

Abstract
We used solid-state deuterium NMR spectroscopy and geometric analysis of labeled alanines to investigate the structure and orientation of a designed synthetic hydrophobic, membrane-spanning α-helical peptide that is anchored within phosphatidylcholine (PC) bilayers using both Trp and Lys side chains near the membrane/water interface. The 23-amino-acid peptide consists of an alternating Leu/Ala core sequence that is expected to be α-helical, flanked by aromatic and then cationic anchors at both ends of the peptide: acetyl-GKALW(LA)6LWLAKA-amide (KWALP23). The geometric analysis of labeled alanines method was elaborated to permit the incorporation and assignment of multiple alanine labels within a single synthetic peptide. Peptides were incorporated into oriented bilayers of dilauroyl- (di-C12:0-), dimyristoyl- (di-C14:0-), or dioleoyl- (di-C18:1c-) PC. In the C12:0 and C14:0 lipids, the 2H-NMR quadrupolar splittings for the set of six core alanines could not be fit to a canonical undistorted α-helix. Rather, we found that a model containing a helical distortion, such as a localized discontinuity or “kink” near the peptide and bilayer center, could fit the data for KWALP23 in these shorter lipids. The suggestion of helix distortion was confirmed by 2H-NMR spectra for KWALP23 in which Leu8 was changed to deuterated Ala8. Further analysis involving reexamination of earlier data led to a similar conclusion that acetyl-GWW(LA)8LWWA-amide (WALP23) is distorted in dilauroyl-PC, allowing significant improvement in the fitting of the 2H-NMR results. In contrast, WALP23 and KWALP23 are well represented as undistorted α-helices in dioleoyl-PC, suggesting that the distortion could be a response to hydrophobic mismatch between peptide and lipids.
INTRODUCTION
Integral membrane proteins are vital to maintaining ion and metabolite balance within a cell via their transport function, and membrane-bound receptors are essential for cell communication. Despite great strides toward understanding their structure and function, progress is limited by the large size of membrane proteins, their hydrophobic nature, and their association with membrane lipids. Amino acid distribution analysis of crystallized membrane proteins has revealed that positively charged amino acids are located preferentially on the cytoplasmic side of a membrane (“positive inside rule”) (1–4). Similar analysis has revealed that large membrane-spanning proteins often have bands of aromatic as well as charged residues at the termini of their membrane-spanning segments (5), forming what is sometimes referred to as a “virtual membrane” at the membrane/water interface (4). The combination of interfacially located aromatic and charged residues raises questions regarding the anchoring properties and preferences of particular side chains.
The WALP model system has proven to be an excellent means for understanding the lipid interactions of membrane-spanning α-helices. This family of single-spanning transmembrane peptides with sequence acetyl-GWW(LA)nWWA-amide, with n ranging from 5 to 12.5, has been used to examine the anchoring properties of tryptophan (6–8). Due to tryptophan's amphiphathic nature, it prefers to be located at the phospholipid headgroup region (6,9,10). In this location at the membrane/water interface, the indole NH can form hydrogen bonds with water or the phospholipid headgroup, and the hydrophobic six-membered ring can interact with the upper acyl-chain region. When the four tryptophans in WALP23 were replaced with positive, negative, or aromatic counterparts, it was concluded that the anchoring residues contribute to peptide/lipid interactions via their interactions with the membrane/water interface (11). When the tryptophans were replaced with lysines, it was revealed that Lys prefers a location ∼3 Å farther from the bilayer center than does Trp (12). Furthermore, a “snorkeling” behavior could be important in allowing the charged terminus of Lys to reach the aqueous phase even when its backbone α-carbon is located some distance from the surface (13).
Transmembrane α-helices in membrane proteins have been observed to exhibit a tilt of typically ∼23° ± 10° relative to the membrane normal (4,14,15). Examples of known tertiary structures that include helical bundles are rhodopsin (16), a potassium channel (17), a chloride channel (18), and others. To facilitate investigations of the principles that govern the helix tilt, methods based on 15N-NMR (19,20) and 2H-NMR (21,22) using macroscopically aligned samples have been developed to characterize the orientations of transmembrane helical domains. A systematic application of the latter, according to the geometric analysis of labeled alanines (GALA) method, has served for analysis of labeled alanines within core transmembrane helical sequences in various WALP model peptides (23). Initially, it was somewhat surprising to discover that the α-helix formed by a straightforward sequence such as WALP19 is not strictly perpendicular to the bilayer membrane; yet the tilting of transmembrane domains is now known to be rather standard. Investigations of WALP19 and WALP23 (Trp-anchored peptides of total length 19 or 23 residues) have revealed a small yet distinctive overall intrinsic tilt of consistently ∼4–5° in several hydrated lipid bilayer membranes having different acyl chain lengths (23,24). Furthermore, these peptides reorient rapidly about the membrane normal but not about their own helix axes. Using similar methods, the Lys-anchored KALP23 has been reported to exhibit a somewhat larger average tilt of ∼8°. Additionally, in bilayers of DLPC (di-C12:0-PC), DMPC (di-C14:0-PC), and DOPC (di-C18:1C-PC), the direction of tilt is substantially different for KALP23 compared to WALP23 (25). In none of these cases was a bend or distortion of a transmembrane WALP-like α-helix reported. This is in line with the expectation that the hydrophobic membrane environment itself could lead to a strengthening of the hydrogen bonds within the helix, and thereby favor “helix reinforcement” (26).
It is feasible, nevertheless, that a distortion or kink of a helix could accompany other possible responses of transmembrane peptides to hydrophobic mismatch (27,28). Distortions such as kinking indeed are characteristic of a number of natural transmembrane helices. For example, the transmembrane domain of Vpu from HIV-1 (human immunodeficiency virus-1) exhibits a tilt that varies with the thickness of the host lipid bilayer; Vpu also is kinked in DOPC, although not in shorter lipids (29,30). In some proteins helix kinks can have important functional implications, such as in the gating hinge of K+ channels. Crystallographic, mutational, and functional experiments all support the concept that a flexible “kink” or “gating hinge” at a particular glycine residue in a pore-lining inner helix is critical for the voltage-dependent opening of several potassium channels (31–33). Transmembrane helices as a group contain more bends than do helices in soluble proteins (34). Although these discontinuities often occur at prolines, mutation or replacement of the proline usually does not restore an ideal straight helix (34). The physicochemical constraints that govern helix distortions therefore remain incompletely understood. It would be of interest to know whether such distortions arise mainly from protein/protein interactions, protein/lipid interactions, or both.
This work reports on a model transmembrane helix that was not expected to distort or “kink”. We sought to examine the anchoring properties of a particular combination of tryptophan and lysine, spaced three residues apart near the ends of a 23-amino acid membrane-spanning α-helical peptide: acetyl-GKALW(LA)6LWLAKA-amide, which we designate “KWALP23”. The spacing between the Trp and Lys anchors was chosen to be consistent with the somewhat different preferred anchoring positions of Trp and Lys with respect to the membrane/water interface (12). We also present methods for incorporating and analyzing multiple deuterated alanines within a single peptide. Table 1 compares the amino acid sequence of KWALP23 with the previously investigated peptides WALP23 and WALP19. KWALP23 and WALP19 share a similar (Leu-Ala)6.5 core sequence between anchoring tryptophans, whereas WALP23 contains a (Leu-Ala)8.5 core sequence. The alanine methyl 2H-quadrupolar splittings will indicate that the KWALP23 α-helix is distinctly bent, “kinked”, or otherwise distorted in the relatively short bilayer lipids DLPC and DMPC but not necessarily in the longer lipid DOPC. To further substantiate this 2H-NMR analysis, we also synthesized the Ala8 mutant of KWALP23 (Table 1). The results suggesting distortion of KWALP23 in short lipids prompted us to reanalyze data for WALP23 and thereby to discover that WALP23 also is significantly distorted in DLPC but not in DMPC or DOPC. The backbone integrity of membrane-spanning α-helices therefore appears to be a function of anchoring residue placement and identity, as well as of hydrophobic matching.
TABLE 1.
Comparison of amino acid sequences for KWALP23, WALP19, and WALP23
| Name | Sequence |
|---|---|
| WALP23 | Ac-GWWLA5LALALALALALALALWWA23-NH2 |
| WALP19 | Ac-GWW3LALALALALALALWWA19-NH2 |
| KWALP23 | Ac-GKALW5LALALALALALALWLAKA23-NH2 |
| A8KW23 | Ac-GKALW5LAAALALALALALWLAKA23-NH2 |
One-letter abbreviations are used. The ends of peptides are blocked with N-acetyl (Ac) and C-amide (NH2). For each peptide, the core (Leu-Ala)n region between the innermost anchoring tryptophans is bold.
MATERIALS AND METHODS
Materials
Rink amide aminomethyl-polystyrene resin and fluorenylmethoxy-carbonyl (Fmoc)*-protected amino acids, including Fmoc-Lys-(tert-butyloxycarbonyl (Boc)-side chain) and Fmoc-Trp-(Boc-side chain), were purchased from Advanced ChemTech (Louisville, KY) and NovaBiochem (San Diego, CA). Deuterated L-alanine-d4 and deuterium-depleted water were purchased from Cambridge Isotope Laboratories (Andover, MA). DLPC, DMPC, and DOPC were purchased from Avanti Polar Lipids (Alabaster, AL). 2,2,2-Trifluororethanol (TFE) was purchased from J. T. Baker (Phillipsburg, NJ), methanol from Burdick and Jackson (Muskegon, MI), trifluoroacetic acid (TFA) from Pierce (Rockford, IL), and triisopropylsilane (TIPS) from Sigma-Aldrich (St. Louis, MO).
Peptide synthesis
Peptides were synthesized using solid-phase methods on a Perkin Elmer/Applied Biosystems (Foster City, CA) 433A synthesizer using “FastMoc” chemistry (35). Before the peptide synthesis, L-alanine-d4 was manually derivatized with an N-terminal Fmoc protecting group, as described by Greathouse et al. (35). The peptide sequence for KWALP23 and the Ala8 mutant are listed in Table 1. One or more alanines in each synthetic peptide were (partially) deuterated by mixing appropriate portions of Fmoc-L-Ala and Fmoc-L-Ala-d4. For each labeled sequence position, a twofold excess of (partially) deuterated Fmoc-L-Ala was coupled to the growing peptide chain, followed by a second coupling using a fivefold excess of unlabeled Fmoc-L-Ala to achieve chemical completion. For each unlabeled sequence position, a fivefold excess of the appropriate Fmoc amino acid was used. Details concerning the design and success of the isotope-labeling strategy are explained in the Results section.
Peptides containing Lys-boc and Trp-boc were deprotected and cleaved from the resins using a cleavage cocktail of 85% TFA, 5% TIPS, 5% H2O, and 5% phenol for 2 h at 22°C, using 50 mg of resin/peptide per 2 mL of cocktail solution. After filtration, the volume was reduced ∼4-fold under N2, and each peptide was precipitated by addition of a 25-fold excess of methyl t-butyl ether/n-hexane (1:1, v/v). Finally, the peptides were lyophilized from 1 mL of acetonitrile/water (1:1, v/v), which yielded a fluffy white powder. Peptide identity and purity were confirmed by mass spectrometry and reversed-phase high-performance liquid chromatography (HPLC). Fig. 1 shows a chromatogram for one of the labeled samples of KWALP23, showing >96% purity.
FIGURE 1.

HPLC trace of KWALP 23. Sample (5 μg peptide) was injected at time zero. Peak 1 is a solvent injection spike. Peak 2 is the peptide signal. Elution from a 4.6 × 50 mm column of 3.5 μm octyl(C8)-silica, using a gradient from 85% to 99% MeOH (including 0.1% TFA) over 5 min, followed by a hold at 99% for 3 min, pumped at 1 mL/min; 22°C, 900 psi.
NMR spectroscopy
Samples for solid-state NMR spectroscopy were prepared based on the stacked glass plate procedure outlined by van der Wel et al. (23). Once optical clarity of the glass plate samples was achieved, denoting liquid-crystalline bilayer alignment, NMR measurements were performed at 46 MHz (deuterium) and 50°C, using Bruker AMX and Bruker Avance spectrometers (Bruker Instruments, Billerica, MA). A quadrupolar echo pulse sequence with full phase cycling (36), 3.7 μs 90° pulse time, and 30–127 μs echo delay were used to record 2H-NMR spectra with the normal to the lipid bilayers aligned either parallel to the magnetic field, β = 0°, or perpendicular, β = 90°. A line-broadening of 100 Hz was applied to all spectra, and quadrupolar splittings were measured as the distances between corresponding peak maxima.
Analysis procedure
According to 2H-NMR theory, a set of two symmetrical peaks will be observed for each deuterated alanine CD3 group incorporated into a peptide. Rapid methyl rotation on the NMR timescale leads to signal averaging and relatively intense peaks. (The backbone Cα deuteron signal, by contrast, is quite weak and often not detectable over the spectral noise.) The frequency difference between the peaks is the quadrupolar splitting, defined by the equation:
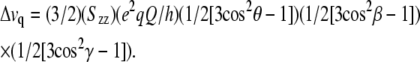 |
(1) |
The quantity Δvq is the quadrupolar splitting magnitude, which is measured experimentally. The following values are known: e2qQ/h is ∼168 kHz, the quadrupolar coupling constant for aliphatic deuterons, γ is 109.5° for the tetrahedral geometry of a methyl group; β depends upon the sample orientation and is the angle between the membrane normal and the magnetic field, either 0° or 90°. The key variable is θ, the angle between the membrane normal (for WALP and KWALP peptides, the long axis for rapid molecular reorientation is coincident not with the helix axis but rather with the membrane normal (23)) and a particular alanine's Cα-Cβ bond, which reports directly on the peptide backbone structure and orientation. Szz is the principal order parameter to account for global (or local) motional averaging, typically ∼0.87 for alanines in WALP peptides (23,24). We found that Szz values between 0.8 and 0.96 led to essentially identical calculated fits to the observed Δvq values.
The GALA method (23,24) was used to determine peptide tilt. An α-helical model of KWALP23 was tilted through an angle τ in a direction defined by an angle ρ relative to a specified reference point at Cα of Gly1, and Δνq was calculated (Eq. 1) from the resulting θ value. Calculated and measured sets of Δνq values were then compared for various (τ, ρ) combinations, and errors were estimated as previously described (23). For the fitting, we stepped τ from 0° to 45° in increments of 0.2°, and ρ from 0° to 359° in increments of 1.0°. In this procedure, the detailed geometry of the alanine CD3 groups is critical, specifically the angle ɛ// between the helix axis and the alanine Cα-Cβ bond. A number of recent data sets have converged upon a rather narrow “fixed” range of ∼58.6° ± 0.5° for ɛ// for alanine in a canonical α-helix (see Results and (23–25)). As will be seen below, when ɛ// is well defined, reasonable estimates of the segmental helix tilt can be deduced from three labeled alanines (although four labeled alanines would be preferred). In initial calculations, ɛ// was stepped from 56.1° to 62.6° in increments of 0.5°; in later calculations using helical segments, ɛ// was often given a fixed value of 58.6°.
To test for the possibility of “bent” or “kinked” helical peptides, different subsets of quadrupolar splittings from subgroups of consecutive Ala labels, within a larger peptide sequence, can be analyzed independently (24). Each such segment is assumed to adopt a regular α-helical conformation. The segmental approach was applied here to a number of data sets, including portions of the previously reported data for WALP23 (24). For WALP23 segments, ɛ// was allowed to converge upon an optimized value, which turned out to be 58.6° for each helical segment.
RESULTS
Multiple labels
Previous methods for determining the intrinsic peptide tilt using the GALA method involved incorporation of one labeled alanine per synthetic peptide (23–25), requiring six to eight different peptides depending on the length of the WALP core helix being investigated. In an attempt to increase data collection efficiency, we devised methods to incorporate and assign multiple deuterated alanines in a single peptide synthesis. This objective was achieved by labeling different alanines in the sequence with different percentages of deuterium. Considerations of the predictable nature of the quadrupolar splittings in a tilted helix can assure optimal positioning of the labels for detection of signals from individual alanines (see below). Fig. 2 shows example 2H-NMR spectra for KWALP23 which has Ala7 100% deuterated and Ala9 50% deuterated.
FIGURE 2.

2H-NMR spectra of labeled alanines in KWALP23 in hydrated DMPC at 40°C and a peptide/lipid molar ratio of 1:20. The sample orientation is β = 90° (A and C) or β = 0° (B and D). A and B show data from a sample in which Ala7 is 100% deuterated and Ala9 is 50% deuterated. (Peaks due to unoriented material are marked with *.) C and D show data from a peptide in which Ala13 is 40% deuterated, Ala15 is 70% deuterated, and Ala17 is 100% deuterated. The numbered arrows in B and D indicate the peak assignments with respect to the alanine positions in the amino acid sequence.
If a transmembrane helix is tilted away from the membrane normal, the alanines on one face of the helix will have different θ-values—and different quadrupolar splittings—from those on the other face of the helix. In the absence of motional averaging due to rotation about the helix axis, the magnitude of the quadrupolar splitting will vary depending upon the radial location of the alanine residue with respect to the direction of tilt. As shown in Fig. 2, two sets of peaks are observed for a single peptide with two labeled residues. Fig. 2 A shows the spectrum for KWALP23 in DMPC at β = 90° with two alanines labeled. The tall outer peaks are from Ala7 (100% deuterated), and the smaller inner peaks are from Ala9 (50% deuterated). These individual resonance assignments were verified using singly labeled samples. Fig. 2 B represents the same sample at β = 0° orientation, the magnitudes of the major splittings being twice those observed at β = 90°. (One smaller pair of peaks in Fig. 2 B represents a signal from unoriented material; we have no explanation for the other pair of small peaks.)
Once it was determined that a double label was successful, a triply labeled peptide was made using 100%, 70%, and 40% deuteration for three successive alanines, namely Ala17, Ala15, and Ala13 in KWALP23. Fig. 2 C shows a spectrum for these alanines in KWALP23/DMPC when β = 90°; Ala17 and Ala13 have similar quadrupolar splittings which overlap and form the tall peaks. Ala15 is assigned to the short peaks. In Fig. 2 D, when β = 0°, one sees the signals from Ala13 projecting as shoulders from the sides of the more intense Ala17 peaks. In subsequent experiments, we found it convenient and productive to label three consecutive alanines, spaced radially by 200° in a WALP-like sequence, to the extent of 100%, 50%, and 75%, respectively. In practical applications of the GALA method, using such a labeling scheme, the Δνq values for the outer alanines of the series may be quite close (e.g., within <1.0 kHz of each other), but in such a case the GALA calculation does not necessarily require that the close splittings be individually distinguished; rather, each of them can be assigned to the average of the two closely similar values. Using the spectra shown in Fig. 2, we were able to measure quadrupolar splittings from five alanines and apply the GALA method. The next step is the investigation of KWALP23 in several different lipids to gain insight into the combined effects of the lysine and tryptophan anchors under hydrophobic mismatch conditions.
Tilt of KWALP23 in several lipids
It has previously been shown that KALP peptides, containing four lysines in place of the tryptophan residues, exhibit an overall tilt of ∼8° in several lipids (25). The finding of a somewhat larger tilt for KALP peptides, as compared to WALP peptides, raised questions as to whether the Lys and Trp anchors could interact and whether one type of residue would dominate the anchoring role. For our peptides, the outer tryptophans of a WALP peptide, positions 2 and 22, were replaced with lysine, and the inner tryptophans were moved to positions 5 and 19, which are farther into the core of the peptide than in WALP23 and comparable to the positions of the “inner” Trps in WALP19.
Quadrupolar splittings at β = 90° and β = 0° were measured independently (not calculated from one another). In agreement with Eq. 1 (above), for each label, the Δνq value when β = 0° is approximately twice the Δνq value when β = 90°, within the measurement uncertainty of ±0.5 kHz. The factor-of-two reduction at β = 90° indicates rapid long-axis reorientation of KWALP23 about the membrane normal. The values observed when β = 0° were used for the GALA method of curve fitting.
Table 2 summarizes the Δνq values for KWALP23 in several lipids. The general pattern is that the quadrupolar splittings for the six core alanines show alternately large and small Δνq values for successive alanines that are two sequence positions apart, corresponding to a radial separation of 200°. The pattern is typical for an α-helix that is tilted with respect to the membrane normal (23). More detailed analysis revealed that the data for KWALP23 in DOPC fit reasonably well to a tilted α-helix. Nevertheless, in DLPC or DMPC we were unable to obtain entirely satisfactory fits for all six core alanines of KWALP23 simultaneously, using a single undistorted α-helix. In each of the latter fits, one of three problems emerged: either a large accumulated error (large root mean-square deviations (rmsds)), an especially large deviation for one or two data points despite fitting the other alanines well, or an unusually large deviation of the Ala side-chain ɛ// angle from the previously observed value of 58.6° ± 0.5° for the WALP19 membrane-spanning α-helix (23); and in some cases a combination of these problems. Fig. 3 A illustrates the problem wherein the deviation for one data point (Δνq for Ala13) is more than 10 times the experimental error. Indeed, Fig. 3 A represents the best GALA fit that we were able to calculate for all six core alanines of KWALP23 in DLPC together. Yet even this “best” fit suffers all of the above three problems: not only is Δνq for Ala13 far from the calculated curve, giving a large accumulated rmsd, but also the global fit to ɛ// in Fig. 3 A is 60.8° instead of 58.6°, suggesting distortion of the helix.
TABLE 2.
Summary of 2H-quadrupolar splittings for labeled alanine methyl groups in KWALP23
| Lipid | Alanine position | Δνq in kHz (β = 90) | Δνq in kHz (β = 0) |
|---|---|---|---|
| DLPC | 7 | 13.8 | 27.8 |
| 9 | 5.2 | 13.6 | |
| 11 | 13.8 | 27.8 | |
| 13 | 7.6 | 15.5 | |
| 15 | 10.6 | 21.5 | |
| 17 | 2.6 | 5.5 | |
| 8* | 6.3 | ||
| 7* | 26.8 | ||
| DMPC | 7 | 10.8 | 22.7 |
| 9 | 2.2 | 4.7 | |
| 11 | 11.5 | 22.6 | |
| 13 | 2.8 | 6.2 | |
| 15 | 8.2 | 17.2 | |
| 17 | 1.4 | 2.7 | |
| 8* | 7.6 | ||
| 7* | 23.1 | ||
| DOPC | 7 | 8.5 | 19.0 |
| 9 | 0.7 | 1.6 | |
| 11 | 8.5 | 18.0 | |
| 13 | 1.5 | 3.3 | |
| 15 | 7.9 | 16.3 | |
| 17 | 1.5 | 3.3 | |
| 8* | 8.2 | ||
| 7* | 16.5 |
Values at β = 90° and β = 0° were measured independently (not calculated from one another). In agreement with Eq. 1 (see text), for each label the Δνq value at β = 0° is approximately twice the Δνq value at β = 90°, within the measurement uncertainty of ±0.5 kHz. The values observed when β = 0° were used for the GALA method of curve fitting.
Quadrupolar splittings observed for Ala7 and Ala8 in the mutant peptide having Ala8 instead of Leu8.
FIGURE 3.

(A) Graph showing experimental 2H-|Δνq| values and the best global GALA fit for six labeled alanines (numbered) in KWALP23, oriented in hydrated bilayers of DLPC. No GALA curve for a standard transmembrane α-helix, tilted with respect to the membrane normal, was able to fit the data for all six alanines satisfactorily. For the best-fit curve, shown, Δνq for Ala13 in the graph deviates from the curve by 7.6 kHz, compared to a measurement error of ±0.5 kHz for Δνq for each alanine. (The measurement error is less than the height of the plotted symbols.) Depending on the exact combination of fit parameters, the deviation can occur at different sequence positions. Furthermore, the global fitted ɛ// was 60.8°, rather than the preferred value of 58.6° ± 0.5° (23). Other values of ɛ// gave poorer fits. (B) Graph showing two GALA curves to fit experimental 2H-|Δνq| values for labeled alanines (numbered) in KWALP23 and [Ala8]KWALP23, oriented in hydrated bilayers of DLPC. The simulated kink occurs at Ala11, whose |Δνq| value falls between the two curves. At Ala11, the direction of the “helix axis” becomes locally undefined. See text for details.
The situation illustrated in Fig. 3 A suggests that the data for KWALP23 in DLPC could be better fit using a bent or distorted helix. What type of distortion would explain these results? The simplest model is based on a subdivision of the peptide into straight helical segments. The initial analysis was done using a fixed ɛ// angle of 58.6° within each segment, as found previously for membrane-spanning WALP19 (23). An analysis of 160 transmembrane helices in multispan proteins has revealed an average rise per residue of 1.5 Å, in agreement with the α-helical geometry proposed by Pauling (37); furthermore, a 1.5 Å rise per residue corresponds to ɛ// of ∼58.5° (Fig. 7 of Özdirekcan et al. (25)). The fixing of ɛ// corresponds to an implicit assumption that each segment of KWALP23 may be nicely helical (with low-energy helical geometry (37)) but that there may be a discontinuity (bend or kink) between the N- and C-terminal segments. Although other types of helical distortion may also be able to fit the data, we find that a single simple discontinuity fits KWALP23 remarkably well. The restriction on ɛ// results in a strong constraint to the overall curve, leading to only one or two solutions in all cases that we tested. The fits also did not vary significantly when Szz was adjusted within the range of 0.82–0.92. When data are available from more than three labeled alanines within a segment, then it is feasible to permit ɛ// to vary and to obtain best-fit values for ɛ//, which generally converge to 58.6° ± 0.5° (see Discussion).
FIGURE 7.

Graphs showing essentially no bend or kink for the WALP23 α-helix in hydrated bilayers of (A) DMPC or (B) DOPC. Within experimental error, the GALA curves for the experimental 2H-|Δνq| values for labeled alanine sets (5,7,9,11) and (13,15,17,19) describe similar magnitude and direction for the tilt of the helix axis. In DMPC, τ = 4.9° ± 0.3°, and ρ − ɛ⊥ = 115° ± 15°. In DOPC, τ = 3.7° ± 0.5°, and ρ − ɛ⊥ = 110° ± 20°. The experimental data are from Strandberg et al. (24).
To ensure three data points for fitting the N-terminal segment, we introduced deuterated Ala8 (Table 1), while at the same time labeling Ala7 as a check of whether Ala8 would perturb the overall structure (another benefit of the multiple label approach). The near invariance of Δνq for Ala7 when Leu8 is changed to Ala8 (Table 2) indicates the near structural equivalence of the “native” and “mutant” forms of KWALP23. Using a segmental approach, in Fig. 3 B one sees that alanines 7, 8, and 9 can be fit nicely to one tilted helical wheel, whereas alanines 13, 15, and 17 fit a somewhat differently tilted helical wheel. The curves in Fig. 3 B imply that the C-terminal segment differs from the N-terminal segment in both the magnitude and direction of its helical tilt (see below). Within the segmental model, Ala11 presents an intermediate case, where the helix axis—and therefore ɛ//—becomes locally “undefined”, such that the Ala11 Δνq value falls in between the sine waves representing the N-terminal and C-terminal helices.
As a further check concerning the curves in Fig. 3 B we prepared error contour plots showing all possible low-rmsd solutions for the N- and C-terminal segments of KWALP23 in DLPC and for fitting all six (or seven) labeled core alanines of KWALP23 (or [Ala8]KWALP23) to a straight, undistorted helix having ɛ// of 58.6°. The results of fitting six or seven alanines were substantially similar, with neither of the fits to an undistorted helix giving any solutions with rmsd below 2.7 kHz (Fig. 4). The N-terminal segment yielded two closely spaced solutions with (τ, ρ; rmsd) values of (14.2°, 264°; 0.5 kHz) and (10.8°, 292°; 0.8 kHz). In similar fashion, the C-terminal segment also yielded two closely spaced solutions with (τ, ρ; rmsd) values of (18.2°, 294°; 0.4 kHz) and (12.2°, 322°; 0.6 kHz). Each of the four possible pairwise combinations of best-fit values for the N- and C-terminal segments implies a kink for KWALP23 in DLPC.
FIGURE 4.

Contour plots of errors for KWALP23 samples in DLPC. Thicker dashed lines in both A and B are contours at 3.0 kHz and 3.5 kHz for fitting an undistorted α-helix to data for all seven core alanines in [Ala8]KWALP23, unkinked. (A) N-terminal segment, fit to Δνq for alanines 7, 8, and 9 in [Ala8]KWALP23. (B) C-terminal segment, fit to Δνq for alanines 13, 15, and 17. ɛ// was maintained at a preferred helix value of 58.6° in these calculations. The thinner contours for the N- and C-terminals are drawn at intervals of 0.5 kHz in rmsd, from 1.0 to 3.5 kHz. Regions with rmsd below 1.0 kHz are shaded gray. For the undistorted helix (thicker dashed contours), no region had rmsd below 2.8 kHz. Curves relating the best (ρ, τ) solutions in A and B (arrows) are depicted in Fig. 3 B.
By how much do the magnitude and direction of tilt for the N- and C-terminal helical segments differ? For KWALP23 in DLPC, all of the solutions in Table 3 correspond to significant τ-values of >10° for the N- and C-terminals. The direction of tilt also varies for three of the pairwise combinations, as indicated by the ρ-values in Table 3. The actual kink angle κ between the two halves is influenced by Δρ (see Appendix), such that cos(κ) = sin(τN) sin(τC) cos(Δρ) + cos(τN) cos(τC). Considering the possible best solutions for the N- and C-terminal segments, κ for KWALP23 in DLPC falls within a range between 6° and 13° (Table 3). A larger value of κ implies a larger localized distortion from a standardized helical conformation. We observed a qualitatively similar pattern when fitting the quadrupolar splittings for KWALP23 in DMPC, namely difficulty with rmsd and ɛ// for the global fit, together with marked improvement using a segmental (domain) approach. Table 3 summarizes the results following the analysis of data for KWALP23 in the three lipids. In the longer DMPC, the data indicate a unique solution of ∼11° for τN, together with a somewhat smaller value of either ∼7° or 9° for τC. Comparing the results for the N- and C-terminals leads one to compute κ in the range of 9–12.5° for KWALP23 in DMPC. Interestingly, the rmsd for attempting to fit an undistorted helix in DMPC (1.7 kHz) is not as unfavorable compared to an undistorted helix in the shorter DLPC (2.7 kHz). A regular undistorted α-helix gives a satisfactory fit to data for KWALP23 in DOPC (Table 3, bottom), with only modest improvement (and possible “overfitting” of the data) when a kinked model is considered.
TABLE 3.
Tilt and kink simulations for KWALP23 and WALP23 in several lipids
| Simulations of segmented helices
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Peptide | Lipid | τN | ρN | rmsd | τC | ρC | rmsd | κ | Δρ |
| KWALP23 | DLPC | 14.2° | 264° | 0.5 kHz | 18.2° | 294° | 0.4 kHz | 9.1° | 30° |
| 10.8° | 292° | 0.8 kHz | 18.2° | 294° | 0.4 kHz | 7.4° | 2° | ||
| 14.2° | 264° | 0.5 kHz | 12.2° | 322° | 0.6 kHz | 12.8° | 58° | ||
| 10.8° | 292° | 0.8 kHz | 12.2° | 322° | 0.6 kHz | 6.1° | 30° | ||
| KWALP23 | DMPC | 11.2° | 252° | 0.4 kHz | 6.8° | 336° | 0.5 kHz | 12.5° | 84° |
| 11.2° | 252° | 0.4 kHz | 9.0° | 304° | 0.4 kHz | 9.0° | 52° | ||
| WALP23* | DLPC | 20.0° | 200° | 0.8 kHz | 6.8° | 153° | 0.2 kHz | 16.1° | 47° |
| Single-helix simulations†
| |||||
|---|---|---|---|---|---|
| Peptide | Lipid | τ | ρ | ɛ// | rmsd |
| KWALP23 | DLPC | 12.0° | 300° | 59.8° | 2.7 kHz |
| KWALP23 | DMPC | 10.0° | 300° | 58.6° | 1.7 kHz |
| KWALP23 | DOPC | 5.8° | 318° | 59.4° | 0.8 kHz |
| WALP23 | DLPC | 8.2° | 143° | 56.9° | 2.2 kHz |
| WALP23 | DMPC | 5.5° | 158° | 58.2° | 0.9 kHz |
| WALP23 | DOPC | 4.5° | 153° | 58.1° | 0.5 kHz |
The (τN, ρN) and (τC, ρC) sets represent the magnitude and direction of tilt of the N- and C-terminal helical segments, on either side of Ala11 of KWALP23. ρ-Values were determined by adding the value of ɛ⊥ for the Ala methyl (43°; see van der Wel et al. (23)) to the maxima in the GALA output graphs (Fig. 3). ρ is referenced relative to Cα of Gly1, with a perspective down the helix axis (see Fig. 1 of van der Wel et al. (23)). The ɛ// value for the Ala methyl side chain was 58.6° ± 0.5° (23), unless otherwise noted. For the fits, τ was searched in 0.2° increments, and ρ was searched in 1.0° increments. The helix kink angle, κ, is computed from cos(κ) = sin(τN) sin(τC) cos(Δρ) + cos(τN) cos(τC), where Δρ indicates the difference in the tilt directions of the segments. (Note that both Δρ and Δτ contribute to κ.) In the segmental analysis, the rmsd values for each segment are listed. There are two closely spaced minima for (τN, ρN) and (τC, ρC) of KWALP23 in DLPC (see also Fig. 4) and for (τC, ρC) in DMPC, with each possible combination giving a distinct kink. All possible values of κ and Δρ for the various combinations of (τN, ρN) and (τC, ρC) are listed.
Existing data for WALP23 (24) were fitted to two comparable segments on either side of Leu12, now maintaining ɛ// at 58.6° ± 0.5°. See Discussion.
Fits to regular straight helices, without kinks, are shown for comparison. In these cases, seven data points were used for alanines, 7, 8, 9, 11, 13, 15, and 17 of KWALP23; and ɛ// was allowed to vary. The fitted values for ɛ// at the lowest values of rmsd are shown. The fit for an unkinked KWALP23 helix in DOPC is within satisfactory limits for rmsd and ɛ//; the data for DMPC and DLPC suggest a local kink or other helix distortion. The results of single-helix simulations for WALP23, from Strandberg et al. (24), are shown for comparison.
The segmental approach represented in Fig. 4 and Table 3 embodies the principle of “least change”. Although it is true that the observed data could be fit by any number of complicated models, e.g., involving differential amounts of local distortion together with local motion, it is striking that one can fit the data for KWALP23 in all three lipids examined using only a single assumption: a unique discontinuity near the center of the membrane-spanning α-helix. Indeed, the same analytical approach, along with an appropriate value of 58.6° for ɛ//, also dramatically improves the fit to 2H-NMR data for WALP23 in DLPC (see Discussion), for which it seemed previously that the GALA method gave an uncharacteristically poor fit. Importantly, the segmental approach preserves several significant molecular parameters: namely, a good helix geometry (consistent ɛ//) within each helical segment, and a single principal order parameter to represent the KWALP core region. Because the intrahelical hydrogen bond distances are shorter for helices in hydrophobic environments (38), it is reasonable to expect that favorable values of ɛ// and Szz should be essentially preserved within each helical segment.
Other conceivable models, of necessity, would need to invoke additional parameters or variables. In using the segmental approach to assess the tilt and kink of KWALP23, it was important to pay careful attention to the value of the Ala side-chain ɛ// angle. For WALP19 it was found that ɛ// should fall within a narrow range of 58.6° ± 0.5° for an undistorted α-helix (23). For the KWALP23 peptide in several lipids, we find that apparent deviations of ɛ// (by up to 2.5°), often accompanied by significant differences between calculated and measured Δνq values (Fig. 3 A), constitute evidence for distortion of the core helix. The segmental model represents a minimalist approach for characterizing the distortion. In this approach, the distortion is localized to a specific kink point at which the helix axis direction and alanine ɛ// value become locally undefined (see Ala11 in Fig. 3 B).
DISCUSSION
The WALP and closely related peptides have been used extensively to study the intricacies of peptide/lipid interactions and in particular hydrophobic matching effects. This is the first report, to our knowledge, that suggests distortion of a member of this class of membrane-spanning peptides from an ideal helical conformation as a function of peptide/lipid hydrophobic mismatch. As our data show no sign of aggregation of the positively charged KWALP peptides (other than conceivable antiparallel dimerization) in response to the mismatch, one deduces that the observed helix distortions are likely caused by peptide/lipid interactions. The question might arise whether the presence of lysine is somehow important for the distortion of the helix, since it has not been reported previously for the Trp-anchored WALP peptides. Nevertheless, a large residual error had been found when fitting either an undistorted α-helix or a segmental model to data for WALP23 in DLPC (24). This system should also experience a significant hydrophobic mismatch, and one might expect also a helix distortion similar to the results for KWALP23. Our current results caused us to reexamine this previously reported poor fit.
WALP23 in DLPC
The previous “best fit” for WALP23 in DLPC, considering eight core alanines in an ideal α-helix, suffered two problems: the rmsd of 2.2 kHz was atypically high, and the alanine side-chain ɛ// angle of 56.9° was unexpectedly low, in comparison to similar analyses for WALP23 and WALP19 in DMPC and DOPC (see van der Wel et al. (23) and Strandberg et al. (24)). A segmental analysis which considered the four C-terminal alanines independent of the four N-terminal alanines, with ɛ// held fixed at 56.9°, yielded little apparent improvement to the fit for WALP23 in DLPC. In consideration of our current results, we reanalyzed these previous data with one important change: in the revised segmental analysis, the 2H-quadrupolar splittings for the C-terminal alanines 13, 15, 17, and 19 were once again considered independently from those for the N-terminal alanines 5, 7, 9, and 11—but this time also allowing ɛ// to “seek” independent best values for the N- and C-terminal segments. With four data points available within each segment, it was feasible to simultaneously fit reliable values of τ, ρ, and ɛ//; we thus were able to allow the alanine side-chain ɛ// angle to assume any value within each segment. The results of the revised analysis (Fig. 5) indicate a significant kink of ∼15° for WALP23 in DLPC, as evidenced by the widely different amplitudes for the two sine waves in Fig. 5. Interestingly, the earlier segmental analysis had revealed a similar kink, but the improvement in the fit was, unfortunately, obscured by the incorrect value of ɛ//, such that the significance of discovering the helix distortion was overlooked. It was the prospect of trying to fit all eight data points to a single sine wave that resulted in a large accumulated error (high rmsd) and at the same time suggested a misleading value of 56.9° for ɛ// (24).
FIGURE 5.

Graph showing two GALA curves to fit experimental 2H-|Δνq| values for labeled alanines (numbered) in WALP23, oriented in hydrated bilayers of DLPC. The experimental data are from Strandberg et al. (24). The WALP23 α-helix appears significantly kinked between Ala11 and Ala13. See Table 3.
When a distortion such as a kink is considered, along with allowing ɛ// to move away from an unfavorable value of 56.9° (in the revised segmental analysis), the individual rmsds for the N- and C-terminals of WALP23 in DLPC are 0.8 kHz and 0.2 kHz, respectively. Furthermore, the alanine side-chain ɛ// angle (remarkably!) converges to 58.6° for both segments of the kinked WALP23 α-helix. The helix discontinuity also explains why different apparent τ-and ρ-values had been deduced when different subsets (other than the N-terminal and C-terminal groupings) of quadrupolar splittings from among the eight WALP23 core alanines were used in calculations of the tilt (24). It is noteworthy that Δνq for Ala11 fits nicely on the curve for the N-terminal segment, whereas the Δνq for Ala13 fits nicely on the curve for the C-terminal segment (Fig. 5), suggesting that the discontinuity is relatively well localized. This model implies that WALP23 is kinked by ∼15° in DLPC (see Table 3) and that the kink position is located very near Leu12, namely between Ala11 and Ala13 (which once again happens to correspond to the exact center of this peptide). In Fig. 6, we show a model to illustrate the kinking of WALP23 in DLPC. This model retains essentially good hydrogen bonds for the helix backbone, with only two hydrogen bonds affected in the vicinity of the kink: the hydrogen bond from Ala9 carbonyl to NH of Ala13 becomes longer by 0.4 Å, whereas the hydrogen bond from Leu10 carbonyl to NH of Leu14 becomes shorter by 0.4 Å. The formation of dimers or low-order aggregates could potentially stabilize these hydrogen bonds (see below).
FIGURE 6.

Illustration of proposed WALP23 backbone structure, as viewed from within the membrane interior (A); and viewing the N- and C-termini along the membrane normal (B and C, respectively). Note that residue Leu12 and all side-chain orientations were not determined. This illustration was prepared using Pymol software.
WALP23 in DMPC and DOPC
Is WALP23 also distorted in longer lipids such as DMPC or DOPC? No. Using similar analysis, when including at least four data points and allowing ɛ// to vary, we find no improvement to the ideal helical wheel fits for WALP23 in DMPC or DOPC when the N- and C-terminal segments are treated independently (Figs. 7, A and B). In these cases, the values of τ and ρ are closely similar for the N- and C-terminals, and the overall rmsd values are low (0.6–0.9 kHz) for the global fits. Furthermore, the alanine side-chain ɛ// values that emerge from the global fits (24) are within the typical range of 58–59°. We conclude that the WALP23 α-helix is not distorted when incorporated into bilayers of DMPC or DOPC.
Location of the helix distortion
For the peptides that are observed to deviate from ideal helical conformations, namely the α-helices of KWALP23 in DMPC and DLPC as well as of WALP23 in DLPC (only), the position of the “kink” or structural distortion can be well fit when a discontinuity is placed essentially at or near the central residue in the peptide sequence. Namely, WALP23 in DLPC is well represented by including a “kink” at Leu12 ± one residue (Fig. 5). In similar fashion, KWALP23 in DMPC or DLPC is well fit when a helix discontinuity is present at Ala11 ± one residue (Fig. 3 B). Each set of results is consistent with a modest yet rather remarkably localized “kink” at the position where the α-helix crosses the bilayer center. Perhaps the proximity to the lipid monolayer/monolayer disjunction could play a role in this localization. Because the hydrogen bonds of transmembrane helices are strengthened by the apolar environment (38), and the (Leu-Ala)n sequence contains no helix breakers, the introduction of a mild discontinuity—with minimal disruption of backbone hydrogen bonds—might preferably occur near the bilayer center. Additionally, if there is any peptide/peptide association (see below), the crossing of helices could correlate also with mild backbone distortions.
Lipid and anchor dependence of kinking and tilting
KWALP23 and WALP23 have the same total length, whereas WALP19 is four residues shorter. When viewed from the core perspective, KWALP23 and WALP19 share the same Trp-flanked core sequence (Table 1), whereas WALP23 contains a four-residue longer helical core. What are the consequences of total length, core length, and presence of lysines? Considering the compilation of numerous experiments performed using bilayers of DLPC, DMPC, and DOPC ((23,24) and this work), we find that WALP19 folds into an ideal α-helix in all three lipids, whereas WALP23 is kinked only in DLPC (and is essentially an undistorted α-helix in the thicker lipids). The somewhat more complex KWALP23 is well described by a modestly kinked α-helix in DLPC or DMPC yet is undistorted in the longer DOPC.
It is apparent that the total helix length and anchor residue identity are both important for lipid/protein hydrophobic matching. Even though we do not know specifically whether KWALP23 remains helical outside of the Trp-flanked core region, it is nevertheless evident that the KWALP23 core helix exhibits a modest distortion, such as a kink, in the shorter lipid bilayers. WALP19, by contrast, remains unkinked and only moderately tilted, ∼4–5° from the bilayer normal in each lipid (23). WALP23, with its longer 17-residue (Leu-Ala)8.5 core helical region, appears kinked significantly in DLPC yet remains remarkably unkinked and tilted similarly to WALP19—∼4.5–5.5°—in DMPC as well as DOPC (24). We therefore attribute the modest kinking of KWALP23 in DMPC to the presence of the outer lysine residues in addition to the total peptide length. It is probable that the lysines seek preferred positions with respect to the lipid phosphate headgroups, to optimize charge/charge interactions, and that such interactions may dictate the overall effective helix length as well as deviations from ideal geometry. It is possible that the radial positions of the opposing lysines (Lys2 and Lys22 of KWALP23) could also be important for the molecular interactions, but we have as yet no information concerning this point.
WALP23 and KWALP23 are both too long to fit as undistorted α-helices in bilayers of DLPC; yet their detailed responses to the hydrophobic mismatch are somewhat different. KWALP23 responds with relatively large values of τ for both its N-terminal segment (11–14°) and C-terminal segment (12–18°). Thus, the DLPC/KWALP23 hydrophobic mismatch is compensated for in part by the resulting κ of 6–12° (Table 3) and in part because both segments of the peptide are tilted significantly with respect to the membrane normal. By contrast, WALP23 responds to the mismatch with a large tilt of its N-terminal (20°) along with a more modest tilt of its C-terminal (7°), such that the apparent resulting kink κ for WALP23 in DLPC is ∼15°. Interestingly, there appears to be only a partial correlation between the peptide/lipid length mismatch and the observed kinking behavior, namely no helix distortions yet observed for WALP19 together with somewhat variable results for the 23-residue peptides. This lack of direct correlation with hydrophobic mismatch could reflect the importance of the orientational positioning of the “anchoring residues” around the helix as well as relative to the membrane environment, which could also be one of the more important factors determining the extent (and importantly, direction) of tilt for the (K)WALP peptides in general. Within this context, it could be significant that Trps near the C-terminal exhibit somewhat different side-chain rotamers and dynamics than Trps near the N-terminal (8).
Several recent molecular dynamics simulations have addressed the tilt of transmembrane WALP and KALP peptides (39–41). In general, the calculations have predicted somewhat larger average tilt angles, τ, than actually observed experimentally to date. Several authors have suggested that peptide oligomerization under experimental conditions could lead to smaller tilt angles observed in experiments than those computed for peptide monomers surrounded by lipids (39–41).
The distortion (or “kinking”) of membrane-spanning KWALP23 or WALP23 helices in short lipids was not predicted. Neither was it predicted that Vpu should kink in DOPC but not in shorter lipids (29). Even though the distortions of helix hydrogen bonds are relatively modest (e.g., ±0.4 Å) for models such as the one in Fig. 6, the detailed molecular interactions that may cause a kink remain obscure. In this context, we cannot exclude that peptide dimerization (or oligomerization) might govern the detailed backbone folding as well as the observed tilt angles. Indeed, peptide/peptide interaction at a midbilayer crossing point could seemingly allow a “shielding” of the discontinuity in the backbone hydrogen bonding. Furthermore, the pyrene fluorescence of end-labeled WALP23 is consistent with the formation of antiparallel dimers—although not of higher order aggregates—in DOPC and to a somewhat greater extent in DMPC (27). For KWALP23, our observation of sharp resonances for samples oriented at β = 90° is consistent with monomers or dimers (or low-order aggregates); but the accompanying requirement for rapid rotational reorientation on the NMR timescale argues against large aggregates.
In summary, both the total helix length and anchor residue identity (and possibly their positioning) influence the tilting and distortion of membrane-spanning α-helices in response to lipid/protein hydrophobic mismatch. Such interactions might have interesting implications for the regulation of membrane protein function by the surrounding lipid bilayer (see Andersen and Koeppe (42)). We have been able to explain one of the few poor fits yet reported using the GALA analysis, the case of WALP23 in DLPC. The GALA method becomes reinforced as a very sensitive and seemingly reliable method, even allowing successful segmental simulations. We also have extended the GALA method through the incorporation of multiple labels to allow more productive use of the synthetic peptides. We note that, analogous to the examination of helical distortions in (K)WALP family peptides using the 2H-GALA method, a helical distortion of Vpu has been detected using the 15N-PISEMA method (29). Further characterization of the structural features of the helix distortions, as well as tilt angle measurements in general, would benefit from more detailed comparisons using these complementary solid-state NMR methods, possibly combined with other structural methods. Site-specific infrared dichroism (43) represents yet another method for addressing these interactions. It has not escaped our attention that additional peptides with different combinations of transmembrane anchoring side chains will be not only useful but indeed essential for further defining the lipid/protein interactions that impact the properties of bilayer-encapsulated α-helices.
Acknowledgments
We thank James Hinton, Christopher Mazzanti, Marvin Leister, Hong Gu, Johanne Froyd-Rankenberg, and Vitaly Vostrikov for helpful discussions. We thank Stanley Opella for encouragement and for reviewing the revised manuscript.
This work was supported in part by the Arkansas Biosciences Institute and by grant RR 15569 from the United States National Center for Research Resources, a component of the National Institutes of Health.
APPENDIX—KINK ANGLE DEFINITION AND CALCULATION
The “kink” angle κ between two helical segments depends upon the relative directions (defined by the respective ρ-angles) as well as the magnitudes of tilt (τ) of the individual segments. When the segments tilt in the same direction (Δρ = 0), their τ-values subtract, whereas if the segments tilt in opposite directions (Δρ = 180°), their τ-values should be added (Fig. 8). Between these extremes, the net “kink” angle can be described using the equation
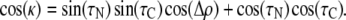 |
FIGURE 8.

Illustration of the influence of N-terminal and C-terminal domain tilt for the kinking of a transmembrane α-helix. For details, see the text of the Appendix.
Patrick C. A. van der Wel's present address is Francis Bitter Magnet Laboratory, Massachusetts Institute of Technology, Cambridge, MA 02139.
Editor: Kathleen B. Hall.
References
- 1.von Heijne, G. 1989. Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature. 341:456–458. [DOI] [PubMed] [Google Scholar]
- 2.Landolt-Marticorena, C., K. A. Williams, C. M. Deber, and R. A. F. Reithmeier. 1993. Non-random distribution of amino acids in the transmembrane segments of human type I single span membrane proteins. J. Mol. Biol. 229:602–608. [DOI] [PubMed] [Google Scholar]
- 3.von Heijne, G. 1992. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J. Mol. Biol. 225:487–494. [DOI] [PubMed] [Google Scholar]
- 4.Ulmschneider, M. B., and M. S. P. Sansom. 2001. Amino acid distributions in integral membrane protein structures. Biochim. Biophys. Acta. 1512:1–14. [DOI] [PubMed] [Google Scholar]
- 5.Schiffer, M., C. H. Chang, and F. J. Stevens. 1992. The functions of tryptophan residues in membrane proteins. Protein Eng. 5:213–214. [DOI] [PubMed] [Google Scholar]
- 6.Killian, J. A., I. Salemink, M. R. R. de Planque, G. Lindblom, R. E. Koeppe 2nd, and D. V. Greathouse. 1996. Induction of non-bilayer structures in diacylphosphatidylcholine model membranes by transmembrane α-helical peptides. Importance of hydrophobic mismatch and proposed role of tryptophans. Biochemistry. 35:1037–1045. [DOI] [PubMed] [Google Scholar]
- 7.Braun, P., and G. von Heijne. 1999. The aromatic residues Trp and Phe have different effects on the positioning of a transmembrane helix in the microsomal membrane. Biochemistry. 38:9778–9782. [DOI] [PubMed] [Google Scholar]
- 8.van der Wel, P. C. A., N. D. Reed, D. V. Greathouse, and R. E. Koeppe 2nd. 2007. Orientation and motion of tryptophan interfacial anchors in membrane-spanning peptides. Biochemistry. 46:7514–7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mazet, J. L., O. S. Andersen, and R. E. Koeppe 2nd. 1984. Single-channel studies on linear gramicidins with altered amino acid sequences. A comparison of phenylalanine, tryptophan, and tyrosine substitutions at positions 1 and 11. Biophys. J. 45:263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Connell, A. M., R. E. Koeppe 2nd, and O. S. Andersen. 1990. Kinetics of gramicidin channel formation in lipid bilayers: transmembrane monomer association. Science. 250:1256–1259. [DOI] [PubMed] [Google Scholar]
- 11.de Planque, M. R. R., J.-W. P. Boots, D. T. S. Rijkers, R. M. J. Liskamp, D. V. Greathouse, and J. A. Killian. 2002. The effects of hydrophobic mismatch between phosphatidylcholine bilayers and transmembrane alpha-helical peptides depend on the nature of interfacially exposed aromatic and charged residues. Biochemistry. 41:8396–8404. [DOI] [PubMed] [Google Scholar]
- 12.de Planque, M. R. R., J. A. Kruijtzer, R. M. Liskamp, D. Marsh, D. V. Greathouse, R. E. Koeppe 2nd, B. de Kruijff, and J. A. Killian. 1999. Different membrane anchoring positions of tryptophan and lysine in synthetic transmembrane alpha-helical peptides. J. Biol. Chem. 274:20839–20846. [DOI] [PubMed] [Google Scholar]
- 13.Strandberg, E., and J. A. Killian. 2003. Snorkeling of lysine side chains in transmembrane helices: how easy can it get? FEBS Lett. 544:69–73. [DOI] [PubMed] [Google Scholar]
- 14.Bowie, J. U. 1997. Helix packing in membrane proteins. J. Mol. Biol. 272:780–789. [DOI] [PubMed] [Google Scholar]
- 15.Spencer, R. H., and D. C. Rees. 2002. The alpha-helix and the organization and gating of channels. Annu. Rev. Biophys. Biomol. Struct. 31:207–233. [DOI] [PubMed] [Google Scholar]
- 16.Blanck, A., D. Oesterhelt, E. Ferrando, E. S. Schegk, and F. Lottspeich. 1989. Primary structure of sensory rhodopsin I, a prokaryotic photoreceptor. EMBO J. 8:3963–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doyle, D. A., J. M. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt-Rose, T., and T. J. Jentsch. 1997. Transmembrane topology of a CLC chloride channel. Proc. Natl. Acad. Sci. USA. 94:7633–7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marassi, F. M., and S. J. Opella. 2000. A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 144:150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang, J., J. Denny, C. Tian, S. Kim, Y. Mo, F. Kovacs, Z. Song, K. Nishimura, Z. Gan, R. Fu, J. R. Quine, and T. A. Cross. 2000. Imaging membrane protein helical wheels. J. Magn. Reson. 144:162–167. [DOI] [PubMed] [Google Scholar]
- 21.Jones, D. H., K. R. Barber, E. W. VanDerLoo, and C. W. M. Grant. 1998. Epidermal growth factor receptor transmembrane domain: 2H NMR implications for orientation and motion in a bilayer environment. Biochemistry. 37:16780–16787. [DOI] [PubMed] [Google Scholar]
- 22.Whiles, J. A., R. Brasseur, K. J. Glover, G. Melacini, E. A. Komives, and R. R. Vold. 2001. Orientation and effects of mastoparan X on phospholipid bicelles. Biophys. J. 80:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Wel, P. C. A., E. Strandberg, J. A. Killian, and R. E. Koeppe 2nd. 2002. Geometry and intrinsic tilt of a tryptophan-anchored transmembrane alpha-helix determined by 2H NMR. Biophys. J. 83:1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strandberg, E., S. Özdirekcan, D. T. S. Rijkers, P. C. A. van der Wel, R. E. Koeppe 2nd, R. M. J. Liskamp, and J. A. Killian. 2004. Tilt angles of transmembrane model peptides in oriented and non-oriented lipid bilayers as determined by 2H solid-state NMR. Biophys. J. 86:3709–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Özdirekcan, S., D. T. S. Rijkers, R. M. J. Liskamp, and J. A. Killian. 2005. Influence of flanking residues on tilt and rotation angles of transmembrane peptides in lipid bilayers. A solid-state 2H NMR study. Biochemistry. 44:1004–1012. [DOI] [PubMed] [Google Scholar]
- 26.Haltia, T., and E. Freire. 1995. Forces and factors that contribute to the structural stability of membrane proteins. Biochim. Biophys. Acta. 1228:1–27. [DOI] [PubMed] [Google Scholar]
- 27.Sparr, E., W. L. Ash, P. V. Nazarov, D. T. S. Rijkers, M. A. Hemminga, D. P. Tieleman, and J. A. Killian. 2005. Self-association of transmembrane alpha-helices in model membranes: importance of helix orientation and role of hydrophobic mismatch. J. Biol. Chem. 280:39324–39331. [DOI] [PubMed] [Google Scholar]
- 28.Killian, J. A., and T. K. Nyholm. 2006. Peptides in lipid bilayers: the power of simple models. Curr. Opin. Struct. Biol. 16:473–479. [DOI] [PubMed] [Google Scholar]
- 29.Park, S. H., and S. J. Opella. 2005. Tilt angle of a transmembrane helix is determined by hydrophobic mismatch. J. Mol. Biol. 350:310–318. [DOI] [PubMed] [Google Scholar]
- 30.Park, S. H., A. A. De Angelis, A. A. Nevzorov, C. H. Wu, and S. J. Opella. 2006. Three-dimensional structure of the transmembrane domain of Vpu from HIV-1 in aligned phospholipid bicelles. Biophys. J. 91:3032–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang, Y., A. Lee, J. Y. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- 32.Magidovich, E., and O. Yifrach. 2004. Conserved gating hinge in ligand- and voltage-dependent K+ channels. Biochemistry. 43:13242–13247. [DOI] [PubMed] [Google Scholar]
- 33.Ding, S., L. Ingleby, C. A. Ahern, and R. Horn. 2005. Investigating the putative glycine hinge in Shaker potassium channel. J. Gen. Physiol. 126:213–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yohannan, S., S. Faham, D. Yang, J. R. Whitelegge, and J. U. Bowie. 2004. The evolution of transmembrane helix kinks and the structural diversity of G protein-coupled receptors. Proc. Natl. Acad. Sci. USA. 101:959–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greathouse, D. V., R. E. Koeppe 2nd, L. L. Providence, S. Shobana, and O. S. Andersen. 1999. Design and characterization of gramicidin channels. Methods Enzymol. 294:525–550. [DOI] [PubMed] [Google Scholar]
- 36.Davis, J. H., K. R. Jeffrey, M. I. Valic, M. Bloom, and T. P. Higgs. 1976. Quadrupolar echo deuteron magnetic resonance spectroscopy in ordered hydrocarbon chains. Chem. Phys. Lett. 42:390–394. [Google Scholar]
- 37.Pauling, L., R. B. Corey, and H. R. Branson. 1951. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA. 37:205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olivella, M., X. Deupi, C. Govaerts, and L. Pardo. 2002. Influence of the environment in the conformation of alpha-helices studied by protein database search and molecular dynamics simulations. Biophys. J. 82:3207–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Im, W., and C. L. Brooks 3rd. 2005. Interfacial folding and membrane insertion of designed peptides studied by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 102:6771–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kandasamy, S. K., and R. G. Larson. 2006. Molecular dynamics simulations of model trans-membrane peptides in lipid bilayers: a systematic investigation of hydrophobic mismatch. Biophys. J. 90:2326–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bond, P. J., J. Holyoake, A. Ivetac, S. Khalid, and M. S. Sansom. 2007. Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J. Struct. Biol. 157:593–605. [DOI] [PubMed] [Google Scholar]
- 42.Andersen, O. S., and R. E. Koeppe 2nd. 2007. Bilayer thickness and membrane protein function: an energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 36:107–130. [DOI] [PubMed] [Google Scholar]
- 43.Torres, J., and I. T. Arkin. 2002. C-deuterated alanine: a new label to study membrane protein structure using site-specific infrared dichroism. Biophys. J. 82:1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]