

Abstract
Aptamer-based drugs represent an attractive approach in pharmacological therapy. The most studied aptamer, thrombin binding aptamer (TBA), folds into a well-defined quadruplex structure and binds to its target with good specificity and affinity. Modified aptamers with improved biophysical properties could constitute a new class of therapeutic aptamers. In this study we show that the modified thrombin binding aptamer (mTBA), 3′GGT5′-5′TGGTGTGGTTGG3′, which also folds into a quadruplex structure, is more stable than its unmodified counterpart and shows a higher thrombin affinity. The stability of the modified aptamer was investigated using differential scanning calorimetry, and the energetics of mTBA and TBA binding to thrombin was characterized by means of isothermal titration calorimetry (ITC). ITC data revealed that TBA/thrombin and mTBA/thrombin binding stoichiometry is 1:2 for both interactions. Structural models of the two complexes of thrombin with TBA and with mTBA were also obtained and subjected to molecular dynamics simulations in explicit water. Analysis of the models led to an improvement of the understanding of the aptamer-thrombin recognition at a molecular level.
INTRODUCTION
α-Thrombin is a serine protease with multiple functions in homeostasis, and it is the only protein capable of catalyzing the cleavage of fibrinogen to produce fibrin clot (1). The multiple functions of α-thrombin in hemostasis concern the procoagulant, anticoagulant, and fibrinolytic pathways and involve a large number of substrates (2). The crystallographic structure of α-thrombin has shown structural features explaining the multiple functions of thrombin (3). Two positively charged regions, termed exosites I and II, are present on the enzyme surface on the opposite sides with respect to the catalytic site. Exosite I is usually referred to as the fibrinogen recognition site and exosite II as the heparin-binding site.
The malfunctioning of thrombin results in hemorrhage, and an excessive coagulation function results in dissemination of the clot in undamaged tissues, causing thrombosis (4). Achieving the ability to specifically inhibit thrombin in vivo with synthetic compounds is an important goal in the prevention of thrombosis.
In 1992, Bock et al. screened a pool of ∼1013 synthetic oligonucleotides (5) and discovered a potent inhibitor of thrombin (6,7) based on a single-stranded 15-mer DNA with the sequence 5′GGTTGGTGTGGTTGG3′ (thrombin binding aptamer, TBA).
The three-dimensional structure of TBA was solved by NMR spectroscopy (8,9). TBA forms a unimolecular quadruplex in solution, arranged in a chair-like structure, consisting of two G-quartets connected by two TT loops and a single TGT loop. TBA does not interact with the active site of thrombin, and a binding site was localized to the anion exosite I (10–13). However, crystallographic studies of TBA complex with thrombin revealed that TBA may interact with both exosites I and II of thrombin (14,15). The three-dimensional structures of thrombin and TBA are preserved in the complex, and specific interactions are found involving the exosites and the loops of the aptamer (15–17).
Recently, the TBA-based oligodeoxynucleotide 3′GGT5′-5′TGGTGTGGTTGG3′ (modified thrombin binding aptamer, mTBA), containing a 5′-5′ site of polarity inversion, was synthesized with the aim to improve the biological and biophysical properties of the thrombin aptamer. The mTBA was studied by circular dichroism (CD) and NMR spectroscopy, and its three-dimensional structure was obtained (18). The structure presents a chair-like conformation characterized by an unusual folding with three strands parallel and one strand oriented in an antiparallel manner.
In this work, we report a differential scanning calorimetric (DSC) study of thermodynamic stability of TBA and mTBA along with an isothermal titration calorimetric (ITC) study to assess the binding stoichiometry and to gain information on the energetics of binding of TBA and modified TBA to the thrombin. To explain the different behavior of the two aptamers we performed molecular dynamics (MD) simulations of TBA and mTBA. In addition, to improve the understanding of the aptamer-thrombin recognition, a model of the complexes was generated by means of docking calculations and was subjected to MD simulations in explicit water. Calorimetric data are also discussed on the basis of the MD simulations results.
MATERIALS AND METHODS
Materials
The oligonucleotides were synthesized as previously reported (18). The solutions were prepared by dissolving solid lyophilized oligonucleotides in the appropriate buffer and were annealed by heating to 90°C for 5 min and slow cooling to room temperature. The buffer used was 10 mM potassium phosphate, 70 mM KCl, 0.1 mM EDTA at pH 7.0. The oligonucleotides concentrations were determined by their adsorption measured at 90°C, in the same buffer, using the same molar extinction coefficient, ɛ(260 nm) = 147,300 M−1cm−1. The molar extinction coefficient was calculated by the nearest neighbor model, assuming that the extinction coefficient of the nucleobases involved in the inversion of the polarity site in the modified aptamer was the same as in the DNA.
Human α-thrombin was purchased from Sigma-Aldrich (St. Louis, MO; product number T6884) and dissolved in the same buffer used for oligonucleotides. Protein solutions were dialyzed against the buffer solution at 4°C by using Spectra Por MW 10,000 membranes. Protein concentration was determined spectrophotometrically using a sequence-based extinction coefficient of 66,612 M−1cm−1 at 280 nm.
Differential scanning calorimetry
The total heat required for the unfolding of the TBA and mTBA was measured with a DSC from Setaram (Caluire, France; Micro-DSC III) interfaced with a personal computer. Linear baselines were drawn for each scan, and molar heat capacity values were generated when the molecular weights of the molecules and the molar concentrations used during each scan were introduced. Since 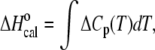 the area of each peak yields a corresponding transition enthalpy, and the peak maximum yields the transition melting temperature. Given
the area of each peak yields a corresponding transition enthalpy, and the peak maximum yields the transition melting temperature. Given 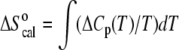 the calorimetric peak was plotted as ΔCp/T versus T to yield a curve, the area of which is ΔS°. The enthalpy and entropy determinations allowed the calculation of the Gibbs free energy via the equation
the calorimetric peak was plotted as ΔCp/T versus T to yield a curve, the area of which is ΔS°. The enthalpy and entropy determinations allowed the calculation of the Gibbs free energy via the equation 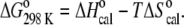 All values reported are an average of at least three repeated measurements.
All values reported are an average of at least three repeated measurements.
Two cells, the sample cell containing 800 μL of TBA or mTBA solution and the reference cell filled with the same volume of buffer solution, were heated from 10°C to 95°C at three different heating rates (0.3°, 0.5°, and 1.0°C min−1). The molecules concentration range was from 8.9 × 10−4 to 1.1 × 10−3 M. This ramp rate over this temperature range led to no hysteresis and thus the complete reversibility of each scan.
Isothermal titration calorimetry
The binding energetics of both TBA and mTBA to thrombin was obtained with a CSC 4200 Calorimeter from Calorimetry Science (Lindon, Utah) at 25°C. Titration experiments were done with successive injections of thrombin into the sample cell (1300 μL) containing 1.6 μM TBA or mTBA until saturation was achieved. Titrations were carried out using a 250 μL syringe with stirring at 297 rpm. The solutions of thrombin and DNA samples were prepared in the same buffer. Control experiments were done by injecting thrombin in buffer to obtain the heat effects for dilution of the protein. The calorimetric enthalpy for each injection was calculated after correction for the heat of thrombin dilution. To obtain the thermodynamic properties of interactions, a nonlinear regression analysis was performed using a single set of identical independent binding sites.
Molecular dynamics simulations
All simulations were performed by means of the GROMACS package (19), employing the all-atom force field parm98 (20). The initial structures of TBA and mTBA were generated by using the coordinates of the NMR structures (TBA Protein Data Bank code: 148D, mTBA Protein Data Bank code: 2IDN), computing the average structure over the 12 and 7 NMR structures, respectively. The mTBA nonstandard residue was created using the Xleap module of the AMBER 7.0 package (21). The molecules were neutralized with 14 Na+ ions (placed following electrostatic potential values) and solvated in a cubic box of size 60 × 60 × 60 Å3 containing ∼6950 TIP3P water molecules (22). Initially, water molecules and ions were relaxed by a first steepest descent energy minimization with positional restraints on solute. The LINCS algorithm (23) was used to constrain the bonds and to carry out an initial simulation, 200 ps long, with the positions of the solute atoms restrained by a force constant of 3000 kJ mol−1nm−2 to let water molecules diffuse around the complex and to equilibrate. Simulations were carried out with periodic boundary conditions at a constant temperature of 300 K. The Berendsen algorithm was applied for temperature and pressure coupling (24), and the particle mesh Ewald method (25) was used for the calculation of electrostatic contribution to nonbonded interactions (grid spacing of 0.12 nm). The trajectory length was 5 ns long for each system.
Docking, complex selection, and simulations
To obtain a model of the complex of TBA with two thrombin molecules, rigid-body docking calculations were carried out by using the program ZDOCK 2.3 (26,27). The structure of the 1:1 complex between the NMR model of TBA and thrombin (hereafter referred to as thrombin A) was kept fixed and, using the TBA molecule as target, a second molecule of thrombin (hereafter referred to as thrombin B) was allowed to rotate and translate around the target. We generated 2000 docked structures, starting from randomly chosen relative orientations. Favorable complexes were selected by a restraint filtering chosen on the basis of the contacts indicated by x-ray data. For TBA the OP atoms of residues G2, G5, T7, and T9 were identified by x-ray to be in contact with the N atoms of the side chains of Arg-233, Lys-236, Lys-240, and Arg-93 of a second thrombin (the residue numeration is according to x-ray structure (15)). An OP-N (TBA-thrombin B) distance below a cutoff of 6 Å was counted as a restraint satisfaction. These restraints were sufficient for an unambiguous complex selection. Indeed, only one of the 2000 docked complexes was passing the filter and presented the thrombin B in a suitable position for binding. This complex was used as the starting structure of MD simulations.
Since structural data concerning the complex were not available in the case of the modified aptamer, to obtain a reasonable starting model of the complex of mTBA with two thrombin proteins the structure of the complex with TBA, obtained by docking, was chosen as a template. The backbone of mTBA was superimposed onto that of TBA complexed with thrombin molecules. The resulting mTBA-thrombin complex model was then energy minimized in vacuo by 1000 steps of the steepest descent method.
The minimizations and successive MD simulations in explicit water were carried out with the protocol described above. The two complexes, consisting of 9232 protein atoms and 488 nucleic acid atoms, were solvated in a box of size 85 × 80 × 115 Å3 containing ∼22,900 TIP3P water molecules. To neutralize the system, six water molecules were replaced by Na+ ions (placed following electrostatic potential values). The trajectory length was 2 ns long for each complex.
RESULTS
DSC melting study
We investigated the unfolding of TBA and mTBA using differential scanning microcalorimetry. The DSC melting curves for the two quadruplexes are shown in Fig. 1, and the corresponding thermodynamic parameters are listed in Table 1. In the experimental conditions, the transitions of TBA and mTBA are reversible, as demonstrated by the recovery of the original signal by rescanning the same sample. Furthermore, the change of the heating rate from 0.3°C to 1.0°C min−1 does not alter the thermodynamic parameters significantly, thereby demonstrating that the studied processes are not kinetically controlled.
FIGURE 1.

DSC profiles for TBA (dashed line) and mTBA (solid line) at 1°C/min. The experiments were performed in a 10 mM potassium phosphate buffer (pH 7.0) supplemented with 70 mM KCl.
TABLE 1.
Thermodynamic parameters for the denaturation process
| Tm °C | ΔH°(Tm) kJ mol−1 | ΔS°(Tm) kJ mol−1K−1 | ΔG°298 K kJ mol−1 | |
|---|---|---|---|---|
| TBA | 53.0 ± 0.5 | 96 ± 5 | 0.30 ± 0.02 | 8 ± 1 |
| mTBA | 57.9 ± 0.5 | 146 ± 5 | 0.43 ± 0.02 | 17 ± 1 |
The DSC curve of TBA shows a symmetric shape with a maximum centered at 53°C. The integration of the denaturation peak gives a ΔH°(Tm) of 96 kJ mol−1. The calorimetric curve of mTBA also shows a symmetric transition and exhibits both higher Tm = 58°C and ΔH°(Tm) = 146 kJ mol−1. The calculated ΔG°(298) values are 8 kJ mol−1 and 17 kJ mol−1 for TBA and mTBA, respectively.
The thermodynamic parameters were calculated assuming a negligible difference in the heat capacity between the initial and final states because, for the investigated transitions, the two states showed comparable heat capacity values. The melting temperatures are in good agreement with those previously obtained by CD melting profiles (18), whereas the enthalpy values are lower than those derived from the van 't Hoff analysis of CD curves, even if the van 't Hoff enthalpy calculated for mTBA by DSC curves (163 kJ mol−1) is in good agreement with the experimental value directly obtained by the area under the curves. For the TBA, analogous thermodynamic parameters were also reported by Macaya et al. (8) and recently by Olsen et al. (28) in a calorimetric study of quadruplexes. The latter authors thought the discrepancy between calorimetric and van 't Hoff enthalpy values was due to aggregation phenomena. On the other hand, a few years ago, to obtain a calorimetric enthalpy value in agreement with the van 't Hoff enthalpy value, Smirnov and Shafer (29) analyzed the TBA calorimetric data, taking into account temperature-dependent heat capacities that vary linearly with T.
The whole set of thermodynamic parameters shows that the mTBA is more stable than its unmodified counterpart. The enthalpy value for mTBA is 50 kJ mol−1 higher. This result suggests the presence of additional intramolecular interactions. Further, entropy change values suggest that the modified aptamer possesses a more rigid structure with respect to TBA.
ITC study
To study the thermodynamics of the interaction of TBA and mTBA with thrombin, we performed ITC experiments. In the upper panels of Fig. 2 are shown the results of the calorimetric titrations of the aptamers into buffered thrombin solution, conducted at 25°C. In both cases, an exothermic heat pulse is observed after each injection of protein solution into the aptamer solution. The area of each exothermic peak is integrated and the obtained heat is divided by the moles of thrombin injected. The binding data were corrected for the dilution heats associated with the addition of the thrombin into buffer, separately determined by injection of either thrombin solution into buffer. The resulting values are plotted as a function of the molar ratio to give the corresponding binding isotherms shown in the lower panels.
FIGURE 2.

(Top) Raw calorimetric data for titration of TBA (on the left) and mTBA (on the right) with serial injections of different volumes of thrombin solution. (Bottom) Binding isotherms resulting from integration of raw calorimetric data after correction for the heat of thrombin dilution.
The corrected data were fitted using the more simplistic model that assumes a single set of equivalent binding sites to determine the stoichiometry (n), binding constant (Kb), and binding enthalpy (ΔbH°). The stoichiometry of both complexes clearly indicates that in solution two thrombin molecules bind to one DNA aptamer. Because the two binding modes do not have distinct energetic profiles, the application of a two-independent-sites model to fit the titration data provides poorer results. The single set of the equivalent sites model is the simplest model to derive K and ΔbH°. These thermodynamic parameters are averaged on all the microstate values. The binding Gibbs energy change, ΔbG°, and entropy change, ΔbS°, are calculated from the equations ΔbG° = −RT lnKb and TΔbS° = ΔbH° − ΔbG°. The thermodynamic parameters are collected in Table 2.
TABLE 2.
Thermodynamic parameters for the interaction with thrombin
| n | Kb M−1 | ΔbH° kJ mol−1 | ΔbS° kJ mol−1 K−1 | ΔbG°298K kJ mol−1 | |
|---|---|---|---|---|---|
| TBA | 1.8 ± 0.1 | (3 ± 1) × 106 | −110 ± 9 | −0.24 ± 0.03 | −37 ± 1 |
| mTBA | 1.9 ± 0.1 | (4 ± 1) × 107 | −160 ± 7 | −0.39 ± 0.02 | −43 ± 1 |
The values of the binding constants and the Gibbs energy changes indicate that the associations are strongly favored, at 25°C, from a thermodynamic point of view. The investigated aptamers bind to thrombin with different affinity. Indeed, the equilibrium constant for the interaction of mTBA with thrombin (Kb = 4 × 107 M−1) is about one order of magnitude greater than that for the TBA-thrombin interaction (Kb = 3 × 106 M−1). The values of ΔbH° and ΔbS° show that, in both cases, the binding processes are enthalpically driven; however, the interaction of mTBA with thrombin is associated with a larger favorable enthalpy (ΔbH° = −160 kJ mol−1) as compared to TBA (ΔbH° = −110 kJ mol−1).
Our results reinforce x-ray data, resolving any doubts about the stoichiometry, and indicate that the interaction of the aptamer with two thrombin molecules is not unique to the crystal complex.
Molecular dynamics of TBA and mTBA
MD simulations of the two aptamers produce stable trajectories, as shown by macroscopic properties of the systems such as density, potential, and total energies (data not shown). The time evolution of the root mean-square deviation (RMSD) values, with respect to the initial energy minimized structure, was calculated for all the atoms in the TBA and mTBA quadruplexes as well as for the guanine base atoms of tetrads (see Fig. 3). It is observed that the all-atoms RMSD gradually increases during the first 1 ns of simulations and then fluctuate around an average value of 2.1 Å for the TBA and 2.4 Å for the mTBA during the last 4 ns. Interestingly, the RMSD of the guanine bases of tetrads oscillates around an average value of 0.6 Å for the TBA and 0.8 Å for the mTBA, indicating that, in both cases, the G-tetrads are rigid and very stable during the entire simulation.
FIGURE 3.

Time dependence of RMSD for all the atoms (top plots) and for the guanine base atoms of tetrads (bottom plots) of TBA (black) and mTBA (shaded) during the MD simulations.
Stereo diagrams of average structures, over the last 4 ns of MD simulations, are shown in Fig. 4. The resulting structures are in good agreement with the NMR structures used as a starting model. In particular, the unusual characteristics of the mTBA, like the anti-anti-anti-syn and syn-syn-syn-anti arrangements of the two tetrads, as well as the peculiar groove widths, are maintained in the theoretical structure. Helix parameters of the two average structures were calculated with the program CURVES (30) (see Supplementary Material). A comparison between the parameters of TBA and mTBA indicates that the base stacking is more efficient in the latter. Indeed, Buckle and Stagger values suggest that the bases within the tetrads of mTBA are more coplanar; and the Rise and Tilt values indicates that the tetrads of mTBA are more parallel to each other in comparison with the tetrads of TBA.
FIGURE 4.

Stereo view representation of the average structures over the last 4 ns of MD simulations of TBA (top) and mTBA (bottom).
Visual inspection of the trajectories clearly shows that the main differences between the two aptamers concern the behavior of loops bases. This different behavior is well emphasized in Fig. 5, where the difference between TBA and mTBA atomic fluctuations (ΔRMSF) is reported. The largest differences in root mean-square fluctuations (RMSF) are associated with the bases T3 (atoms 77–90), T7 (atoms 207–209), and T9 (atoms 272–285) of TBA. The smaller fluctuations of T3 of mTBA in comparison to T3 of TBA could be ascribed to the diverse base orientation that could confer less mobility. Analysis of the trajectory reveals that the T7 of mTBA, which folds back into a groove, forms long time-residence intramolecular H-bonds with the guanine bases of the tetrads. In particular, three H-bonds were observed: O2(T7)⋯N2(G6), O4(T7)⋯N2(G11), and N3(T7)⋯N3(G11), with an H-bond distance of <3.4 Å for more than 80% of the simulated time for the first one and more than 50% for the last two. These interactions are not present in the TBA. Visual inspection of the MD simulation also reveals that the T9 of mTBA remains coplanar and parallel to the adjacent G-tetrad, maintaining the stacking interactions during the entire simulation, whereas the T9 of TBA fluctuates toward the solvent.
FIGURE 5.

Difference between the atomic fluctuations of TBA and mTBA. The largest differences in RMSF (ΔRMSF) are associated with the atoms 77–90 (T3 base), atoms 207–209 (T7 base), and atoms 272–285 (T9 base) of TBA.
Docking and MD simulations of complexes
We performed ZDOCK calculations with the thrombin molecule and the 1:1 TBA-thrombin complex solved by x-ray. One of the 2000 docked complexes satisfied the filter and presented the thrombin in a suitable binding position (see Materials and Methods). For this complex, 2 ns of MD simulations were performed, and the final structure was used for further analysis. The resulting model of the 2:1 thrombin-TBA complex is reported in Fig. 6 A. The time evolution of the RMSD values with respect to the starting structures indicates that the molecular components of the complex have individually reached stable structures during the 2 ns long simulation (Fig. 7 A). Table 3 shows the interactions between TBA and the exosites I and II of thrombin A and thrombin B, respectively. During the MD simulations a large number of contacts are conserved and additional interactions are formed. In particular, the hydrophobic interactions between T3 and Tyr-76 and those between T12 and the residues Ile-24, His-71, Ile-79, and Tyr-117, indicated by Padmanabhan and Tulinsky (15), remain unaltered. In contrast, the more weak interaction between T3 and Ile-82 is lost. Visual inspection of the complex reveals that additional interactions concerning the exosite I of thrombin A and the nucleotides of the TT loops of TBA are present: a), the side chain of Arg-67 forms an ion pair with O2P atom of T4; b), the NE and NH2 atoms of Arg-75 make two hydrogen bonds with O2 of T4 and O4 of T13, respectively; c), the side chain of Glu-77 forms a hydrogen bond with the nitrogen atom of T12; d), the side chain of Arg-77A forms a hydrogen bond with O2 of T13; and e), the NH2 group of Asn-78 forms a hydrogen bond with the phosphate oxygen atom of T13. It is worth noting that His-71, Arg-75, Tyr-76, and Arg-77A were reported to be important for inhibition by the thrombin aptamer (10,12).
FIGURE 6.

Structural models of the complex of (A) TBA or (B) mTBA with two thrombin molecules. The interacting residues of exosite I (green) and exosite II (cyan) are shown.
FIGURE 7.

Time evolution of RMSD for DNA (black lines), thrombin A (shaded lines), and thrombin B (light shaded lines) of the complex with TBA (A) and the complex with mTBA (B).
TABLE 3.
Interactions between thrombin and aptamers
| TBA
| ||||
|---|---|---|---|---|
| DNA | Thrombin A | Interaction | ||
| T3 | Tyr-76 | Hydrophobic | ||
| T4 | O2P | Arg-67 | NH1 | Ion pair |
| T4 | O2 | Arg-75 | NE | Hydrogen bond |
| T12 | Ile-24 | Hydrophobic | ||
| T12 | Ile-79 | Hydrophobic | ||
| T12 | His-71 | Hydrophobic | ||
| T12 | Tyr-117 | Hydrophobic | ||
| T12 | N3 | Glu-77 | OE2 | Hydrogen bond |
| T13 | O4 | Arg-75 | NH2 | Hydrogen bond |
| T13 | O2 | Arg-77A | NH2 | Hydrogen bond |
| T13 | O2P | Asn-78 | ND2 | Hydrogen bond |
| DNA | Thrombin B | Interaction | ||
| G2 | O1P | Lys-236 | NZ | Ion pair |
| G5 | O1P | Lys-236 | NZ | Ion pair |
| T7 | O1P | Lys-240 | NZ | Ion pair |
| T9 | O1P | Arg-93 | NH1 | Ion pair |
| mTBA
| ||||
| DNA
|
Thrombin A
|
Interaction
|
||
| T3 | Tyr-76 | Hydrophobic | ||
| T3 | O4 | Arg-67 | NH2 | Hydrogen bond |
| T4 | O2 | Arg-75 | NE | Hydrogen bond |
| T4 | O4 | Arg-77A | NE | Hydrogen bond |
| T12 | Ile-24 | Hydrophobic | ||
| T12 | Ile-79 | Hydrophobic | ||
| T12 | O2 | Asn-78 | ND2 | Hydrogen bond |
| T13 | O4 | Arg-75 | NH2 | Hydrogen bond |
| DNA | Thrombin B | Interaction | ||
| G2 | O2P | Arg-233 | NE | Ion pair |
| G2 | O2P | Arg-233 | NH2 | Ion pair |
| T4 | O1P | Lys-236 | NZ | Ion pair |
| G5 | O1P | Lys-236 | NZ | Ion pair |
| G6 | O1P | Lys-240 | NZ | Ion pair |
| T9 | O1P | Arg-93 | NH1 | Ion pair |
Exosite II of thrombin B, as also suggested by Padmanabhan and Tulinsky (15), interacts through numerous ion pairs and hydrogen bonds with TBA residues. In the starting structure, the side chain of Arg-233 forms an ion pair with the O2P atom of G2. During the simulation this contact is lost and the O1P atom of G2 gains interaction with the side chain of Lys-236, which, at the same time, forms another ion pair with the O1P atom of G5. The side chains of Lys-240 and Arg-93 form good ion pairs with the O1P atoms of T7 and T9, respectively. These contacts persist for the entire simulation.
MD simulations of the complex of mTBA with two thrombin molecules also produced stable trajectories as shown by RMSD of the components of the complex (Fig. 7 B). The structural model of the 2:1 thrombin-mTBA complex is reported in Fig. 6 B. The contacts between mTBA and the exosites of the two proteins are reported in Table 3. The comparison between the patterns obtained for the complex with TBA and the complex with mTBA indicates that, even though some contacts are conserved, several differences occur. In particular, for the exosite I of thrombin A, residues Ile-24, Arg-75, Tyr-76, and Ile-79 make similar interactions with mTBA. On the other hand, the interactions with the side chains of residues His-71, Glu-77, and Tyr-117 are lost in the complex with mTBA. Residues Arg-67, Arg-77A, and Asn-78 interact with the modified aptamer but in a different manner. Indeed, the side chain of Arg-67 forms a hydrogen bond with the T3 base, the side chain of Arg-77A forms a hydrogen bond with O4 of T4, and the NH2 group of Asn-78 forms a hydrogen bond with the O2 atom of T12. It is worth noting that Arg-75, identified as a key determinant for interaction with TBA (10,13), makes two identical hydrogen bonds in both models.
Differences also occur in the case of exosite II of thrombin B. Although Arg-93 interacts with mTBA in the same manner as with TBA, Lys-236 and Lys-240 have a different behavior. Indeed, the side chain of Lys-236 makes, simultaneously, two ion pairs with O1P atoms of T4 and G5; and Lys-240 forms an ion pair with the O1P atom of G6. In addition, the side chain of Arg-233 makes two hydrogen bonds with phosphate oxygen atoms of G2. These interactions are not present in the complex with TBA.
DISCUSSION
We investigated the thermodynamic stability of TBA and mTBA by DSC and the energetics of binding of these molecules to thrombin by ITC. DSC data indicate that mTBA is more stable than TBA. Inspection of Table 1 reveals that the introduction of a 5′-5′ inversion of polarity increases the melting temperature of ∼5°C. The Tm trend reflects the thermodynamic stability as shown by the Gibbs energy values. The enthalpy value for the unmodified aptamer is in agreement with the values previously reported for the same molecule (8,28). The ΔH°(Tm) value for the mTBA is 50 kJ mol−1 higher than TBA. These results suggest the presence of additional intramolecular interactions in the modified aptamer. Moreover, the entropy change values suggest that mTBA possesses a more rigid structure with respect to TBA. These findings can be rationalized by the results of MD simulations: a), in the mTBA the T7 forms intramolecular hydrogen bonds with the guanine bases of the tetrads that are entirely lost in TBA; b), mTBA presents stronger stacking interactions between the G-tetrads as compared to TBA; and c), the T9 base of mTBA, differently from TBA, remains perfectly stacked with the adjacent G-tetrad during the entire simulation. These supplementary interactions of the thymine bases and the strong stacking interactions of the neighboring guanine tetrads may justify the higher enthalpy change observed for the denaturation of mTBA in comparison with TBA. Moreover, the bases T3, T7, and T9 of TBA show larger fluctuations than those of mTBA. The lower mobility of the bases of mTBA loops, in comparison to TBA, suggests that the modified aptamer possesses a more rigid structure with a lower entropy. This could justify the major entropic gain observed for the denaturation process of this molecule.
ITC measurements demonstrate that the binding of TBA and mTBA to thrombin is exothermic in nature and that both aptamers bind with a stoichiometry of 1:2 (aptamer/protein). The latter finding is consistent with the crystallographic studies of the TBA complex with thrombin that revealed that TBA was sandwiched between two thrombin molecules, contacting exosite I on one thrombin molecule and exosite II on the other.
ITC data do not permit a distinction between the two sites because the titration data do not have distinct energetic profiles; however, the thermodynamic data reveal that mTBA has, on average, a higher affinity for thrombin and that its interaction is associated with a larger favorable enthalpy change (Table 2). The ranking orders of binding constants and enthalpies found in this calorimetric study are not in agreement with the biological activity previously measured in vitro where TBA showed a more marked effect over mTBA (18). Moreover, Tsiang et al. (12), studying the ability of TBA to inhibit mutant thrombins, have proposed that TBA does not bind to exosite II or that exosite II binding is noninhibitory. Wang et al. (17) additionally showed that the substitution of an abasic residue for either T4 or T13 bases significantly reduces the inhibition of thrombin. In our structural models, both T4 and T13 bases form hydrogen bonds with residues of exosite I.
The overall picture that emerged from this thermodynamic analysis, also taking into account the findings of other research groups and our MD simulations results, is that the aptamers interact with both exosites I and II and that the binding to exosite I is the only one with an inhibitory effect. This could justify not only the binding stoichiometry but also the disagreement between thermodynamic results and biological activity. In fact, mTBA binds to thrombin with an increased affinity, but this enhancement could be due to better interaction with the not-inhibiting exosite II.
MD simulations of complexes, which allow an analysis of the binding mode at the molecular level, corroborate this hypothesis. Indeed, the molecular models show that the residues of exosite I make more contacts with TBA than mTBA, and, conversely, the residues of exosite II make more interactions with the modified aptamer (see Table 3).
Although mTBA forms less and different interactions with the exosite I of thrombin A, it is interesting to note that the interaction mode of Arg-75, identified as fundamental for aptamer binding and inhibition, is also preserved in this case. Other four residues were identified by mutagenesis studies (12) as being important for inhibition by a thrombin aptamer: Lys-70, His-71, Tyr-76, and Arg-77A. In our models, Tyr-76 forms the same electrostatic interaction in both complexes, whereas Arg-77A interacts with the modified aptamer in a different manner, and His-71 does not interact with mTBA. These two differences could explain the decreasing of inhibitory activity. It is also interesting to emphasize that, in both models, the side chain of Lys-70 does not interact with the aptamers but is involved in interactions with other amino acids of exosite I. In particular, Lys-70 forms a hydrogen bond with the backbone oxygen atom of Arg-75 and two ion pairs with side chains of Glu-77 and Glu-80 (see Supplementary Material). A mutation of Lys-70 breaks these interactions, allowing the negatively charged side chains of Glu-77 and Glu-80 to move, preventing the binding of the aptamer.
In conclusion, the stability of TBA and of a modified TBA containing an inversion of polarity and the binding energetics of these molecules to thrombin were investigated experimentally. MD simulation studies of the aptamers provided information about the different stability behavior of the molecules and the structural models of complexes with thrombin molecules provided a detailed view of the interactions occurring between each aptamer and its target. Biophysical studies presented here show that a fine analysis of the molecular recognition process is fundamental for a rational aptamer design.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Editor: Jonathan B. Chaires.
References
- 1.Davie, E. W., K. Fujikawa, and W. Kisiel. 1991. The coagulation cascade: initiation, maintenance, and regulation. Biochemistry. 30:10363–10370. [DOI] [PubMed] [Google Scholar]
- 2.Huntington, J. A. 2005. Molecular recognition mechanisms of thrombin. J. Thromb. Haemost. 3:1861–1872. [DOI] [PubMed] [Google Scholar]
- 3.Stubbs, M. T., and W. Bode. 1993. A player of many parts: the spotlight falls on thrombin's structure. Thromb. Res. 69:1–58. [DOI] [PubMed] [Google Scholar]
- 4.Degen, S. J. F., and W. Y. Sun. 1998. The biology of prothrombin. Crit. Rev. Eukaryot. Gene Expr. 8:203–224. [DOI] [PubMed] [Google Scholar]
- 5.Bock, L. C., L. C. Griffin, J. A. Latham, E. H. Vermaas, and J. J. Toole. 1992. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature. 355:564–566. [DOI] [PubMed] [Google Scholar]
- 6.Griffin, L. C., G. F. Tidmarsh, L. C. Bock, J. J. Toole, and L. L. Leung. 1993. In vivo anticoagulant properties of a novel nucleotide-based thrombin inhibitor and demonstration of regional anticoagulation in extracorporeal circuits. Blood. 81:3271–3276. [PubMed] [Google Scholar]
- 7.Li, W. X., A. V. Kaplan, G. W. Grant, J. J. Toole, and L. L. Leung. 1994. A novel nucleotide-based thrombin inhibitor inhibits clot-bound thrombin and reduces arterial platelet thrombus formation. Blood. 83:677–682. [PubMed] [Google Scholar]
- 8.Macaya, R. F., P. Schultze, F. W. Smith, J. A. Roe, and J. Feigon. 1993. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc. Natl. Acad. Sci. USA. 90:3745–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang, K. Y., S. McCurdy, R. G. Shea, S. Swaminathan, and P. H. Bolton. 1993. A DNA aptamer which binds to and inhibits thrombin exhibits a new structural motif for DNA. Biochemistry. 32:1899–1904. [DOI] [PubMed] [Google Scholar]
- 10.Wu, Q., M. Tsiang, and J. E. Sadler. 1992. Localization of the single-stranded DNA binding site in the thrombin anion-binding exosite. J. Biol. Chem. 267:24408–24412. [PubMed] [Google Scholar]
- 11.Paborsky, L. R., S. N. McCurdy, L. C. Griffin, J. J. Toole, and L. L. Leung. 1993. The single-stranded DNA aptamer-binding site of human thrombin. J. Biol. Chem. 268:20808–20811. [PubMed] [Google Scholar]
- 12.Tsiang, M., A. K. Jain, K. E. Dunn, M. E. Rojas, L. L. Leung, and C. S. Gibbs. 1995. Functional mapping of the surface residues of human thrombin. J. Biol. Chem. 270:16854–16863. [DOI] [PubMed] [Google Scholar]
- 13.Tsiang, M., C. S. Gibbs, L. C. Griffin, K. E. Dunn, and L. L. Leung. 1995. Selection of a suppressor mutation that restores affinity of an oligonucleotide inhibitor for thrombin using in vitro genetics. J. Biol. Chem. 270:19370–19376. [DOI] [PubMed] [Google Scholar]
- 14.Padmanabhan, K., K. P. Padmanabhan, J. D. Ferrara, J. E. Sadler, and A. Tulinsky. 1993. The structure of alpha-thrombin inhibited by a 15-mer single-stranded DNA aptamer. J. Biol. Chem. 268:17651–17654. [DOI] [PubMed] [Google Scholar]
- 15.Padmanabhan, K., and A. Tulinsky. 1996. An ambiguous structure of a DNA 15-mer thrombin complex. Acta Crystallogr. D Biol. Crystallogr. 52:272–282. [DOI] [PubMed] [Google Scholar]
- 16.Kelly, J. A., J. Feigon, and T. O. Yeates. 1996. Reconciliation of the x-ray and NMR structures of the thrombin-binding aptamer d(GGTTGGTGTGGTTGG). J. Mol. Biol. 256:417–422. [DOI] [PubMed] [Google Scholar]
- 17.Wang, K. Y., S. H. Krawczyk, N. Bischofberger, S. Swaminathan, and P. H. Bolton. 1993. The tertiary structure of a DNA aptamer which binds to and inhibits thrombin determines activity. Biochemistry. 32:11285–11292. [DOI] [PubMed] [Google Scholar]
- 18.Martino, L., A. Virno, A. Randazzo, A. Virgilio, V. Esposito, C. Giancola, M. Bucci, G. Cirino, and L. Mayol. 2006. A new modified thrombin binding aptamer containing a 5′-5′ inversion of polarity site. Nucleic Acids Res. 34:6653–6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91:43–56. [Google Scholar]
- 20.Cheatham, T. E., 3rd, P. Cieplak, and P. A. Kollman. 1999. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 16:845–862. [DOI] [PubMed] [Google Scholar]
- 21.Pearlman, D., D. Case, J. Caldwell, W. Ross, I. Cheatham, S. Debolt, D. Ferguson, G. Seibel, and P. Kollman. 1995. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 91:1–41. [Google Scholar]
- 22.Jorgensen, W., J. Chandrasekhar, J. Madura, R. Impey, and M. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 23.Hess, B., H. Bekker, H. J. C. Berendsen, and J. Fraaije. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18:1463–1472. [Google Scholar]
- 24.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. Dinola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 25.Darden, T., L. Perera, L. Li, and L. Pedersen. 1999. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure. 7:55–60. [DOI] [PubMed] [Google Scholar]
- 26.Chen, R., L. Li, and Z. Weng. 2003. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 52:80–87. [DOI] [PubMed] [Google Scholar]
- 27.Fanelli, F., and S. Ferrari. 2006. Prediction of MEF2A-DNA interface by rigid body docking: a tool for fast estimation of protein mutational effects on DNA binding. J. Struct. Biol. 153:278–283. [DOI] [PubMed] [Google Scholar]
- 28.Olsen, C. M., W. H. Gmeiner, and L. A. Marky. 2006. Unfolding of G-quadruplexes: energetic, and ion and water contributions of G-quartet stacking. J. Phys. Chem. B. 110:6962–6969. [DOI] [PubMed] [Google Scholar]
- 29.Smirnov, I., and R. H. Shafer. 2000. Effect of loop sequence and size on DNA aptamer stability. Biochemistry. 39:1462–1468. [DOI] [PubMed] [Google Scholar]
- 30.Lavery, R., and H. Sklenar. 1988. The definition of generalized helicoidal parameters and of axis curvature for irregular nucleic acids. J. Biomol. Struct. Dyn. 6:63–91. [DOI] [PubMed] [Google Scholar]