

Abstract
Peroxisomal matrix protein import requires PEX12, an integral peroxisomal membrane protein with a zinc ring domain at its carboxy terminus. Mutations in human PEX12 result in Zellweger syndrome, a lethal neurological disorder, and implicate the zinc ring domain in PEX12 function. Using two-hybrid studies, blot overlay assays, and coimmunoprecipitation experiments, we observed that the zinc-binding domain of PEX12 binds both PEX5, the PTS1 receptor, and PEX10, another integral peroxisomal membrane protein required for peroxisomal matrix protein import. Furthermore, we identified a patient with a missense mutation in the PEX12 zinc-binding domain, S320F, and observed that this mutation reduces the binding of PEX12 to PEX5 and PEX10. Overexpression of either PEX5 or PEX10 can suppress this PEX12 mutation, providing genetic evidence that these interactions are biologically relevant. PEX5 is a predominantly cytoplasmic protein and previous PEX5-binding proteins have been implicated in docking PEX5 to the peroxisome surface. However, we find that loss of PEX12 or PEX10 does not reduce the association of PEX5 with peroxisomes, demonstrating that these peroxins are not required for receptor docking. These and other results lead us to propose that PEX12 and PEX10 play direct roles in peroxisomal matrix protein import downstream of the receptor docking event.
Keywords: PTS1 receptor, PEX5, PEX10, Zellweger syndrome, peroxisome biogenesis disorder
Subcellular organelles are a defining feature of eukaryotes and make essential contributions to virtually all aspects of cellular metabolism. Although each organelle is physically and chemically unique, there are several common aspects to the biogenesis of all membrane-limited compartments. One is that all or most of their protein content must be imported. The protein uptake mechanisms used in the biogenesis of each organelle must account for the recognition of newly synthesized proteins that are destined for each organelle, the transport of these proteins to the surface of the appropriate compartment, and the vectorial translocation of these proteins across one or more lipid bilayers ( Schatz and Dobberstein 1996).
Several paradigms for protein import into organelles have emerged from studies of the ER ( Matlack et al. 1998), the mitochondrion ( Pfanner et al. 1997), and the nucleus ( Mattaj and Englmeier 1998). However, the process of peroxisomal protein import does not conform to any of these paradigms ( Subramani 1993): proteins are posttranslationally imported into the peroxisome but cotranslationally inserted into the ER; peroxisomes can import completely folded proteins, internally cross-linked isopeptide complexes, and even gold particles, whereas the ER and mitochondria can only accommodate unfolded precursors; and peroxisomes lack detectable pores, whereas the nucleus utilizes large gated pores to allow entry of folded import substrates. Thus, the elucidation of peroxisomal protein import mechanisms will undoubtedly reveal new concepts in organelle biogenesis.
Peroxisomal proteins are encoded by nuclear genes, synthesized in the cytoplasm, and imported posttranslationally into peroxisomes ( Lazarow and Fujiki 1985). Peroxisomal matrix proteins contain either of two peroxisomal targeting signals, the PTS1 ( Gould et al. 1989) and PTS2 ( Swinkels et al. 1991). Although either of these signals is sufficient to direct proteins into the peroxisome matrix, the vast majority of peroxisomal matrix proteins utilize the COOH-terminal PTS1 rather than the NH2-terminal PTS2 ( Subramani 1993). These signals are recognized by PEX5 and PEX7, the physical receptors for the PTS1 and PTS2, respectively ( McCollum et al. 1993; Marzioch et al. 1994; Distel et al. 1996). In contrast, integral peroxisomal membrane proteins (PMPs)1 do not use either PTS1 or PTS2 motifs for targeting and are efficiently imported into peroxisomes independently of PEX5 and PEX7 ( Subramani 1993; Dyer et al. 1996; Chang et al. 1999).
Current models of peroxisomal matrix protein import are heavily influenced by the fact that PEX5 is a predominantly cytoplasmic protein in several species ( Dodt et al. 1995; Wiemer et al. 1995; Elgersma et al. 1996; Gould et al. 1996) and appears to cycle between the cytoplasm and peroxisomes in mammalian cells ( Dodt and Gould 1996). The dynamic distribution of the PTS1 receptor implies that peroxisomal matrix protein import involves overlapping cytoplasmic and peroxisomal events in addition to the actual translocation process ( Braverman et al. 1995; Rachubinski and Subramani 1995). Before translocation, import of PTS1-containing proteins (ligands) is likely to involve recognition by PEX5 in the cytoplasm, followed by transport of the receptor–ligand complex to the peroxisome surface and binding of the receptor–ligand complex to docking factors on the peroxisome membrane. After ligand translocation, additional factors are thought to mediate the recycling of PEX5 back to the cytoplasm. PEX14, PEX13, and PEX17 have been implicated in receptor docking ( Elgersma et al. 1996; Erdmann and Blobel 1996; Gould et al. 1996; Albertini et al. 1997; Huhse et al. 1998; Girzalsky et al. 1999), and PEX4 has been reported to act in the recycling of PEX5 from the peroxisome to the cytoplasm ( van der Klei et al. 1998). Other PEX genes such as PEX1 ( Portsteffen et al. 1997; Reuber et al. 1997; Chang et al. 1999), PEX2 ( Shimozawa et al. 1992; Chang et al. 1999), PEX6 ( Yahraus et al. 1996; Chang et al. 1999), PEX10 ( Kalish et al. 1995; Warren et al. 1998; Chang et al. 1999), and PEX12 ( Kalish et al. 1996; Chang et al. 1997; Chang and Gould 1998; Chang et al. 1999) have also been implicated in peroxisomal matrix protein import. However, their roles in matrix protein import are only poorly understood and other reports have suggested that PEX1 and PEX6 may instead participate in the biogenesis of peroxisome membranes ( Erdmann et al. 1991; Spong and Subramani 1993; Heyman et al. 1994; Titorenko et al. 1997; Faber et al. 1998; Titorenko and Rachubinski 1998a, Titorenko and Rachubinski 1998b).
Although most PEX genes were originally identified in yeast, our understanding of peroxisome biogenesis has also been advanced by the analysis of this process in humans and how it is disrupted in the peroxisome biogenesis disorders (PBDs) ( Lazarow and Moser 1995). Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease represent a phenotypic continuum of diseases within the PBDs that we refer to as the Zellweger spectrum. Their clinical phenotypes include developmental delay, multiple neural, hepatic, and renal defects, and pronounced mental retardation. These clinical phenotypes are most severe in Zellweger syndrome patients, who rarely survive their first year, are milder in neonatal adrenoleukodystrophy, and even less pronounced in infantile Refsum disease. At the cellular level, virtually all Zellweger spectrum patients display defects in the import of both PTS1 and PTS2 proteins, though there are rare patients who are defective only in PTS1 protein import ( Slawecki et al. 1995). There is significant genetic heterogeneity within the Zellweger spectrum and recent studies have identified the genes that are mutated in >95% of PBD patients and nine of the eleven known PBD complementation groups. These include PEX1 ( Portsteffen et al. 1997; Reuber et al. 1997), PEX2 ( Shimozawa et al. 1992), PEX5 ( Dodt et al. 1995), PEX6 ( Fukuda et al. 1996; Yahraus et al. 1996), PEX10 ( Okumoto et al. 1998a; Warren et al. 1998), PEX12 ( Chang et al. 1997; Okumoto et al. 1998b), PEX13 ( Liu et al. 1999; Shimozawa et al. 1999), PEX16 ( Honsho et al. 1998; South and Gould 1999), and PEX19 ( Matsuzono et al. 1999). Variations in the clinical and cellular phenotypes within the Zellweger spectrum are related primarily to the severity of the affected alleles, with mild phenotypes arising from PEX gene mutations that only partially reduce PEX gene function ( Reuber et al. 1997; Chang and Gould 1998; Collins and Gould 1999). In contrast to the Zellweger spectrum of diseases, rhizomelic chondrodysplasia punctata is caused by mutations in PEX7, which encodes the PTS2 receptor ( Braverman et al. 1997; Motley et al. 1997; Purdue et al. 1997).
We initially identified PEX12 in the yeast Pichia pastoris as a gene that is required for normal peroxisome biogenesis ( Kalish et al. 1996). More recent studies have described the human homologue of this gene and reported that mutations in PEX12 are responsible for complementation group 3 of the PBDs ( Chang et al. 1997; Okumoto and Fujiki 1997; Chang and Gould 1998; Okumoto et al. 1998b). Both yeast and human PEX12 encode an integral peroxisomal membrane protein with a zinc ring motif at its COOH terminus ( Kalish et al. 1996; Okumoto and Fujiki 1997). These studies also established that PEX12 spans the peroxisome membrane twice and extends its NH2 and COOH termini into the cytoplasm. In this report, we investigate the function of human PEX12 in peroxisome biogenesis. We find that PEX12 interacts with PEX5 and PEX10 via its COOH-terminal zinc-binding domain, that these interactions are biologically significant, and that PEX12 and PEX10 participate in an aspect of peroxisomal matrix protein import that occurs downstream of receptor docking.
Materials and Methods
Cell Lines, Transfections, and Indirect Immunofluorescence
Skin fibroblast cell lines from patients with peroxisome biogenesis disorders are referred to by their PBD numbers and were cultured in complete medium (DME supplemented with 10% FBS and penicillin/streptomycin). Transfections were performed by electroporation using the protocol outlined by Chang et al. 1997 and were processed 2 d later for immunofluorescence. Immunofluorescence experiments were performed essentially as described ( Slawecki et al. 1995). In brief, cells were grown on cover glasses, fixed, permeabilized, washed, incubated with the primary antibodies, washed extensively, incubated with the secondary antibodies, washed extensively, and then mounted on glass slides. Standard permeabilization was for 5 min with 1% Triton X-100, which permeabilizes both plasma and peroxisome membranes. Differential permeabilization was for 2–4 min with 25 μg/ml digitonin, which permeabilizes the plasma membrane but does not permeabilize the peroxisome membrane. Hence, only cytoplasmically exposed antigens can be detected under these conditions. Differential permeabilization experiments were generally performed with additional controls to ensure that the incubation in digitonin had not permeabilized any intracellular membranes. Rabbit polyclonal antibodies against the PTS1 tripeptide ser-lys-leu-COOH have been described ( Gould et al. 1990) and the anti-human PEX5 antibodies were generated against bacterially expressed forms of the protein. Mouse mAbs against the c-myc epitope were obtained from the tissue culture supernatant of the hybridoma 1-9E10 ( Evan et al. 1985). Rabbit polyclonal antibodies against c-myc, sheep anti-human catalase antibodies, and fluorescently labeled secondary antibodies were obtained from commercial sources.
Mutation Detection
We previously described a mutation detection strategy for human PEX12 ( Chang et al. 1997; Chang and Gould 1998). In brief, the entire 2.3-kb PEX12 gene was amplified from the total human genomic DNA using the primers Chang-21 and Chang-20 (Table ). The coding regions and intron/exon junctions were sequenced directly from the PCR product using the primers used for amplification as well as several additional gene specific primers (Table , Chang-17, Chang-24, and Chang-30). Total genomic DNA was isolated from PBD054 cells using the PureGene kit (Gentra Systems, Inc.).
Table 1.
Sequence of Primers Used in this Article
| Chang-10 | 5′-CCCGGATCCGTTCTCAGGGGAGTAGAGTTTA- ATC-3′ |
|---|---|
| Chang-17 | 5′-AAAATCGTTTCCAGCGGG-3′ |
| Chang-20 | 5′-CCAGGATCCGTGAGGATAAGACATGATTCCC-3′ |
| Chang-21 | 5′-CAAGGTACCAAGTGAAAGCCAGTACACGCAG-3′ |
| Chang-24 | 5′-CACAAGTCTCAGAGATTGGC-3′ |
| Chang-30 | 5′-ATGATGCAGCAACCAGCCAG-3′ |
| 10.2HC5′ | 5′-CCCGTCGACGAGGCAGCGGCAGCGAGCCAGG-3′ |
| 10.2H3′ | 5′-CCAAGCGGCCGCCGTAGAGGTCATCTGTGTCC-3′ |
| 12.2HC5′ | 5′-CCCGTCGACGCAGTTCCTTGACTGGTGGTCC-3′ |
| 12.2H3′ | 5′-CCAAGCGGCCGCTCAGTTCTCAGGGGAGTAGAG-3′ |
| 14.2H5′ | 5′-CCCGTCGACGGCGTCCTCGGAGCAGGCAGAG- CAG-3′ |
| 14.2H3′ | 5′-CCAAGCGGCCGCAGCTCCTCCTCCACTGAG-3′ |
Plasmids
Most of the plasmids used for yeast two-hybrid studies were based on pPC62, a LEU2-based GAL4 DNA–binding domain fusion protein expression vector and pPC86, a TRP1-based GAL4 transcriptional activation domain (AD) fusion protein expression vector ( Chevray and Nathans 1992). These plasmids contain two PvuI sites in symmetric positions, and the PvuI fragments of these plasmids were switched to create pJL59 ( Vidal et al. 1996) and pPC86/L2. The plasmid pJL59 is identical to pPC62 except that it contains the TRP1 gene in place of the LEU2 gene, and pPC86/L2 is identical to pPC86 except that it contains the LEU2 gene in place of the TRP1 gene. The plasmid pGAD424 (CLONTECH Laboratories) was also used for expression of some GAL4-AD fusion proteins.
The plasmid pJL59-PEX12C was created by amplifying a subregion of PEX12 using the primers 12.2HC5′ and 12.2H3′ (Table ) and pcDNA3-PEX12 as the template ( Chang et al. 1997), cleaving the resulting 370-bp product with SalI and NotI, and inserting this fragment between the SalI and NotI sites of pJL59. This plasmid is designed to express a fusion between the GAL4 DNA–binding domain and amino acids 260–359 of human PEX12. The plasmid pJL59-PEX12C/S320F was created using the same cloning strategy except that pcDNA3-PEX12/S320F (see below) was used as the template. The plasmid pPC86/L2-PEX10C was created by amplifying a subregion of PEX10 using the primers 10.2HC5′ and 10.2H3′ (Table ) and pcDNA3-PEX10 ( Warren et al. 1998) as the template, cleaving the resulting 327-bp product with SalI and NotI, and inserting this fragment between the SalI and NotI sites of pJL59. This plasmid is designed to express a fusion between the COOH-terminal 87 amino acids of human PEX10 and the GAL4 activation domain. The plasmid pPC86/L2-PEX14 encodes a fusion between the GAL4 activation domain and full-length PEX14 and was created by amplifying the entire PEX14 open reading frame (ORF) using the primers 14.2H5′ and 14.2H3′ (Table ) and total human cDNA as a template, cleaving the resulting product with SalI and NotI, and inserting this fragment between the SalI and NotI sites of pPC86/L2. The PEX5 two-hybrid plasmids were created as follows. The entire PEX5S ORF was excised from plasmid pGD100 ( Dodt et al. 1995) by cleavage with NcoI, after which the ends were made blunt with the Klenow fragment of DNA PolI and dNTPs, and then cleavage with BglII. The resulting 2-kb PEX5S fragment was isolated and inserted between the SmaI and BglII sites of pGAD424, downstream of and in-frame with the GAL4 transcription activation domain. The resulting plasmid, pGAD424-PEX5S, encodes a fusion between the GAL4 activation domain and full-length PEX5S. The plasmid pGAD424-PEX5L was created by cleaving pPEX5L ( Braverman et al. 1998) with NcoI, making the ends blunt with the Klenow fragment of DNA PolI and dNTPs, cleaving this DNA with BglII, isolating the resulting 2-kb PEX5L fragment, and inserting it between the SmaI and BglII sites of pGAD424. The plasmid pGAD424-PEX5S/ΔN encodes a fusion protein between the GAL4 transcriptional activation domain and the COOH-terminal 317 amino acids of PEX5. It was created by excising the NH2 terminally truncated PEX5 fragment from pGD105 ( Dodt et al. 1995) with NcoI (after which the ends were made blunt with the Klenow fragment of DNA PolI and dNTPs) and BglII and inserting the resulting 1-kb PEX5 fragment between the SmaI and BglII sites of pGAD424.
Bacterial expression vectors were based on a derivative of pMAL-c2 (New England Biolabs Inc.), pMBP differs from pMAL-c2 in that it contains a SalI site downstream of the EcoRI site (GAATTCAAGTCGAC, EcoRI and SalI sites underlined), and a NotI site upstream of the HindIII site (GCGGCCGCAAGCTT, NotI and HindII sites underlined). pMBP-PEX12C was created by excising the PEX12 SalI-NotI fragment from pJL59-PEX12C and inserting it between the SalI and NotI sites of pMBP. pMBP-PEX12C/S320F was created by excising the PEX12 SalI-NotI fragment from pJL59-PEX12C/S320F and inserting it between the SalI and NotI sites of pMBP.
All mammalian expression vectors are based on pcDNA3 (Invitrogen Corp.). We have previously described the expression vectors pcDNA3-PEX5S (pGD100; Dodt et al. 1995), pcDNA3-PEX5L (pPEX5L; Braverman et al. 1998), pcDNA3-PEX10 ( Warren et al. 1998), and pcDNA3-PEX12 ( Chang et al. 1997). To create pcDNA3-PEX12/3xmyc, the PEX12 ORF was amplified using the oligonucleotides Chang-21 and Chang-10 (Table ) and pcDNA3-PEX12 as a template. These primers append an Asp718 site upstream of the ORF and replace the stop codon with a BamHI site. The resulting PCR fragment was cleaved with Asp718 and BamHI and cloned upstream of the triple c-myc tag in pcDNA3-3xmyc ( Geisbrecht et al. 1998). To create the plasmid pcDNA3-PEX12/S320F, we first amplified the PEX12 ORF from PBD054 cDNA. Total RNA was extracted from PBD054 cells using the PureScript kit (Gentra Systems, Inc.) and PEX12 cDNA was synthesized as described ( Chang and Gould 1998). The PEX12 ORF was amplified from the first strand PBD054 PEX12 cDNA using the primers Chang-21 and Chang-20 (Table ), cleaved with Asp718 and BamHI, and cloned between the Asp718 and BamHI sites of pcDNA3, generating pcDNA3-PEX12/S320F. The sequence of the final plasmid was confirmed to ensure the presence of the S320F mutation and the absence of any undesired mutations. The plasmid pcDNA3-3xHA has a 114-bp DNA insert between the Asp718 and XbaI sites of pcDNA3, which contains a BglII site (AGATCT) immediately upstream of short ORF encoding three repeats of the HA epitope tag (GRIFYPYDVPDYAGYPYDVPDYAGSYPYDVPDYALSTOP, the HA epitopes are underlined). To create pcDNA3-PEX10/3xHA, we excised the PEX10 ORF (lacking its stop codon) from pcDNA-PEX10myc ( Warren et al. 1998) using the restriction enzymes Asp718 and BamHI, excised the 3xHA tag from pcDNA3-3xHA by cleavage with BglII and XbaI, and inserted these fragments in tandem between the Asp718 and XbaI sites of pcDNA3.
The regions of all plasmids that were generated by PCR were sequenced to confirm the absence of any unintended mutations. Any plasmids that did contain undesired mutations were discarded and additional clones were characterized until one with the desired sequence was obtained.
Two-Hybrid Analysis
The Saccharomyces cerevisiae two-hybrid reporter strain BY3168 was used for all experiments ( Vidal et al. 1996). All strains were grown overnight on a nitrocellulose filter membrane (Schleicher & Schuell, Inc.) that was placed on a plate with minimal medium lacking tryptophan and leucine (Sc-W-L). The cells were lysed by submersion in liquid nitrogen, and activity of the two-hybrid reporter gene β-galactosidase was assessed by placing the filter membrane onto a filter paper saturated with 0.1% 5-bromo-4-chloro-3-indoyl β-d-galactopyranoside (X-gal) in 100 mM potassium phosphate buffer, pH 7.0. The filters were photographed after color development.
Protein extracts from BY3168 carrying either pJL59-PEX12C or pJL59-PEX12C/S320F were prepared according to established protocols ( Adams et al. 1997). In brief, the yeast were grown in 4 ml Sc-W-L medium to an OD600 of 2. The cells were transferred to a solution containing 50 mM Tris, pH 7.5, and 10 mM sodium azide, and then pelleted by centrifugation (5,000 g) for 10 min. The pellets were resuspended in 30 μl of ESB (80 mM Tris, pH 6.8, 1.5% DTT, 2% SDS, 10% glycerol, and 0.1 mg/ml bromophenol blue), and immediately boiled for 3 min. The tubes were cooled on ice, and then mixed vigorously with 0.1 g of 425–600-micron glass beads (Sigma Chemical Co.) for 2 min to lyse the cells. The resulting lysates were added to an additional 70 μl of ESB and boiled for 1 min. Equal amounts of each sample were separated by SDS-PAGE, transferred to Immobilon-P membranes (Millipore), and probed with antibodies raised against the COOH-terminal 17 amino acids of human PEX12 (a segment that is not present in yeast PEX12).
Protein Purification, In Vitro Translation of Proteins and Overlay Assays
A 1-liter culture of DH10B cells ( Grant et al. 1990) carrying pMBP-PEX12C was grown to an OD600 of 0.4, induced with 1 mM isopropyl-β-d-thiogalactopyranoside, and grown overnight at 18°C. These cells were harvested and resuspended in column buffer (20 mM Tris, pH 7.4, 200 mM NaCl, 1 mM EDTA, and 10 mM β-mercaptoethanol) plus 0.5 mg/ml lysozyme. The cells were frozen in liquid nitrogen, thawed, and lysed by sonication. The lysate was centrifuged for 20 min at 14,000 g, and the supernatant was loaded onto a column containing 10 ml of amylose resin. The column was washed with 12-bed volumes of column buffer and the protein was eluted with 10 mM maltose. The eluant was collected in 1.5-ml fractions and the purity was assessed using SDS-PAGE. MBP-PEX12C/S320F and MBP-LacZα fusion proteins were purified following the same protocol.
Filter binding experiments were performed as follows. 10 μg of purified MBP-PEX12C and 10 μg of MBP-LacZα were spotted separately onto Immobilon-P membranes (Millipore). Two identical membranes were prepared, one for overlay with HsPEX5S and one for HsPEX5L. The membranes were allowed to dry for 15 min at room temperature, and then washed in methanol for 30 s followed by Milli-Q H2O for 1 min. The membranes were transferred to buffer A (50 mM Tris-HCl, pH 7.5, 100 mM potassium acetate, 150 mM NaCl, 1 mM DTT, 5 mM MgCl2, 1 mM EDTA, 0.3% [vol/vol] Tween 20, 100 μM ZnCl2, 5% [wt/vol] nonfat milk, and 100 mM methionine; Fransen et al. 1998) and incubated with shaking for 1 h at room temperature. Radiolabled HsPEX5S and HsPEX5L were made using the TNT T7-Quick Coupled in vitro transcription/translation kit (Promega Corp.) and [35S]methionine (NEN Life Science Products) according to the manufacturer's protocols. 20 μl of the in vitro transcription/translation reaction was mixed with 5 ml of buffer A plus 10 μg/ml BSA and incubated with the membrane for 1 h at 37°C with shaking. The membranes were washed twice for 5 min at room temperature with buffer A, dried, and placed on film.
For assessing the PEX10-PEX12 interaction, 10 μg of purified MBP-PEX12C and 10 μg of MBP-LacZα were separated by SDS-PAGE and transferred to Immobilon-P membranes. After transfer, the proteins were renatured for 2 h at 4°C in buffer A. Radiolabeled PEX10 was synthesized using the TNT T7-Quick Coupled system and [35S]methionine as described above. The membranes were incubated overnight at 4°C with shaking in 5 ml of buffer A containing 25 μl of the in vitro transcription/translation reaction. After washing twice with buffer A at room temperature, the membranes were dried, and bound [35S]PEX10 was detected by autoradiography.
Immunoprecipitations
Normal human skin fibroblasts were transfected with pcDNA3-PEX12 and pcDNA3-PEX12/3xmyc. 2 d after transfection, cell lysates were prepared from each of the transfected populations by scraping the cells into TBSN buffer (10 mM Tris, pH 7.8, 150 mM NaCl, 1% NP-40, 5 mM benzamidine, 0.2 mg/ml NaF, 25 μg/ml aprotinin, and 62.5 μg/ml leupeptin). Each cell lysate was mixed with 1 μg of rabbit polyclonal antibodies against c-myc (Santa Cruz Biotechnology) for 1 h at 4°C. Protein A agarose beads were preincubated with 1% BSA, and then incubated with the lysate–antibody mixture for 1 h at 4°C with gentle agitation. The agarose beads were collected by centrifugation (1,000 g), washed four times with 1 ml of TBSN buffer, and resuspended in 30 μl SDS-PAGE sample buffer. Equal amounts of each sample were immunoblotted using anti–PEX5 antibodies. For assessing the coimmunoprecipitation between PEX10 and PEX12, cells were cotransfected with pcDNA3-PEX12 and pcDNA3-PEX10/3xHA or pcDNA3-PEX12/3xmyc and pcDNA3-PEX10/3xHA. After preparation of lysates and immunoprecipitation with anti-myc polyclonal antibodies, levels of PEX10/3xHA were determined by immunoblot using the 12CA5 monoclonal anti–HA antibody (Boehringer Mannheim Corp.). Equivalent levels of PEX5 and PEX10/3xHA in the crude lysates were confirmed by standard immunoblotting techniques.
Cell Fractionation and Protease Protection Assay
Skin fibroblast cells were grown to 90% confluency in 100-mm dishes, removed from the plate by trypsinization, washed, resuspended in lysis buffer, and lysed in a ball bearing homogenizer as previously described ( Dodt and Gould 1996). Postnuclear supernatants were prepared by successive 1,500 g spins, and then separated into organelle pellets and cytosolic supernatants by centrifugation at 15,000 g for 10 min. To determine the relative levels of PEX5 in the cytoplasm and peroxisome of human fibroblasts, organelle pellets and cytosolic supernatants were prepared as above, transferred to membranes, and probed with polyclonal anti–PEX5 antibodies. For protease protection experiments, organelle preparations were derived from PBD006 and PBD054 cells in the same manner and were each split into eight equal fractions of 8 μg of protein. Triton X-100 was added to four of the eight tubes to a final concentration of 1%. We placed the samples on ice and added 0, 15, 30, or 60 μg of a trypsin preparation (Calbiochem-Novabiochem) to the four samples lacking detergent and the four samples containing detergent. These mixtures were incubated on ice for 20 min. Reactions were terminated by adding a twofold excess of bovine trypsin inhibitor (Sigma Chemical Co.). Equal amounts of each sample were processed for immunoblot with anti–PEX5 antibodies.
Results
The Zinc-binding Domain Is Required for PEX12 Function
We have identified an array of PEX12 mutations that cause Zellweger syndrome and the milder phenotypic variants of neonatal adrenoleukodystrophy and infantile Refsum disease ( Chang et al. 1997; Chang and Gould 1998). To better understand the regions of PEX12 that are important for its role in peroxisome biogenesis, we compared the deduced products of the PEX12 alleles in severely and mildly affected patients ( Fig. 1). Severe defects in PEX12 activity were associated with mutations that truncated PEX12 upstream of the cytoplasmically exposed zinc ring domain. Furthermore, all moderately and mildly affected patients expressed at least one PEX12 allele capable of encoding a protein that contained the COOH-terminal zinc ring domain. This phenotype–genotype correlation suggested that the COOH-terminal zinc-binding domain is critical for PEX12 function. This hypothesis is also supported by the results from directed mutagenesis experiments on PEX12 in both yeast ( Kalish et al. 1996) and mammalian cells ( Okumoto et al. 1998b).
Figure 1.

The deduced PEX12 products of seven PBD patients. The diagram shows the predicted protein product of each PEX12 allele. The zinc ring domain is indicated by a black box and each of two transmembrane domains is indicated by the cross-hatched boxes. Straight lines show the length of additional amino acids that are appended as a result of frameshifting mutations. ZS, Zellweger syndrome; NALD, neonatal adrenoleukodystrophy; and IRD, infantile Refsum disease.
PEX12 Binds PEX5, the PTS1 Receptor
Previous studies have suggested that zinc ring domains may mediate protein–protein interactions ( Borden 1998), and the important role of this domain in PEX12 suggested that it may mediate interactions between PEX12 and other proteins that are involved in peroxisome biogenesis. We employed the yeast two-hybrid system to search for such proteins. A fusion between the GAL4 DNA–binding domain and the COOH-terminal 100 amino acids of PEX12 was used as bait to screen a library of fusions between the GAL4-activating domain and all known human peroxins. We detected a strong interaction between the zinc-binding domain of PEX12 and PEX5, the PTS1 receptor ( Fig. 2 A). Recent studies ( Braverman et al. 1998; Otera et al. 1998) have established that two isoforms of PEX5, PEX5S and PEX5L, are synthesized in roughly equivalent levels in human cells, but we did not observe any difference in the interaction between PEX12 and PEX5S or PEX5L.
Figure 2.

PEX12 interacts with PEX5. (A) Results of two-hybrid studies between PEX12 and PEX5. Two-hybrid reporter strains expressing the indicated fusion proteins were transferred to a nitrocellulose filter, submerged in liquid nitrogen to lyse the cells, and assayed for β-galactosidase activity. AD, GAL4 activation domain; and BD, GAL4-binding domain. (B) Filter binding experiments with PEX12 and PEX5. Equal amounts of MBP-LacZα and MBP-PEX12C were spotted on membranes and probed with [35S]PEX5S (upper panel) or [35S]PEX5L (lower panel). (C) PEX5 coimmunoprecipitates with PEX12/3xmyc. Lysates were prepared from fibroblasts that had been transfected with either pcDNA3-PEX12 or pcDNA3-PEX12/3xmyc. After immunoprecipitation with anti–myc antibodies, the immunoprecipitates were analyzed by immunoblot with anti-PEX5 antibodies (upper panel). In addition, equal amounts of the crude lysate before immunoprecipitation were assayed for PEX5 levels by immunoblot (lower panel). (D) PEX12 interacts with the PTS1-binding domain of PEX5. Two-hybrid reporter strains expressing the indicated fusion proteins were transferred to a nitrocellulose filter, submerged in liquid nitrogen to lyse the cells, and assayed for β-galactosidase activity.
Next, we used a protein binding assay to independently assess the interaction between PEX12 and PEX5. We generated a recombinant fusion protein between maltose-binding protein (MBP) and the COOH-terminal 100 amino acids of PEX12, which includes its zinc-binding domain. The resulting protein, MBP-PEX12, was tested for its ability to bind PEX5 using a filter binding assay. Equal amounts of purified MBP-PEX12 and an MBP-LacZα fusion protein were spotted onto membranes and subsequently probed with 35S-labeled PEX5 that had been synthesized in vitro in a rabbit reticulocyte lysate. PEX5 was bound by MBP-PEX12 but not by MBP-LacZα, indicating that PEX12 was capable of binding PEX5 ( Fig. 2 B).
To determine whether the physical interaction between PEX5 and the zinc-binding domain of PEX12 reflected an association between these proteins in vivo, we tested whether PEX12 and PEX5 formed a complex of sufficient stability to withstand coimmunoprecipitation from cell lysates. Human fibroblasts were transfected with either of two plasmids, pcDNA3-PEX12 or pcDNA3-PEX12/3xmyc. 2 d after transfection the cells were lysed, the lysates were subjected to immunoprecipitation with anti-myc polyclonal antibodies, and the immunoprecipitates were separated by SDS-PAGE and probed with anti–PEX5 antibodies. Equal amounts of PEX5 were present in both crude lysates, but PEX5 was immunoprecipitated only from the lysate of cells expressing PEX12/3xmyc ( Fig. 2 C).
We next mapped the PEX12-binding domain of PEX5 by expressing different regions of PEX5 in the yeast two-hybrid system and assaying their interaction with the PEX12 zinc-binding domain. These fragments of PEX5 were also assayed for their ability to interact with PEX14, a known docking factor for PEX5 ( Albertini et al. 1997; Fransen et al. 1998; Schliebs et al. 1999) ( Fig. 2 D). The PTS1 binding site of PEX5 is contained within its COOH-terminal half, a region that contains seven tetratricopeptide repeats ( Dodt et al. 1995; Terlecky et al. 1995). A fragment of PEX5 containing little more than the PTS1-binding domain of PEX5 retained full binding to PEX12. However, it was unable to bind PEX14, as expected from the recent study by Schliebs et al. 1999 in which the PEX14 binding sites were localized to the NH2-terminal half of PEX5. Additional truncation mutants failed to define a smaller PEX12-binding site within PEX5 (data not shown).
The Zinc-binding Domain of PEX12 Binds PEX10
In addition to the interaction between PEX12 and PEX5, the yeast two-hybrid screen also revealed an interaction between PEX12 and PEX10, an integral PMP that is required for peroxisomal matrix protein import ( Fig. 3 A). Like PEX12, human PEX10 contains a cytoplasmically exposed zinc ring domain ( Warren et al. 1998) and the interaction we detected between these two proteins was mediated through their COOH-terminal zinc ring domains (amino acids 240–326 of PEX10 and 260–359 of PEX12). Independent biochemical evidence for physical interaction between PEX12 and PEX10 was obtained using blot overlay experiments. Equal amounts of purified, recombinant MBP-PEX12 and MBP-LacZα were resolved by SDS-PAGE, immobilized on membranes, and probed with 35S-labeled PEX10 that had been synthesized in vitro in rabbit reticulocytes ( Fig. 3 B). PEX10 was bound by the MBP-PEX12 fusion protein but not by MBP-LacZα, suggesting specific binding between PEX12 and PEX10. To assess whether these proteins were present in a complex in vivo, we transfected normal human fibroblasts with plasmids designed to express tagged forms of these proteins, PEX12/3xmyc and PEX10/HA, or PEX10/HA and an untagged version of PEX12. 2 d after transfection, lysates were prepared from the two sets of transfected cells, subjected to immunoprecipitation using an anti-myc polyclonal antibody, and then blotted with a monoclonal anti–HA antibody. Equivalent amounts of PEX10/HA were detected in both crude lysates, but PEX10/HA was only detected in the immunoprecipitate from cells expressing PEX12/3xmyc. Thus, PEX12 and PEX10 do appear to be present in a complex in vivo ( Fig. 3 C). Control experiments revealed that these tagged forms of PEX12 and PEX10 have normal activity in vivo. This region of PEX12 failed to interact with any of the remaining 12 human peroxins in the yeast two-hybrid assay.
Figure 3.
The COOH-terminal domain of PEX12 interacts with the COOH-terminal domain of PEX10 in the yeast two-hybrid system. Two-hybrid reporter strains expressing the indicated fusion proteins were transferred to a nitrocellulose filter, submerged in liquid nitrogen to lyse the cells, and assayed for β-galactosidase activity. AD, GAL4 activation domain; and BD, GAL4-binding domain. (B) PEX10 binds immobilized PEX12C in vitro. 10 μg of purified MBP-LacZα and purified MBP-PEX12C were resolved by SDS-PAGE, transferred to membranes, and probed with 35S-labeled PEX10 (upper panel). A duplicate gel was stained before transfer with Coomassie blue (lower panel). (C) Coimmunoprecipitation of PEX10 with PEX12. Lysates from cells cotransfected with either pcDNA3-PEX12 and pcDNA3-PEX10/3xHA (left lane) or pcDNA3-PEX12/3xmyc and pcDNA3-PEX10/3xHA (right lane) were immunoprecipitated with anti–myc antibodies and analyzed by immunoblot with anti–HA antibodies. Aliquots of the crude lysates before immunoprecipitation were also assayed for PEX10/3xHA levels by immunoblot.


Genetic Interactions between PEX12, PEX10, and PEX5
The simplest explanation for the physical association of PEX12 with both PEX5 and PEX10 is that these interactions contribute to the biogenesis of peroxisomes. In such an instance, we might expect that high dosage, allele-specific extragenic suppression could be observed among the corresponding three genes. Therefore, we tested whether overexpression of any one of these genes could suppress mutations in either of the other two genes. In brief, fibroblast cell lines with mild or severe mutations in PEX12, PEX10, or PEX5 were transfected separately with expression vectors designed to express these genes, as well as a vector control. 2 d later, each cell population was processed for indirect immunofluorescence using antibodies specific for a peroxisomal matrix protein marker, catalase. The relative rescue activity of each gene in each cell line was calculated by comparing the frequency of cells importing catalase in each set of transfected cells.
All three patients from complementation group 2 of the PBDs have mutations in PEX5 ( Dodt et al. 1995; Slawecki et al. 1995). Two of these patients (PBD018 and PBD093) are homozygous for a PEX5-N489K/N526K mutation (N489/N586 refers to the position of this asparagine in PEX5S and PEX5L, respectively). The other PEX5-deficient patient, PBD005, is homozygous for a PEX5 nonsense mutation, R390ter/R427ter, which inactivates PEX5. PBD018 and PBD005 cells were transfected with the plasmids pcDNA3, pcDNA3-PEX5, pcDNA3-PEX12, and pcDNA3-PEX10, incubated for 2 d, and then processed for immunofluorescence using antibodies specific for peroxisomal catalase. Although expression of PEX5 efficiently rescued catalase import in both cell lines, PEX12 and PEX10 were unable to restore catalase import in these lines (data not shown).
We have also characterized eight complementation group 3 PBD patients, all of whom are mutated in PEX12 ( Chang et al. 1997; Chang and Gould 1998). Fibroblast cell lines from each of these patients were transfected with the above vectors, and the import of peroxisomal matrix proteins was determined in each population of transfected cells. As expected, pcDNA3-PEX12 efficiently rescued the peroxisomal protein import defects in all of these cell lines. However, we failed to observe any evidence for extragenic suppression in the CG3 cells that were transfected with the PEX10 or PEX5 expression vectors (data not shown).
Our previous work has established that PEX10 is the gene defective in two patients from complementation group 7 (CG7), PBD052, and PBD100 ( Warren et al. 1998). Although we have yet to identify the PEX10 mutations in four other CG7 patients, we used fibroblasts from all six available CG7 patients for our studies. Expression of PEX10 rescued peroxisomal matrix protein import in all six CG7 cell lines, whereas PEX12 and PEX5 failed to have any effect on the cytosolic catalase distribution in five of these cell lines. However, expression of PEX12 clearly led to the restoration of catalase import in PBD054 cells ( Fig. 4, A–F). Expression of PEX5 also rescued peroxisomal matrix protein import in this cell line, though to a lesser extent (Table ).
Figure 4.

Peroxisomal protein import in PBD054 cells is restored by expression of PEX10 or PEX12. PBD054 cells were transfected with pcDNA3 (A and D), pcDNA3-PEX10 (B and E), or pcDNA3-PEX12 (C and F). 2 d after transfection, the cells were fixed, permeabilized with Triton X-100, and processed for double indirect immunofluorescence using anticatalase antibodies (A–C) and anti–PMP70 antibodies (D–F). Bar, 25 μm.
Table 2.
Suppression of Catalase Import Defect in PBD054 Cells by Overexpression of PEX5 and PEX10
| PEX gene | Percent relative activity to restore catalase import in PBD054 cells* |
|---|---|
| HsPEX12 | 100 |
| HsPEX10 | 40 ± 7 |
| HsPEX5S | 19 ± 9 |
| HsPEX5L | 13 ± 3 |
* n = 3.
To better understand the molecular basis of the apparent suppression of PEX10 allele(s) by overexpression of PEX12 or PEX5, we sequenced the PEX10 gene from PBD054 cells. Surprisingly, we failed to detect any alteration to the gene in this patient. This result, combined with the fact that PEX12 was more effective than PEX10 at rescuing peroxisomal protein import in PBD054 cells (Table ), led us to consider whether PEX12, rather than PEX10, might be mutated in PBD054. The PEX12 gene was amplified from PBD054 genomic DNA and all coding portions of the gene were sequenced directly from the PCR products. We detected a missense mutation, S320F, in the PEX12 gene from this patient and no evidence of the wild-type sequence, suggesting that this patient was homozygous for this mutation in PEX12 ( Fig. 5 A). Although S320 is a conserved residue of PEX12 from yeast to humans (always a serine or threonine), missense mutations may be silent. We engineered the S320F mutation into the PEX12 expression vector and used a functional complementation assay ( Chang et al. 1997; Chang and Gould 1998) to assess the effects of this mutation. PEX12-deficient cells were transfected with pcDNA3, pcDNA3-PEX12, or pcDNA3-PEX12/S320F, and 2 d after transfection the percentage of cells importing matrix proteins into peroxisomes was determined by immunofluorescence using antibodies to the peroxisomal matrix protein catalase. In each of two trials the PEX12/S320F cDNA displayed 10–15% of the rescue activity of the wild-type PEX12 cDNA (data not shown). The fact that the S320F mutation reduced but did not eliminate PEX12 function was consistent with the relatively mild cellular and clinical phenotypes of this patient (PBD054 was diagnosed with neonatal adrenoleukodystrophy, and a previous study has demonstrated that PBD054 cells are able to import small amounts of some peroxisomal matrix proteins [ Dodt and Gould 1996]).
Figure 5.

PBD054 is mutated in PEX12. (A) Sequence chromatographs of PEX12 genomic DNA showing the wild-type sequence (top) and PBD054 sequence (bottom) in the region corresponding to nucleotides 950–968 of the PEX12 ORF. Note the C to T transition mutation, S320F, which changes the serine codon TCT to the phenylalanine codon TTT (underlined). (B) The S320F mutation attenuates the interaction between PEX12 and PEX10. Two-hybrid reporter strains expressing the indicated fusion proteins were assayed for β-galactosidase activity (top). To assess the effects of this mutation by blot overlay assay, purified MBP-LacZα, MBP-PEX12C/S320F, and MBP-PEX12C were separated by SDS-PAGE, transferred to membranes, and probed with 35S-labeled PEX10 (upper half of lower panel). These proteins were also separated by SDS-PAGE and stained with Coomassie blue (lower half of lower panel). (C) The S320F mutation attenuates the PEX12-PEX5 interaction in the yeast two-hybrid assay. Two-hybrid reporter strains expressing the listed proteins were assayed for β-galactosidase activity (top). The three strains expressing AD-5S were also lysed and assayed for BD-PEX12 levels by immunoblot with anti–PEX12 antibodies.
The ability of PEX10 and PEX5 to suppress the PEX12/S320F mutation was a clear example of allele-specific suppression rather than bypass suppression since neither PEX10 nor PEX5 were capable of rescuing peroxisomal protein import in any cells with severe mutations in PEX12. This finding, together with the fact that the PEX12-S320F mutation lies within the zinc-binding domain of PEX12, suggested that this mutation might reduce the interaction between the zinc ring domain of PEX12 and either PEX10 or PEX5. Using the two-hybrid assay and the blot overlay assay, we observed that the S320F mutation led to a marked reduction in the PEX12–PEX10 interaction ( Fig. 5 B). Similarly, the PEX12/S320F mutation appeared to reduce the interaction between PEX12 and PEX5 in the yeast two-hybrid assay ( Fig. 5 C). It is interesting to note that the placement of PBD054 cells into CG7 of the PBDs was based upon its noncomplementation with PBD052 cells. PBD052 cells are mutated in PEX10 and express one allele with a missense mutation (H290Q) in the PEX10 zinc-binding domain ( Warren et al. 1998). These results indicate that the combination of the PEX12/S320F and PEX10/H290Q alleles may have a deleterious effect on peroxisomal matrix protein import even in the presence of normal alleles of each gene.
PEX12 Appears to Act Downstream of Receptor Docking
A variety of earlier studies have established that loss of PEX12 or PEX10 results in a severe defect in peroxisomal matrix protein import ( Kalish et al. 1995; Liu et al. 1995; Slawecki et al. 1995; Kalish et al. 1996; Chang et al. 1997; Okumoto et al. 1998a, Okumoto et al. 1998b; Warren et al. 1998). It is also known that PEX12 and PEX10 are not required for synthesis of peroxisome membranes or import of peroxisomal membrane proteins ( Kalish et al. 1995, Kalish et al. 1996; Chang et al. 1997, Chang et al. 1999; Chang and Gould 1998; Warren et al. 1998). Furthermore, the morphological abnormalities that have been reported for peroxisomes in cells lacking PEX12 or PEX10 are indistinguishable from those of peroxisomes in PEX5-deficient cells and appear to be a secondary effect of the metabolic deficiencies that are caused by the matrix protein import defects in these cells ( Baes et al. 1997; Chang et al. 1999). These results, together with our observation that PEX12 interacts with both PEX5 and PEX10, suggest that PEX12 and PEX10 participate in peroxisomal matrix protein import.
Current models suggest that there may be several steps of peroxisomal matrix protein import that are limited to the peroxisome membrane and could, therefore, involve PEX12 or PEX10. These include docking of receptor–ligand (matrix protein) complexes to the peroxisome, matrix protein translocation across the peroxisome membrane, and receptor recycling ( Braverman et al. 1995; Rachubinski and Subramani 1995). To distinguish between these different possibilities, we first tested whether PEX12 or PEX10 were required for docking of the PTS1 receptor, PEX5, to peroxisomes. Postnuclear supernatants were prepared from normal human fibroblasts and from a fibroblast cell line which appears to lack PEX12 activity altogether (PBD006 [ Chang and Gould 1998]). Peroxisomes were pelleted from each postnuclear supernatant by centrifugation, and the relative amount of PEX5 in the cytosolic supernatant and organelle pellet was determined by immunoblot. Levels of peroxisome-associated PEX5 were not reduced in the absence of PEX12 or PEX10 and actually appeared to be slightly elevated in the pex10 and pex12 mutants ( Fig. 6 A). This was also evident in immunofluorescence experiments in which the staining for peroxisome-associated PEX5 appeared to be greater in the pex12 and pex10 mutants than in wild-type cells ( Fig. 6, B–G). These results argue against roles for PEX12 and PEX10 in PEX5 docking and suggest that they participate in a downstream step of peroxisomal matrix protein import.
Figure 6.

PEX12 and PEX10 are not required for PEX5 docking. (A) Postnuclear supernatants were separated into cytosolic supernatants (S) and organelle pellets (P). Equal proportions of the two fractions from each cell line were assayed for PEX5 levels by immunoblot. Immunofluorescence experiments were performed with wild-type (B and C), PEX12-deficient PBD006 (D and E), and PEX10-deficient PBD100 (F and G) cells using anti–PEX5 antibodies (B, D, and F) and anti–PMP70 antibodies (C, E, and G). Bar, 25 μm.
A Missense Mutation in PEX12 Results in PEX5 Import
Taken together, the properties of PEX12 indicate that it may participate in peroxisomal matrix protein translocation rather than receptor docking or recycling (as described in Discussion, the phenotypes of PEX12-deficient cells are not consistent with a role for PEX12 in receptor recycling). One prediction of this hypothesis is that mild mutations in PEX12 might alter the properties of the peroxisomal matrix protein translocation apparatus without eliminating translocation altogether. Before our discovery that PBD054 cells are mutated in PEX12, we reported that PBD054 cells import PEX5 into the peroxisome lumen and also import small amounts of some peroxisomal matrix proteins into peroxisomes ( Dodt and Gould 1996). We revisited the issue of PEX5 distribution in PBD054 cells and also compared it to the distribution of PEX5 in PBD006 cells. By immunofluorescence studies in which all cellular membranes are permeabilized, both PBD006 and PBD054 cells contain detectable levels of peroxisome-associated PEX5 ( Fig. 7, A–D). However, differential permeabilization experiments (in which antibodies only have access to cytoplasmically exposed antigens) revealed that cytoplasmically exposed PEX5 could still be detected in PBD006 cells but not in PBD054 cells ( Fig. 7, E–H). Similar results were observed in each of three trials.
Figure 7.
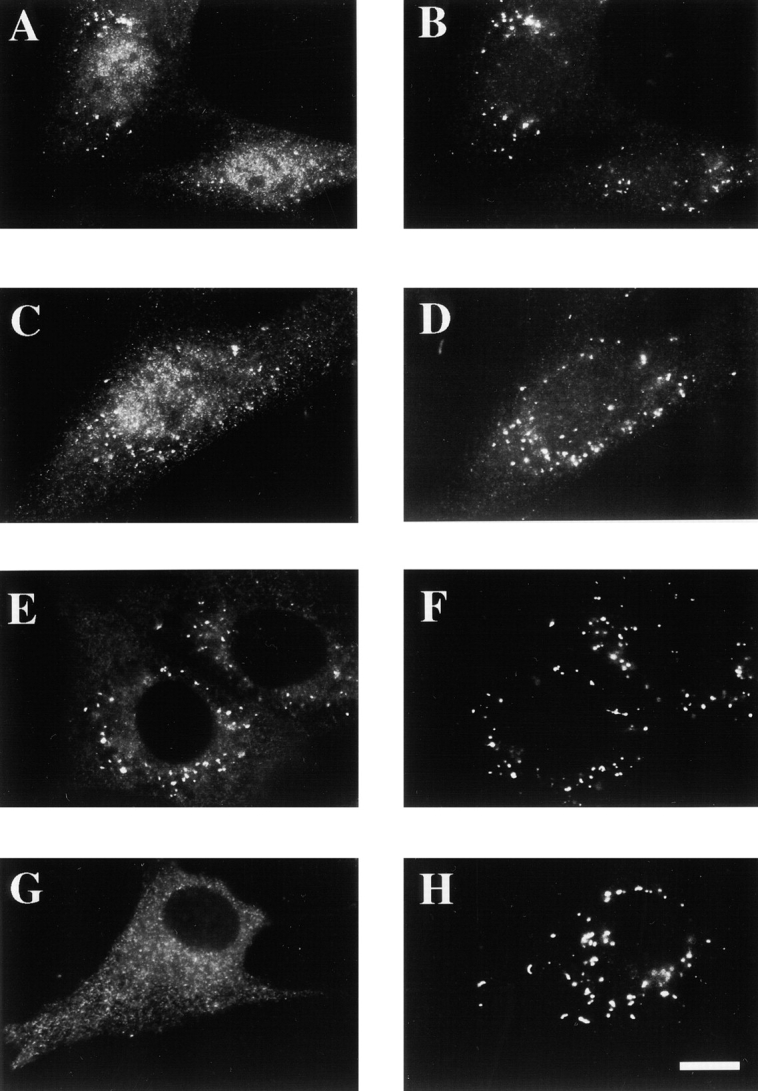
Distribution of PEX5 in PBD006 and PBD054 cells. PBD006 (A, B, E, and F) and PBD054 (C, D, G, and H) cells were fixed, permeabilized with either Triton X-100 (A–D) or digitonin (E–H), and processed for double indirect immunofluorescence using anti–PEX5 antibodies (A, C, E, and G) and anti–PMP70 antibodies (B, D, F, and H). Bar, 25 μm.
These differential permeabilization experiments indicated that PBD006 cells contained more cytoplasmically exposed PEX5 than PBD054 cells and that PBD054 cells imported PEX5 into the peroxisome lumen. However, there are two potential caveats to these experiments. First, immunofluorescence experiments can be influenced greatly by conformational changes in the antigen, in this case PEX5. Second, they do not address the question of whether some of the peroxisomal PEX5 in PBD006 cells may also be protected from antibodies in the differential permeabilization experiments. Therefore, we performed protease protection experiments on organelle preparations from PBD006 and PBD054 cells. Postnuclear supernatants were prepared from each cell line, and peroxisomes and other large organelles were recovered by differential centrifugation. These organelle pellets were resuspended and incubated with various amounts of protease in the presence or absence of detergent, the reactions were quenched, and each sample was assayed for levels of PEX5 by immunoblot ( Fig. 8A and Fig. B). PBD054 cells appeared to contain more protease-resistant PEX5 than PBD006 cells, which may reflect import of PEX5 into the peroxisome lumen of these cells. Similar results were obtained in each of three trials.
Figure 8.

Protease protection analysis of peroxisomal PEX5 in PBD006 and PBD054 cells. Organelle fractions were prepared from PBD006 (A) and PBD054 (B) cells and incubated with protease in the absence or presence of detergent, and then assayed for PEX5 by immunoblot binding. (C) Postnuclear supernatants from PBD006 and PBD054 cells were separated into cytosolic supernatants (S) and organelle pellets (P). Equal proportions of each were assayed for PEX5 levels by immunoblot.
If PBD054 cells actually do import PEX5 into the peroxisome lumen, we might expect that peroxisomes of these cells would contain more PEX5 than those of PBD006 cells. We tested this hypothesis by cell fractionation studies. Postnuclear supernatants were prepared from wild-type, PBD006 and PBD054 fibroblasts, peroxisomes were separated from cytosol by differential centrifugation, and the levels of PEX5 in the cytosolic supernatant and organelle pellet fractions were determined by immunoblot. PBD054 cells contained more peroxisome-associated PEX5, as predicted ( Fig. 8 C).
Discussion
In this paper we investigated the role of PEX12 in peroxisome biogenesis by examining the phenotypes of PEX12-deficient cells and identifying peroxins that physically and genetically interact with PEX12. Previous studies have established that loss of PEX12 results in the absence of detectable peroxisomal matrix protein import, but has virtually no effect on the synthesis of peroxisomes or the import of peroxisomal membrane proteins ( Kalish et al. 1996; Chang et al. 1997, Chang et al. 1999; Chang and Gould 1998; Okumoto et al. 1998b). Such a phenotype alone points to a role for PEX12 in peroxisomal matrix protein import. However, the data presented in this report advance this hypothesis by demonstrating physical and genetic interaction between PEX12 and PEX5, the receptor for newly synthesized peroxisomal matrix proteins. We detected interactions between PEX12 and PEX5 using the yeast two-hybrid system, by filter binding assay, in coimmunoprecipitation experiments, and by genetic suppression studies.
A Role for PEX12 in Peroxisomal Matrix Protein Import
It is generally accepted that PEX5 is the receptor for newly synthesized PTS1-containing proteins ( McCollum et al. 1993; Dodt et al. 1995; Fransen et al. 1995; Terlecky et al. 1995; Elgersma et al. 1996) and is a predominantly cytoplasmic protein in mammalian cells ( Dodt et al. 1995; Dodt and Gould 1996) and several fungal species ( van der Klei et al. 1995; Elgersma et al. 1996; Gould et al. 1996). Studies in human cells have suggested that PEX5 shuttles between the cytoplasm and peroxisome ( Dodt and Gould 1996), and several models predict that PEX5 moves through a variety of steps as it catalyzes peroxisomal matrix protein import. The proportion of PEX5 that resides in the cytoplasm at steady state probably reflects the needs of each cell to efficiently capture newly synthesized peroxisomal matrix proteins (ligands) from the cytoplasm. However, once these ligands are bound by PEX5 many additional events must occur. These may be grouped into the general processes of (1) the transport to and docking of PEX5–ligand complexes with the peroxisome membrane, (2) the translocation of ligands into the peroxisome lumen, and (3) the recycling of receptors back to the cytoplasm ( Braverman et al. 1995; Rachubinski and Subramani 1995). To distinguish which of these events may involve PEX12, we considered the phenotypes that are expected for a mutant in each process and compared them to the phenotypes of PEX12-deficient cells.
Several studies have implicated the integral peroxisomal membrane proteins PEX13 and PEX14 in docking of PTS receptors to the peroxisome membrane ( Elgersma et al. 1996; Erdmann and Blobel 1996; Gould et al. 1996; Albertini et al. 1997; Girzalsky et al. 1999). The defining features of these docking factors are as follows: (1) their ability to bind PEX5 and PEX7, either directly or indirectly, and (2) the fact that loss of PEX13 or PEX14 results in a significant reduction in the amount of peroxisome-associated PEX5. The fact that loss-of-function mutations in PEX12 do not result in any detectable reduction in the levels of peroxisome-associated PEX5 argues strongly against the hypothesis that PEX12 participates in receptor docking. In fact, our data indicate that the loss of PEX12 may actually increase the levels of peroxisome-associated PEX5. This observation, together with the fact that receptor docking is the first peroxisome-localized step of peroxisomal matrix protein import, demonstrates that PEX12 acts downstream of the docking event. Thus, PEX12 appears to be the first known PEX5-binding protein that is not required for docking the PTS1 receptor to the peroxisome.
Of the two remaining aspects of peroxisomal matrix protein import, protein translocation and receptor recycling, there is no report of a bona fide protein translocation factor, but there is one report that proposes a role for PEX4 in receptor recycling ( van der Klei et al. 1998). This conclusion was based in part on the phenotypes of pex4 mutants, which display a very mild defect in peroxisomal matrix protein import and can be suppressed by overexpression of PEX5. However, we observed that cells lacking human PEX12 display a severe defect in peroxisomal matrix protein import that cannot be suppressed by overexpression of PEX5. Thus, PEX12 does not have the properties we might expect of a factor that is required for receptor recycling.
The remaining aspect of peroxisomal matrix protein import to consider is the protein translocation process. Actually, a role for PEX12 in peroxisomal matrix protein translocation would fit well with the known properties of this protein. First, PEX12 has the appropriate physical characteristics for such a role: it is an integral peroxisomal membrane protein that spans the membrane twice and extends its NH2 and COOH termini toward the cytoplasm where they may interact with other protein import factors ( Kalish et al. 1996; Chang et al. 1997; Okumoto and Fujiki 1997). Second, it utilizes its COOH-terminal zinc-binding domain to interact with PEX5, the PTS1 receptor. Third, cells with inactivating mutations in PEX12 are unable to import peroxisomal matrix proteins but do synthesize peroxisomes and import integral peroxisomal membrane proteins ( Kalish et al. 1996; Chang et al. 1997, Chang et al. 1999; Chang and Gould 1998). Fourth, PEX12 interacts with PEX10, another integral peroxisomal membrane protein that displays a specific defect in the import of peroxisomal matrix proteins ( Kalish et al. 1995; Warren et al. 1998) and yet does not appear to participate in receptor docking or recycling. Fifth, a missense mutation in PEX12, S320F, appears to affect the specificity of the translocation apparatus, resulting in the import of PEX5 into the peroxisome lumen.
Although there is no established in vitro protein translocation assay that can be used to test this hypothesis directly, it is useful to consider possible roles for PEX12 in the translocation process. The main function of a matrix protein translocon would be to move matrix proteins from the cytoplasmic side of the peroxisome membrane to the lumenal side. Given that most of these proteins arrive at the peroxisomes in a complex with PEX5 and that studies of Yarrowia lipolytica PEX5 strongly suggest that PEX5 participates in the matrix protein translocation process ( Szilard et al. 1995), we favor a model for matrix protein translocation that includes PEX5. Such a model may involve: (1) acceptance of PEX5–ligand complexes from PEX14, the primary PEX5 docking site; (2) retention of PEX5 at the translocation apparatus; (3) opening of the matrix protein translocation pathway; (4) PEX5–ligand dissociation and ligand translocation; (5) closure of the translocation pathway; and (6) release of the unoccupied receptor from the translocation apparatus. The ability of PEX12 to bind PEX5 suggests that PEX12 may contribute to retaining PEX5 at the translocation apparatus. Some additional support for this hypothesis comes from the fact that PEX5 appears to be imported into the peroxisome as a result of the PEX12/S320F mutation, which reduces the interaction between PEX12 and PEX5.
This model predicts that PEX5 enters the peroxisomal compartment during the normal course of peroxisomal matrix protein import. However, it also predicts that PEX5 should normally be retained at the translocation apparatus rather than being released to move freely through the peroxisome lumen. A low rate of PEX5 release from the translocation apparatus into the lumen could explain the detection of intraperoxisomal PEX5 in Hansenula polymorpha ( van der Klei et al. 1995). Y. lipolytica PEX5, which is detected only on or in the peroxisome, could also function within such a model, provided that it participates in just the peroxisome-limited steps of matrix protein import ( Szilard et al. 1995).
Acknowledgments
We thank James Morrell for excellent technical assistance in plasmid construction. We also thank Dr. Gabrielle Dodt (Ruhr-Universitat, Bochum, Germany) for providing polyclonal anti–PEX5 antibodies that were used in some of the experiments.
This work was supported by grants from the National Institutes of Health to S.J. Gould (DK45787 and HD10987).
Footnotes
1.used in this paper: MBP, maltose-binding protein; ORF, open reading frame; PBD, peroxisome biogenesis disorder; PMP, peroxisomal membrane protein
References
- Adams A., Gottschling D.E., Kaiser C.A., Stearns T. Methods in Yeast Genetics 1997. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: pp. 115–116 [Google Scholar]
- Albertini M., Rehling P., Erdmann R., Girzalsky W., Kiel J.A., Veenhuis M., Kunau W.-H. Pex14p, a peroxisomal membrane protein binding both receptors of the two PTS-dependent import pathways. Cell. 1997;89:83–92 . doi: 10.1016/s0092-8674(00)80185-3. [DOI] [PubMed] [Google Scholar]
- Baes M., Gressens P., Baumgart E., Carmeliet P., Casteels M., Fransen M., Evrard P., Fahimi D., Declercq P.E., Collen D., van Veldhoven P.P., Mannaerts G.P. A mouse model for Zellweger syndrome. Nat. Genet. 1997;17:49–57 . doi: 10.1038/ng0997-49. [DOI] [PubMed] [Google Scholar]
- Borden K.L. RING fingers and B-boxeszinc-binding protein-protein interaction domains. Biochem. Cell Biol. 1998;76:351–358 . doi: 10.1139/bcb-76-2-3-351. [DOI] [PubMed] [Google Scholar]
- Braverman N., Dodt G., Gould S.J., Valle D. Disorders of peroxisome biogenesis. Hum. Mol. Genet. 1995;4:1791–1798 . doi: 10.1093/hmg/4.suppl_1.1791. [DOI] [PubMed] [Google Scholar]
- Braverman N., Steel G., Obie C., Moser A., Moser H., Gould S.J., Valle D. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat. Genet. 1997;15:369–376 . doi: 10.1038/ng0497-369. [DOI] [PubMed] [Google Scholar]
- Braverman N., Dodt G., Gould S.J., Valle D. An isoform of Pex5p, the human PTS1 receptor, is required for the import of PTS2 proteins into peroxisomes. Hum. Mol. Genet. 1998;7:1195–1205 . doi: 10.1093/hmg/7.8.1195. [DOI] [PubMed] [Google Scholar]
- Chang C.-C., Gould S.J. Phenotype-genotype relationships in complementation group 3 of the peroxisome-biogenesis disorders. Am. J. Hum. Genet. 1998;63:1294–1306 . doi: 10.1086/302103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.-C., Lee W.-H., Moser H.W., Valle D., Gould S.J. Isolation of the human PEX12 gene, mutated in group 3 of the peroxisome biogenesis disorders. Nat. Genet. 1997;15:385–388 . doi: 10.1038/ng0497-385. [DOI] [PubMed] [Google Scholar]
- Chang C.-C., South S., Warren D., Jones J., Moser A.B., Moser H.W., Gould S.J. Metabolic control of peroxisome abundance. J. Cell Sci. 1999;112:1579–1590 . doi: 10.1242/jcs.112.10.1579. [DOI] [PubMed] [Google Scholar]
- Chevray P.M., Nathans D. Protein interaction cloning in yeastidentification of mammalian protein that reacts with the leucine zipper of Jun. Proc. Natl. Acad. Sci. USA. 1992;89:5789–5793 . doi: 10.1073/pnas.89.13.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C.S., Gould S.J. Identification of a common mutation in severely affected PEX1-deficient patients. Hum. Mutat. 1999;14:45–53 . doi: 10.1002/(SICI)1098-1004(1999)14:1<45::AID-HUMU6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Distel B., Erdmann R., Gould S.J., Blobel G., Crane D.I., Cregg J.M., Dodt G., Fujiki Y., Goodman J.M., Just W.W. A unified nomenclature for peroxisome biogenesis. J. Cell Biol. 1996;135:1–3 . doi: 10.1083/jcb.135.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt G., Gould S.J. Multiple PEX genes are required for proper subcellular distribution and stability of Pex5p, the PTS1 receptorevidence that PTS1 protein import is mediated by a cycling receptor. J. Cell Biol. 1996;135:1763–1774 . doi: 10.1083/jcb.135.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt G., Braverman N., Wong C., Moser A., Moser H.W., Watkins P., Valle D., Gould S.J. Mutations in the PTS1 receptor gene, PXR1, define complementation group 2 of the peroxisome biogenesis disorders. Nat. Genet. 1995;9:115–124 . doi: 10.1038/ng0295-115. [DOI] [PubMed] [Google Scholar]
- Dyer J.M., McNew J.A., Goodman J.M. The sorting sequence of the peroxisomal integral membrane protein PMP47 is contained within a short hydrophilic loop. J. Cell Biol. 1996;133:269–280 . doi: 10.1083/jcb.133.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgersma Y., Kwast L., Klein A., Voorn-Brouwer T., van den Berg M., Metzig B., America T., Tabak H.F., Distel B. The SH3 domain of the Saccharomyces cerevisiae peroxisomal membrane protein Pex13p functions as a docking site for Pex5p, a mobile receptor for the import of PTS1-containing proteins. J. Cell Biol. 1996;135:97–109 . doi: 10.1083/jcb.135.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann R., Blobel G. Identification of Pex13p, a peroxisomal membrane receptor for the PTS1 recognition factor. J. Cell Biol. 1996;135:111–121 . doi: 10.1083/jcb.135.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann R., Wiebel F.F., Flessau A., Rytka J., Beyer A., Frohlich K.U., Kunau W.-H. PAS1, a yeast gene required for peroxisome biogenesis, encodes a member of a novel family of putative ATPases. Cell. 1991;64:499–510 . doi: 10.1016/0092-8674(91)90234-p. [DOI] [PubMed] [Google Scholar]
- Evan G.E., Lewis G.K., Ramsay G., Bishop J.M. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 1985;5:3610–3616 . doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber K.N., Heyman J.A., Subramani S. Two AAA family proteins, PpPex1p and PpPex6p, interact with each other in an ATP-dependent manner and are associated with different subcellular membranous structures distinct from peroxisomes. Mol. Cell. Biol. 1998;18:936–943 . doi: 10.1128/mcb.18.2.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen M., Brees C., Baumgart E., Vanhooren J.C.T., Baes M., Mannaerts G.P., van Veldhoven P.P. Identification and characterization of the putative human peroxisomal terminal targeting signal import receptor. J. Biol. Chem. 1995;270:7731–7736 . doi: 10.1074/jbc.270.13.7731. [DOI] [PubMed] [Google Scholar]
- Fransen M., Terlecky S.R., Subramani S. Identification of a human PTS1 receptor docking protein directly required for peroxisomal protein import. Proc. Natl. Acad. Sci. USA. 1998;95:8087–8092 . doi: 10.1073/pnas.95.14.8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda S., Shimozawa N., Suzuki Y., Zhang Z., Tomatsu S., Tsukamoto T., Hashiguchi N., Osumi T., Masuno M., Imaizumi K. Human peroxisome assembly factor-2 (PAF-2)a gene responsible for group C peroxisome biogenesis disorder in humans. Am. J. Hum. Genet. 1996;59:1210–1220 . [PMC free article] [PubMed] [Google Scholar]
- Geisbrecht B.V., Collins C.S., Reuber B.E., Gould S.J. Disruption of a PEX1-PEX6 interaction is the most common cause of the neurologic disorders Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease. Proc. Natl. Acad. Sci. USA. 1998;95:8630–8635 . doi: 10.1073/pnas.95.15.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girzalsky W., Rehling P., Stein K., Kipper J., Blank L., Kunau W.H., Erdmann R. Involvement of Pex13p in Pex14p localization and peroxisomal targeting signal 2–dependent protein import into peroxisomes. J. Cell Biol. 1999;144:1151–1162 . doi: 10.1083/jcb.144.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould S.J., Keller G.A., Hosken N., Wilkinson J., Subramani S. A conserved tripeptide sorts proteins to peroxisomes. J. Cell Biol. 1989;108:1657–1664 . doi: 10.1083/jcb.108.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould S.J., Krisans S., Keller G.A., Subramani S. Antibodies directed against the peroxisomal targeting signal of firefly luciferase recognize multiple mammalian peroxisomal proteins. J. Cell Biol. 1990;110:27–34 . doi: 10.1083/jcb.110.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould S.J., Kalish J.E., Morrell J.C., Bjorkman J., Urquhart A.J., Crane D.I. An SH3 protein in the peroxisome membrane is a docking factor for the PTS1 receptor. J. Cell Biol. 1996;135:85–95 . doi: 10.1083/jcb.135.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant S.G., Jessee J., Bloom F.R., Hanahan D. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. USA. 1990;87:4645–4649 . doi: 10.1073/pnas.87.12.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyman J.A., Mononsov E., Subramani S. Role of the PAS1 gene of Pichia pastoris in peroxisome biogenesis. J. Cell Biol. 1994;127:1259–1273 . doi: 10.1083/jcb.127.5.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honsho M., Tamura S., Shimozawa N., Suzuki Y., Kondo N., Fujiki Y. Mutation in PEX16 is causal in the peroxisome-deficient Zellweger syndrome of complementation group D. Am. J. Hum. Genet. 1998;63:1622–1630 . doi: 10.1086/302161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huhse B., Rehling P., Albertini M., Blank L., Meller K., Kunau W.H. Pex17p of Saccharomyces cerevisiae is a novel peroxin and component of the peroxisomal protein translocation machinery. J. Cell Biol. 1998;140:49–60 . doi: 10.1083/jcb.140.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalish J.E., Theda C., Morrell J.C., Berg J.M., Gould S.J. Formation of the peroxisome lumen is abolished by loss of Pichia pastoris Pas7p, a zinc-binding integral membrane protein of the peroxisome. Mol. Cell. Biol. 1995;15:6406–6419 . doi: 10.1128/mcb.15.11.6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalish J.E., Keller G.A., Morrell J.C., Mihalik S.J., Smith B., Cregg J.M., Gould S.J. Characterization of a novel component of the peroxisomal protein import apparatus using fluorescent peroxisomal proteins. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:3275–3285 . [PMC free article] [PubMed] [Google Scholar]
- Lazarow P.B., Fujiki Y. Biogenesis of peroxisomes. Annu. Rev. Cell Biol. 1985;1:489–530 . doi: 10.1146/annurev.cb.01.110185.002421. [DOI] [PubMed] [Google Scholar]
- Lazarow P.B., Moser H.W. Disorders of peroxisome biogenesis. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York : 1995. pp. 2287–2324. [Google Scholar]
- Liu H., Tan X., Russell K.A., Veenhuis M., Cregg J.M. PER3, a gene required for peroxisome biogenesis in Pichia pastoris, encodes a peroxisomal membrane protein involved in protein import. J. Biol. Chem. 1995;270:10940–10951 . doi: 10.1074/jbc.270.18.10940. [DOI] [PubMed] [Google Scholar]
- Liu Y., Bjorkman J., Urquhart A., Wanders R.J.A., Crane D., Gould S.J. PEX13 is mutated in complementation group 13 of the peroxisome biogenesis disorders. Am. J. Hum. Genet. 1999;65:621–634 . doi: 10.1086/302534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzioch M., Erdmann R., Veenhuis M., Kunau W.-H. PAS7 encodes a novel yeast member of the WD-40 protein family essential for import of 3-oxoacyl-CoA thiolase, a PTS2-containing protein, into peroxisomes. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:4908–4918 . doi: 10.1002/j.1460-2075.1994.tb06818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlack K.E., Mothes W., Rapoport T.A. Protein translocationtunnel vision. Cell. 1998;92:381–390 . doi: 10.1016/s0092-8674(00)80930-7. [DOI] [PubMed] [Google Scholar]
- Matsuzono Y., Kinoshita N., Tamura S., Shimozawa N., Hamasaki M., Ghaedi K., Wanders R.J., Suzuki Y., Kondo N., Fujiki Y. Human PEX19cDNA cloning by functional complementation, mutation analysis in a patient with Zellweger syndrome, and potential role in peroxisomal membrane assembly. Proc. Natl. Acad. Sci. USA. 1999;96:2116–2121 . doi: 10.1073/pnas.96.5.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattaj I.W., Englmeier L. Nucleocytoplasmic transportthe soluble phase. Annu. Rev. Biochem. 1998;67:265–306 . doi: 10.1146/annurev.biochem.67.1.265. [DOI] [PubMed] [Google Scholar]
- McCollum D., Monosov E., Subramani S. The pas8 mutant of Pichia pastoris exhibits the peroxisomal protein import deficiencies of Zellweger syndrome cells. The PAS8 protein binds to the COOH-terminal tripeptide peroxisomal targeting signal and is a member of the TPR protein family. J. Cell Biol. 1993;121:761–774 . doi: 10.1083/jcb.121.4.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley A.M., Hettema E.H., Hogenhout E.M., Brittes P., ten Asbroek A.L., Wijburg F.A., Baas F., Heijmans H.S., Tabak H.F., Wanders R.J.A., Distel B. Rhizomelic chondrodysplasia punctata is a peroxisomal protein targeting disease caused by a non-functional PTS2 receptor. Nat. Genet. 1997;15:377–380 . doi: 10.1038/ng0497-377. [DOI] [PubMed] [Google Scholar]
- Okumoto K., Fujiki Y. PEX12 encodes an integral membrane protein of peroxisomes. Nat. Genet. 1997;17:265–266 . doi: 10.1038/ng1197-265. [DOI] [PubMed] [Google Scholar]
- Okumoto K., Itoh R., Shimozawa N., Suzuki Y., Tamura S., Kondo N., Fujiki Y. Mutations in PEX10 is the cause of Zellweger peroxisome deficiency syndrome of complementation group B Hum. Mol. Genet. 7 1998. 1399 1405a [DOI] [PubMed] [Google Scholar]
- Okumoto K., Shimozawa N., Kawai A., Tamura S., Tsukamoto T., Osumi T., Moser H., Wanders R.J.A., Suzuki Y., Kondo N., Fujiki Y. PEX12, the pathogenic gene of group III Zellweger syndromecDNA cloning by functional complementation on a CHO cell mutant, patient analysis, and characterization of Pex12p Mol. Cell. Biol. 18 1998. 4324 4336b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H., Okumoto K., Tateishi K., Ikoma Y., Matsuda E., Mishimura M., Tsukamoto T., Osumi T., Ohashi K., Higuchi O., Fujiki Y. Peroxisome targeting signal type-1 (PTS1) receptor is involved in import of both PTS1 and PTS2studies with PEX5-defective CHO cell mutants. Mol. Cell. Biol. 1998;18:388–399 . doi: 10.1128/mcb.18.1.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfanner N., Craig E.A., Honlinger A. Mitochondrial preprotein translocase. Annu. Rev. Cell Dev. Biol. 1997;13:25–51 . doi: 10.1146/annurev.cellbio.13.1.25. [DOI] [PubMed] [Google Scholar]
- Portsteffen H., Beyer A., Becker E., Epplen C., Pawlak A., Kunau W.-H., Dodt G. Human PEX1 is mutated in complementation group 1 of the peroxisome biogenesis disorders. Nat. Genet. 1997;17:449–452 . doi: 10.1038/ng1297-449. [DOI] [PubMed] [Google Scholar]
- Purdue P.E., Zhang J.W., Skoneczny M., Lazarow P.B. Rhizomelic chondrodysplasia punctata is caused by deficiency of human PEX7 a homologue of the yeast PTS2 receptor. Nat. Genet. 1997;15:381–384 . doi: 10.1038/ng0497-381. [DOI] [PubMed] [Google Scholar]
- Rachubinski R.A., Subramani S. How proteins penetrate peroxisomes. Cell. 1995;83:525–528 . doi: 10.1016/0092-8674(95)90091-8. [DOI] [PubMed] [Google Scholar]
- Reuber B.E., Germain Lee E., Collins C.S., Morrell J.C., Ameritunga R., Moser H.W., Valle D., Gould S.J. Mutations in PEX1 are the most common cause of the peroxisome biogenesis disorders. Nat. Genet. 1997;17:445–448 . doi: 10.1038/ng1297-445. [DOI] [PubMed] [Google Scholar]
- Schatz G., Dobberstein B. Common principles of protein translocation across membranes. Science. 1996;271:1519–1526 . doi: 10.1126/science.271.5255.1519. [DOI] [PubMed] [Google Scholar]
- Schliebs W., Saidowsky J., Agianian B., Dodt G., Herberg F.W., Kunau W.H. Recombinant human peroxisomal targeting signal receptor PEX5. Structural basis for interaction of PEX5 with PEX14. J. Biol. Chem. 1999;274:5666–5673 . doi: 10.1074/jbc.274.9.5666. [DOI] [PubMed] [Google Scholar]
- Shimozawa N., Tsukamoto T., Suzuki Y., Orii T., Shirayoshi Y., Mori T., Fujiki Y. A human gene responsible for Zellweger syndrome that affects peroxisome assembly. Science. 1992;255:1132–1134 . doi: 10.1126/science.1546315. [DOI] [PubMed] [Google Scholar]
- Shimozawa N., Suzuki Y., Zhang Z., Imamura A., Toyama R., Mukai S., Fujiki Y., Tsukamoto T., Osumi T., Orii T., Wanders R.J.A., Kondo N. Nonsense and temperature-sensitive mutations in PEX13 are the cause of complementation group H of peroxisome biogenesis disorders. Hum. Mol. Genet. 1999;8:1077–1083 . doi: 10.1093/hmg/8.6.1077. [DOI] [PubMed] [Google Scholar]
- Slawecki M., Dodt G., Steinberg S., Moser A.B., Moser H.W., Gould S.J. Identification of three distinct peroxisomal protein import defects in patients with peroxisomal biogenesis disorders. J. Cell Sci. 1995;108:1817–1829 . doi: 10.1242/jcs.108.5.1817. [DOI] [PubMed] [Google Scholar]
- South S., Gould S.J. Peroxisome synthesis in the absence of preexisting peroxisomes. J. Cell Biol. 1999;144:255–266 . doi: 10.1083/jcb.144.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spong A.P., Subramani S. Cloning and characterization of PAS5a gene required for peroxisome biogenesis in the methylotrophic yeast Pichia pastoris . J. Cell Biol. 1993;123:535–548 . doi: 10.1083/jcb.123.3.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramani S. Protein import into peroxisomes and biogenesis of the organelle. Annu. Rev. Cell Biol. 1993;9:445–478 . doi: 10.1146/annurev.cb.09.110193.002305. [DOI] [PubMed] [Google Scholar]
- Swinkels B.W., Gould S.J., Bodnar A.G., Rachubinski R.A., Subramani S. A novel, cleavable peroxisomal targeting signal at the amino-terminus of the rat 3-ketoacyl-CoA thiolase. EMBO (Eur. Mol. Biol. Organ.) J. 1991;10:3244–3262 . doi: 10.1002/j.1460-2075.1991.tb04889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilard R.K., Titorenko V.I., Veenhuis M., Rachubinski R.A. Pay32p of the yeast Yarrowia lipolytica is an intraperoxisomal component of the matrix protein translocation machinery. J. Cell Biol. 1995;131:1453–1469 . doi: 10.1083/jcb.131.6.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlecky S.R., Nuttley W.M., McCollum D., Sock E., Subramani S. The Pichia pastoris peroxisomal protein Pas8p is the receptor for the C-terminal tripeptide peroxisomal targeting signal. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:3627–3634 . doi: 10.1002/j.1460-2075.1995.tb00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titorenko V.I., Rachubinski R.A. The endoplasmic reticulum plays an essential role in peroxisome biogenesis TIBS (Trends Biochem. Sci.). 23 1998. 231 233a [DOI] [PubMed] [Google Scholar]
- Titorenko V.I., Rachubinski R.A. Mutants of the yeast Yarrowia lipolytica defective in protein export exit from the endoplasmic reticulum are also defective in peroxisome biogenesis Mol. Cell. Biol. 18 1998. 2789 2803b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titorenko V.I., Ogrydziak D.M., Rachubinski R.A. Four distinct secretory pathways serve protein secretion, cell surface growth, and peroxisome biogenesis in the yeast Yarrowia lipolytica . Mol. Cell. Biol. 1997;17:5210–5226 . doi: 10.1128/mcb.17.9.5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Klei I.J., Hibrands R.E., Swaving G.J., Waterham H.R., Vrieling E.G., Titorenko V.I., Cregg J.M., Harder W., Veenhuis M. The Hansenula polymorpha PER3 gene is essential for the import of PTS1 proteins into the peroxisome matrix. J. Biol. Chem. 1995;270:17229–17236 . doi: 10.1074/jbc.270.29.17229. [DOI] [PubMed] [Google Scholar]
- van der Klei I.J., Hibrands R.E., Kiel J.A., Rasmussen S.W., Cregg J.M., Veenhuis M. The ubiquitin-conjugating enzyme Pex4p of Hansenula polymorpha is required for efficient functioning of the PTS1 import machinery. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3608–3618 . doi: 10.1093/emboj/17.13.3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M., Brachman R.K., Fattaey A., Harlow E., Boeke J.D. Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc. Natl. Acad. Sci. USA. 1996;93:10315–10320 . doi: 10.1073/pnas.93.19.10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren D.S., Morrell J.C., Moser H.W., Valle D., Gould S.J. Identification of PEX10, the gene defective in complementation group 7 of the peroxisome-biogenesis disorders. Am. J. Hum. Genet. 1998;63:347–359 . doi: 10.1086/301963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemer E.A.C., Nuttley W.M., Bertolaet B.L., Li X., Franke U., Wheelock M.J., Anne W.K., Johnson K.R., Subramani S. Human peroxisomal targeting signal-1 receptor restores peroxisomal protein import in cells from patients with fatal peroxisomal disorders. J. Cell Biol. 1995;130:51–65 . doi: 10.1083/jcb.130.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahraus T., Braverman N., Dodt G., Kalish J.E., Morrell J.C., Moser H.W., Valle D., Gould S.J. The peroxisome biogenesis disorder group 4 gene, PXAAA1, encodes a cytoplasmic ATPase required for stability of the PTS1 receptor. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:2914–2923. [PMC free article] [PubMed] [Google Scholar]