

Abstract
p53 is mutated in ∼50% of human cancers, whereas mutations of the related p73 gene are rare. p73 can activate p53-responsive promoters and induce apoptosis when overexpressed in certain p53-deficient tumor cells. We show that p73 isoforms, p73α and p73β, can each induce permanent growth arrest with markers of replicative senescence when overexpressed in a tetracycline-regulatable manner in human cancer cells lacking functional p53. Human homologue of mouse double minute 2 gene product (hMDM2), but not an NH2-terminal deletion mutant, coimmunoprecipated with p73α or p73β, and inhibited p73 transcriptional activity as with p53. In contrast to p53, ectopically expressed hemagglutinin (HA)-tagged p73 proteins were not stabilized by treatment with several DNA damaging agents. Furthermore, unlike normal p53, which increases in response to DNA damage due to enhanced protein stability in MCF7 cells, endogenous p73 protein levels were not increased in these cells under the same conditions. Thus, although p73 has an ability, comparable to that of p53, to suppress tumor cell growth in p53-deficient cells, p73 induction is regulated differently from p53. These findings suggest that the selective pressures for p53 rather than p73 inactivation in tumors may reflect their differential responses to stresses such as DNA damage, rather than their capacities to induce permanent growth arrest or apoptosis programs.
Keywords: p53, p73, tumor suppression, replicative senescence, DNA damage
The p53 gene is the most frequently inactivated tumor suppressor identified in human tumors. Approximately 50% of all human cancers lack a wild-type p53 allele, and thus fail to produce a normal version of the p53 protein ( Nigro et al. 1989; Harris and Hollstein 1993; Hollstein et al. 1994; Levine et al. 1995). Wild-type p53 limits cellular proliferation by inducing either a transient cell cycle block, apoptosis, or senescence, depending on the cellular context ( Levine 1997). These activities have been linked to the ability of p53 to bind to specific DNA sequences and activate transcription ( Gottlieb and Oren 1996; Ko and Prives 1996; Levine 1997). The p53 protein accumulates dramatically in response to genotoxic stress induced by DNA damage, hypoxia, and depletion of ribonucleotide triphosphate pools ( Levine 1997). The induction of p53 is mainly regulated at the level of protein stability. p53 is both neutralized and degraded by the mouse double minute 2 gene product ([MDM2] oncoprotein) (human homologue of MDM2 [hMDM2] in human) ( Momand et al. 1992; Oliner et al. 1993; Haupt et al. 1997; Kubbutat et al. 1997), which itself is a p53 target gene.
Recently, several members of the p53 family have been identified ( Jost et al. 1997; Kaghad et al. 1997; Osada et al. 1998; Trink et al. 1998; Yang et al. 1998). One member, p73, has been shown to encode two differently spliced products, p73α and p73β. p73β, which is 499 amino acids (aa) in length with a unique pentamer at its extreme COOH terminus, is 137 aa shorter than p73α, which has 636 aa. In addition, p73 not only shares a high degree of similarity with p53 in primary sequence (∼60% identity in the core DNA binding domain, 29% identity in NH2-terminal transactivation domain, and 42% identity in the COOH-terminal oligomerization domain), but also seems to exhibit similar functions. Like p53, both p73α and p73β can bind to DNA and activate transcription ( Jost et al. 1997; Marin et al. 1998; Zhu et al. 1998). It is currently unknown whether these proteins bind to DNA as homooligomers or heterooligomers. However, it has been reported that p73β can bind to itself and bind weakly to p73α in yeast two-hybrid assays ( Kaghad et al. 1997). When overproduced, both p73α and p73β can block cell proliferation and induce apoptosis in cells, irrespective of their p53 status ( Jost et al. 1997; Kaghad et al. 1997).
It is well-established that oncoproteins encoded by certain DNA tumor viruses inhibit the function of p53 ( Fields et al. 1996). The adenovirus E1B 55-kD ( Yew and Berk 1992; Debbas and White 1993) protein and SV-40 T antigen ( Lane and Crawford 1979; Linzer and Levine 1979; Bargonetti et al. 1992; Mietz et al. 1992; Jiang et al. 1993) bind to p53 and sequester it in an inactive complex. The human papillomavirus E6 protein interacts with p53 and promotes its ubiquitin-dependent degradation ( Scheffner et al. 1990; Werness et al. 1990; Mietz et al. 1992; Band et al. 1993; Hoppe-Seyler and Butz 1993). However, none of these viral oncoproteins appear to interact with p73 ( Marin et al. 1998; Roth et al. 1998). Unlike p53, which is widely mutated in human cancers, to date intragenic p73 mutations have been identified in only three lung cancer cell lines ( Yoshikawa et al. 1999). In this report, we developed a tetracycline (tet)-regulatable system for p73 overexpression in human EJ bladder cancer cells in order to compare the regulation and effector functions of these related proteins in tumor cells that selectively lost p53 function in the course of their evolution.
Materials and Methods
Cell Culture
The EJ human bladder carcinoma cell line, 293T human embryonic kidney cell line, and MCF7 human breast carcinoma cell line were maintained in DMEM supplemented with 10% FBS (GIBCO BRL). H1299 human lung carcinoma cells were maintained in RPMI 1640 supplemented with 10% FBS. Both EJ-p53 and EJ-p73 cells were maintained in DMEM supplemented with 10% FBS, penicillin-streptomycin (50 U/ml), hygromycin (100 μg/ml), and geneticin (750 μg/ml). To repress the expression of p73α, p73β, or p53, tet was added to the medium every 3 d to a final concentration of 1 μg/ml. To induce p73 expression, cells were washed three times with PBS and seeded directly in culture medium without tet.
Plasmid Construction and DNA Transfection
The NH2-terminal hemagglutinin (HA)-tagged coding sequence of p73α (or p73β) (obtained from M. Kaghad, Sanofi Recherche, Innopole, France) was released with BamHI and StuI from pcDNA3-p73α (or p73β) and then ligated with pBluescript SK digested with BamHI and EcoRV. The resulting plasmid pBluescript-p73α (or p73β) was then digested with BamHI and SalI, and the fragment encoding p73α (or p73β) was cloned downstream of the tet-regulated promoter into pUHD10-3 (generously provided by H. Bujard, Universitat Heidelberg, Heidelberg, Germany), resulting in plasmid pTet-p73α (or p73β). EJ-tTA cells, generated as described previously ( Sugrue et al. 1997), were transfected with pTet-p73α (or p73β) using the standard calcium phosphate method. Transfectants were doubly selected in the presence of hygromycin (100 μg/ml) and geneticin (750 μg/ml). Individual clones of stable transfectants, designated EJ-p73α or EJ-p73β, were selected for further analysis.
Immunoblot Analysis
Cells cultured in the presence or absence of tet were washed twice with ice-cold PBS with 2 mM sodium vanadate and lysed in EBC lysis buffer as described previously ( Fang et al. 1999). Lysates were cleared by centrifugation at 14,000 rpm for 20 min at 4°C. Protein concentrations were determined using the BCA protein assay kit (Pierce). About 40 μg of cellular protein per sample were subjected to 12 (for p73) or 7.5% (for hMDM2) SDS-PAGE and transferred to Immobilon (Millipore) polyvinylidene difluoride filter. p73 was detected using an HA polyclonal antibody (Santa Cruz Biotechnology) or Ab-2 mAb (clone ER-15; NeoMarkers), and wild-type as well as mutant hMDM2 were detected using Ab-1 mAb (Oncogene Science), followed by ECL detection system (Amersham Pharmacia Biotech).
Immunoprecipitation Analysis
293T cells were transiently cotransfected with p73α or p73β and mdm2 using Fugene 6 (Boehringer Mannheim). Cell lysates were made using EBC lysis buffer and 200 μg of cellular protein was incubated with HA or MDM2 antibody at 4°C for 1 h, followed by another hour incubation with protein A beads. Immunoprecipitation complexes were washed three times with NET-N buffer ( Fang et al. 1999), and subjected to SDS-PAGE followed by immunoblot with the reciprocal antibody.
Senescence-associated β-Galactosidase (SA-β-gal) Staining
Cells were cultured in the presence or absence of tet for the indicated times, washed in PBS, and fixed with 2% formaldehyde/0.2% glutaraldehyde in PBS for 5 min at room temperature. The method for senescence-associated β-galactosidase (SA-β-gal) (pH 6.0) staining was performed as described ( Dimri et al. 1995).
Cell Cycle Analysis
Subconfluent cultures were pulse labeled for 30 min with 10 μM 5-bromo-2′-deoxyuridine (BrdU) (Sigma). Both adherent and floating cells were harvested, fixed in 70% ethanol, and then double stained with fluorescein isothiocyanate-conjugated anti-BrdU antibody (Becton Dickinson) and 5 μg/ml propidium iodide (Sigma Chemical Co.). Cell cycle analysis was performed on a fluorescence-activated cell sorter (FACScan; Becton Dickinson). Data were analyzed using Elite software (Becton Dickinson).
Treatment with DNA Damaging Agents
EJ-p53, EJ-p73α, and EJ-p73β cells were seeded in the presence of 2 ng/ml tet to induce submaximal levels of either p53 or p73. Cells were then treated with 2 or 5 μg/ml mitomycin C, 0.02 or 0.1 μg/ml doxorubicin, or 5 or 10 ng/ml actinomycin D for 24 h. MCF7 cells were treated under the same condition. Cell lysates were prepared and aliquots containing 40 μg of cell protein were subjected to SDS-PAGE followed by immunoblot analysis with 1801 mAb for p53, HA polyclonal antibody, or ER-15 mAb for p73.
Luciferase Assays
Plasmid DNA was transiently transfected into H1299 cells using Fugene 6 (Boehringer Mannheim). Approximately 2 × 106 cells were cotransfected with plasmids as indicated. Cells were harvested 48 h after transfection, and luciferase activity was measured using a luciferase assay kit (Promega). The assay was normalized by cotransfection of a pCMV–β-gal plasmid and measurement of β-galactosidase activities.
Results
Inducible Expression of p73α or p73β in EJ Cells
It has been shown that p53 can induce growth arrest, apoptosis, or senescence depending on the cell context ( Levine 1997). Since p73 is a homologue of p53, we attempted to examine the biological consequences of induced p73 overexpression in EJ cells, which lack functional p53 due to a mutation in exon 5 ( Sharma et al. 1993; Rieger et al. 1995). To obtain tightly regulated p73α or p73β expression, the tet-regulatable expression system ( Gossen and Bujard 1992) was used. EJ-tTA cells, which contain the transactivator with a hygromycin-resistant marker, were transfected with either pTet-p73α or pTet-p73β containing HA-tag and a neomycin-resistant marker. Stable clones were isolated by double selection. More than 10 tet-regulatable clones for each gene were selected and two independent clones were analyzed in detail. The phenotypes in each case (designated EJ-p73α or EJ-p73β) were similar. As shown in Fig. 1 A, there was no detectable amount of HA-p73α in the presence of 1 μg/ml tet as determined by immunoblot analysis ( Fig. 1 A, lane 1). Within 24 h of tet removal, p73α was induced and became readily detectable ( Fig. 1 A, lane 2), with p73α levels further increasing to a steady-state level by 48 h ( Fig. 1 A, lane 3). To test whether p73α induction was reversible, tet was added back to the medium after induction for 24 h, and p73α levels examined 24 h later. It was apparent that p73α returned to an undetectable level (compare lanes 6 and 2 in Fig. 1 A) under these conditions, indicating that p73α expression was fully reversible. Similar results were obtained with EJ-p73β as shown in Fig. 1 B.
Figure 1.

Tet regulation of p73α and p73β expression and induction of target genes in EJ-p73α (A) and EJ-p73β (B) cells. Immunoblot analysis of HA-tagged p73α and p73β as well as endogenous p21 and mdm2 expression in (+) tet (lane 1) or (−) tet for 1 d (lane 2), 2 d (lane 3), 3 d (lane 4), 4 d (lane 5), or in (−) tet for 1 d followed by the addition of tet for 1 d (lane 6).
It can also been observed that two p53 transcriptional target genes, p21 and mdm2, were induced by both p73α and p73β ( Fig. 1). The kinetics of the induction paralleled that of p73, and was also reversible following readdition of tet ( Fig. 1A and Fig. B, lanes 6).
p73 Induces Irreversible Growth Arrest Associated with Senescence-like Morphology
In response to p73 induction in EJ-p73α or EJ-p73β cells, we observed profound alterations in both cell proliferative capacity and morphology. Whereas EJ-p73 cells grew as small, rounded, refractile cells in the presence of tet and reached confluence, similar to parental EJ cells, the induction of p73 expression caused cells to stop growth and exhibit increased size and flattened morphology as well as enlarged nuclei ( Fig. 2). Of note, there were no characteristics of apoptosis detected in these cells as determined by 4′,6-diamidino-2′-phenylindole dihydrochloride nuclear staining (data not shown). To examine the reversibility of p73-induced growth arrest in EJ cells, we performed a colony formation assay. EJ-p73α or EJ-p73β cells were seeded at about 100 cells per 60-mm plate and maintained in the absence of tet for varying time periods followed by tet readdition. Cultures were subsequently maintained in the presence of tet for another 2 wk, followed by fixation and Giemsa staining. The number of colonies were counted and plotted as shown in Fig. 3. Maintenance of the cells in the absence of tet for three or more days resulted in a marked reduction of the ability to form colonies. Indeed, the kinetics of permanent inhibition of colony formation by p73α or p73β was comparable to that observed with p53 ( Sugrue et al. 1997). These experiments demonstrated that induced expression of either p73α or p73β in EJ cells causes irreversible growth arrest.
Figure 2.

Morphological changes induced by overexpression of p73α or p73β in EJ-p73α and EJ-p73β cells. EJ-p73α or EJ-p73β cells were seeded directly in (+) tet or (−) tet medium at 105 cells per 100-mm plate. At day 2, 4, 6, and 8, cells were photographed using Nikon Eclipse TE200 microscope (100×).
Figure 3.

Irreversible growth inhibition of EJ-p73α and EJ-p73β cells after induction of p73α or p73β. EJ-p73α or EJ-p73β cells were seeded directly in (+) tet or (−) tet at 100 cells per 60-mm plate for indicated time period. After readdition of tet for another 2 wk, colonies visualized by Giemsa staining were counted and plotted. The percentage of recoverable colonies for each condition was normalized to the number in (+) tet. All time points were performed in triplicate.
Expression of p73 Induces G1 and G2 Cell Cycle Arrest
To investigate in which specific cell cycle stage(s) p73 arrested EJ cells, we performed fluorescence-activated cell sorting analysis using EJ-p73α or EJ-p73β cell. EJ-p73 cells were maintained in the presence or absence of tet for varying time periods, followed by analysis using simultaneous flow cytometry for both DNA content and DNA synthesis, with propidium iodide staining and BrdU labeling, respectively. After tet removal, EJ-p73 cells exhibited a dramatic reduction in BrdU incorporation within 3 d, with the population of S phase cells declining from 45.2 and 51.7% in (+) tet to 5.9 and 12.4% in (−) tet for EJ-p73α or EJ-p73β, respectively ( Fig. 4). Conversely, the percentage of cells in both G1 and G2/M phases increased from 34.1 and 20.2% in (+) tet to 60.5 and 33.6% in (−) tet for EJ-p73α, and from 31.5 and 16.0% in (+) tet to 56.3 and 26.5% in (−) tet for EJ-p73β by 3 d, respectively. Thus, induced expression of p73α or p73β arrested EJ cells in both G1 and G2/M phases. Induction of both G1 and G2/M arrest has also been observed with p53 overexpression in EJ-p53 cells ( Sugrue et al. 1997). Of note, there was no evidence of a sub-G1 population as usually seen in apoptosis in either EJ-p73α or β cells.
Figure 4.

Cell cycle analysis of EJ-p73α and EJ-p73β cells after induction of p73α or p73β. Cell cycle analysis was performed as described in Materials and Methods. BrdU uptake, as measured by FITC fluorescence, is depicted on the y-axis. DNA content, as measured by propidium iodide fluorescence, is depicted on the x-axis. Populations of cells in different phases of the cell cycle are gated: G0/G1 population (R1), S-phase population (R2), and G2/M population (R3). The percentage of cells in each gate is indicated for each sample in the table. (A) EJ-p73α, (+) tet. (B) EJ-p73α, (−) tet, 3 d. (C) EJ-p73β, (+) tet. (D) EJ-p73β, (−) tet, 3 d.
Expression of a Senescence-specific Marker after p73 Induction
It has been shown that senescent but not presenescent, quiescent, or terminally differentiated cells express a SA-β-gal, which can be detected by incubating cells at pH 6.0 with 5-bromo-4-chloro-3-indolyl β-d-galactoside (X-gal) ( Dimri et al. 1995). Since a striking feature of induced p73 expression was a morphological change characteristic of senescent cells, we examined whether EJ-p73 cells expressed this senescent-specific marker after p73 induction. As shown in Fig. 5, >90% of EJ-p73 cells became positive for SA-β-gal staining within 7 d after induction of p73α or p73β, whereas EJ-p73 cells grown in the presence of tet over the entire time course of the experiment showed no staining (only [+] tet 7 d of EJ-p73α is shown). These results indicated that expression of p73 can promote a senescence-like program in EJ cells.
Figure 5.

SA-β-gal staining after p73α or p73β induction. EJ-p73α or EJ-p73β cells were maintained in (+) tet or (−) tet medium. Cells were washed, fixed, and stained at pH 6.0 as described in Materials and Methods. Cells were photographed using a Nikon Eclipse TE200 microscope (200×). (A) EJ-p73α cells, (+) tet, 7 d. (B) EJ-p73α cells, (−) tet, 7 d. (C) EJ-p73β cells, (−) tet, 7 d.
Wild-Type but Not NH2-Terminal Deleted hMDM2 Interacts with p73α or p73β and Inhibits Their Transcriptional Activity
It has been reported that the product of mdm2, a p53 transcriptional response gene, can interact with p53 and target it for degradation ( Haupt et al. 1997; Kubbutat et al. 1997). Since p73 shares high homology with p53, we sought to investigate whether mdm2 also interacted with p73. 293T cells were cotransfected with p73α or p73β, together with wild-type or a mutant human MDM2 with the first 58 aa deleted (ΔN-hMDM2). This deletion is known to abolish MDM2's ability to interact with p53 ( Brown et al. 1993). Reciprocal coimmunoprecipitation was performed using anti-mdm2 or HA antibody, followed by Western blot analysis. As shown in Fig. 6 B, p73α or p73β was detected in the immunocomplexes precipitated by the anti-mdm2 antibody; similarly, hMDM2 was also detected in the immunocomplexes precipitated by the anti-HA antibody. However, there was no detectable p73α or p73β associated with the mutant hMDM2; similarly, mutant hMDM2 was not detected in the immunocomplexes associated with p73α or p73β. These experiments demonstrated that p73α and p73β interact with wild-type but not NH2-terminal deleted hMDM2, despite the comparable expression level of these proteins ( Fig. 6 A).
Figure 6.

Comparison of wild-type and ΔN-hMDM2 interactions with p73α or p73β. 293T cells were transfected with HA-tagged p73α or p73β, together with pcDNA3 vector, hMDM2, or ΔN-hMDM2 as indicated at the top of each lane. Western blot analysis was performed either directly with total lysates (A) or after immunoprecipitation with antibodies as indicated (B).
Next, we attempted to investigate whether the interaction between hMDM2 and p73 had any effects on p73's transcriptional activity. To do so, p73 or vector was cotransfected into H1299 cells along with a luciferase reporter plasmid that contains the genomic sequence from the p21 promoter. As shown in Fig. 7, p53, p73α, and p73β increased the luciferase activity by 15-, 18-, and 38-fold compared with vector, respectively. Neither hMDM2 nor ΔN-hMDM2 alone had any effect on the luciferase activity. When wild-type mdm2 was cotransfected with p53, p73α, or p73β, the luciferase activity decreased three-, four-, and sixfold, respectively. However, when ΔN-hMDM2 was cotransfected, there was no significant change in the p21 promoter response. These experiments demonstrated that wild-type hMDM2 interacts with p73 and specifically inhibits its transcriptional activation of the p21 promoter, consistent with recent reports ( Balint et al. 1999; Dobbelstein et al. 1999; Zeng et al. 1999).
Figure 7.

Wild-type but not NH2-terminal deleted hMDM2 inhibits transcriptional activities of p73α and p73β. H1299 cells in 60-mm plates were cotransfected with the plasmids indicated, as well as the p21 genomic promoter fused to a luciferase reporter and a β-galactosidase expression vector. At 48 h after transfection, luciferase activity was measured and the relative units were plotted. Results of one representative experiment of three independently performed assays are shown. Determinations were made in duplicate for each experimental point.
p73 Is Not Induced at a Posttranslational Level by DNA Damaging Agents
It has been shown that p53 is induced by various stresses such as DNA damage, hypoxia, or nucleotide pool depletion ( Ko and Prives 1996; Levine 1997), and this induction is mainly regulated at the level of p53 protein stability ( Haupt et al. 1997; Kubbutat et al. 1997). To investigate whether p73 could also be stabilized by DNA damaging agents, we titrated the amount of tet required to induce moderate increases in tet-regulatable p53 or HA-p73 levels, and treated the cells with different concentrations of mitomycin C (MMC), doxorubicin (Dox.), or actinomycin D (Act.D) to activate DNA damage checkpoints, followed by immunoblot analysis with p53 or HA antibodies. As shown in Fig. 8 A, p53 levels increased in response to each DNA damaging agent, consistent with previous reports ( Di Leonardo et al. 1994; Nelson and Kastan 1994). In striking contrast, the levels of both p73α and p73β did not increase after exposure to any of these DNA damaging agents. Since the HA-tagged p73 was transcriptionally active, it is unlikely that it would respond differently from endogenous p73 to DNA damage, although we cannot exclude this possibility. Thus, we next tested whether endogenous p73 behaved similarly.
Figure 8.
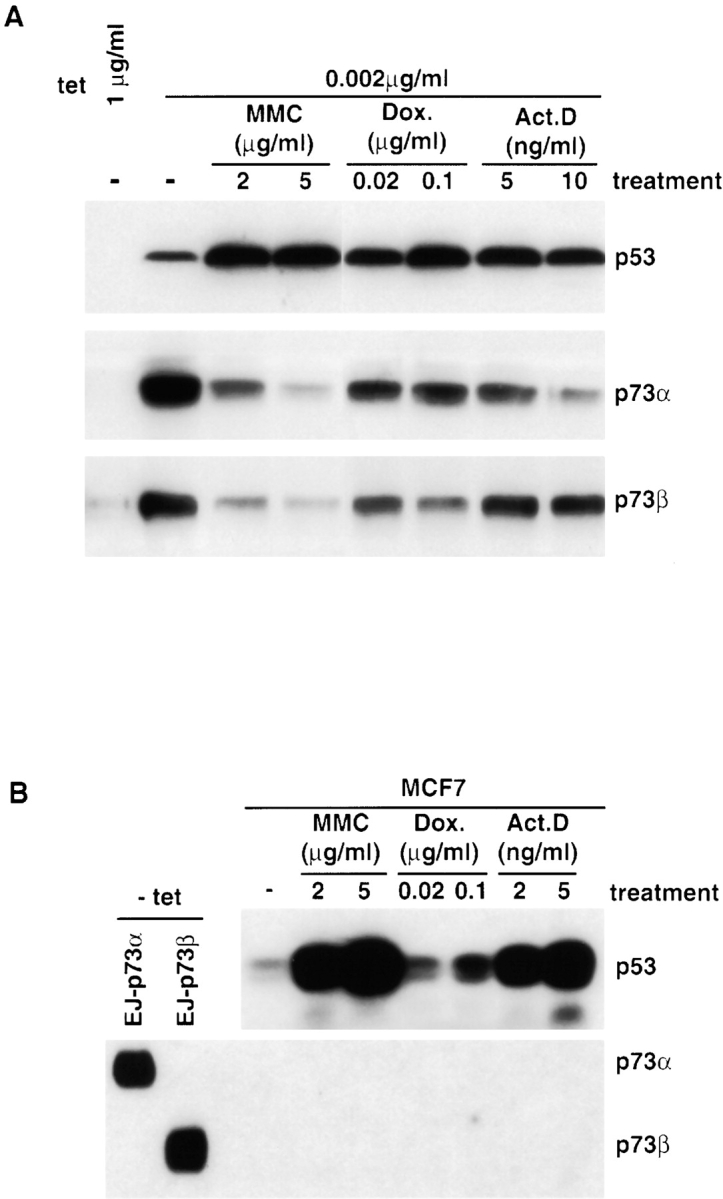
p73α and p73β proteins are not stabilized by DNA damage. (A) EJ-p53, EJ-p73α, and EJ-p73β cells were seeded in medium containing indicated tet concentrations. After 8 h, cells were treated with DNA damaging agents as indicated at the top of each lane. 24 h later, lysates were prepared and subjected to SDS-PAGE. Western blot analysis was performed using anti-p53 and HA antibodies. (B) MCF7 cells were treated with DNA damaging agents as indicated at the top of each lane. 24 h later, lysates were subjected to SDS-PAGE. Western blots were performed using anti-p53 or p73 antibodies. EJ-p73α and EJ-p73β in (−) tet were used as positive controls.
MCF7 cells were treated with different concentrations of DNA damaging agents, followed by immunoblot analysis with p53 or p73 antibodies. As shown in Fig. 8 B, p53 levels increased after each treatment. However, the levels of both p73α and p73β did not increase in response to any of the DNA damaging agents tested. These results suggested that unlike p53, p73 protein stability was not increased in response to several different genotoxic agents.
Discussion
These studies demonstrate that in tumor cells lacking functional p53, the induced overexpression of either p73α or p73β, an alternative product of the p73 gene, promoted a cellular response leading to irreversible growth arrest with markers of replicative senescence. This conclusion is supported by the following observations: induction of a flattened, enlarged cell morphology, commonly observed with senescent fibroblasts; and SA-β-gal staining (pH 6.0), a specific biochemical marker of senescent cells ( Dimri et al. 1995). The commitment to senescence became irreversible within 3 d and no longer required p73 expression. Similar results have been observed with overexpression of p53 or p21, an effector of both p53 and p73, in these same cells ( Fang et al. 1999; Sugrue et al. 1997). p73 has been reported to induce apoptosis when overexpressed in some tumor cells, independent of p53 status ( Jost et al. 1997; Kaghad et al. 1997; Zhu et al. 1998). In our studies, there were no findings consistent with apoptosis in response to p73, p53, or p21 overexpression in any of the assays used ( Fang et al. 1999; Sugrue et al. 1997), suggesting that p73, like p53, induces apoptosis in a cell context–dependent manner.
We also found that p73 can induce mdm2 and p21, two known transcriptional targets of p53, consistent with previous studies ( Di Como et al. 1999; Zhu et al. 1998). It has been shown that p53 interacts with the NH2-terminal 58 aa of hMDM2, since removal of this segment abolishes this interaction ( Brown et al. 1993). Similarly, we showed that hMDM2 coimmunoprecipitates with both p73α and p73β, and this interaction was also disrupted by deletion of the NH2-terminal 58 aa residues of hMDM2, indicating that p73 interacts through the same NH2-terminal 58 residues. We further observed that hMDM2 inhibited the transcriptional response from the p21 promoter in response to p73, as has been reported for p53 ( Haupt et al. 1997; Kubbutat et al. 1997). All of these findings indicate striking similarities in several aspects of p53 and p73 biology.
Unlike the case with p53, hMDM2 interaction did not target p73 for degradation, since p73 protein levels did not decrease ( Fig. 6 A). These results indicate that although hMDM2 can interact with both p53 and p73, its inhibition of p73 transcriptional activity is not mediated by a mechanism involving p73 protein degradation. Similar findings have been reported recently by Zeng et al. 1999.
We observed another major difference in p53 and p73 biology. In EJ tumor cells in which p53 function had been inactivated, exogenously expressed p53 but not p73 showed increased protein level in response to several different DNA damaging agents. Since transcription of each gene was under the control of the same tet-regulatable promoter, these findings likely reflect p53 protein stabilization in response to genotoxic stress by mechanisms that remained intact in these tumor cells. The lack of response of p73 to the same agents further implies differential regulation of these genes at the level of protein stabilization in these tumor cells. These findings could help to explain a selective pressure for inactivation of p53 but not p73 function in the evolution of this tumor despite their comparable ability of inducing permanent growth arrest in these cells.
We also observed that in MCF7 breast cancer cells with intact p53, neither endogenous p73α nor p73β was induced by DNA damaging agents under conditions in which p53 overexpression was readily observed, consistent with a previous report ( Kaghad et al. 1997). Recent studies have indicated that in mouse embryo fibroblasts, certain other DNA damaging agents such as cisplatin were able to induce protein stabilization through a mechanism involving c-Abl ( Agami et al. 1999; Gong et al. 1999; Yuan et al. 1999). We have also observed variation in responsiveness among different tumor cell lines to p73 induction by DNA damaging agents (our unpublished observations). Thus, cell context or the specific agent may be critical determinants of p73 induction in response to DNA damage. Our present findings that p73 can induce permanent growth arrest, in combination with previous studies that p73 can induce apoptosis in other cells ( Jost et al. 1997) imply that p73 can mimic two major p53 effector functions used in its role of guardian of the genome. Thus, the paucity of p73 mutations in human tumors may reflect its lack of responsiveness to genotoxic stresses which commonly induce p53, or to a more restricted tissue expression pattern ( Senoo et al. 1998).
Acknowledgments
We thank Dr. Mourad Kaghad for kindly providing p73 plasmids and Dr. Zhenqiang Pan for critical reading of the manuscript.
This work was supported in part by National Institutes of Health grant CA66654 and the T.J. Martell Foundation for Leukemia Cancer and AIDS Research (to S.A. Aaronson), and National Institutes of Health grants CA78356 and CA82211 (to S.W. Lee). L. Fang is a recipient of Forchheimer Foundation Fellowship.
Footnotes
Abbreviations used in this paper: aa, amino acid(s); BrdU, 5-bromo-2′-deoxyuridine; HA, hemagglutinin; hMDM2, human homolgue of MDM2; MDM2, mouse double minute 2 gene product; SA-β-gal, senescence-associated β-galactosidase; tet, tetracycline.
References
- Agami R., Blandino G., Oren M., Shaul Y. Interaction of c-Abl and p73α and their collaboration to induce apoptosis. Nature. 1999;399:809–813 . doi: 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- Balint E., Bates S., Vousden K.H. Mdm2 binds p73α without targeting degradation. Oncogene. 1999;18:3923–3929 . doi: 10.1038/sj.onc.1202781. [DOI] [PubMed] [Google Scholar]
- Band V., Dalal S., Delmolino L., Androphy E.J. Enhanced degradation of p53 protein in HPV-E6 and BPV-1 E6 immortalized human mammary epithelial cells. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:1847–1852 . doi: 10.1002/j.1460-2075.1993.tb05833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargonetti J., Reynisdottir I., Friedman P.N., Prives C. Site-specific binding of wild-type p53 to cellular DNA is inhibited by SV40 T antigen and mutant p53. Genes Dev. 1992;6:1886–1898 . doi: 10.1101/gad.6.10.1886. [DOI] [PubMed] [Google Scholar]
- Brown D.R., Deb S., Munoz M., Subler M.A., Deb S.P. The tumor suppressor p53 and the oncoprotein simian virus 40 T antigen bind to overlapping domains on the MDM2 protein. Mol. Cell. Biol. 1993;13:6849–6857 . doi: 10.1128/mcb.13.11.6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debbas M., White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 1993;7:546–554 . doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- Di Como C.J., Gaiddon C., Prives C. p73 function is inhibited by tumor derived p53 mutants in mammalian cells. Mol. Cell. Biol. 1999;19:1438–1449 . doi: 10.1128/mcb.19.2.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leonardo A., Linke S.P., Clarkin K., Wahl G.M. DNA damage triggers a prolonged p53-dependent G1 arrest and long term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994;8:2540–2551 . doi: 10.1101/gad.8.21.2540. [DOI] [PubMed] [Google Scholar]
- Dimri G.P., Lee X., Basile G. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 1995;92:9363–9367 . doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbelstein M., Wienzek S., Konig C., Roth J. Inactivation of the p53-homologue p73 by the mdm2-oncoprotein. Oncogene. 1999;18:2101–2106 . doi: 10.1038/sj.onc.1202512. [DOI] [PubMed] [Google Scholar]
- Fang L., Igarashi M., Leung J., Sugrue M., Lee S., Aaronson S. p21Waf1/Cip1/Sdi1 induces permanent growth arrest with markers of replicative senescence in tumor cells lacking functional p53. Oncogene. 1999;18:2789–2797 . doi: 10.1038/sj.onc.1202615. [DOI] [PubMed] [Google Scholar]
- Fields B.N., Knipe D.M., Howley P.M. Virology 1996. J.B. Lippincott, Co; Philadelphia, PA: pp. 3,216 pp [Google Scholar]
- Gong J.G., Costanzo A., Yang H.Q., Melino G., Kaelin W.G.J., Levrero M., Wang J.Y.J. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–809 . doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- Gossen M., Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA. 1992;89:5547–5551 . doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb T.M., Oren O. p53 in growth control and neoplasia. Biochim. Biophys. Acta. 1996;1287:77–102 . doi: 10.1016/0304-419x(95)00019-c. [DOI] [PubMed] [Google Scholar]
- Harris C.C., Hollstein M. Clinical implication of the p53 tumor-suppressor gene. N. Engl. J. Med. 1993;329:1318–1327 . doi: 10.1056/NEJM199310283291807. [DOI] [PubMed] [Google Scholar]
- Haupt Y., Maya R., Kazaz A., Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299 . doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Hollstein M., Rice K., Greenblatt M.S., Soussi T., Fuchs R., Sorlie T., Hovig E., Smith-Sorensen B., Montesano R., Harris C.C. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994;22:3551–3555 . [PMC free article] [PubMed] [Google Scholar]
- Hoppe-Seyler F., Butz K. Repression of endogenous p53 transactivation function in Hela cervical carcinoma cells by human papillomavirus type 16 E6, human mdm2, and mutant p53. J. Virol. 1993;67:3111–3117 . doi: 10.1128/jvi.67.6.3111-3117.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D., Srinivasan A., Lozano G., Robbins P.D. SV40 T antigen abrogates p53-mediated transcriptional activity. Oncogene. 1993;8:2805–2812 . [PubMed] [Google Scholar]
- Jost C.A., Marin M.C., Kaelin W.G.J. p73 is a human p53-related protein that can induce apoptosis. Nature. 1997;389:191–194 . doi: 10.1038/38298. [DOI] [PubMed] [Google Scholar]
- Kaghad M., Bonnet H., Yang A., Creancier L., Biscan J.C., Valent A., Minty A., Chalon P., Lelias J.M., Dumont X. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–819 . doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- Ko L.J., Prives C. p53puzzle and paradigm. Genes Dev. 1996;10:1054–1072 . doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- Kubbutat M.H., Jones S.N., Vousden K.H. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303 . doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Lane D.P., Crawford L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–263 . doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- Levine A.J. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331 . doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Levine A.J., Wu M.C., Chang A., Silver A., Attiyeh E., Lin J., Epstein C.B. The spectrum of mutations at the p53 locus. Evidence for tissue-specific mutagenesis, selection of mutant alleles, and a “gain of function” phenotype. Ann. N.Y. Acad. Sci. 1995;768:111–128 . doi: 10.1111/j.1749-6632.1995.tb12115.x. [DOI] [PubMed] [Google Scholar]
- Linzer D.I., Levine A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43–52 . doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- Marin M.C., Jost C.A., Irwin M.S., DeCaprio J.A., Caput D., Kaelin W.G.J. Viral oncoproteins discriminate between p53 and the p53 homolog p73. Mol. Cell. Biol. 1998;18:6316–6324 . doi: 10.1128/mcb.18.11.6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mietz J.A., Unger T., Huibregtse J.M., Howley P.M. The transcriptional transactivation function of wild-type p53 is inhibited by SV40 large T-antigen and by HPV-16 E6 oncoprotein. EMBO (Eur. Mol. Biol. Organ.) J. 1992;11:5013–5020 . doi: 10.1002/j.1460-2075.1992.tb05608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momand J., Zambetti G.P., Olsen D.C., George D., Levine A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245 . doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- Nelson W.G., Kastan M.B. DNA strand breaksthe DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol. Cell. Biol. 1994;14:1815–1823 . doi: 10.1128/mcb.14.3.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro J.M., Baker S.J., Preisinger A.C., Jessup J.M., Hostetter R., Cleary K., Bigner S.H., Davidson N., Baylin S., Devilee P. Mutations in the p53 gene occur in diverse human tumor types. Nature. 1989;342:705–708 . doi: 10.1038/342705a0. [DOI] [PubMed] [Google Scholar]
- Oliner J.D., Pietenpol J.A., Thiagalingam S., Gyuris J., Kinzler K.W., Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumor suppressor p53. Nature. 1993;362:857–860 . doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- Osada M., Ohba M., Kawahara C., Ishioka C., Kanamaru R., Katoh I., Ikawa Y., Nimura Y., Nakagawara A., Obinata M., Ikawa S. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat. Med. 1998;4:839–843 . doi: 10.1038/nm0798-839. [DOI] [PubMed] [Google Scholar]
- Rieger K.M., Little A.F., Swart J.M., Kastrinakis W.V., Fitzgerald J.M., Hess D.T., Libertino J.A., Summerhayes I.C. Human bladder carcinoma cell lines as indicators of oncogenic change relevant to urothelial neoplastic progression. Br. J. Cancer. 1995;72:683–690 . doi: 10.1038/bjc.1995.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J., Konig C., Wienzek S., Weigel S., Ristea S., Dobbelstein M. Inactivation of p53 but not p73 by adenovirus type 5 E1B 55-kilodalton and E4 34-kilodalton oncoproteins. J. Virol. 1998;72:8510–8516 . doi: 10.1128/jvi.72.11.8510-8516.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M., Werness B.A., Huibregtse J.M., Levine A.J., Howley P.M. The E6 oncoprotein encoded by human papillomaviruses types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136 . doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Senoo M., Seki N., Ohira M., Sugano S., Watanabe M., Tachibana M., Tanaka T., Shinkai Y., Kato H. A second p53-related protein, p73L, with high homology to p73. Biochem. Biophys. Res. Commun. 1998;248:603–607 . doi: 10.1006/bbrc.1998.9013. [DOI] [PubMed] [Google Scholar]
- Sharma S., Schwarte Waldhoff I., Oberhuber H., Schafer R. Functional interaction of wild-type and mutant p53 transfected into human tumor cell lines carrying activated ras genes. Cell Growth Differ. 1993;4:861–869 . [PubMed] [Google Scholar]
- Sugrue M.M., Shin D.Y., Lee S.W., Aaronson S.A. Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc. Natl. Acad. Sci. USA. 1997;94:9648–9653 . doi: 10.1073/pnas.94.18.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trink B., Okami K., Wu L., Sriuranpong V., Jen J., Sidransky D. A new human p53 homologue. Nat. Med. 1998;4:747–748 . doi: 10.1038/nm0798-747. [DOI] [PubMed] [Google Scholar]
- Werness B.A., Levine A.J., Howley P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–79 . doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- Yang A., Kaghad M., Wang Y., Gillett E., Fleming M.D., Dotsch V., Andrews N.C., Caput D., McKeon F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell. 1998;2:305–316 . doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- Yew P.R., Berk A.J. Inhibition of p53 transactivation required for transformation by adenovirus early E1B protein. Nature. 1992;357:82–85 . doi: 10.1038/357082a0. [DOI] [PubMed] [Google Scholar]
- Yoshikawa H., Nagashima M., Khan M.A., McMenamin M.G., Hagiwara K., Harris C.C. Mutational analysis of p73 and p53 in human cancer cell lines. Oncogene. 1999;18:3415–3421 . doi: 10.1038/sj.onc.1202677. [DOI] [PubMed] [Google Scholar]
- Yuan Z.M., Shioya H., Ishiko T., Sun X.G., Gu J.J., Huang Y.Y., Lu H., Kharbanda S., Weichselbaum R., Kufe D. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature. 1999;399:814–817 . doi: 10.1038/21704. [DOI] [PubMed] [Google Scholar]
- Zeng X.Y., Chen L.H., Jost C.A., Maya R., Keller D., Wang X.J., Kaelin W.G.J., Oren M., Chen J.D., Lu H. MDM2 suppress p73 function without promoting p73 degradation. Mol. Cell. Biol. 1999;19:3257–3266 . doi: 10.1128/mcb.19.5.3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Jiang J., Zhou W., Chen X. The potential tumor suppressor p73 differentially regulates cellular p53 target genes. Cancer Res. 1998;58:5061–5065. [PubMed] [Google Scholar]