved in hepatocyte growth factor–induced cell scattering of MDCK cells (Royal and Park 1995; Royal et al. 1997). Such focal complexes are under the control of Rac, which has been identified as a downstream target of PI 3-kinase (Reif et al. 1996). The focal complex localization of p85 indicates that PI 3-kinase is directly involved in signaling through these complexes and not just as an upstream regulator.
After 500 nM EGF stimulation, GFP-p85 is rapidly translocated to the EGFR/erbB-3 receptor dimers which then form patches. No other cellular relocation was observed. In C18 cells, the patches appear to remain at the cell membrane with no obvious receptor internalization occurring. The patches were also not associated with clathrin, which suggests that the receptors were not being internalized into endosomal vesicles. Heregulin is slowly internalized and degraded when it is added to cells that express high levels of erbB-3 and erbB-2 receptors (Baulida and Carpenter 1997). Experiments using a similar EGFR/erbB-3 chimera in NIH-3T3 cells have also shown that internalization is very limited and the half-life of the receptor remains unchanged after the addition of EGF (Baulida et al. 1996). EGFR is the only member of the type 1 growth factor receptor family that can internalize normally (Baulida et al. 1996), and in A431 cells certain individual cells did show signs of receptor/p85 internalization. The difference in receptor trafficking between the cell lines could be due to their receptor status. C18 cells have 60 times more (chimeric) erbB-3 receptor than EGFR. Upon EGF stimulation the predominant receptor present in the receptor/GFP-p85 clusters will be erbB-3, which slows down the dimer internalization. In A431 cells, there is 10 times more EGFR present than erbB-3. After EGF stimulation of these cells, the predominant receptor in the clusters will be EGFR, which might explain why internalization appears to occur after 1 h of ligand stimulation. Note that this is slower than expected for EGFR expressed alone, but as PI 3-kinase binds only to erbB-3, we are presumably only following internalization of erbB-3/EGFR heterodimers.
p85 remains visible in the clusters at the cell membrane for up to 1 h. After 45 min, the erbB-3 chimera is fully dephosphorylated and mitogenic signaling should be attenuated (Prigent and Gullick 1994). PI 3-kinase has been implicated in the trafficking of receptors in mammalian cells; indeed, the presence of p85 binding sites on the platelet-derived growth factor receptor is required for its trafficking and degradation (Joly et al. 1995), and p85 has been reported to be internalized with the receptor (Kapeller et al. 1993). Wortmannin, a PI 3-kinase inhibitor (Okada et al. 1994), can also affect other processes, such as transferrin receptor recycling (Martys et al. 1996) and transcytosis (Hansen et al. 1995). It can also induce changes in the morphology of endosomes (Shpetner et al. 1996). PI 3-kinase lies upstream of early endosomal antigen 1, which binds to endosomes (Patki et al. 1997), and upstream of Rab5, which regulates early endosome fusion (Li et al. 1995). These data suggest that a continuous p85 presence may be involved in the trafficking of the erbB-3 receptor, which is probably recycled rather than degraded (Waterman et al. 1998).
The translocation of p85 to the receptor is not dependent on PI 3-kinase activity or the actin cytoskeleton; thus, its binding to erbB-3 is more likely due to random collision rather than active relocation. However, it is dependent on the tyrosine phosphorylation of the cytoplasmic domain of erbB-3, which allows SH2 interactions. It is generally believed that the two SH2 domains are all that are required for receptor binding (McGlade et al. 1992). However, the results presented here suggest that the NH2-terminal portion of the protein (containing the SH3, Bcr, and proline-rich domains) is also important in localizing p85 to the erbB-3 receptor, possibly due to self association allowing the recruitment of additional copies to the complex, or by stabilization of p85 at the receptor by binding to other signaling molecules.
PI 3-kinase itself has many functions in different intracellular systems. Our results demonstrate that in NIH-3T3 cells it exists at focal complexes and probably as an inactive cytoplasmic pool which is drawn to the cell surface by growth factor receptors in response to extracellular signals. Direct visualization of the subcellular location of second messengers using this technique will allow similar experiments in other cell types and in response to other stimuli to determine if these are common effects. In these environments, low affinity interactions, possibly not measurable by biochemical techniques, may be observed with activators, substrates, and downstream molecules. Finally, since ligand binding does not accelerate internalization of c-erbB-3 (or c-erbB-2 and c-erbB-4), it is not clear why receptor clustering occurs in this system. It is also not known if ligand-unoccupied as well as occupied receptors are present in these patches. It is possible that such behavior may control signal sensitivity by mechanisms such as those described recently by Bray et al. 1998. It is also possible that aggregation of second messenger molecules or the juxtaposition of different second messengers may permit combinatorial signaling. Light-based systems should be helpful in exploring such models. Moreover, they might be utilized to act as screens for promising pharmacological activators or inhibitors.
Acknowledgments
We thank Ivan Gout for his generous gift of the p85α cDNA, Dave Shima for the GFP antibody, and Stanley Falkow for the MUT2 GFP cDNA. We are very grateful to Debbie Lyon for performing the microinjections, Jonathan Wilding for his advice on computing, and Mark Hudson for his help with the figure presentations.
Footnotes
1.used in this paper: EGFR, epidermal growth factor receptor; GFP, green fluorescent protein; PI 3-kinase, phosphatidylinositol 3-kinase; SH, src homology domain
References
- Anderson K.E., Coadwell J., Stephens L.R., Hawkins P.T. Translocation of PDK-1 to the plasma membrane is important in allowing PDK-1 to activate protein kinase B. Curr. Biol. 1998;8:684–691. doi: 10.1016/s0960-9822(98)70274-x. [DOI] [PubMed] [Google Scholar]
- Auger K., Serunian L., Soltoff S., Libby P., Cantley L. PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell. 1989;57:167–175. doi: 10.1016/0092-8674(89)90182-7. [DOI] [PubMed] [Google Scholar]
- Backer J.M., Myers M.G., Shoelson S.E., Chin D.J., Sun X.J., Miralpeix M., Hu P., Margolis B., Skolnik E.Y., Schlessinger J., White M.F. Phosphatidylinositol 3-kinase is activated by association with IRS-1 during insulin stimulation. EMBO (Eur. Mol. Biol. Organ.) J. 1992;11:3469–3479. doi: 10.1002/j.1460-2075.1992.tb05426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulida J., Carpenter G. Heregulin degradation in the absence of rapid receptor-mediated internalization. Exp. Cell Res. 1997;232:167–172. doi: 10.1006/excr.1997.3515. [DOI] [PubMed] [Google Scholar]
- Baulida J., Kraus M.H., Alimandi M., Di Fiore P.P., Carpenter G. All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J. Biol. Chem. 1996;271:5251–5257. doi: 10.1074/jbc.271.9.5251. [DOI] [PubMed] [Google Scholar]
- Bergman M., Joukov V., Virtanen I., Alitalo K. Overexpressed CSK tyrosine kinase is localized in focal adhesions, causes reorganization of alpha(V)beta(5) integrin, and interferes with HeLa cell spreading. Mol. Cell. Biol. 1995;15:711–722. doi: 10.1128/mcb.15.2.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockholt S.M., Burridge K. Cell spreading on extracellular matrix proteins induces tyrosine phosphorylation of tensin. J. Biol. Chem. 1993;268:14565–14567. [PubMed] [Google Scholar]
- Bray D., Levin M.D., Morton-Firth C.J. Receptor clustering as a mechanism to control sensitivity. Nature. 1998;393:85–88. doi: 10.1038/30018. [DOI] [PubMed] [Google Scholar]
- Brown W.J., DeWald D.B., Emr S.D., Plutner H., Balch W.E. Role for phosphatidylinositol 3-kinase in the sorting and transport of newly synthesized lysosomal enzymes in mammalian cells. J. Cell Biol. 1995;130:781–796. doi: 10.1083/jcb.130.4.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K., Fath K., Kelly T., Nuckolls G., Turner C. Focal adhesions-transmembrane junctions between the extracellular matrix and the cytoskeleton. Annu. Rev. Cell Biol. 1988;4:487–525. doi: 10.1146/annurev.cb.04.110188.002415. [DOI] [PubMed] [Google Scholar]
- Burridge K., Turner C.E., Romer L.H. Tyrosine phosphorylation of paxillin and pp125(fak) accompanies cell-adhesion to extracellular matrixa role in cytoskeletal assembly. J. Cell Biol. 1992;119:893–903. doi: 10.1083/jcb.119.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley L.C., Auger K.R., Carpenter C., Duckworth B., Graziani A., Kapeller R., Soltoff S. Oncogenes and signal transduction. Cell. 1991;64:281–302. doi: 10.1016/0092-8674(91)90639-g. [DOI] [PubMed] [Google Scholar]
- Carpenter C.L., Cantley L.C. Phosphoinositide kinases. Curr. Opin. Cell Biol. 1996;8:153–158. doi: 10.1016/s0955-0674(96)80060-3. [DOI] [PubMed] [Google Scholar]
- Chantry D., Vojtek A., Kashishian A., Holtzman D.A., Wood C., Gray P.W., Cooper J.A., Hoekstra M.F. p110δ, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J. Biol. Chem. 1997;272:19236–19241. doi: 10.1074/jbc.272.31.19236. [DOI] [PubMed] [Google Scholar]
- Chen H.-C., Guan J.-L. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA. 1994;91:10148–10152. doi: 10.1073/pnas.91.21.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark E.A., Brugge J.S. Integrins and signal transduction pathwaysthe road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Cormack B.P., Valdivia R.H., Falkow S. Facs-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- Fincham V.J., Unlu M., Brunton V.G., Pitts J.D., Wyke J.A., Frame M.C. Translocation of src kinase to the cell periphery is mediated by the actin cytoskeleton under the control of the Rho family of small G proteins. J. Cell Biol. 1996;135:1551–1564. doi: 10.1083/jcb.135.6.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullick W.J., Downward D.J.H., Waterfield M.D. Antibodies to the autophosphorylation sites of the epidermal growth factor receptor protein tyrosine kinase as probes of structure and function. EMBO (Eur. Mol. Biol. Organ.) J. 1985;4:2869–2877. doi: 10.1002/j.1460-2075.1985.tb04016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hansen S.H., Olsson A., Casanova J.E. Wortmannin, an inhibitor of phosphoinositide 3-kinase, inhibits transcytosis in polarized epithelial cells. J. Biol. Chem. 1995;270:28425–28432. doi: 10.1074/jbc.270.47.28425. [DOI] [PubMed] [Google Scholar]
- Hemmings B.A. Akt signallinglinking membrane events to life and death decisions. Science. 1997;275:628–630. doi: 10.1126/science.275.5300.628. [DOI] [PubMed] [Google Scholar]
- Herman P.K., Emr S.D. Characterization of VPS34, a gene required for vacuolar protein sorting and vacuolar segregation in Saccharomyces cerevisiae . Mol. Cell. Biol. 1990;10:6742–6754. doi: 10.1128/mcb.10.12.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L.K.F., Liu D.X., Rozycka M., Brown R.A., Fry M.J. Identification of four novel human phosphoinositide 3-kinases defines a multi-isoform subfamily. Biochem. Biophys. Res. Commun. 1997;235:130–137. doi: 10.1006/bbrc.1997.6747. [DOI] [PubMed] [Google Scholar]
- Hu Q., Klippel A., Muslin A.J., Fantl W.J., Williams L.T. Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol 3-kinase. Science. 1995;268:100–102. doi: 10.1126/science.7701328. [DOI] [PubMed] [Google Scholar]
- Janmey P.A. Protein regulation by phosphatidylinositol lipids. Chem. Biol. 1995;2:61–65. doi: 10.1016/1074-5521(95)90276-7. [DOI] [PubMed] [Google Scholar]
- Jhun B.H., Rose D.W., Seely B.L., Rameh L., Cantley L., Saltiel A.R., Olefsky J.M. Microinjection of the SH2 domain of the 85-kilodalton subunit of phosphatidylinositol 3-kinase inhibits insulin-induced DNA synthesis and c-fos expression. Mol. Cell. Biol. 1994;14:7466–7475. doi: 10.1128/mcb.14.11.7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly M., Kazlauskas A., Corvera S. Phosphatidylinositol 3-kinase activity is required at a postendocytic step in platelet-derived growth factor receptor trafficking. J. Biol. Chem. 1995;270:13225–13230. doi: 10.1074/jbc.270.22.13225. [DOI] [PubMed] [Google Scholar]
- Kapeller R., Cantley L.C. Phosphatidylinositol 3-kinase. Bioessays. 1994;16:565–576. doi: 10.1002/bies.950160810. [DOI] [PubMed] [Google Scholar]
- Kapeller R., Chakrabarti R., Cantley L., Fay F., Corvera S. Internalization of activated platelet-derived growth factor receptor-phosphatidylinositol 3-kinase complexespotential interactions with the microtubule cytoskeleton. Mol. Cell. Biol. 1993;13:6052–6063. doi: 10.1128/mcb.13.10.6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippel A., Escobedo J.A., Fantl W.J., Williams L.T. The C-terminal SH2 domain of p85 accounts for the high affinity and specificity of the binding of phosphatidylinositol 3-kinase to phosphorylated platelet-derived growth factor beta receptor. Mol. Cell. Biol. 1992;12:1451–1459. doi: 10.1128/mcb.12.4.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., D'Souza-Schorey C., Barbieri M.A., Roberts R.L., Klippel A., Williams L.T., Stahl P.D. Evidence for phosphatidylinositol 3-kinase as a regulator of endocytosis via activation of Rab5. Proc. Natl. Acad. Sci. USA. 1995;92:10207–10211. doi: 10.1073/pnas.92.22.10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.Q., Marengere L.E.M., Koch C.A., Pawson T. The v-src SH3-domain binds phosphatidylinositol 3-kinase. Mol. Cell. Biol. 1993;13:5225–5232. doi: 10.1128/mcb.13.9.5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martys J.L., Wjasow C., Gangi D.M., Kielian M., McGraw T.E., Backer J.M. Wortmannin-sensitive trafficking pathways in Chinese hamster ovary cells. Differential effects on endocytosis and lysosomal sorting. J. Biol. Chem. 1996;271:10953–10962. doi: 10.1074/jbc.271.18.10953. [DOI] [PubMed] [Google Scholar]
- McGlade C.J., Ellis C., Reedijk M., Anderson D., Mbamalu G., Reith A.D., Panayotou G., End P., Bernstein A., Kazlauskas A. SH2 domains of the p85α subunit of phosphatidylinositol 3-kinase regulate binding to growth factor receptors. Mol. Cell. Biol. 1992;12:991–997. doi: 10.1128/mcb.12.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto T., Sakai R., Ozawa K., Yazaki Y., Hirai H. Direct binding of C-terminal region of p130(cas) to SH2 and SH3 domains of Src kinase. J. Biol. Chem. 1996;271:8959–8965. doi: 10.1074/jbc.271.15.8959. [DOI] [PubMed] [Google Scholar]
- Nakanishi H., Brewer K.A., Exton J.H. Activation of the ζ isozyme of protein kinase C by phosphatidylinositol 3,4,5-triphosphate. J. Biol. Chem. 1993;263:13–16. [PubMed] [Google Scholar]
- Ohmichi M., Decker S.J., Saltiel A.R. Activation of phosphatidylinositol-3 kinase by nerve growth factor involves direct coupling of the Trk protooncogene with Src homology-2 domains. Neuron. 1992;9:769–777. doi: 10.1016/0896-6273(92)90039-g. [DOI] [PubMed] [Google Scholar]
- Ojaniemi M., Martin S.S., Dolfi F., Olefsky J.M., Vuori K. The proto-oncogene product p120cbl links c-src and phosphatidylinositol 3-kinase to the integrin signaling pathway. J. Biol. Chem. 1997;272:3780–3787. doi: 10.1074/jbc.272.6.3780. [DOI] [PubMed] [Google Scholar]
- Okada T., Sakuma L., Fukui Y., Hazeki O., Ui M. Blockage of chemotactic peptide-induced stimulation of neutrophils by wortmannin as a result of selective inhibition of phosphatidylinositol 3-kinase. J. Biol. Chem. 1994;269:3563–3567. [PubMed] [Google Scholar]
- Otsu M., Hiles I., Gout I., Fry M.J., Ruiz L.F., Panayotou G., Thompson A., Dhand R., Hsuan J., Totty N. Characterization of two 85kd proteins that associate with receptor tyrosine kinases, middle-T/pp60src complexes, and PI3-kinase. Cell. 1991;65:91–104. doi: 10.1016/0092-8674(91)90411-q. [DOI] [PubMed] [Google Scholar]
- Patki V., Virbasius J., Lane W.S., Toh B.H., Shpetner H.S., Corvera S. Identification of an early endosomal protein regulated by phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA. 1997;94:7326–7330. doi: 10.1073/pnas.94.14.7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiman C.M., Hertz W.M., Cambier J.C. Activation of phosphatidylinositol 3-kinase by src-family kinase SH3 binding to the p85 subunit. Science. 1994;263:1609–1612. doi: 10.1126/science.8128248. [DOI] [PubMed] [Google Scholar]
- Ponzetto C., Bardelli A., Maina F., Longati P., Panayotou G., Dhand R., Waterfield M.D., Comoglio P.M. A novel recognition motif for phosphatidylinositol 3-kinase binding mediates its association with the hepatocyte growth factor scatter factor receptor. Mol. Cell. Biol. 1993;13:4600–4608. doi: 10.1128/mcb.13.8.4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigent S.A., Gullick W.J. Identification of c-erbB-3 binding sites for phosphatidylinositol 3-kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:2831–2841. doi: 10.1002/j.1460-2075.1994.tb06577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif K., Nobes C.D., Thomas G., Hall A., Cantrell D.A. Phosphatidylinositol 3-kinase signals activate a selective subset of Rac/Rho-dependent effector pathways. Curr. Biol. 1996;6:1445–1455. doi: 10.1016/s0960-9822(96)00749-x. [DOI] [PubMed] [Google Scholar]
- Royal I., Park M. Hepatocyte growth factor–induced scatter of Madin-Darby canine kidney cells requires phosphatidylinositol 3-kinase. J. Biol. Chem. 1995;270:27780–27787. doi: 10.1074/jbc.270.46.27780. [DOI] [PubMed] [Google Scholar]
- Royal I., Fournier T.M., Park M. Differential requirement of Grb2 and PI 3-kinase in HGF/SF-induced cell motility and tubulogenesis. J. Cell. Physiol. 1997;173:196–201. doi: 10.1002/(SICI)1097-4652(199711)173:2<196::AID-JCP20>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Schlaepfer D.D., Hanks S.K., Hunter T., Vanderdeer P. Integrin-mediated signal transduction linked to ras pathway by Grb2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Shpetner H., Joly M., Hartely D., Corvera S. Potential sites of PI-3 kinase function in the endocytic pathway revealed by the PI-3 kinase inhibitor wortmannin. J. Cell Biol. 1996;132:595–605. doi: 10.1083/jcb.132.4.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierke S.L., Cheng K., Kim H.-H., Koland J.G. Biochemical characterization of the protein tyrosine kinase homology domain of the ErbB-3 (HER3) receptor protein. Biochem. J. 1997;322:757–763. doi: 10.1042/bj3220757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler C., Beguinot L., Carpenter G. Individual epidermal growth factor receptor autophosphorylation sites do not stringently define association motifs for several SH2-containing proteins. J. Biol. Chem. 1994;269:12320–12324. [PubMed] [Google Scholar]
- Soltoff S.P., Cantley L.C. p120cbl is a cytosolic adapter protein that associates with phosphoinositide 3-kinase in response to epidermal growth factor in PC12 and other cells. J. Biol. Chem. 1996;271:563–567. doi: 10.1074/jbc.271.1.563. [DOI] [PubMed] [Google Scholar]
- Soltoff S.P., Carraway K.-L., Prigent S.A., Gullick W.J., Cantley L.C. ErbB-3 is involved in the activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol. Cell. Biol. 1994;14:3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack J.H., Emr S.D. VPS34P required for yeast vacuolar protein sorting is a multiple specificity kinase that exhibits both protein kinase and phosphatidylinositol-specific PI-3–kinase activities. J. Biol. Chem. 1994;269:31552–31562. [PubMed] [Google Scholar]
- Stoyanov B.S., Volinia T., Hanck I., Rubio M., Loubtchenkov D., Malek S., Stiyanova B., Vanaesebroeck R., Dhand B., Nurnberg P. Cloning and characterization of a G-protein–activated human phosphoinositide-3 kinase. Science. 1995;269:690–693. doi: 10.1126/science.7624799. [DOI] [PubMed] [Google Scholar]
- Toker A.M., Meyer K., Reddy J.R., Falck R., Aneja R., Aneja S., Parra A., Burns L.M., Cantley L.C. Activation of protein kinase C members by the novel polyphosphoinositdes PtdIns-3,4-P2 and PtdIns-3,4,5-P3. J. Biol. Chem. 1994;269:32358–32367. [PubMed] [Google Scholar]
- Vuori K., Hirai H., Aizawa S., Ruoslahti E. Induction of p130(cas) signalling complex-formation upon integrin-mediated cell-adhesiona role for src family kinases. Mol. Cell. Biol. 1996;16:2606–2613. doi: 10.1128/mcb.16.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman H., Sabanai I., Geiger B., Yarden Y. Alternative intracellular routing of erbB receptors may determine signalling potency. J. Biol. Chem. 1998;273:13819–13827. doi: 10.1074/jbc.273.22.13819. [DOI] [PubMed] [Google Scholar]
- Weng Q.-P., Andrabi K., Kipple A., Kozlowski M.T., Williams L.T., Avruch J. Phosphatidylinositol 3-kinase signals activation of p70 S6 kinase in situ through site-specific p70 phosphorylation. Proc. Natl. Acad. Sci. USA. 1995;92:5744–5748. doi: 10.1073/pnas.92.12.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]