

Abstract
During apoptosis, an important pathway leading to caspase activation involves the release of cytochrome c from the intermembrane space of mitochondria. Using a cell-free system based on Xenopus egg extracts, we examined changes in the outer mitochondrial membrane accompanying cytochrome c efflux. The pro-apoptotic proteins, Bid and Bax, as well as factors present in Xenopus egg cytosol, each induced cytochrome c release when incubated with isolated mitochondria. These factors caused a permeabilization of the outer membrane that allowed the corelease of multiple intermembrane space proteins: cytochrome c, adenylate kinase and sulfite oxidase. The efflux process is thus nonspecific. None of the cytochrome c-releasing factors caused detectable mitochondrial swelling, arguing that matrix swelling is not required for outer membrane permeability in this system. Bid and Bax caused complete release of cytochrome c but only a limited permeabilization of the outer membrane, as measured by the accessibility of inner membrane-associated respiratory complexes III and IV to exogenously added cytochrome c. However, outer membrane permeability was strikingly increased by a macromolecular cytosolic factor, termed PEF (permeability enhancing factor). We hypothesize that PEF activity could help determine whether cells can recover from mitochondrial cytochrome c release.
Keywords: apoptosis, Bid, Bax, cytochrome c, mitochondrial outer membrane
The translocation of cytochrome c from the mitochondrial intermembrane space to the cytoplasm plays a crucial role in apoptosis (reviewed by Reed 1997). In cell-free systems derived from mammalian somatic cells ( Liu et al. 1996) or Xenopus eggs ( Kluck et al. 1997a, Kluck et al. 1997b), and in whole cells ( Liu et al. 1996; Kluck et al. 1997a; Yang et al. 1997; Bossy-Wetzel et al. 1998), cytochrome c is released from mitochondria during apoptosis. Once in the cytoplasm, cytochrome c switches on the cell-dismembering caspases by complexing with Apaf-1, procaspase-9, and dATP ( Li et al. 1997b). In some cell types the cytochrome c molecule can be both necessary and sufficient for the activation of an apoptotic pathway, as the introduction of cytochrome c into cells by microinjection ( Li et al. 1997a; Zhivotovsky et al. 1998) is capable of initiating apoptosis, and microinjection of cytochrome c neutralizing antibody abrogates apoptosis ( Neame et al. 1998; Juin et al. 1999). Furthermore, in knockout mice lacking either caspase-9 ( Hakem et al. 1998; Kuida et al. 1998) or Apaf-1 ( Cecconi et al. 1998; Yoshida et al. 1998), reduced apoptosis in the brain can be linked to the absence of a program normally activated by cytosolic cytochrome c.
A major unanswered question concerns how cytochrome c leaves mitochondria during apoptosis. One potential mechanism involves mitochondrial swelling, either due to opening of the permeability transition pore in the inner membrane ( Bernardi 1996; Susin et al. 1997, Susin et al. 1999b) or to mitochondrial hyperpolarization ( Vander Heiden et al. 1999, Vander Heiden et al. 1997). However, it is questionable whether such mitochondrial swelling occurs in most instances of apoptosis or is required for cytochrome c release. Electron micrographs of apoptotic cells frequently contain apparently intact unswollen mitochondria ( Searle et al. 1975; Mancini et al. 1997; Zhuang et al. 1998; Martinou et al. 1999). Further, it has been reported that the pro-apoptotic proteins Bid ( Luo et al. 1998) and Bax ( Eskes et al. 1998; Jurgensmeier et al. 1998; Finucane et al. 1999) can release cytochrome c from isolated mitochondria in the absence of detectable mitochondrial swelling.
Exactly how Bid, Bax, and related proteins function to cause cytochrome c release is unclear. In cells given certain apoptotic stimuli, Bid or Bax can translocate to mitochondria ( Wolter et al. 1997; Luo et al. 1998; Desagher et al. 1999) to initiate the release of cytochrome c ( Jurgensmeier et al. 1998; Li et al. 1998; Luo et al. 1998; Narita et al. 1998; Rosse et al. 1998). In some cases, translocation of these proteins may require changes in their conformation ( Desagher et al. 1999; Nechushtan et al. 1999). The formation of ion channels in synthetic lipid bilayers by some members of the Bcl-2 family such as Bax ( Antonsson et al. 1997; Schlesinger et al. 1997), Bcl-xL ( Minn et al. 1997), and Bcl-2 ( Schendel et al. 1997), suggests that pro-apoptotic members of this family might interact directly with the outer mitochondrial membrane to allow efflux of cytochrome c. Ion channel formation by anti-apoptotic members of the Bcl-2 family has been proposed to prevent hyperpolarization ( Vander Heiden et al. 1997) and thus indirectly counteract closure of the voltage-dependent anion channel (VDAC) on the outer membrane ( Vander Heiden et al. 1999), although other studies report that the outer membrane is freely permeable to small ions ( Mannella 1992). Alternatively, Bax has recently been reported to interact directly with VDAC on the outer membrane to release cytochrome c ( Narita et al. 1998; Shimizu et al. 1999) or with ANT on the inner membrane to initiate the permeability transition, indirectly leading to cytochrome c release ( Marzo et al. 1998). More recently, intriguing studies report that Bax may cause instability in artificial lipid membranes, suggesting another mechanism by which Bax may permeabilize the outer mitochondrial membrane ( Basanez et al. 1999). An important caveat in this regard is that Bcl-xL did not prevent these effects, whereas it is known to block cytochrome c release from intact mitochondria. The three-dimensional structures of Bid ( Chou et al. 1999; McDonnell et al. 1999) and Bcl-xL ( Muchmore et al. 1996; Sattler et al. 1997) are similar, suggesting that Bid may also form ion channels. However, studies from Desagher et al. 1999 have suggested that Bid requires the cooperation of relatives such as Bax for its effect.
Here we examine the mitochondrial changes associated with apoptotic cytochrome c efflux in the cytosol-based Xenopus cell-free system. Bid and Bax, as well as pro-apoptotic factors endogenous to the Xenopus system, initiated the release of multiple intermembrane space proteins, namely cytochrome c, adenylate kinase (AK) and sulfite oxidase (SOx). Despite these alterations in outer membrane permeability, both the size and ultrastructural morphology of mitochondria remained unchanged, results inconsistent with a mechanism for release of cytochrome c requiring swelling of the mitochondrial matrix and rupture of the outer membrane.
Interestingly, we found that Bid and Bax by themselves produced only a small permeabilization of the outer mitochondrial membrane. However, a much greater permeabilization occurred when mitochondria were incubated with Bid or Bax in cytosol. This secondary permeability change is the result of a novel cytosolic activity, termed permeability enhancing factor (PEF). We propose that the presence or regulation of PEF activity may help decide whether mitochondria can recover from the activation of an apoptotic program.
Materials and Methods
Materials
Horse heart cytochrome c was obtained from Sigma Chemical Co. or from Amersham. SOx was purified from rat and chicken and anti–rat SOx antibodies raised in rabbits as previously described ( Barber and Neame 1990; Neame et al. 1998). Ac-DEVD-CHO and zVAD-fmk were purchased from Biomol and dissolved in DMSO.
Xenopus Egg Extracts and Incubations
Xenopus egg extracts were prepared as described ( Newmeyer 1998; Newmeyer et al. 1994). The final egg cytosolic extracts were approximately two parts cytoplasm and one part buffer A (250 mM sucrose, 20 mM Hepes/KOH, pH 7.5, 50 mM KCl, 2.5 mM MgCl2, 1 mM DTT, 5 μg/ml cytochalasin B, and 50 μg/ml cycloheximide). Incubations were comprised of either crude egg extract that contained mitochondria, or of isolated Xenopus egg mitochondria (1 mg/ml as protein) incubated in either buffer or Xenopus egg cytosol. All cell-free incubations, including those with buffer only, were supplemented with an ATP regenerating system (10 mM phosphocreatine, 2 mM ATP, and 150 μg/ml creatine phosphokinase).
The buffer used in incubations was buffer A supplemented to 100 mM KCl (buffer B), as we found that 80 mM KCl was required for complete dissociation of cytochrome c (but not AK) from mitochondrial membranes after hypotonic lysis or exposure to either Bid or Bax (not shown; Margoliash and Bosshard 1983; Gaikwad et al. 1991; Eskes et al. 1998).
Measurement of DEVDase Activity
To measure caspase-3-like activation, extract aliquots (2 μl) were incubated with DEVD-pNA (N-acetyl-Asp-Glu-Val-Asp-p-nitroanilide, 40 μM; Biomol) in 200 μl of a buffer (250 mM sucrose, 20 mM Hepes/KOH pH 7.5, 50 mM KCl, 2.5 mM MgCl2, and 1 mM DTT) similar to that used to make the egg extracts. Incubations were kept at 22°C and A405 development monitored over 30 min (SPECTRAmax 250 microplate spectrophotometer).
Isolation and Incubation of Rat Liver Mitochondria
Male Sprague Dawley rats (300–500 gm body weight) were killed with carbon dioxide and their livers were removed and placed in ice-cold sucrose solution (300 mM sucrose, 10 mM Tris-HCl pH 7.4, and 1 mM EDTA) for transport. All subsequent procedures were performed at 0 or 4°C. Tissue was placed in ice-cold buffer C (60 mM sucrose, 210 mM mannitol, 10 mM KCl, 0.5 mM DTT, 10 mM succinate, 10 mM Hepes/KOH, pH 7.5, 5 mM EGTA, 1 mM PMSF, 0.1% BSA, 10 μg/ml aprotinin, and 10 μg/ml leupeptin), diced, and aliquots transferred to fresh buffer in a 10-ml Wheaton Potter-Elvehjem tissue grinder. Using a drill-driven Teflon-coated pestle, aliquots were homogenized by 3–5 strokes. Total homogenate was diluted to 100 ml with buffer C and centrifuged twice at 2,000 g for 5 min to remove unbroken cells. Mitochondria were then pelleted from the supernatant by 10 min at 8,500 g (Sorvall RC5C), and washed twice before a final wash in buffer C without PMSF or BSA, then resuspension in a minimal volume. Protein concentration was estimated by the Biuret method.
Light Scatter
Mitochondrial swelling was assessed by measuring the decrease in light scattering at 520 nm (see Fig. 1). Light scatter measurements of reconstituted extracts needed to be performed on aliquots diluted with buffer A (which already constitutes 1/3 of the extract), as A520 of undiluted extract was in the nonlinear range (>3). Aliquots (40 μl) of reconstituted extracts were diluted with 260 μl buffer B and added to a 1-ml cuvette for measurement of A520 in a Hitachi 2000 spectrophotometer. As a positive control, mitochondria were swollen by incubation with the membrane disrupting peptide mastoparan from wasp venom (Sigma Chemical Co.), sometimes used as an inducer of permeability transition ( Pfeiffer et al. 1995).
Figure 1.

Absence of gross mitochondrial swelling with apoptotic cytochrome c release as indicated by light scatter. (A) Xenopus egg mitochondria were incubated in Xenopus egg cytosol at either 0°C or 22°C for 4 h. As positive controls for swelling, mastoparan was added at 200, 300, and 400 μg/ml. At the times indicated, light scatter of mitochondria was assayed by diluting 40 μl of extract with 260 μl buffer B and measuring the average A520 over 1 min. (B) Mitochondria were recovered from the samples measured in A and assessed for cytochrome c translocation by Western blot.
Transmission Electron Microscopy
Samples of extract containing mitochondria (5 μl) were placed in an Eppendorf tube and fixative (5 μl of 4% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4) layered on top, so that after a few minutes on ice the pellets formed a congealed plug. After full fixation (2 h), samples were washed in cacodylate buffer, followed by secondary fixation in 4% osmium tetroxide in 0.1 M cacodylate buffer pH 7.4. Samples were then washed in double distilled water and stained en bloc with 2% aqueous uranyl acetate overnight. Dehydration was carried out stepwise in 30, 50, 70, 90, 95, and 100% acetone/water solutions before embedment in Durcopan (EMS). Thin sections ∼100 nm thick were cut and imaged with transmission electron microscopy on either a JEOL 100CX or Philips 410 microscope. Cross-sectional area measurements were performed using NIH Image program (1.61) and presented as mean ± SD, with statistical significance determined using the paired Student's t test.
Field Emission In-Lens Scanning Electron Microscopy
High resolution scanning electron microscopy was performed using the recent technology afforded by high-brightness sources used in in-lens field emission instruments (FEISEM), which provide resolution to 1 nm ( Allen et al. 1996). Reconstituted extracts were incubated at 22°C with BaxΔTM or alamethicin (2 μg/ml; Sigma Chemical Co.) to induce cytochrome c release (confirmed by DEVDase activation and Western blot). Aliquots (10 μl) were mixed with buffer B (400 μl) to wash the mitochondria, and then placed on 5-mm-square silicon chips in chambers constructed from Eppendorf tubes. Mitochondria were spun onto the chips (5 min, 14,000 g), followed by removal of supernatant, and fixation in 100 μl 150 mM sucrose, 80 mM Pipes-KOH, pH 6.8, 1 mM MgCl2, 2% paraformaldehyde, 0.25% glutaraldehyde on ice, followed by postfixation in OsO4 in 0.2 M cacodylate buffer, pH 7.4. After 10 min in 1% aqueous uranyl acetate, the specimens were dehydrated through an ethanol series, and critical point dried from CO2 using Arklone (Freon112; ICI), as intermediate solvent. The chips were then sputter coated with 4 nm chromium, and examined in a Topcon ISI DS 130F field emission, in-lens SEM at 30 kV accelerating voltage, with digital image acquisition.
Western Blotting
To determine mitochondrial and cytosolic content of various proteins, incubation aliquots were centrifuged (12,000 g) to pellet mitochondria and the supernatant carefully removed. For SDS-PAGE, supernatant samples (10 μl) were mixed with 2× loading buffer (10 μl) and mitochondrial pellets were resuspended in a volume of 1× loading buffer equivalent to the initial volume of the centrifuged aliquot. Supernatant (20 μl) and mitochondrial (10 μl) samples were heated at 95°C for 5 min, loaded onto 12% SDS–polyacrylamide gels for electrophoresis, and then transferred to nitrocellulose (BioRad) membranes. Membranes were then blocked for 1 h in TBST (25 mM Tris-HCl, pH 7.4, 140 mM NaCl, 27 mM KCl, and 0.02% Tween 20) containing 5% nonfat dried milk. Membranes were probed with monoclonal anti–cytochrome c antibody (clone 7H8.2C12; PharMingen), rabbit antiserum to rat SOx, mouse anti-mtHsp70 antibody (kindly provided by Richard Morimoto), or rabbit anti-serum against bovine heart Rieske's iron-sulfur protein (MW 21406; kindly provided by Diego Gonzalez, Northwestern University, Evanston, IL). Recognized proteins were detected using horseradish peroxidase-labeled secondary antibodies (Amersham) and enhanced chemiluminescence (Amersham).
Adenylate Kinase Activity
Mitochondrial AK activity was measured by a modification of the method of Schmidt et al. 1984. Extract aliquots (50 μl) containing ∼0.05 mg mitochondrial protein were pelleted at 12,000 g for 3 min, and the pellet washed twice in 800 μl buffer D (60 mM sucrose, 210 mM mannitol, 10 mM KCl, 0.5 mM DTT, 10 mM succinate, 10 mM Hepes/KOH, pH 7.5, and 5 mM EGTA) to remove contaminating cytosolic AKs. The mitochondrial pellets were lysed with 50 μl of 1% Triton X-100 in buffer D to release remaining AK and the sample stored at –80°C. AK activity was measured in a mixture composed of 1 ml of 130 mM KCl, 6 mM MgSO4, 100 mM Tris-HCl, pH 7.5, 15 μl of 0.1 M NADH, and 5 μl each of 0.1 M ATP, 100 mM phosphoenol pyruvate, 1 mM rotenone, 1.5 mM oligomycin, a mixture of pyruvate kinase and lactate dehydrogenase (80 U/ml each), and 0.15 M AMP. 200 μl of buffer mix was added to 6 μl of sample in a microtiter plate. The absorbance decrease of NADH was measured at 366 nm in a microtiter plate reader (SPECTRAmax 250) for 10 min at 22°C. The rates were calculated (SOFTmax PRO) and calibrated against chicken muscle myokinase (Sigma) activity. Rates obtained in the presence of the inhibitor di-adenosine pentaphosphate (400 μM; Sigma) were minimal and subtracted as background.
Complex IV Accessibility (Cytochrome c Oxidase Latency Assay)
To examine the intactness of the outer mitochondrial membrane we measured the accessibility of cytochrome c oxidase to exogenous cytochrome c. The assay (modified from Wojtczak et al. 1972) involved the addition of extract (2 μl) containing mitochondria (final protein concentration ∼9 μg/ml) to 450 μl assay solution in a 1-ml cuvette. The assay solution consisted of 100 μM horse heart cytochrome c, 60 mM KCl, 125 mM sucrose, 20 mM Tris-HCl, pH 7.4, 1 μΜ each of carbonyl cyanide m-chlorophenylhydrazone (CCCP), rotenone and antimycin, and sufficient sodium dithionite crystals to obtain near fully reduced cytochrome c (A550/A565 between 6 and 10). On addition of mitochondria, samples were quickly mixed and A550 recorded every 6 s (Hitachi 2000). The rate of cytochrome c oxidation at 550 nm integrated over 30 s reflected complex IV accessibility to the solution. Maximal complex IV accessibility was obtained by premixing 2 μl extract with 2 μl Triton X-100 (2%) and used to calculate percent complex IV accessibility.
Complex III Accessibility (Quinol–Cytochrome c Assay)
An additional method used to assess permeability of the mitochondrial outer membrane was the reduction of exogenous cytochrome c by complex III (cytochrome bc1; Degli Esposti et al. 1982). Mitochondria from treated extracts (250 μl) were washed by dilution in 1.25 ml buffer E (125 mM sucrose, 60 mM KCl, 20 mM Tris-HCl, pH 7.4), followed by pelleting for 5 min at 12,000 g. The mitochondrial pellet was resuspended in 250 μl buffer E and 25 μl (final concentration 0.15 mg/ml) added to 300 μl buffer E containing 2 mM potassium cyanide (to block oxidation by complex IV). Decyl benzoquinol (DBH2, 5 μl of 3.5 mM stock in ethanol) was added and the sample sealed with parafilm and mixed. Finally, 10 μl of 2.5 mM ferricytochrome c (horse heart; Sigma Chemical Co.) was added with a Hamilton syringe and the rate of cytochrome c reduction at 550 nm was integrated over 30 s.
Production of Recombinant Bcl-2 Family Proteins
Bcl-2 and the control β-galactosidase were expressed in baculovirus-infected Sf-9 cells, and lysates were prepared as described ( Newmeyer et al. 1994). Human Bid (hBid) cDNA was amplified with PCR and subcloned into the BamHI and NotI sites in pGEX4T-1 (Pharmacia). BL21(DE3) cells were transformed and GST-hBid induced with 1 mM isopropyl β-d-thiogalactopyranoside (IPTG) at 37°C for 4 h. The induced protein was incubated with glutathione sepharose (Pharmacia) and eluted with 20 mM reduced glutathione. Eluted GST-hBid was digested with thrombin (Pharmacia) to cleave GST, which was subsequently removed by incubation with a new batch of glutathione sepharose. The hBid product was >90% pure with minor contamination of GST. Human caspase-8 lacking the prodomain in PET15b vector (Novagen) was expressed in BL21(DE3) cells by induction with 1 mM IPTG at 37°C for 4 h and affinity-purified with Ni++-NTA (Qiagen). The protein was eluted with 50 mM and 250 mM imidazole, and the 250 mM imidazole eluate further purified on a 1 ml Q-Sepharose FF HiTrap column (Pharmacia) using a 0–0.6 M KCl linear gradient. The product was fully processed and electrophoretically >90% pure. tBid was made by incubation of recombinant human Bid with active recombinant caspase-8 at 37°C for 2–4 h in 25 mM Hepes-KOH, pH 7.5, 80 mM KCl, and 10 mM DTT. (The tBid preparation was used without removing caspase-8; however, control experiments showed that caspase-8 alone did not release cytochrome c from mitochondria.) COOH-terminally deleted Bax (BaxΔTM) was produced as previously described ( Finucane et al. 1999). Bak BH3 domain (NH2-GQVGRQLAIIGDDINR-COOH) and mutant (NH2-GQVGRQAAIIGDDINR-COOH) peptides were kindly provided by S. Kataoka and Y. Tokoro (Kirin Brewery Co., Japan).
Characterization of Cytosolic PEF
Cytosol was size-fractionated using Millipore Ultrafree-MC centrifugal ultrafiltration membranes. Cytosol was dialyzed against 4 changes of buffer B (500 ml each) over 24 h at 4°C in 3500 MW cutoff dialysis tubing (Spectra/Por). Cytosolic proteins were hydrolyzed by incubation with 0.1 mg/ml proteinase K or 1.0 mg/ml trypsin (Sigma Chemical Co.) for 30 min at 22°C, followed by addition of phenylmethylsulfonyl fluoride (1 mM). Heat-denatured cytosol was prepared by heating at 50°C for 15 min or 95°C for 1 min, followed by clearing at 12,000 g for 5 min.
Results
Absence of Gross Mitochondrial Swelling Accompanying Cytochrome c Release from Xenopus Mitochondria
To determine if mitochondrial swelling might participate in the efflux of mitochondrial cytochrome c during apoptosis in the Xenopus cell-free system, we assessed mitochondrial swelling by three methods. First, we measured the light scatter of mitochondrial suspensions. As mitochondria swell, their refractive index decreases, resulting in a drop in spectrophotometric absorbance at higher wavelengths ( Knight et al. 1981). Xenopus mitochondria were induced to release cytochrome c by incubation in Xenopus cytosol for 4 h at 22°C. We found that these mitochondria did not exhibit decreased light scattering ( Fig. 1) despite complete cytochrome c release. As a control to determine if mitochondrial swelling could be measured with Xenopus mitochondria using this technique, we used mastoparan, a membrane disrupting peptide that causes mitochondrial swelling associated with permeability transition ( Nicolay et al. 1994; Pfeiffer et al. 1995; Fig. 2) and cytochrome c release ( Ellerby et al. 1997). As shown in Fig. 1, mastoparan at 300 and 400 μg/ml rapidly induced both a decrease in light scattering and loss of mitochondrial cytochrome c. We conclude that the constitutive pro-apoptotic factor present in the Xenopus cell-free system initiates cytochrome c release by a mechanism independent of mitochondrial swelling, as measured by light scatter.
Figure 2.
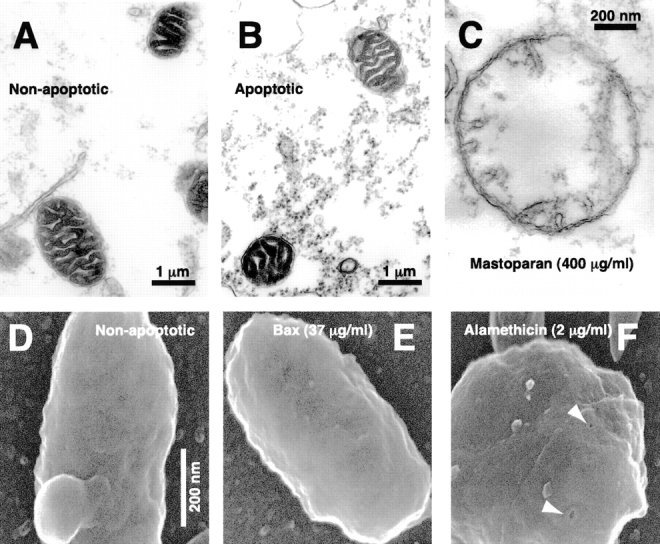
Apoptotic mitochondria exhibit normal size and morphology on electron microscopy. (A–C) Crude extracts were incubated at 0°C as a non-apoptotic control (A) or incubated at 22°C to induce spontaneous cytochrome c release (B). Immediately after cytochrome c release in the sample taken at 2.75 h, aliquots were fixed and examined by transmission electron microscopy. (C) A swollen mitochondrion taken from a further sample treated with mastoparan (400 μg/ml). (D–F) FEISEM of reconstituted extracts incubated at 0°C for 3 h, 40 min (D, non-apoptotic) or 22°C in the presence of 35 μg/ml BaxΔTM for 1 h, 40 min (E). (F) Alamethicin addition (2 μg/ml, 40 min) caused both cytochrome c release and the formation of 10–13-nm pores (arrows). Bar, (D–F) 200 nm.
Second, we used transmission electron microscopy to compare the size and morphology of apoptotic and non-apoptotic mitochondria ( Fig. 2A and Fig. B). Crude egg extracts were incubated at 22°C to induce mitochondrial cytochrome c loss. In the Xenopus system, essentially all mitochondrial cytochrome c becomes cytosolic ∼30 min before the detection of DEVDase activation ( Kluck et al. 1997a, Kluck et al. 1997b). Samples fixed immediately after full DEVDase activation exhibited no obvious mitochondrial swelling or morphological changes when compared with (non-apoptotic) extracts that had been incubated on ice. Quantitative assessment of mitochondrial cross-sectional area indicated no difference (P = 0.32, n = 38) between apoptotic (0.75 ± 0.25, arbitrary units) and non-apoptotic mitochondria (0.77 ± 0.26). The integrity of the outer membrane appeared equal in apoptotic and non-apoptotic samples. Comparison of several individual mitochondria by three-dimensional electron tomography (Renken, C., and G. Perkins, unpublished data) also failed to reveal obvious changes in outer membrane ultrastructure. Mitochondria fixed at 3 h after cytochrome c release also were unswollen (not shown), indicating that neither activated caspases nor other cytosolic factors cause gross morphological mitochondrial changes.
The majority of mitochondria in Xenopus extracts was observed to be in the condensed form (dark matrix and wide cristae), rather than the orthodox configuration (light matrix and narrower cristae). A condensed matrix is thought to reflect low respiration ( Hackenbrock 1968; Ord and Smith 1982). Thus, the condensed morphology of Xenopus extract mitochondria may indicate low respiratory rates in response to the high external energy status caused by addition of the ATP regenerating system (see Materials and Methods). Alternatively, unfertilized Xenopus eggs may contain a factor similar to that implicated in the condensed mitochondrial morphology of unfertilized sea urchin eggs ( Innis et al. 1976). Condensed mitochondria have recently been observed in association with apoptosis ( Mancini et al. 1997; Zhuang et al. 1998). With the opening of the permeability transition pore, mitochondria in the condensed state are converted irreversibly (in the presence of sucrose) to the orthodox conformation. Thus, simply on ultrastructural grounds, as both apoptotic and non-apoptotic Xenopus mitochondria are condensed, it is unlikely that a permeability transition had taken place.
To examine further the ultrastructure of apoptotic mitochondria, we used a third approach, a high-resolution form of scanning electron microscopy (FEISEM; Allen and Goldberg 1993; Allen et al. 1996). As Fig. 2 shows, FEISEM revealed that mitochondria depleted of cytochrome c were of similar size and appearance to control mitochondria. Outer membrane discontinuities were not observed. This technique did reveal outer membrane pores, 10–13 nm in diameter, in mitochondria treated with the antibiotic peptide alamethicin ( Fig. 2 F), which also induced complete release of cytochrome c (Kluck and Newmeyer, unpublished). However, no such pores were observed in mitochondria depleted of cytochrome c by the factors present in Xenopus cytosol, or by Bid or Bax, suggesting that if these factors form pores, they are either transient or considerably smaller than those formed by alamethicin ( Woolley and Wallace 1992). In summary, multiple approaches ( Fig. 1 and Fig. 2) demonstrated the absence of gross ultrastructural changes in mitochondria accompanying cytochrome c release in the Xenopus cell-free system.
Adenylate Kinase Loss from the Intermembrane Space Precedes Caspase Activation
To continue investigating the mitochondrial events leading to cytochrome c release, we next determined whether other soluble proteins present in the intermembrane space were released during apoptosis. One such protein is adenylate kinase (25,200 Da). The amount of mitochondrial adenylate kinase (AK) released into the cytosol is difficult to measure because cytosol already contains ∼50% of the cell's AK activity (not shown). However, the AK activity remaining in the mitochondrial pellet can readily be assayed. We incubated mitochondria in cytosol for various times, washed them free of cytosol, and measured adenylate kinase content in the final pellet. As Fig. 3 shows, with apoptosis induced by Xenopus cytosol, almost all AK activity was lost from mitochondria before DEVDase activation, with kinetics similar to those observed for cytochrome c.
Figure 3.

Adenylate kinase and sulfite oxidase loss from mitochondria, like loss of cytochrome c, precedes DEVDase activation. Xenopus mitochondria were incubated in either Xenopus cytosol or buffer, at either 0 or 22°C for 4 h. At the times indicated, 100-μl aliquots were centrifuged and the mitochondrial pellets washed twice, lysed in 1% Triton X-100, then assayed for adenylate kinase activity (A). Data are means and SD of triplicates. (B) At the times indicated, aliquots (2 μl) were assessed for DEVDase activity to determine the time of cytochrome c release. Data are representative of 6 experiments. (C) Rat liver mitochondria (5 mg/ml) were incubated in Xenopus egg cytosol at either 0 or 22°C for 8 h. At the indicated times, aliquots were assessed for cytochrome c and sulfite oxidase translocation by Western blot. Data are representative of three experiments.
Sulfite Oxidase Is Coreleased with Cytochrome c from Rat Liver Mitochondria
Another mitochondrial protein, sulfite oxidase (SOx), is found only in the intermembrane space. As antisera raised to rat SOx failed to recognize the Xenopus protein, we determined whether rat liver mitochondria incubated in Xenopus extracts coreleased SOx with cytochrome c. Many of the apoptotic features of the Xenopus egg cell-free system were duplicated if rat liver mitochondria were incubated in Xenopus cytosol. That is, mitochondrial cytochrome c was released after a period of incubation at 22°C, but not at 0°C ( Fig. 3). (It should be noted that, for reasons unknown, some preparations of rat liver mitochondria failed to exhibit cytochrome c release in response to incubation with Bid, Bax or Xenopus egg cytosol.) Near the time of rat cytochrome c release, Xenopus cytosolic DEVDases became activated, and coincubated rat liver nuclei exhibited chromatin condensation, margination and beading, with subsequent nuclear disintegration (not shown) typical of apoptosis in the Xenopus system. Swelling of the rat liver mitochondria was not observed by cross-sectional area measurements of transmission electron micrographs (P = 0.5, n = 21), with non-apoptotic mitochondria showing an average area of 2.45 ± 1.1 (arbitrary units), compared with 2.45 ± 1.0 for apoptotic mitochondria. Light scatter measurements (not shown) also demonstrated a lack of swelling. Despite this absence of swelling, we found that SOx (a dimer of 52 kD subunits) was coreleased from the intermembrane space with cytochrome c ( Fig. 3) and with AK (not shown). Loss of these proteins from mitochondria occurred over a period of under 1 h.
As SOx is normally found only in the intermembrane space, it was of interest to examine whether the presence of this protein in cytosol altered the pro-apoptotic activity of cytochrome c. Purified rat or chicken SOx added to Xenopus cytosol, either in the presence or absence of cytochrome c, did not alter DEVDase activation (not shown), indicating that SOx does not affect caspase activation. (It is unlikely that released mitochondrial AK has any effect on the activation of caspases, as much of the cellular AK is already present is cytosol.) Western blot analysis of rat SOx released from mitochondria suggested that this protein was degraded in apoptotic cytosol, but not in either buffer ( Fig. 3) or an ultrafiltrate obtained by passing cytosol through a 10-kD cutoff membrane (not shown). Chicken SOx was cleaved to a 45-kD fragment after a 1-h incubation with either caspase-3 or cytochrome c–activated cytosol, and this cleavage was blocked by the caspase inhibitor Ac-DEVD-CHO (1 μM, not shown). However, while chicken SOx appeared to be a good caspase substrate, rat SOx was cleaved in a much less efficient manner to several products of different molecular mass. In summary, the corelease of cytochrome c, AK and SOx with apoptosis, argues against an outer membrane transport channel specific for cytochrome c, and is consistent with the formation of a nonspecific opening or protein channel in the outer mitochondrial membrane.
Complex IV Accessibility Changes Coincide with the Release of Cytochrome c and Are Prevented by Bcl-2
To examine in more detail the apoptotic changes occurring in mitochondrial outer membrane permeability, we measured the accessibility of exogenous cytochrome c to complex IV (cytochrome c oxidase), present on the outside face of the inner mitochondrial membrane. When detergent-treated or mechanically disrupted mitochondria are suspended in a solution containing reduced cytochrome c, this exogenous cytochrome c becomes gradually oxidized by complex IV, leading to a steady change in the absorption spectrum of cytochrome c. In intact mitochondria, this change does not occur, because the permeability barrier of the outer membrane prevents the interaction of exogenous cytochrome c with the catalytic site exposed within the intermembrane space ( Wikstrom and Casey 1985). The rate of change of absorbance at 550 nm is taken as a measure of the accessibility of complex IV, and hence the permeability of the outer membrane. The values obtained were expressed as percentages of the rate of change of A550 observed after exposure of mitochondria to 1% Triton X-100, as this treatment was assumed to cause complex IV to be completely accessible. Carefully isolated Xenopus egg and rat liver mitochondrial preparations exhibited <1% of this value, while osmotic lysis of the outer membrane gave complex IV accessibility values close to 100%.
We used this assay to examine the permeability changes occurring in mitochondria incubated in Xenopus cytosol. With the onset of apoptosis, complex IV accessibility increased at around the time of cytochrome c release ( Fig. 4), indicating that outer membrane permeability to cytochrome c in apoptotic mitochondria (in the presence of cytosol) is bidirectional. The addition of Bcl-2 to the extracts blocked both the increase in complex IV accessibility and the release of endogenous cytochrome c.
Figure 4.

Outer mitochondrial membrane permeability to cytochrome c is bidirectional in the Xenopus cell-free system. Xenopus mitochondria were incubated in cytosol at 0 or 22°C for 4 h in the presence of Sf-9 cell lysates (2% vol/vol) containing either β-galactosidase or Bcl-2 expressed after baculovirus infection. At the times indicated, 2-μl aliquots were assayed for either DEVDase activity (top) or complex IV accessibility (bottom).
Both Bid and Bax Induce Mitochondrial Cytochrome c Release without Gross Mitochondrial Swelling
Next, we examined the effects of two known pro-apoptotic human proteins, Bid and Bax. Addition of recombinant Bid or Bax to reconstituted Xenopus egg extracts (mitochondria mixed with cytosol) caused the rapid release of mitochondrial cytochrome c ( Fig. 5) and the activation of DEVDases (not shown). Cytochrome c release was complete and highly synchronous. Recombinant Bax exhibited a 1,000-fold lower specific activity than recombinant Bid (not shown); this may be due to the absence of the Bax transmembrane domain or to undefined differences in the way these proteins are expressed in bacteria. Cleavage of full-length Bid by caspase-8 (see Materials and Methods) to give truncated Bid (tBid) consistently enhanced its potency (not shown), as reported by others ( Li et al. 1998; Luo et al. 1998).
Figure 5.

In the absence of cytosol, Bid and Bax release cytochrome c and adenylate kinase, but do not develop high complex IV accessibility. Xenopus egg mitochondria were incubated in buffer B or cytosol alone, or with added tBid (170 ng/ml) or Bax (37 μg/ml) for 4 h at 22°C. At the indicated times, aliquots were measured for (A) mitochondrial cytochrome c content, (B) mitochondrial adenylate kinase activity, and (C) complex IV accessibility. Data are single measurements except those for adenylate kinase (mean and SD of triplicates), and are representative of two experiments. (D) PEF acts subsequent to tBid, and tBid effects on the outer membrane are sustained. Xenopus mitochondria were first incubated at 22°C in buffer B with or without tBid (170 ng/ml) for 2 h to allow tBid to cause complete cytochrome c release (not shown). Mitochondria were then washed in buffer B and resuspended in fresh cytosol, and aliquots were tested every 15 min for complex IV accessibility.
Transmission electron microscopy of mitochondria depleted of cytochrome c by treatment with Bax or caspase-activated Bid revealed no significant changes in mitochondrial size (P = 0.44, n = 21 and P = 0.33, n = 21 for comparisons of control mitochondria with Bid- and Bax-treated mitochondria, respectively). The mean cross-sectional areas of control, Bid- and Bax-treated mitochondria, were 0.684 ± 0.2, 0.70 ± 0.2, and 0.71 ± 0.2, respectively (arbitrary units). Thus, Bid and Bax, like the factors present in Xenopus egg cytosol, induced cytochrome c release without gross changes in mitochondrial morphology.
Adenylate Kinase Is Coreleased with Cytochrome c after Treatment with Bid and Bax
As cytosol-treated mitochondria coreleased AK with cytochrome c ( Fig. 3), we examined whether this was also the case for tBid- and Bax-treated mitochondria. Indeed, tBid and Bax both released AK with kinetics similar to the release of cytochrome c ( Fig. 5). This release occurred in the presence of either buffer or cytosol. The constitutive presence of large amounts of AK activity in the cytosol prevented us from measuring the AK released from mitochondria incubated in cytosol; however, for mitochondria incubated in buffer, the amount of AK released into the supernatant of tBid and Bax-treated mitochondria accounted for that lost from the mitochondrial pellet (not shown), indicating that the protein was translocated, rather than inactivated. These data support the hypothesis that similar cytochrome c–releasing mechanisms are used by various cytochrome c–releasing proteins.
Cytosol Greatly Increases Complex IV Accessibility Induced by Bid and Bax
Cytochrome c release from Xenopus mitochondria incubated in Xenopus cytosol is associated with high accessibility of the inner membrane protein cytochrome c oxidase (complex IV) to exogenous cytochrome c ( Fig. 4). This high accessibility is also seen in Xenopus mitochondria treated with tBid and Bax in the presence of Xenopus cytosol ( Fig. 5 C). It was surprising to note, however, that mitochondria treated with tBid and Bax in buffer displayed an almost undetectable level of accessibility of complex IV to exogenous cytochrome c ( Fig. 5 C), despite their complete loss of endogenous cytochrome c ( Fig. 5 A). These results imply that a novel cytosolic activity, which we call Permeability Enhancing Factor, or PEF, is responsible for a striking increase in complex IV accessibility beyond that produced by factors such as tBid or Bax.
PEF Can Act Downstream of tBid
PEF might function either simultaneously with factors like tBid or subsequent to their action. To address this question, we did the following experiment. Mitochondria were first depleted of cytochrome c by an initial incubation with tBid in buffer, and then washed and resuspended in cytosol for a second incubation. As Fig. 5 D shows, complex IV accessibility rapidly increased during this second incubation in cytosol. This is consistent with data showing that tBid stays strongly associated with mitochondria after washing (Kuwana, O., T. von Ahsen, and D.D. Newmeyer, unpublished results), but further implies that the effect of tBid is sustained. Some samples were incubated in fresh buffer before reincubation with cytosol, in an attempt to encourage maximum repair of the tBid-induced permeability. However, this intermediate incubation had no effect (not shown). We conclude from this experiment that PEF can function subsequent to tBid-induced cytochrome c translocation. This experiment also demonstrates that PEF activity is not dependent on downstream effects of cytochrome c release, because the cytosol used in Fig. 5 D contained neither cytochrome c (nor any other soluble proteins from the mitochondrial intermembrane space) nor active caspases.
Cytosol Increases Complex III Accessibility
It could have been argued that the quantitative differences in accessibility of complex IV reflected some effect specific for that enzyme complex, rather than changes in the permeability of the outer membrane. To rule out this possibility, we measured the accessibility of another mitochondrial inner membrane component, complex III. This assay is similar in principle to the complex IV assay, except that cytochrome c reduction rather than oxidation is measured, and an electron donor (DBH2) is added to ensure saturating substrate (see Materials and Methods). These experiments showed that cytosol-induced cytochrome c release was accompanied by an increase in the accessibility of both complexes III and IV ( Fig. 6, lane 4), confirming the bidirectional permeability of the outer membrane under these conditions. Again, however, in the presence of buffer alone, tBid induced a nearly undetectable accessibility of complexes III and IV ( Fig. 6, lane 2), which was greatly enhanced in the presence of cytosol ( Fig. 6, lanes 6 and 7). The ability of cytosol to increase outer membrane permeability was not due to a feedback effect of cytochrome c–activated caspases, as Ac-DEVD-CHO (10 μM) did not prevent the development of high complex III and IV accessibility (not shown).
Figure 6.

Cytosol (PEF) increases both complex III and IV accessibility after tBid-induced cytochrome c release. Mitochondria were incubated with or without tBid in either buffer B or cytosol, at 0 or 22°C. At suitable times (1.7, 2.7, or 3.7 h), mitochondria were pelleted and analyzed by Western blot for cytochrome c translocation (bottom) or washed and resuspended in buffer E for measurement of complex III (top) and IV (middle) accessibility (arbitrary units). For comparison between assays, mitochondria were lysed by incubation in water for 30 min on ice. These osmotically lysed mitochondria retain their cytochrome c due to the low salt content (see Materials and Methods). Data are mean and SD for duplicate measurements and are representative of three experiments. Note: The time course of permeabilization by PEF is slower in Fig. 6 than Fig. 5; this merely reflects a normal variation between Xenopus extracts in time of onset of apoptotic changes ( Newmeyer et al. 1994; Farschon et al. 1997).
In summary, the enhancing effect of cytosol on accessibility of complexes III and IV ( Fig. 4 Fig. 5 Fig. 6) is explained by the presence of a factor in Xenopus cytosol, which we have named permeability enhancing factor (PEF). PEF does not release cytochrome c by itself, but rather enhances outer membrane permeability after a pro-apoptotic factor has caused the release of endogenous cytochrome c.
tBid and PEF Do Not Permeabilize the Mitochondrial Inner Membrane
To address the possibility that PEF increases accessibility of complexes III and IV by grossly perturbing the integrity of mitochondria, we examined whether a soluble mitochondrial matrix protein, mtHsp70, is released after the actions of tBid and PEF. First, to show that mtHsp70 can be released if the inner mitochondrial membrane is permeabilized, we treated mitochondria with increasing concentrations of digitonin and followed the redistribution of mtHsp70 from the mitochondrial pellet fraction to the soluble supernatant. Fig. 7 A shows that cytochrome c is released at a low concentration (0.1%) of digitonin, but that mtHsp70 becomes soluble at higher digitonin concentrations (0.2%). Only at still higher concentrations (0.6%) did the membrane-associated Rieske protein become solubilized, showing that at the intermediate concentrations sufficient to release mtHsp70, digitonin did not solubilize the mitochondrial membranes, but merely permeabilized them. Next, we examined the behavior of mtHsp70 in mitochondria induced to release cytochrome c by cytosol or by tBid (in the presence or absence of cytosol). As seen in Fig. 7 B, mtHsp70 remained in the mitochondrial pellet fraction under all conditions, even in the presence of cytosol containing active PEF. We conclude that cytosolic factors and tBid can permeabilize the outer mitochondrial membrane while leaving the inner membrane intact.
Figure 7.

tBid and PEF preserve the integrity of the mitochondrial matrix compartment, as measured by retention of a soluble mitochondrial matrix protein, mtHsp70. (A) mtHsp70 is released when mitochondria are treated with intermediate concentrations of digitonin. Xenopus egg mitochondria were treated with increasing concentrations of digitonin to selectively permeabilize the outer, then the inner mitochondrial membranes. After two hours incubation, the contents of the pellet and supernatant fractions were analyzed by immunoblotting (left panel) with antibodies to cytochrome c, mtHsp70, and the inner membrane-associated Rieske protein (part of complex III). Also, accessibility of complex IV was measured as described above (right panel). Note that mtHsp70 is released by digitonin at 0.2%, a concentration insufficient to solubilize the Rieske protein, but more than sufficient to release cytochrome c. (B) mtHsp70 is not released by tBid and PEF, despite full release of cytochrome c and full accessibility of complex IV. Xenopus mitochondria were treated with 170 ng/ml tBid, in either the presence or absence of cytosol, and either at 0 or 22°C, as indicated. DEVD-CHO (10 μM) was added to cytosol incubations to minimize PEF degradation and thus maximize its activity. After 3 h of incubation, the contents of the mitochondrial pellet were analyzed by immunoblotting with antibodies to cytochrome c, mtHsp70 and Rieske protein (left), and the accessibility of complex IV (right). The samples were taken well after full cytochrome c release from mitochondria, which occurred in this experiment at 1 h in the tBid-treated sample and at 2 h in cytosol at 22°C (not shown). Note that mtHsp70 is retained in the pellet fraction despite full cytochrome c release and increased complex IV accessibility (reflecting PEF activity). (The bands stained by anti-Rieske antibody in cytosol are nonmitochondrial cross-reacting proteins.)
PEF Characteristics
To begin characterizing PEF, we treated cytosol in various ways before incubating it with Xenopus mitochondria and tBid ( Fig. 8). Treatment with proteinase K ( Fig. 8), trypsin, or heat (50°C, 15 min, or 95°C, 1 min; not shown) greatly diminished or abolished cytosol's ability to increase complex IV accessibility, indicating that PEF is proteinaceous. PEF activity was not observed in the ultrafiltrate obtained by passing cytosol through a 300-kD cutoff membrane, and was not lost on dialysis, suggesting that PEF is macromolecular and possibly present in a large complex. The complex IV accessibility produced by PEF accumulates over a few hours to approach that seen in mitochondria treated with Triton X-100 (see Materials and Methods) and digitonin (not shown). PEF activity is reduced substantially on dilution of cytosol. A PEF-like activity may also be present in mammalian cells, as a lysate (40 mg/ml) of human Jurkat cells caused an increase in complex IV accessibility of isolated Xenopus mitochondria (not shown). This Jurkat cell PEF-like activity was observed in the presence or absence of tBid, suggesting that the Jurkat cell lysate contained endogenous cytochrome c–releasing factors.
Figure 8.

PEF is large and proteinaceous. Mitochondria were incubated with tBid (170 ng/ml) in the presence of either buffer B, ultra- or micro-filtered cytosol (10-, 50-, 300-kD, and 0.22-μm filters) normal cytosol, cytosol diluted with buffer B, proteinase K–treated cytosol, or dialyzed cytosol. The control (first sample) did not contain tBid. Ac-DEVD-CHO (10 μM) was added to all samples to maximize PEF activity (see Fig. 9). At the indicated times, aliquots were either pelleted and analyzed for mitochondrial cytochrome c content (bottom) or tested for complex IV accessibility (top).
PEF Is Destroyed by Caspases
To address the possibility that PEF is a substrate of caspases, we examined the effect of cytochrome c–activated cytosol on fresh Xenopus mitochondria. Fresh cytosol was first incubated for 3 h with a low level (0.5 μM) of cytochrome c to activate cytosolic caspases (DEVDases). Subsequent addition of mitochondria to this activated cytosol resulted in rapid loss of mitochondrial cytochrome c and AK (by 1 h, not shown). The factor responsible for this cytochrome c efflux is likely to be a caspase-activated Bid-like factor (Kuwana, 1998), rather than caspases themselves, as addition of caspase inhibitors just before the addition of mitochondria did not block cytochrome c release. Strikingly, however, cytochrome c–activated cytosol did not produce high complex IV accessibility, even though it caused the complete release of cytochrome c from mitochondria ( Fig. 9). Thus, this cytochrome c–treated cytosol contained activated DEVDases, inactivated PEF, and activated Bid-like activity.
Figure 9.

PEF is inactivated by caspases. Mitochondria were incubated with either normal cytosol at 0 or 22°C (left), or with cytosol that had been activated by preincubation (3 h) with 0.5 μM horse heart cytochrome c (HHCc) (right). Samples in activated cytosol contained either vehicle (0.5% DMSO), Ac-DEVD-CHO (10 μM) or zVAD-fmk (100 μM). One sample (mock-activated), had Ac-DEVD-CHO (10 μM) added before incubation with HHCc. At the indicated times, aliquots (2 μl) were assessed for complex IV accessibility. Mitochondria in the three activated cytosol samples had lost their cytochrome c by 1 h due to tBid-like activity (not shown). Data are representative of three experiments.
Further, we found that the addition of caspase inhibitors to cytosol substantially enhanced and prolonged PEF activity. Typically, in the presence of cytosol, complex IV accessibility increased over 1–2 h after cytochrome c release and then reached a plateau ( Fig. 9). If caspase inhibitors were added, however, this plateau was surpassed and complex IV accessibility approached 100% of that seen with Triton-treated mitochondria ( Fig. 9). In summary ( Fig. 10), cytosolic PEF begins to act on mitochondria once they have lost their cytochrome c, and over 2–3 h, progressively allows a higher permeability of the outer membrane to exogenous cytochrome c. If caspases are activated by the released cytochrome c, PEF activity is inactivated after perhaps 1–2 h. Addressing whether this inactivation by caspases might be direct or indirect awaits the molecular identification of PEF.
Figure 10.

Model of two types of mitochondrial outer membrane permeability observed in Xenopus extracts during apoptosis. Mitochondria treated with tBid or Bax in buffer undergo efflux of endogenous cytochrome c (12 pmol/μl mitochondria) across the outer membrane, with limited permeability to exogenous cytochrome c. Additional exposure to cytosol (PEF) allows high permeability to exogenous cytochrome c (60 nmol/μl mitochondria per min, as measured by the rate of the complex IV reaction). Thus, this increased exchange of endogenous cytochrome c is at least ∼5,000 times the rate of cytochrome c efflux after tBid or Bax treatment, assuming that all of the mitochondrial cytochrome c is released over 1 min. PEF begins to act immediately after tBid–induced permeabilization, but is inactivated over 1–2 h by caspase activation. The inner membrane appears unaffected.
Discussion
We have investigated how mitochondrial cytochrome c is released during apoptosis in a cell-free system based on Xenopus egg extracts ( Kluck et al. 1997a, Kluck et al. 1997b; Kuwana et al. 1998). Four pro-apoptotic cytochrome c–releasing activities were examined: (a) the spontaneous cytochrome c–releasing activity of Xenopus cytosol, (b) the Bid-like activity of cytochrome c–activated Xenopus cytosol, (c) recombinant human Bid activated with caspase-8, and (d) recombinant Bax. These factors all caused a permeability of the outer membrane that allowed the corelease of AK (and SOx, where measurable) with cytochrome c. None of these cytochrome c–releasing factors caused detectable mitochondrial swelling ( Fig. 1 and Fig. 2). From this and other data, we conclude that neither mitochondrial permeability transition nor hyperpolarization occur in this apoptotic system.
A potentially attractive mechanism for the release of multiple proteins from the intermembrane space would involve swelling of the mitochondrial matrix, leading to mechanical disruption of the outer mitochondrial membrane. A single break in the outer mitochondrial membrane is theoretically sufficient to allow cytochrome c to diffuse rapidly out of mitochondria down its concentration gradient (10 mM in intermembrane space and cristae). Ruptures in the outer membrane ensue if the matrix swells past the point that the outer membrane can expand. Such matrix swelling is proposed to occur during apoptosis in response either to the formation of a 1.5-kD permeability transition pore in the inner membrane ( Bernardi 1996; Petit et al. 1996; Susin et al. 1997) or to hyperpolarization of the inner membrane ( Vander Heiden et al. 1997, Vander Heiden et al. 1999).
However, mitochondrial swelling has not been observed as a frequent accompaniment to apoptosis. In the studies reported here, light scatter measurements ( Fig. 1) and transmission and scanning electron microscopy ( Fig. 2) all failed to provide evidence of mitochondrial swelling in association with apoptotic cytochrome c efflux from Xenopus mitochondria. Another group has reported that, in whole cells of Xenopus embryos treated with gamma irradiation, apoptotic mitochondria display normal size and morphology ( Hensey and Gautier 1997). In other systems also, mitochondrial swelling has not been observed with apoptosis ( Searle et al. 1975; Mancini et al. 1997; Zhuang et al. 1998), and in some apoptotic cells mitochondria even shrink ( Martinou et al. 1999). Previously, Bid was reported to release cytochrome c from isolated mitochondria without swelling ( Luo et al. 1998). Bax-mediated cytochrome c release has been associated in some reports with mitochondrial swelling ( Narita et al. 1998) or permeability transition ( Pastorino et al. 1998), but in other reports with an absence of swelling ( Jurgensmeier et al. 1998; Martinou et al. 1999). The presence of swollen mitochondria in some apoptotic systems, including whole cells, may reflect either secondary necrosis or a fundamentally distinct apoptotic cytochrome c release mechanism.
Cytochrome c efflux is not a specific export process, as AK (25,200 Da) was also released by each of the four pro-apoptotic factors we studied. Furthermore, SOx (104 kD) was released from rat liver mitochondria incubated in cytosol ( Fig. 3). Several intermembrane space proteins, including AK ( Kohler et al. 1999; Samali et al. 1999; Single et al. 1998), a cytochrome c–GFP fusion protein ( Heiskanen et al. 1999), AIF ( Susin et al. 1997, Susin et al. 1999b), and certain caspases ( Mancini et al. 1998; Susin et al. 1999a) have been reported to be coreleased with cytochrome c in other systems, often in the absence of a permeability transition.
Whereas proteins such as Bid, Bax, and certain endogenous Xenopus egg cytosolic factors produced only a limited outer membrane permeability, we detected another cytosolic activity that caused a much stronger permeabilization effect. We termed this novel cytosolic activity PEF, as it enhanced, rather than initiated, outer membrane permeabilization ( Fig. 5 Fig. 6 Fig. 7 Fig. 8 Fig. 9 Fig. 10). The initial permeability caused by cytochrome c releasing factors is limited as it allows a relatively small cytochrome c flux across the outer membrane. This flux is sufficient to allow the loss of endogenous intermembrane space proteins, but not the rapid exchange of exogenous cytochrome c measured as accessibility of complexes III and IV ( Fig. 5). However, if fresh cytosol (PEF) is present when cytochrome c–releasing factors act on mitochondria, an additional mitochondrial change occurs which strikingly enhances the permeability of the outer membrane to exogenous cytochrome c. From measurements of complex IV accessibility such as those presented in Fig. 4, we calculate that 1 μl of apoptotic mitochondria can oxidize ∼60 nmol of cytochrome c per minute. As the same volume of Xenopus egg mitochondria contains ∼12 pmol of cytochrome c, we conclude that the rate of cytochrome c exchange across the outer membrane produced by PEF is at least 5,000-fold greater than that needed for complete efflux of endogenous cytochrome c per minute.
A potential explanation for the PEF activity we observed is that PEF could be an analogue of Bax or Bid that is present in cytosol at higher concentrations than those we used with recombinant Bid and Bax. However, this notion can be ruled out for the following reasons: (a) when cytosol activated by pretreatment with cytochrome c is subsequently mixed with mitochondria, rapid release of cytochrome c is observed, likely due to the cleavage and activation of endogenous Xenopus Bid. This activated cytosol, however, has lost PEF activity rather than gained it; (b) recombinant tBid was added at five-fold higher concentrations (170 ng/ml) than those required for cytochrome c release (not shown); and (c) (most compelling) PEF-containing cytosol does not possess the ability to cause rapid release of cytochrome c, and therefore cannot contain Bid or Bax activity greater than the amounts of recombinant protein we added. Further dilution of PEF-containing cytosol results in diminution of PEF activity, but rapid cytochrome c release is never seen. Thus, PEF does not behave like Bax and Bid.
The high permeability caused by PEF is consistent with a major rearrangement of the outer mitochondrial membrane, as a similar permeability (complex III or IV accessibility) was obtained with Triton X-100 ( Fig. 7 Fig. 8 Fig. 9), osmotic breakage of the outer membrane ( Fig. 6), and with digitonin ( Fig. 7) in the absence of cytosol. The permeability increase by PEF was not, however, accompanied by observable changes in mitochondrial ultrastructure ( Fig. 2B and Fig. E). Furthermore, evidence suggests that the inner mitochondrial membrane remains intact even after PEF has acted to permeabilize the outer membrane. We observed that the mtHsp70 protein was released from the matrix of Xenopus mitochondria treated with certain concentrations of the detergent digitonin but was not released with apoptosis ( Fig. 7), in agreement with another report that the inner membrane remains impermeable to proteins ( Kohler et al. 1999). In addition, our earlier data ( Kluck et al. 1997a) show that inner membrane potential (as measured by mitochondrial retention of the dye, DiOC6) remains high for at least 3 h after cytochrome c release, a finding inconsistent with inner membrane permeabilization. Thus, the effects of tBid, Bax, and PEF are confined to the outer membrane, rather than reflecting a global alteration in mitochondrial integrity.
The outer membrane permeability changes we observed were not due to a feedback effect of cytochrome c–activated caspases. Caspase inhibitors failed to block cytochrome c and AK efflux induced by Bid, Bax and the pro-apoptotic proteins in Xenopus cytosol. Moreover, Bid and Bax induced cytochrome c and AK release from mitochondria incubated in buffer alone ( Fig. 5). Caspase inhibitors also did not block the permeabilizing activity of PEF, but rather enhanced it ( Fig. 9).
Preliminary characterization showed that PEF is proteinaceous and apparently large (>300 kD). While the slow kinetics of PEF action may suggest enzymatic behavior, PEF is not a caspase, as it was not inhibited by caspase inhibitors. Indeed, PEF is inactivated by caspases, because cytosol in which caspases had been activated by the addition of cytochrome c contained no PEF activity, and caspase inhibitors prolonged the effects of PEF in fresh cytosol. A PEF-like activity was also detected in human Jurkat cell extracts (not shown). It is possible that PEF is active in whole cells, as a similar assay monitoring oxygen consumption (complex IV activity) in permeabilized cells found that apoptosis induced high oxygen consumption in the presence of exogenous reduced cytochrome c ( Vander Heiden et al. 1997).
There are several mechanisms by which pro-apoptotic Bcl-2 family members might induce outer membrane permeability without affecting the inner membrane. These membrane-associated proteins may form channels large enough for the passage of soluble proteins (reviewed by Reed 1997) or cooperate with channels already present ( Mannella 1992) such as VDAC ( Shimizu et al. 1999; Vander Heiden et al. 1999). Alternate mechanisms might be similar to the manner in which 15-lipoxygenase can interact specifically with organellar membranes to cause visible changes (3–10 nm) in the lipid structure and permeability to high molecular mass proteins ( van Leyen et al. 1998), or to that in which Bax causes instabilities in artificial membranes ( Basanez et al. 1999). Also unclear is the mechanism of PEF action, in particular how PEF is permitted to function only after a prior permeabilization of the outer membrane by factors such as Bid and Bax. One possibility is that PEF could interact with a target located on the lumenal side of the outer membrane. In this case, PEF would only gain access to its sites of action when the outer membrane becomes permeable to some degree. Alternatively, PEF could require the cooperation of a nonsoluble cofactor that is located within the intermembrane space (the experiment in Fig. 5 D rules out the involvement of a soluble intermembrane space protein). Yet another possibility is that PEF interacts directly with channels produced by tBid or Bax.
It is thought that eukaryotic mitochondria originate from a symbiotic relationship between primitive host cells and primitive gram-negative bacteria, which allowed the host cells to use oxygen to produce energy ( Brown and Doolittle 1997; Martin and Muller 1998). It can be argued that pressures to maintain this symbiotic relationship may have led to the utilization of death pathways involving mitochondria, i.e., apoptosis via cytochrome c or necrosis via reactive oxygen species ( Kobayashi 1998; Blackstone and Green 1999). From this perspective, it is interesting to consider whether the Bcl-2 family might have evolved from primitive antibiotics produced by the host cell. All known effects of the Bcl-2 family members, with the exception of Bcl-2 cell cycle effects ( Huang et al. 1997), can be linked to their regulation of cell death, often via effects on mitochondria. Indeed, the structures of Bcl–xL ( Muchmore et al. 1996; Sattler et al. 1997) and Bid ( Chou et al. 1999; McDonnell et al. 1999) reveal similarities to those of the pore-forming domains of bacterial toxins. Moreover, like bacterial toxins, Bcl-2 proteins form channels in synthetic lipid membranes.
Other factors, derived from the host cell, might also have evolved to destroy mitochondrial integrity. Such products include defensins ( White et al. 1995), granulysin ( Stenger et al. 1998), bactericidal permeability-increasing proteins ( Beamer et al. 1998), and channel-forming peptides ( Nicolay et al. 1994; Bechinger 1997), such as mastoparan and alamethicin, both of which we have shown can release mitochondrial cytochrome c. PEF can now be added to this list of molecules affecting mitochondrial permeability.
What are the implications of PEF activity for a cell? If PEF is present, attempts to rescue doomed cells by using caspase inhibitors would likely be futile, and may even enhance mitochondrial damage by leaving PEF fully active. Some cells given an apoptotic stimulus undergo necrotic death instead of apoptosis when caspase inhibitors are present ( Green and Reed 1998), and in such cases PEF might be responsible for ensuring that mitochondria are irreversibly damaged. On the other hand, in Apaf-1 and caspase-9 knockout mice, where the cytochrome c–initiated caspase activation pathway is missing, many neurons survive despite likely mitochondrial cytochrome c release ( Cecconi et al. 1998; Hakem et al. 1998; Kuida et al. 1998; Yoshida et al. 1998). Moreover, in the presence of caspase inhibitors, trophic factor-deprived primary sympathetic neurons survive for many days despite mitochondrial cytochrome c translocation ( Deshmukh and Johnson 1998; Martinou et al. 1999). In such saved cells, neuronal mitochondria can survive the loss of cytochrome c and, upon restoration of trophic support, actually recover their content of cytochrome c after 24 h ( Martinou et al. 1999). It has not yet been determined whether the recovery of mitochondrial cytochrome c in these neurons is due to multiplication of a few intact mitochondria, or to the reimport of (apo-)cytochrome c into mitochondria that recover from their permeabilization. We hypothesize that in most nonneural tissues, PEF provides a backup mechanism for killing the cell in the event of a disabled caspase pathway. This kind of redundancy in death pathways might be desirable in lymphocytes and Xenopus eggs, but not in irreplaceable cells such as neurons in the adult organism.
Acknowledgments
We thank Mike Yaffe for advice on the complex IV accessibility assay and Beni Wolf for Jurkat extract. Caspase-8 cDNA was a gift from V. Dixit (Genentech Inc., San Francisco) and hBid cDNA from H. Yoshida (Kirin Brewery Co., Japan).
This work was supported by grants from the National Institutes of Health (NIH) to D.D. Newmeyer (GM50284), M.J. Barber (GM32696), and D.R. Green (AI40646, CA69381). Some of the work included here was conducted at the National Center for Microscopy and Imaging Research, San Diego, which is supported by NIH grant RR04050 to M.H. Ellisman. E. Bossy-Wetzel was supported by the Swiss National Science Foundation (823A-046638) and subsequently by NIH training grant (CI-2-05999). C. Renken acknowledges support from Grant-in-Aid (98-256A) from the Western States Affiliate of the American Heart Association to T.G. Frey. This is publication no. 311 from La Jolla Institute for Allergy and Immunology.
Footnotes
Dr. Degli Esposti is on leave from the Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Vic 3169, Australia.
. Abbreviations used in this paper: AK, adenylate kinase; FEISEM, field emission in-lens scanning electron microscopy; PEF, permeability enhancing factor; SOx, sulfite oxidase; tBid, truncated Bid; VDAC, voltage-dependent anion channel.
References
- Allen T.D., Goldberg M.W. High-resolution SEM in cell biology. Trends. Cell Biol. 1993;3:205–208 . doi: 10.1016/0962-8924(93)90215-m. [DOI] [PubMed] [Google Scholar]
- Allen T.D., Bennion G.R., Rutherford S.A., Reipert S., Ramalho A., Kiseleva E., Goldberg M.W. Accessing nuclear structure for field emission, in lens, scanning electron microscopy (FEISEM) Scanning Microsc. Suppl. 1996;10:149–163 . [PubMed] [Google Scholar]
- Antonsson B., Conti F., Ciavatta A., Montessuit S., Lewis S., Martinou I., Bernasconi L., Bernard A., Mermod J.J., Mazzei G. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277:370–372 . doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- Barber M.J., Neame P.J. A conserved cysteine in molybdenum oxotransferases. J. Biol. Chem. 1990;265:20912–20915 . [PubMed] [Google Scholar]
- Basanez G., Nechushtan A., Drozhinin O., Chanturiya A., Choe E., Tutt S., Wood K.A., Hsu Y., Zimmerberg J., Youle R.J. Bax, but not Bcl-xL, decreases the lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations. Proc. Natl. Acad. Sci. USA. 1999;96:5492–5497 . doi: 10.1073/pnas.96.10.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beamer L.J., Carroll S.F., Eisenberg D. The BPI/LBP family of proteinsa structural analysis of conserved regions. Protein Sci. 1998;7:906–914 . doi: 10.1002/pro.5560070408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechinger B. Structure and functions of channel-forming peptidesmagainins, cecropins, melittin and alamethicin. J. Membr. Biol. 1997;156:197–211 . doi: 10.1007/s002329900201. [DOI] [PubMed] [Google Scholar]
- Bernardi P. The permeability transition pore. Control points of a cyclosporin A-sensitive mitochondrial channel involved in cell death. Biochim. Biophys. Acta. 1996;1275:5–9 . doi: 10.1016/0005-2728(96)00041-2. [DOI] [PubMed] [Google Scholar]
- Blackstone N.W., Green D.R. The evolution of a mechanism of cell suicide. Bioessays. 1999;21:84–88 . doi: 10.1002/(SICI)1521-1878(199901)21:1<84::AID-BIES11>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E., Newmeyer D.D., Green D.R. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:37–49 . doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J.R., Doolittle W.F. Archaea and the prokaryote-to-eukaryote transition. Microbiol. Mol. Biol. Rev. 1997;61:456–502 . doi: 10.1128/mmbr.61.4.456-502.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecconi F., Alvarez-Bolado G., Meyer B.I., Roth K.A., Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737 . doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- Chou J.J., Li H., Salvesen G.S., Yuan J., Wagner G. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell. 1999;96:615–624 . doi: 10.1016/s0092-8674(00)80572-3. [DOI] [PubMed] [Google Scholar]
- Degli Esposti M., Lenaz G., Izzo G., Papa S. Kinetics and sidedness of ubiquinol-cytochrome c reductase in beef-heart mitochondria. FEBS Lett. 1982;146:101–105 . doi: 10.1016/0014-5793(82)80713-8. [DOI] [PubMed] [Google Scholar]
- Desagher S., Osen-Sand A., Nichols A., Eskes R., Montessuit S., Lauper S., Maundrell K., Antonsson B., Martinou J.C. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 1999;144:891–901 . doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh M., Johnson E.M., Jr. Evidence of a novel event during neuronal deathdevelopment of competence-to-die in response to cytoplasmic cytochrome c . Neuron. 1998;21:695–705 . doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- Ellerby H.M., Martin S.J., Ellerby L.M., Naiem S.S., Rabizadeh S., Salvesen G.S., Casiano C.A., Cashman N.R., Green D.R., Bredesen D.E. Establishment of a cell-free system of neuronal apoptosiscomparison of premitochondrial, mitochondrial, and postmitochondrial phases. J. Neurosci. 1997;17:6165–6178 . [PMC free article] [PubMed] [Google Scholar]
- Eskes R., Antonsson B., Osen-Sand A., Montessuit S., Richter C., Sadoul R., Mazzei G., Nichols A., Martinou J.C. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J. Cell Biol. 1998;143:217–224 . doi: 10.1083/jcb.143.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farschon D.M., Couture C., Mustelin T., Newmeyer D.D. Temporal phases in apoptosis defined by the actions of Src Homology 2 domains, ceramide, Bcl-2, interleukin-1β converting enzyme family proteases, and a dense membrane fraction. J. Cell Biol. 1997;137:1117–1125 . doi: 10.1083/jcb.137.5.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane D.M., Bossy-Wetzel E., Waterhouse N.J., Cotter T.G., Green D.R. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J. Biol. Chem. 1999;274:2225–2233 . doi: 10.1074/jbc.274.4.2225. [DOI] [PubMed] [Google Scholar]
- Gaikwad A.S., Ramasarma T., Ramakrishna Kurup C.K. Brown adipose tissue mitochondria are cytochrome c-subsaturated. Mol. Cell. Biochem. 1991;105:119–125 . doi: 10.1007/BF00227751. [DOI] [PubMed] [Google Scholar]
- Green D.R., Reed J.C. Mitochondria and Apoptosis. Science. 1998;281:1309–1312 . doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Hackenbrock C.R. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. II. Electron transport-linked ultrastructural transformations in mitochondria. J. Cell Biol. 1968;37:345–369 . doi: 10.1083/jcb.37.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakem R., Hakem A., Duncan G.S., Henderson J.T., Woo M., Soengas M.S., Elia A., de la Pompa J.L., Kagi D., Khoo W. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352 . doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- Heiskanen K.M., Bhat M.B., Wang H.W., Ma J., Nieminen A.L. Mitochondrial depolarization accompanies cytochrome c release during apoptosis in PC6 cells. J. Biol. Chem. 1999;274:5654–5658 . doi: 10.1074/jbc.274.9.5654. [DOI] [PubMed] [Google Scholar]
- Hensey C., Gautier J. A developmental timer that regulates apoptosis at the onset of gastrulation. Mech. Dev. 1997;69:183–195 . doi: 10.1016/s0925-4773(97)00191-3. [DOI] [PubMed] [Google Scholar]
- Huang D.C., O'Reilly L.A., Strasser A., Cory S. The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:4628–4638 . doi: 10.1093/emboj/16.15.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innis M.A., Beers T.R., Craig S.P. Mitochondrial regulation in sea urchins. I. Mitochondrial ultrastructure transformations and changes in the ADP:ATP ratio at fertilization. Exp Cell Res. 1976;98:47–56 . doi: 10.1016/0014-4827(76)90461-4. [DOI] [PubMed] [Google Scholar]
- Juin P., Hueber A.O., Littlewood T., Evan G. c-myc-induced sensitization to apoptosis is mediated through cytochrome c release. Genes Dev. 1999;13:1367–1381 . doi: 10.1101/gad.13.11.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgensmeier J.M., Xie Z., Deveraux Q., Ellerby L., Bredesen D., Reed J.C. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA. 1998;95:4997–5002 . doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel E., Green D.R., Newmeyer D.D. The release of cytochrome c from mitochondriaa primary site for Bcl-2 regulation of apoptosis Science 275 1997. 1132 1136a [DOI] [PubMed] [Google Scholar]
- Kluck R.M., Martin S.J., Hoffman B.M., Zhou J.S., Green D.R., Newmeyer D.D. Cytochrome c activation of CPP32-like proteolysis plays a critical role in a Xenopus cell-free apoptosis system EMBO (Eur. Mol. Biol. Organ.) J. 16 1997. 4639 4649b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight V.A., Wiggins P.M., Harvey J.D., O'Brien J.A. The relationship between the size of mitochondria and the intensity of light that they scatter in different energetic states. Biochim. Biophys. Acta. 1981;637:146–151 . doi: 10.1016/0005-2728(81)90220-6. [DOI] [PubMed] [Google Scholar]
- Kobayashi I. Selfishness and deathraison d'être of restriction, recombination and mitochondria. Trends Genet. 1998;14:368–374 . doi: 10.1016/s0168-9525(98)01532-7. [DOI] [PubMed] [Google Scholar]
- Kohler C., Gahm A., Noma T., Nakazawa A., Orrenius S., Zhivotovsky B. Release of adenylate kinase 2 from the mitochondrial intermembrane space during apoptosis. FEBS Lett. 1999;447:10–12 . doi: 10.1016/s0014-5793(99)00251-3. [DOI] [PubMed] [Google Scholar]
- Kuida K., Haydar T.F., Kuan C.Y., Gu Y., Taya C., Karasuyama H., Su M.S., Rakic P., Flavell R.A. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337 . doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Kuwana T., Smith J.J., Muzio M., Dixit V., Newmeyer D.D., Kornbluth S. Apoptosis induction by caspase-8 is amplified through the mitochondrial release of cytochrome c . J. Biol. Chem. 1998;273:16589–16594 . doi: 10.1074/jbc.273.26.16589. [DOI] [PubMed] [Google Scholar]
- Liu X., Kim C.N., Yang J., Jemmerson R., Wang X. Induction of apoptotic program in cell-free extractsrequirement for dATP and cytochrome c . Cell. 1996;86:147–157 . doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Li F., Srinivasan A., Wang Y., Armstrong R.C., Tomaselli K.J., Fritz L.C. Cell-specific induction of apoptosis by microinjection of cytochrome c. Bcl-xL has activity independent of cytochrome c release J. Biol. Chem. 272 1997. 30299 30305a [DOI] [PubMed] [Google Scholar]
- Li P., Nijhawan D., Budihardjo I., Srinivasula S.M., Ahmad M., Alnemri E.S., Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade Cell 91 1997. 479 489b [DOI] [PubMed] [Google Scholar]
- Li H., Zhu H., Xu C.J., Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501 . doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Luo X., Budihardjo I., Zou H., Slaughter C., Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490 . doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Mancini M., Anderson B.O., Caldwell E., Sedghinasab M., Paty P.B., Hockenbery D.M. Mitochondrial proliferation and paradoxical membrane depolarization during terminal differentiation and apoptosis in a human colon carcinoma cell line. J. Cell Biol. 1997;138:449–469 . doi: 10.1083/jcb.138.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini M., Nicholson D.W., Roy S., Thornberry N.A., Peterson E.P., Casciola-Rosen L.A., Rosen A. The caspase-3 precursor has a cytosolic and mitochondrial distributionimplications for apoptotic signaling. J. Cell Biol. 1998;140:1485–1495 . doi: 10.1083/jcb.140.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannella C.A. The ‘ins’ and ‘outs’ of mitochondrial membrane channels. Trends Biochem. Sci. 1992;17:315–320 . doi: 10.1016/0968-0004(92)90444-e. [DOI] [PubMed] [Google Scholar]
- Margoliash E., Bosshard E.R. Guided by electrostatics, a textbook protein comes of age. Trends Biochem. Sci. 1983;8:316–320 . [Google Scholar]
- Martin W., Muller M. The hydrogen hypothesis for the first eukaryote. Nature. 1998;392:37–41 . doi: 10.1038/32096. [DOI] [PubMed] [Google Scholar]
- Martinou I., Desagher S., Eskes R., Antonsson B., André E., Fakan S., Martinou J.-C. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J. Cell Biol. 1999;144:883–889 . doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I., Brenner C., Zamzami N., Jurgensmeier J.M., Susin S.A., Vieira H.L., Prevost M.C., Xie Z., Matsuyama S., Reed J.C., Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031 . doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- McDonnell J.M., Fushman D., Milliman C.L., Korsmeyer S.J., Cowburn D. Solution structure of the proapoptotic molecule BIDa structural basis for apoptotic agonists and antagonists. Cell. 1999;96:625–634 . doi: 10.1016/s0092-8674(00)80573-5. [DOI] [PubMed] [Google Scholar]
- Minn A.J., Velez P., Schendel S.L., Liang H., Muchmore S.W., Fesik S.W., Fill M., Thompson C.B. Bcl-x(L) forms an ion channel in synthetic lipid membranes. Nature. 1997;385:353–357 . doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- Muchmore S.W., Sattler M., Liang H., Meadows R.P., Harlan J.E., Yoon H.S., Nettesheim D., Chang B.S., Thompson C.B., Wong S.-L., Ng S.-C., Fesik S.W. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341 . doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- Narita M., Shimizu S., Ito T., Chittenden T., Lutz R.J., Matsuda H., Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA. 1998;95:14681–14686 . doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neame S.J., Rubin L.L., Philpott K.L. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J. Cell Biol. 1998;142:1583–1593 . doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechushtan A., Smith C.L., Hsu Y.T., Youle R.J. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:2330–2341 . doi: 10.1093/emboj/18.9.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmeyer, D.D. 1998. Analysis of apoptotic events using a cell-free system based on Xenopus laevis eggs. Weir's Handbook of Experimental Immunology. Vol. III. L.A. Herzenberg, C. Blackwell, and D.M. Weir, editors. Blackwell Scientific. 127.121–127.126.
- Newmeyer D.D., Farschon D.M., Reed J.C. Cell-free apoptosis in Xenopus egg extractsinhibition by Bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell. 1994;79:353–364 . doi: 10.1016/0092-8674(94)90203-8. [DOI] [PubMed] [Google Scholar]
- Nicolay K., Laterveer F.D., van Heerde W.L. Effects of amphipathic peptides, including presequences, on the functional integrity of rat liver mitochondrial membranes. J. Bioenerg. Biomembr. 1994;26:327–334 . doi: 10.1007/BF00763104. [DOI] [PubMed] [Google Scholar]
- Ord M.J., Smith R.A. Correlation of mitochondrial form and function in vivomicroinjection of substrate and nucleotides. Cell Tissue Res. 1982;227:129–137 . doi: 10.1007/BF00206336. [DOI] [PubMed] [Google Scholar]
- Pastorino J.G., Chen S.T., Tafani M., Snyder J.W., Farber J.L. The overexpression of Bax produces cell death upon induction of the mitochondrial permeability transition. J. Biol. Chem. 1998;273:7770–7775 . doi: 10.1074/jbc.273.13.7770. [DOI] [PubMed] [Google Scholar]
- Petit P.X., Susin S.A., Zamzami N., Mignotte B., Kroemer G. Mitochondria and programmed cell deathback to the future. FEBS Lett. 1996;396:7–13 . doi: 10.1016/0014-5793(96)00988-x. [DOI] [PubMed] [Google Scholar]
- Pfeiffer D.R., Gudz T.I., Novgorodov S.A., Erdahl W.L. The peptide mastoparan is a potent facilitator of the mitochondrial permeability transition. J. Biol. Chem. 1995;270:4923–4932 . doi: 10.1074/jbc.270.9.4923. [DOI] [PubMed] [Google Scholar]
- Reed J.C. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–776 . doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- Rosse T., Olivier R., Monney L., Rager M., Conus S., Fellay I., Jansen B., Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c . Nature. 1998;391:496–499 . doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- Samali A., Cai J., Zhivotovsky B., Jones D.P., Orrenius S. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of Jurkat cells. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:2040–2048 . doi: 10.1093/emboj/18.8.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M., Liang H., Nettesheim D., Meadows R.P., Harlan J.E., Eberstadt M., Yoon H.S., Shuker S.B., Chang B.S., Minn A.J. Structure of Bcl-xL-Bak peptide complexrecognition between regulators of apoptosis. Science. 1997;275:983–986 . doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- Schendel S.L., Xie Z., Montal M.O., Matsuyama S., Montal M., Reed J.C. Channel formation by antiapoptotic protein Bcl-2. Proc. Natl. Acad. Sci. USA. 1997;94:5113–5118 . doi: 10.1073/pnas.94.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger P.H., Gross A., Yin X.M., Yamamoto K., Saito M., Waksman G., Korsmeyer S.J. Comparison of the ion channel characteristics of proapoptotic BAX and antiapoptotic BCL-2. Proc. Natl. Acad. Sci. USA. 1997;94:11357–11362 . doi: 10.1073/pnas.94.21.11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt B., Wachter E., Sebald W., Neupert W. Processing peptidase of neurospora mitochondria. Two-step cleavage of imported ATPase subunit 9. Eur. J. Biochem. 1984;144:581–588 . doi: 10.1111/j.1432-1033.1984.tb08505.x. [DOI] [PubMed] [Google Scholar]
- Searle J., Lawson T.A., Abbott P.J., Harmon B., Kerr J.F. An electron-microscope study of the mode of cell death induced by cancer-chemotherapeutic agents in populations of proliferating normal and neoplastic cells. J. Pathol. 1975;116:129–138 . doi: 10.1002/path.1711160302. [DOI] [PubMed] [Google Scholar]
- Shimizu S., Narita M., Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487 . doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- Single B., Leist M., Nicotera P. Simultaneous release of adenylate kinase and cytochrome c in cell death. Cell Death Differ. 1998;5:1001–1003 . doi: 10.1038/sj.cdd.4400462. [DOI] [PubMed] [Google Scholar]
- Stenger S., Hanson D.A., Teitelbaum R., Dewan P., Niazi K.R., Froelich C.J., Ganz T., Thoma-Uszynski S., Melian A., Bogdan C. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science. 1998;282:121–125 . doi: 10.1126/science.282.5386.121. [DOI] [PubMed] [Google Scholar]
- Susin S.A., Zamzami N., Castedo M., Daugas E., Wang H.G., Geley S., Fassy F., Reed J.C., Kroemer G. The central executioner of apoptosismultiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J. Exp. Med. 1997;186:25–37 . doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Brenner C., Larochette N., Prévost M.C., Alzari P.M., Kroemer G. Mitochondrial release of Caspase-2 and -9 during the apoptotic process J. Exp. Med. 189 1999. 381 394a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Snow B.E., Brothers G.M., Mangion J., Jacotot E., Costantini P., Loeffler M., Larochette N., Goodlett D.R., Aebersold R., Siderovski D.P., Penninger J.M., Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor Nature 397 1999. 441 446b [DOI] [PubMed] [Google Scholar]
- van Leyen K., Duvoisin R.M., Engelhardt H., Wiedmann M. A function for lipoxygenase in programmed organelle degradation. Nature. 1998;395:392–395 . doi: 10.1038/26500. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Chandel N.S., Williamson E.K., Schumacker P.T., Thompson C.B. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637 . doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Chandel N.S., Schumacker P.T., Thompson C.B. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol. Cell. 1999;3:159–167 . doi: 10.1016/s1097-2765(00)80307-x. [DOI] [PubMed] [Google Scholar]
- White S.H., Wimley W.C., Selsted M.E. Structure, function, and membrane integration of defensins. Curr. Opin. Struct. Biol. 1995;5:521–527 . doi: 10.1016/0959-440x(95)80038-7. [DOI] [PubMed] [Google Scholar]
- Wikstrom M., Casey R. The oxidation of exogenous cytochrome c by mitochondria. Resolution of a long-standing controversy. FEBS Lett. 1985;183:293–298 . doi: 10.1016/0014-5793(85)80796-1. [DOI] [PubMed] [Google Scholar]
- Wojtczak L., Zaluska H., Wroniszewska A., Wojtczak A.B. Assay for the intactness of the outer membrane in isolated mitochondria. Acta Biochim. Pol. 1972;19:227–234 . [PubMed] [Google Scholar]
- Wolter K.G., Hsu Y.T., Smith C.L., Nechushtan A., Xi X.G., Youle R.J. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1997;139:1281–1292 . doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley G.A., Wallace B.A. Model ion channelsgramicidin and alamethicin. J. Membr. Biol. 1992;129:109–136 . doi: 10.1007/BF00219508. [DOI] [PubMed] [Google Scholar]
- Yang J., Liu X., Bhalla K., Kim C.N., Ibrado A.M., Cai J., Peng T.-I., Jones D.P., Wang X. Prevention of apoptosis by Bcl-2release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132 . doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Kong Y.Y., Yoshida R., Elia A.J., Hakem A., Hakem R., Penninger J.M., Mak T.W. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750 . doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Zhivotovsky B., Orrenius S., Brustugun O.T., Doskeland S.O. Injected cytochrome c induces apoptosis. Nature. 1998;391:449–450 . doi: 10.1038/35060. [DOI] [PubMed] [Google Scholar]
- Zhuang J., Dinsdale D., Cohen G.M. Apoptosis, in human monocytic THP.1 cells, results in the release of cytochrome c from mitochondria prior to their ultracondensation, formation of outer membrane discontinuities and reduction in inner membrane potential. Cell Death Differ. 1998;5:953–962. doi: 10.1038/sj.cdd.4400440. [DOI] [PubMed] [Google Scholar]