

Abstract
Electroporation is generally used to transfect cells in suspension, but the technique can also be applied to load a defined zone of adherent cells with substances that normally do not permeate the plasma membrane. In this case a pulsed high-frequency oscillating electric field is applied over a small two-wire electrode positioned close to the cells. We compared unipolar with bipolar electroporation pulse protocols and found that the latter were ideally suited to efficiently load a narrow longitudinal strip of cells in monolayer cultures. We further explored this property to determine whether electroporation loading was useful to investigate the extent of dye spread between cells coupled by gap junctions, using wild-type and stably transfected C6 glioma cells expressing connexin 32 or 43. Our investigations show that the spatial spread of electroporation-loaded 6-carboxyfluorescein, as quantified by the standard deviation of Gaussian dye spread or the spatial constant of exponential dye spread, was a reliable approach to investigate the degree of cell-cell coupling. The spread of reporter dye between coupled cells was significantly larger with electroporation loading than with scrape loading, a widely used method for dye-coupling studies. We conclude that electroporation loading and dye transfer is a robust technique to investigate gap-junctional coupling that combines minimal cell damage with accurate probing of the degree of cell-cell communication.
INTRODUCTION
Electroporation is an efficient and versatile technique to bring plasma membrane impermeable compounds into biological cells. It has been extensively applied for gene transfer, as an alternative to viral vectors (1), and it currently experiences a renewed and vigorous interest for its use as a safe method to increase the efficiency of antitumor treatments (“electrochemotherapy”) (2) or a promising tool for gene therapy (“electroporation gene therapy”) (3,4). The spectrum of biological preparations to which electroporation has been applied ranges from single cells (5) through adherent cells (6), brain slice preparations (7), embryonic tissues (8,9), skin (10), up to whole tissues such as liver, lung, and muscle (11–14). Electroporation is also well suited to specifically load a geometrically defined zone in two-dimensional cell arrays, e.g., monolayer cell cultures or viable tissue slices, with membrane-impermeable intracellular messengers and inhibitor substances to investigate intracellular signaling (15,16). The advantage of this technique is the availability of a nonloaded zone in which control cellular responses can be recorded and compared with responses in the loaded zone, according to a paired experimental paradigm. Loading of a restricted zone is done with a special two-wire electrode positioned close to cells (illustrated in Fig. 1) to which a pulsed high-frequency (50 kHz) oscillating electric field (as opposed to a single high-amplitude pulse) is applied so as to combine high efficiency and cell survival (15,17,18). The efficiency of cell loading is dependent on many factors including the time course and magnitude of the electric field, the composition of the solutions bathing the cells, and the cell type and geometry (19). Pulsed high-frequency electric fields can in principle be applied with or without an accompanying net direct current (DC) component, according to a unipolar or bipolar protocol, respectively (20). Unipolar electroporation can improve the loading of charged substances by electrophoresis (21,22) but can also introduce toxicity by current flow and subsequent cell heating (23) or the formation of electrolytic side products such as oxygen gas and metal ions released from the electrode (24). Bipolar electroporation may, on the other hand, be more efficient because it results in poration of the plasma membrane at two opposing sides of the cell, whereas the unipolar protocol porates preferentially at the side where the net DC electric field sums with the local field of the membrane potential (25). Uni- and bipolar electroporation have been compared for efficiency and cell survival in transfection experiments (18) and cell suspensions (25,26), but no information is available for its use with monolayer cell cultures and acute loading with reporter dyes or intracellular messengers for functional studies. Although some degree of cell death may be acceptable for transfection purposes (cells will recover and divide during the expression interval), cell death must be extremely low for functional studies. In addition, other parameters such as the surface area of loaded cells are important when electroporation performance is studied in adherent cell layers. Most studies performed hitherto with pulsed high-frequency electroporation of cell monolayers have been performed with the unipolar protocol (15–17,27–29).
FIGURE 1.

Electroporation setup. (A) General setup with the electroporation electrode positioned above the cell monolayer. (B) Illustration of a 6-CF loaded zone in confluent ECV304 cells. (C) Pulse protocol for unipolar electroporation. (D) Bipolar pulse protocol. The voltage amplitude was expressed as the peak value measured relative to the zero potential, as indicated by the vertical arrows in C and D.
The purpose of this study was to compare uni- and bipolar electroporation of monolayer cell cultures in terms of loading efficiency, size of the loaded zone, and cell death in this zone. Our results show that bipolar electroporation combines the best loading efficiency with the lowest degree of cell death. In addition, the bipolar protocol made it possible to safely load a narrow strip of cells within a culture, and we further explored this property to load a restricted zone of cells with a gap-junction-permeable dye to study dye transfer between the cells via gap junctions. Electroporation loading and dye transfer (ELDT) appeared to be a promising method as an alternative to the widely used scrape loading and dye transfer (SLDT) (30,31). The advantage of both ELDT and SLDT is that they quantify dye coupling over a large population of cells as opposed to fluorescence recovery after photobleaching (FRAP) (32) or local activation of a molecular fluorescent probe (LAMP) (33), which investigate dye coupling in a single cell (or a small group of cells). ELDT has the advantage that it is associated with minimal cell death, as opposed to SLDT, which involves mechanical disruption of plasma membranes to get the fluorescent reporter dye into the cells. Our results demonstrate that the level of dye coupling determined with ELDT is significantly above the level monitored with SLDT and may thus better reflect the normal degree of cell-cell coupling.
MATERIALS AND METHODS
Cell cultures
Initial experiments aimed at finding the best electroporation settings were done on ECV304 cells (European Collection of Animal Cell Cultures, Salisbury, UK) grown as monolayers on Nunclon four-well plates (NUNC, Roskilde, Denmark). Cultures were maintained in Medium-199 (Gibco, Merelbeke, Belgium) with 10% fetal bovine serum and 2 mM glutamine. Experiments were done on confluent cultures (cell density 2100 ± 50 cells/mm2; mean ± SE; n = 4) with the cells bathed in Hanks' balanced salt solution buffered with 25 mM HEPES (HBSS-HEPES, pH 7.4) at room temperature (20°C). Further experiments on dye coupling were performed on C6 glioma cells either as wild-type (WT) or stably transfected with Cx32 (34) or 43 (35). C6 was grown to confluency in DMEM-Ham's F12 (1:1) with 10% fetal bovine serum and 2 mM glutamine.
Electroporation procedure
Electroporation loading was done with the cells on the microscope stage and using a special electrode consisting of two parallel wires (Pt-Ir) supported by a holder (Fig. 1). All electroporation experiments were done at room temperature. Cultures were washed twice with a special electroporation buffer with low electrical conductivity (300 mM sorbitol, 4.2 mM KH2PO4, 10.8 mM K2HPO4, 1 mM MgCl2, and 2 mM HEPES, pH 7.20), and after complete removal of this solution, a small volume (10 μl) of fluorescent dye solution in electroporation buffer was pipetted on the electrode wires. The applied solution settled between the electrode wires and the cells just underneath by capillary forces. The dyes used were 6-carboxyfluorescein (6-CF, 0.376 kDa) and dextran fluoresceins of 3, 10, and 40 kDa, all from Molecular Probes (Invitrogen). The molarity of the dextran fluoresceins was expressed in terms of dye concentration (not dextran concentration). The net charge of the applied probes was −2 for 6-CF and −1 for the dextran fluoresceins. Unipolar or bipolar electroporation pulses were applied to the electrode as illustrated in Fig. 1. The electroporation signal was a 50-kHz signal either going in positive direction only, resulting in a net DC component, or alternating between positive and negative polarity without a net DC component. The choice of this particular frequency was based on the study performed by Tekle et al. (18) and on follow-up work with similar settings in the study of Boitano et al. (15). The 50-kHz signal was applied to the electrode in a gated manner, delivering repeated trains of 2-ms pulses as depicted in Fig. 1, C and D. The voltage amplitude of the electroporation pulses was expressed as the peak value measured relative to the zero level (Fig. 1). After electroporation, the cultures were washed with HBSS-HEPES, and images were taken after 15 min unless otherwise indicated in the figure legends. Images were acquired on a Nikon TE300 inverted microscope equipped with a ×2 (CFI Plan Apo, numerical aperture (NA) 0.1) or ×10 objective (CFI Fluor, NA 0.5) and DS-5M camera head (all from Nikon Belux, Brussels, Belgium) and were analyzed with custom-developed software written in Visual C++ (Fluoroframes program).
Cell viability after electroporation was assayed 10 min after application of the procedure by incubating the cells for another 10 min in a 0.4% trypan blue solution (Sigma, Bornem, Belgium) followed by three washes in HBSS-HEPES and counting the number of trypan-positive (dead) cells in four images of 0.49 by 0.37 mm (transillumination, Nikon ×10 objective, CFI Fluor, NA 0.5). The number of trypan-blue-positive cells was zero in nonelectroporated cell zones.
The electronic drive circuits to deliver uni- or bipolar electroporation signals are schematically depicted in Fig. 2. Detailed circuit diagrams are available on request; these devices are not standard commercially available items (a review of various drive circuits can be found in Puc et al. (36)). Fig. 2 illustrates a generator circuit built around an NE555 timer configured as a gatable astable multivibrator delivering trains of 50 kHz, 50% duty cycle pulses. Positive, negative, and bipolar outputs are obtained by weighted subtraction of the 50 kHz input signal. The gate is controlled by one of the output lines of the LPT port from a PC, with a small C-program running under Windows (with all interrupts disabled to allow correct timing) functioning as a programmable pulse generator (software available on request). Two amplifier circuits were used in the experiments described in this article: a true DC high-voltage amplifier and a fully symmetrical MOSFET amplifier with a toroidal step-up transformer at the output stage (Fig. 2). The transformer-based amplifier has the advantage of a floating high-voltage output and a true mean zero signal over the pulse trains so as to minimize the DC component applied to the electrode. The frequency responses of the two amplifiers were characterized by −3 dB points at 90 kHz and 55 kHz, respectively (electrode connected and immersed into electroporation solution).
FIGURE 2.

Schematic overview of the generator and amplifier setup used for unipolar and bipolar electroporation. The scheme in the upper half demonstrates a true DC high-voltage amplifier used for unipolar electroporation (FB signifies feedback circuit). This amplifier can in principle be used for bipolar electroporation, but to minimize the DC component, we developed an amplifier with a transformer output, depicted in the lower half. The inset illustrates the first two periods of the bipolar output of the transformer-based amplifier; the peak amplitude was 50 V (100 V total swing), and the scale bar delineates 10 μs. Pulse flanks had an exponential time course characterized by a time constant of 1.75 μs on average for the unipolar amplifier and 2.86 μs for the bipolar amplifier.
SLDT
Confluent monolayer cultures were washed two times with nominally calcium-free HBSS-HEPES (CF-HBSS-HEPES, same calcium composition as in electroporation buffer). Cells were incubated for 1 min in CF-HBSS-HEPES containing 400 μM 6-CF; a linear scratch (one per culture) was made across the cell layer using a syringe needle, and the cells were left for another minute in the same solution. Cultures were then washed four times with HBSS-HEPES and left for 15 min at room temperature, and images were taken as described for electroporation loading. For the combined ELDT/SLDT experiments (see Fig. 7 F), cells were washed two times with electroporation buffer, and a scratch was applied after removal of the buffer, immediately followed by electroporation of the culture with 400 μM 6-CF.
FIGURE 7.

Comparison of SLDT and ELDT. (A) Examples of exponential diffusion profiles obtained in WT and connexin-expressing C6 cells after scrape loading (n = 10 for each curve). (B) Diffusion profiles obtained with electroporation loading (n = 8 for each curve). The shaded curves are copies of the curves illustrated in A, added for comparison. (C and D) Summary graphs of the spatial constant of dye spread λ determined in the experiments illustrated in A and B, respectively. Stars give significancies as compared with C6-WT (n = 10 and 8, respectively). (E) Increase of λ in connexin-expressing C6 cells as compared with WT with the two loading protocols. Dye spread was more than twice as large in ELDT as compared with SLDT experiments. Stars show significances as compared with SLDT. (F) Decrease of λ determined with ELDT in cultures that received an SLDT scratch before electroporation. The picture illustrates the horizontal ELDT strip and the vertical SLDT scratch applied just before electroporation. Dye spread decreased to the level observed in SLDT when ELDT was done in cultures that received the SLDT scratch. Stars indicate significances as compared with ELDT (n = 20).
FRAP
Confluent monolayer cultures were grown on glass-bottom microwells (MatTek, Ashland, MA) and were loaded with 5-CFDA (10 μM) in HBSS-HEPES for 45 min at room temperature. Fluorescence within a single cell was photobleached by spot exposure to the 488-nm line of an argon laser, and imaging (again at 488 nm excitation) was done with a custom-made video-rate confocal laser scanning microscope (37) with a 40× oil immersion objective (CFI Plan Fluor, NA 1.4, Nikon Belux); recovery was then measured over a 5-min time period after photobleaching. In the experiments where supernatant of scraped cultures was used, the supernatant was obtained 1 min after the scratch (conditions as described above), and acceptor cultures were exposed for 10 min before the start of the FRAP experiments.
Western blotting
Cell protein lysates were extracted by treatment of confluent cultures with radioimmunoprecipitation assay buffer (25 mM Tris, 50 mM NaCl, 0.5% NP40, 0.5% deoxycholate, 0.1% SDS, 0.055 g/ml β-glycerophosphate, 1 mM DTT, 20 μl/ml phosphatase inhibitor cocktail, 20 μl/ml mini-EDTA-free protease inhibitor cocktail) and sonicated by three 10-s pulses. Separation of Triton X-100-soluble and -insoluble material was done essentially according to the method described by Cooper and Lampe (38). Cells from 75 cm2 culture flasks were washed three times with phosphate-buffered saline (PBSD; 137 mM NaCl, 2.68 mM KCl, 0.90 mM CaCl2, 0.334 mM MgCl2·6H2O, 1.47 mM KH2PO4, 6.46 mM Na2HPO4·2H2O, pH 7.4) and harvested in ice-cold 1% Triton X-100 in PBSD supplemented with 50 mM NaF, 1 mM Na3VO4, 1% protease inhibitor cocktail (Sigma), 1% phosphatase inhibitor cocktail 1 and 2 (Sigma), and 1× mini-EDTA-free protease inhibitor cocktail (Roche Diagnostics, Penzberg, Germany). Samples were separated into Triton X-100-soluble and -insoluble fractions by centrifugation at 16,000 × g for 10 min. Triton-insoluble fractions (pellet) were resuspended in 1× Laemmli sample buffer (10% glycerol, 2.3% SDS, 125 mM Tris, pH 6.8) (39) and sonicated by five 10-s pulses. Total protein was determined with a BioRad (Nazareth, Belgium) DC protein assay kit and a plate reader. Proteins were separated on a 10% polyacrylamide gel and transferred to a nitrocellulose membrane (GE Healthcare, Little Chalfont, UK). Blots were probed with a rabbit polyclonal anti-rat Cx43 antibody (Sigma, 1/10,000), a rabbit polyclonal anti-rat Cx32 (Sigma, 1/500), or a rabbit polyclonal anti-rat β-tubulin (Abcam, Cambridge, UK; 1/5000; loading control) followed by alkaline phosphatase-conjugated goat anti-rabbit IgG (Sigma, 1/8000), and detection was done with nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate reagent (Zymed, Invitrogen).
Immunocytochemistry
Cells grown on glass coverslips were fixed with 4% paraformaldehyde in PBSD (10 min, room temperature) and subsequently permeabilized with 0.2% Triton X-100 in PBSD (10 min, room temperature). Cells were incubated for 1 h with primary antibody, rabbit polyclonal anti-rat Cx32 (Sigma, 1/1000), and a rabbit polyclonal anti-rat Cx43 (Sigma, 1/10,000) diluted in blocking solution (0.4% gelatin in PBSD), washed three times with PBSD, and incubated for 1 h with the secondary antibody coupled to an Alexa fluorophore 488 (goat anti-rabbit IgG antibody; Molecular Probes, Invitrogen). Cell nuclei were stained with DAPI (1 μg/ml), and the preparations were then mounted in Vectashield (Vector Laboratories, Orton Southgate, UK) to prevent photobleaching. Images were acquired as described for electroporation-treated cultures, but with a 100× oil immersion objective (Fluor, NA 1.3, Nikon Belux instead.
Data analysis and statistics
For measurements of the fluorescence intensity in the electroporation zone, images were first thresholded. The threshold corresponded to the upper level of the background noise and was determined from the fluorescence histogram of nonelectroporated cells (cells outside the electroporation zone) as the fluorescence intensity at the right flank of the distribution where the frequency was half-maximal. The average value of above-threshold pixels was then calculated. This value was subsequently converted to a relative fluorescence intensity by dividing it by the fluorescence intensity of a thin layer of the loading solution sandwiched between two coverslips. The thickness of the layer was adjusted to approximate the thickness of monolayer cell cultures, which was 20 ± 2.2 μm (mean ± SE, n = 9) as determined by inspection of fluo-3-loaded ECV304 cultures with a confocal laser scanning microscope. The size of the electroporation zone was determined by applying a threshold to the images (as described above) and calculating the surface area of above-threshold pixels.
Fluorescence intensity profiles perpendicular to the main axis of the electroporation-loaded strip were determined by averaging all picture lines in this direction into an array of line width, using software written in Visual C++. Curve fitting of the Gaussian intensity profiles was done with the program Prism 3.0 (GraphPad Software, San Diego, CA) according to the following equation:
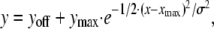 |
where y is the fluorescence intensity, yoff the y-offset, ymax the peak value minus yoff, x the distance, xmax the x-coordinate of the peak, and σ the standard deviation of the Gaussian relation. The spatial constant lambda (λ) of dye spread was determined from the exponentially decreasing portion of the diffusion profiles obtained in ELDT or SLDT experiments. A single λ value was determined from both the right and left flanks in each experiment.
The data are expressed as mean ± SE with n denoting the number of experiments on different cell cultures. Comparison of two groups was done using a one-tailed unpaired t-test with a p value <0.05 indicating significance. Comparison of more than two groups was done with one-way ANOVA and a Bonferroni posttest. Statistical significance is indicated in the figures with a single symbol (stars or #) for p < 0.05, two symbols for p < 0.01, and three symbols in case p < 0.001.
RESULTS
Electroporation of confluent ECV304 monolayer cell cultures was performed with an electrode consisting of two parallel wires placed on the microscope stage and positioned such that the electrode wires were in close apposition to the cells (Fig. 1 A). Application of the electroporation procedure in the presence of the fluorescent reporter dye 6-CF (100 μM) resulted in the loading of a band-like cell zone beneath the electrode wires (Fig. 1 B). To quantify the efficiency of cell loading with the reporter dye, we determined the size of the electroporation-loaded zone and the average fluorescence intensity within this zone. The fluorescence intensity was expressed relative to the fluorescence intensity of a layer of cell thickness of the loading solution, thus giving an estimate of the efficiency of the loading procedure. The size of the loaded zone was determined by measuring the surface area of cells that displayed a fluorescence intensity above the background level determined in nonloaded cell zones within the same culture (see Materials and Methods).
Experiments with a low-molecular-mass dye
We investigated the fluorescence intensity and the size of the loaded zone with 6-CF as a reporter dye (0.376 kDa), making use of uni- and bipolar electroporation. The pulse protocols for the two methods are illustrated in Fig. 1, C and D, and the performance for both methods is depicted in Fig. 3. Field strengths below 400 V/cm (i.e., 20 V peak amplitude applied over electrode wires separated over 0.5 mm; see Fig. 1) did not result in significant cell loading. Above this field strength, both uni- and bipolar electroporation resulted in loading efficiencies that increased with the field strength (Fig. 3 A). Unipolar electroporation gave loading efficiencies in the order of 20–25% in the 1200- to 1800-V/cm range, and similar values were obtained for bipolar electroporation in the 1000- to 1300-V/cm range. None of the bars in these two voltage ranges showed significant differences on comparison with analysis of variance (average loading efficiency of 21.4% over the former and 19.8% over the latter range). Increasing the field strength of unipolar electroporation in the 1200- to 1800-V/cm range did not result in better loading efficiencies and was associated with increased cell death (Fig. 3 B). For bipolar electroporation, increasing the field strength above 1300 V/cm further increased the loading efficiency, and at 1400 V/cm the efficiency was ∼38% (Fig. 3 A), significantly above the efficiency observed for unipolar electroporation with any of the applied field strengths (p < 0.001). At this setting, cell death associated with the bipolar protocol attained values in the order of what was observed in unipolar mode (∼90 cells on a total of 2100 cells/mm2 in the electroporated zone, i.e., ∼4% of the electroporated cells). Thus, for the same degree of cell death, bipolar electroporation allowed more efficient loading.
FIGURE 3.

Comparison of uni- and bipolar electroporation in ECV304 cells. (A) Loading efficiency for 6-CF electroporation in function of the electric field strength. The three star symbols indicate a significant difference with p < 0.001 compared with all other bars in this graph; n = 13 for unipolar and 12 for bipolar electroporation. (B) Cell death in function of electric field strength, as determined with trypan blue. Stars indicate significances compared with all the other unipolar data (other significant differences are given in the text); n = 16 for unipolar and 12 for bipolar electroporation. (C) Size of the electroporation-loaded cell zone in function of electric field strength. The star symbol represents a significant difference with p < 0.05 compared with all bipolar data; n = 13 for both uni- and bipolar electroporation.
Analysis of the size of the electroporation-loaded zone gave a somewhat different picture. The surface area of loaded cells increased with the field strength for both uni- and bipolar electroporation, but the unipolar protocol yielded larger loading zones than the bipolar protocol (Fig. 3 C). For unipolar electroporation, the loaded zone measured around 2 mm2 at 1600–1800 V/cm, corresponding to ∼40% of the surface area delineated by the electrode wires. At maximum, the surface area of loaded cells was ∼40% larger with unipolar than with bipolar electroporation (Fig. 3 C). Bipolar electroporation was, on the other hand, better suited to load a narrow strip of cells, with a full-width at half-maximum in the order of 250 μm (for an electrode wire separation of 500 μm).
Experiments with higher-molecular-mass dyes
The electroporation performance was further investigated with fluorescent probes of 3 and 40 kDa (3- and 40-kDa dextran-fluorescein dyes applied at 100 μM). Field strengths determined for the low-molecular-weight fluorescent reporter in the previous section do not necessarily apply for these higher-molecular-weight probes; we chose 1200 V/cm for both unipolar and bipolar electroporation because this setting combined a low degree of cell death with acceptable loading efficiency. With this setting, cell death in the electroporation zone averaged 28 ± 4 cells/mm2 (n = 21) for unipolar and 18 ± 3 cells/mm2 (n = 16) for bipolar electroporation (Fig. 3 B), corresponding to 1.3% and 0.8% of the total number of cells in this zone, respectively (both values significantly different from each other with p < 0.05).
The fluorescence intensity and the size of the electroporation-loaded zone as functions of the molecular mass of the reporter dye are illustrated in Fig. 4 for the two protocols. The experiments demonstrate decreased loading performance for probes with larger molecular weights, as expected because of slower diffusion. With the electroporation settings chosen, unipolar pulses were better than bipolar pulses in terms of loading efficiencies and size of the loaded zones for the various fluorescent probes used. The larger loading zone observed with the 0.376 kDa 6-CF (Fig. 4 B) was not a result of spreading of the dye via gap junctions between cells (allowing the passage of substances below 1–1.5 kDa) because the ECV304 cell batch used in these experiments did not express the major connexins 32 or 43 (Fig. 5 A).
FIGURE 4.

Uni- and bipolar electroporation performance for various fluorescent reporter dyes in ECV304. (A) Electroporation loading efficiency as a function of molecular mass. (B) Size of the electroporation-loaded cell zone as a function of molecular mass. Electroporation was done with a field strength of 1200 V/cm for both unipolar and bipolar protocols. Stars indicate significances as compared with the corresponding unipolar condition; number signs indicate significances compared with the corresponding bar for 0.376 kDa; n = 10 for unipolar and 6 for bipolar electroporation.
FIGURE 5.

Western blot and immunocytochemical analysis of connexin expression. (A) Western blots illustrating connexin 43 and 32 expression patterns in ECV304 and the C6 cells used in this study. (B and C) Western blots displaying connexin expression in total lysates and Triton X-100-soluble and -insoluble fractions. The majority of connexins were present in the Triton X-100-insoluble phase, indicating incorporation into gap junctions. Antitubulin bands were added as a loading control (almost absent in the Triton X-100-insoluble fractions as expected). (D) Immunocytochemistry for connexin 43 and 32. Numerous immunopositive plaques and dots were observed at cell-cell interfaces; nuclei were stained with DAPI. The arrows point to large gap junction plaques, arrowheads to immunopositive dots. Scale bar represents 100 μm.
Experiments on cells coupled by gap junctions
We further performed experiments with bipolar electroporation (1200 V/cm field strength) to investigate dye spread from the loaded cell strip to nonloaded zones in cells that are coupled by gap junctions. We used C6 glioma cells stably transfected with connexin 32 (C6-Cx32) or 43 (C6-Cx43) to this purpose. Both cell lines displayed significantly stronger expression of their respective connexins as compared with wild-type C6 cells (C6-WT) in Western blot analysis (Fig. 5, B and C). The majority of the connexins were present in the Triton X-100-insoluble fraction, which corresponds to the fraction that contains the gap junctions (40,41). Immunocytochemistry showed immunopositive dots and plaques at cell-cell interfaces (Fig. 5 D).
In FRAP experiments, C6-Cx32 and C6-Cx43 showed significantly faster recovery as compared with C6-WT, as illustrated in Table 1. Accordingly, electroporation with the gap-junction-permeable 6-CF (400 μM) resulted in a significantly larger fluorescent zone in C6-Cx32 and 43 as compared with C6-WT (Fig. 6 A), illustrating dye transfer from the electroporation-loaded zone to neighboring cells via gap junction channels. Electroporation of C6-Cx32 and C6-Cx43 with the gap junction impermeable 3- or 10-kDa dextran fluoresceins (400 μM) yielded very small zones of loaded cells as illustrated in Fig. 6 A, but because the loading efficiency of these higher-molecular-weight probes was intrinsically different from 6-CF (see Fig. 4), these data cannot be directly compared with the 6-CF data. We evaluated another parameter indicative of dye spread that is independent of the loading efficiency of the fluorescent reporter. The profile of the fluorescence intensity along an axis perpendicular to the length axis of the loaded strip was bell-shaped, as illustrated in Fig. 6 B, and could reliably be fitted by a Gaussian curve with standard deviation σ. The σ is an intrinsic parameter of dye spread and corresponds to the distance over which 68.3% of the dye molecules have diffused in both directions from the initial loading strip. Fig. 6 C demonstrates that σ for 6-CF spread was significantly larger in the C6-Cx32 and C6-Cx43 as compared with C6-WT. Treatment of the cells with the gap junction blocker carbenoxolone (25 μM, 30 min) significantly reduced σ. The value of σ was also strongly reduced when dye spread was probed with the gap-junction-impermeable 3- or 10-kDa dextran fluoresceins (Fig. 6 C).
TABLE 1.
Fluorescence recovery after photobleaching (FRAP) in C6 cells
| Cell line | Fluorescence recovery after 5 min (n = 20) |
|---|---|
| C6-WT | 17.6 ± 3.5% |
| C6-Cx43 | 59.6 ± 5.3%* |
| C6-Cx32 | 63.3 ± 4.4%* |
Significantly different from C6-WT with p < 0.001.
FIGURE 6.

Bipolar electroporation loading and dye transfer via gap junctions in C6 glioma cells. (A) Size of the 6-CF-loaded cell zone in WT and connexin-transfected C6 cells, 30 min after electroporation. The size of the loaded zone in C6-WT was not different from that in ECV304, as can be appreciated by comparison with Fig. 4 B. In C6-Cx43 and C6-Cx32, the loaded zone was significantly larger than that in C6-WT. Electroporation loading with the gap-junction-impermeable dyes 3 or 10 kDa dextran fluorescein (DF) resulted in very small zones, because the efficiency of loading these high-molecular-weight probes into the cells was much lower than that for 6-CF (see Fig. 4 A). (B) Example traces of the fluorescence intensity profile for the conditions indicated, 30 min after electroporation. The fluorescence intensity in the ordinate was normalized to the peak of the intensity profile, and each curve is the average of eight different experiments. The image on top of the graph illustrates a representative example of 6-CF loading in C6-WT. (C) Averaged data for the standard deviation of dye spread σ as determined by fitting individual intensity profiles to a Gauss equation. The σ of 6-CF spread was significantly larger in C6-Cx43 and C6-Cx32 than in C6-WT. Carbenoxolone (CBX) treatment (25 μM, 30 min) significantly reduced σ, and an even lower σ was observed when the cells (C6-Cx32) were loaded with the gap-junction-impermeable 3- or 10-kDa dextran fluoresceins. Stars give significances as compared with C6-WT; n = 8 for A and C.
In situ electroporation loading thus appeared to be useful for investigating gap junctional coupling between cells, referred to above as ELDT, analogous to the scrape loading and dye transfer (SLDT) method. In SLDT, a linear scratch is made over the culture with a needle, followed by dye uptake in disrupted cells and dye transfer via gap junctions to neighboring cells (30,31). The dye diffusion profiles obtained in SLDT experiments are difficult to fit by a Gaussian relation because of the presence of a central dip where the cells are removed as a result of scraping. We therefore compared ELDT and SLDT by analyzing the spatial constant λ of dye spread, as determined by fitting the flanks of the diffusion profiles by a monoexponentially decaying function (Fig. 7). The λ corresponds to the distance where the dye fluorescence has decreased by a factor 1/e and is, like σ, an intrinsic parameter of dye spread not affected by the absolute fluorescence levels. Fig. 7, A–D, illustrates a significantly larger λ in cells transfected with Cx32 or 43 as compared with WT, as expected. Moreover, the λ obtained in ELDT experiments was approximately twice as large as that given by the SLDT procedure. This difference may in part be related to the fact that the flanks of the electroporation-loaded profiles are less sharply defined as those obtained with scrape loading, as a consequence of a gradual fall-off of the electric field strength away from the electrode. However, a closer look at Fig. 7 B shows that the change in diffusion profile from WT to connexin-expressing cells is much more apparent in ELDT as compared with SLDT, and this is also clear from Fig. 7, C and D (note that the ordinates have different calibrations in C and D). The increase of λ in connexin-expressing cells as compared with WT cells, symbolized by λCx − λWT, is depicted in Fig. 7 E, illustrating that this parameter is more than twice as large when probed with ELDT as compared with SLDT (40–50 μm range for ELDT and <20 μm for SLDT). These data indicate that the degree of coupling determined with ELDT is significantly larger than that seen with SLDT. Cell scraping as performed in SLDT involves strong mechanical stimulation with plasma membrane rupture, leakage of cell contents into the medium, and activation of various signaling cascades. We tested whether dye spread measured with ELDT was influenced by the SLDT procedure by applying the SLDT scrape immediately before the start of ELDT, as illustrated in Fig. 7 F. These experiments showed that λCx − λWT determined from ELDT experiments was significantly lower in cultures that had received the SLDT scratch. In a further step, we tested whether supernatant obtained from SLDT-treated cultures influenced dye coupling in acceptor cultures as measured in FRAP experiments. FRAP recovery in supernatant-exposed cultures was mildly but not significantly depressed in C6-Cx43 and not affected in C6-Cx32 (data not shown), indicating that decreased dye coupling observed with SLDT is caused by intracellular factors rather than by factors released into the medium.
DISCUSSION
Electroporation is a technique that uses electrical pulses to create transient aqueous pores in the plasma membrane that act as a conduit for the delivery of cell-impermeable substances (reviewed in Gehl (42)). The molecular processes taking place during permeabilization are currently not fully understood, but various aspects of pore formation have been experimentally investigated and mathematically modeled (43–46). Pore formation happens on a time scale of microseconds and is followed by membrane resealing that is very much slower and proceeds on a time scale of minutes (42). If the field strength becomes too strong, pore resealing may be hampered, resulting in cell death. Recent work indicates that pore formation depends on the time during which the electroporation pulses exceed a certain threshold value (26,47). For this reason, care should be taken in comparing unipolar and bipolar electroporation protocols as applied in the study presented here. Several differences between the two protocols make comparisons at the level of field strength intensity difficult: first, the unipolar pulses had a duty cycle of 50%, i.e., the pulse was on for 10 μs and off for the next 10 μs in the 50-kHz bursts, whereas the off period was lacking in the bipolar protocol (which switched from 10 μs of positive deflection to 10 μs of negative deflection without an off period). Second, small differences in the response time of the two amplifiers may result in slightly different above-threshold voltage profiles and thus loading efficiencies. In addition, the reported field strengths should be taken only as indicative numbers: electroporation behavior depends on numerous influences, with cell geometry, electrode position, temperature, and electroporation solution as some of the most obvious influences that may vary substantially among different setups. Notwithstanding these considerations, the aim of the work presented here was to establish whether in situ electroporation with oscillating electric fields could be applied for functional studies on cell-cell dye coupling via gap junctions. All previous work (from our group and others) with in situ electroporation of adherent cell monolayers has been done with unipolar pulse protocols (15–17,27–29). Comparison of uni- and bipolar electroporation has been done in the context of cell transfection in suspension cultures (18,25) but not for functional studies where cell death is an important issue.
Taken over the range of applied electric field strengths, the efficiency of bipolar loading of the low-molecular-mass probe 6-CF was on average comparable to unipolar loading (Fig. 3 A). At 1400 V/cm, the bipolar protocol gave a significantly higher loading efficiency in the order of almost 40%. Unipolar electroporation loaded the largest zone of cells (Fig. 3 C) but was associated with sharply increasing cell death (Fig. 3 B). Cell death with the bipolar protocol was comparable with unipolar electroporation, but for the same amount of cell death, bipolar electroporation gave better loading efficiency at 1400 V/cm. A field strength of 1200 V/cm combined good loading with an acceptable degree of cell death for the unipolar and bipolar protocols. Electroporation loading of higher-molecular-weight dyes with these settings showed superior loading in terms of efficiency and size with the unipolar as compared with the bipolar protocol. Efficiencies approximately halved between 0.376 and 40 kDa molecular mass (Fig. 4 A), and the decline was much more pronounced for the size of the loaded zone (Fig. 4 B).
Few researchers have compared uni- and bipolar protocols for in situ electroporation of monolayer cultures, for reasons delineated by Kotnik et al. (24,25), mainly related to the lack of commercially available devices that generate a bipolar output. These authors compared uni- and bipolar protocols on cell suspensions, but the applied electroporation signals were pulses rather than a high-frequency oscillating signal. Nevertheless, bipolar electroporation yielded more efficient uptake of a reporter dye but no gain at the level of cell death (25). Tekle et al. (18) reported data obtained with high-frequency oscillating fields applied to adherent cells. These authors found bipolar electroporation to be more efficient than its unipolar variant for DNA transfection of NIH 3T3 fibroblasts, but the conditions investigated were much more extreme, as cell death levels ranging from 3% up to 99% were reported. Clearly, cell death may be extensive for transfection experiments, as the cells will recover and grow over the hours or days after which the expression efficiency is evaluated. The goal of electroporation for transfection purposes is indeed to maximize efficiency, whereas the goal in our experiments was to identify conditions combining an absolute minimum of cell death with a realistic efficiency, so as to allow subsequent functional studies.
Bipolar electroporation was an excellent method to load a narrow (∼250 μm wide) strip-like cell zone within a confluent monolayer, and we further investigated whether this loading method could be used to estimate the degree of functional coupling and dye transfer between cells via gap junctions. Analysis of a simple parameter such as the size of the zone with suprathreshold fluorescence was sufficient to distinguish connexin-expressing cells from WT noncoupled cells (Fig. 6 A). More sophisticated analysis was necessary to interpret the data obtained with higher-molecular-weight, nongap-junction-permeant probes because of intrinsically different loading properties of higher-molecular-weight molecules. The standard deviation of the Gaussian dye spread σ proved to be a useful and robust parameter (independent of the actual fluorescence levels) to evaluate the transfer of various reporter dyes between cells in the monolayer (Fig. 6, B and C). SLDT is a well-known and widely used method to investigate dye coupling via gap junctions (30,31), and we compared the performance of ELDT and SLDT in terms of dye spread quantified by the spatial constant λ. Both methods clearly demonstrated increased dye coupling in transfected and coupled cells as compared with WT cells. The most prominent difference was, however, that the change of λ from WT to coupled cells was more than twice as large in ELDT as in SLDT (Fig. 7 E), indicating an underestimation of the degree of dye coupling with SLDT. An obvious reason for this difference may reside in the nature of the cell-loading protocol. SLDT is done by applying a linear scrape (“scratch”) whereby plasma membranes are disrupted and the reporter dye leaks into the damaged cells. Cell death is one major difference between the two loading protocols, and the first part of this study was devoted to adjusting the electroporation settings to obtain minimal cell death in the loading zone. A consequence of traumatic cell loading is the release of cellular constituents into the medium and the activation of intracellular signals that may influence the degree of cell-cell coupling via gap junctions. Application of the SLDT scratch followed by ELDT showed that ELDT dye spread was significantly reduced to the level observed with SLDT (Fig. 7 F). The FRAP experiments on cells that received supernatant from scraped cultures indicate that an extracellular factor is probably not the major cause of decreased coupling in SLDT. Presumably, SLDT triggers intracellular signals that act to decrease transfer via gap junctions. One possibility is the generation of large intercellular calcium waves that increase the cytoplasmic calcium concentration (mostly by releasing calcium from the endoplasmic reticulum (48)) to a level that depresses gap junctional coupling (33,49).
We conclude from this study that bipolar pulsed high-frequency electroporation combines good loading performance with safety in terms of cell death and is the preferred method to load a narrow longitudinal strip of cells for subsequent studies on the temporal and spatial spread of reporter dyes via gap junctions. ELDT is an alternative for SLDT associated with low levels of cell death and better reflects the true level of cell-cell coupling via gap junctions. In comparison with FRAP and LAMP, which represent point measurements of cell-cell coupling, ELDT has the advantage of reporting an average degree of coupling in a large population of cells. Studies on the evolution of the dye spread parameter σ as a function of time in principle make it possible to establish an apparent diffusion constant (σ2 = 2·D·t) for the movement of reporter dye molecules in and between cells. Future work will be directed to fully exploiting the possibilities of this method.
Acknowledgments
We are very grateful to Bart Blanckaert, electronic engineer, for the helpful discussions on the circuit diagrams of electroporation amplifiers. Bipolar electroporation equipment is available from L.L.
Our work is financially supported by the Fund for Scientific Research Flanders, Belgium (F.W.O., grant Nos. 3G023599, 3G001201, G.0335.03, G.0354.07, and G.0140.08 to L.L.), Ghent University (B.O.F., grant Nos. 01115099, 01107101, and 01113403 to L.L.), the Queen Elisabeth Medical Foundation (grant No. 365B5602 to L.L.), and the Interuniversity Attraction Poles Program (Belgian Science Policy, project P6/31).
Editor: David A. Eisner.
References
- 1.Golzio, M., M. P. Rols, and J. Teissie. 2004. In vitro and in vivo electric field-mediated permeabilization, gene transfer, and expression. Methods. 33:126–135. [DOI] [PubMed] [Google Scholar]
- 2.Gothelf, A., L. M. Mir, and J. Gehl. 2003. Electrochemotherapy: results of cancer treatment using enhanced delivery of bleomycin by electroporation. Cancer Treat. Rev. 29:371–387. [DOI] [PubMed] [Google Scholar]
- 3.Wells, D. J. 2004. Gene therapy progress and prospects: electroporation and other physical methods. Gene Ther. 11:1363–1369. [DOI] [PubMed] [Google Scholar]
- 4.Li, S. 2004. Electroporation gene therapy: new developments in vivo and in vitro. Curr. Gene Ther. 4:309–316. [DOI] [PubMed] [Google Scholar]
- 5.Olofsson, J., K. Nolkrantz, F. Ryttsen, B. A. Lambie, S. G. Weber, and O. Orwar. 2003. Single-cell electroporation. Curr. Opin. Biotechnol. 14:29–34. [DOI] [PubMed] [Google Scholar]
- 6.Teruel, M. N., T. A. Blanpied, K. Shen, G. J. Augustine, and T. Meyer. 1999. A versatile microporation technique for the transfection of cultured CNS neurons. J. Neurosci. Methods. 93:37–48. [DOI] [PubMed] [Google Scholar]
- 7.Murphy, R. C., and A. Messer. 2001. Gene transfer methods for CNS organotypic cultures: a comparison of three nonviral methods. Mol. Ther. 3:113–121. [DOI] [PubMed] [Google Scholar]
- 8.Krull, C. E. 2004. A primer on using in ovo electroporation to analyze gene function. Dev. Dyn. 229:433–439. [DOI] [PubMed] [Google Scholar]
- 9.Inoue, T., and R. Krumlauf. 2001. An impulse to the brain–using in vivo electroporation. Nat. Neurosci. 4(Suppl):1156–1158. [DOI] [PubMed] [Google Scholar]
- 10.Denet, A. R., R. Vanbever, and V. Preat. 2004. Skin electroporation for transdermal and topical delivery. Adv. Drug Deliv. Rev. 56:659–674. [DOI] [PubMed] [Google Scholar]
- 11.Mir, L. M., M. F. Bureau, J. Gehl, R. Rangara, D. Rouy, J. M. Caillaud, P. Delaere, D. Branellec, B. Schwartz, and D. Scherman. 1999. High-efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc. Natl. Acad. Sci. USA. 96:4262–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aihara, H., and J. Miyazaki. 1998. Gene transfer into muscle by electroporation in vivo. Nat. Biotechnol. 16:867–870. [DOI] [PubMed] [Google Scholar]
- 13.Matsuno, Y., H. Iwata, Y. Umeda, H. Takagi, Y. Mori, A. Kosugi, K. Matsumoto, T. Nakamura, and H. Hirose. 2003. Hepatocyte growth factor gene transfer into the liver via the portal vein using electroporation attenuates rat liver cirrhosis. Gene Ther. 10:1559–1566. [DOI] [PubMed] [Google Scholar]
- 14.Dean, D. A., D. Machado-Aranda, K. Blair-Parks, A. V. Yeldandi, and J. L. Young. 2003. Electroporation as a method for high-level nonviral gene transfer to the lung. Gene Ther. 10:1608–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boitano, S., E. R. Dirksen, and M. J. Sanderson. 1992. Intercellular propagation of calcium waves mediated by inositol trisphosphate. Science. 258:292–295. [DOI] [PubMed] [Google Scholar]
- 16.Braet, K., C. Mabilde, L. Cabooter, G. Rapp, and L. Leybaert. 2004. Electroporation loading and photoactivation of caged InsP3: tools to investigate the relation between cellular ATP release in response to intracellular InsP3 elevation. J. Neurosci. Methods. 132:81–89. [DOI] [PubMed] [Google Scholar]
- 17.Leybaert, L., and M. J. Sanderson. 2001. Intercellular calcium signaling and flash photolysis of caged compounds. A sensitive method to evaluate gap junctional coupling. Methods Mol. Biol. 154:407–430. [DOI] [PubMed] [Google Scholar]
- 18.Tekle, E., R. D. Astumian, and P. B. Chock. 1991. Electroporation by using bipolar oscillating electric field: an improved method for DNA transfection of NIH 3T3 cells. Proc. Natl. Acad. Sci. USA. 88:4230–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cegovnik, U., and S. Novakovic. 2004. Setting optimal parameters for in vitro electrotransfection of B16F1, SA1, LPB, SCK, L929 and CHO cells using predefined exponentially decaying electric pulses. Bioelectrochemistry. 62:73–82. [DOI] [PubMed] [Google Scholar]
- 20.Chang, D. C. 1989. Cell poration and cell fusion using an oscillating electric field. Biophys. J. 56:641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muller, K. J., M. Horbaschek, K. Lucas, U. Zimmermann, and V. L. Sukhorukov. 2003. Electrotransfection of anchorage-dependent mammalian cells. Exp. Cell Res. 288:344–353. [DOI] [PubMed] [Google Scholar]
- 22.Satkauskas, S., M. F. Bureau, M. Puc, A. Mahfoudi, D. Scherman, D. Miklavcic, and L. M. Mir. 2002. Mechanisms of in vivo DNA electrotransfer: respective contributions of cell electropermeabilization and DNA electrophoresis. Mol. Ther. 5:133–140. [DOI] [PubMed] [Google Scholar]
- 23.Pliquett, U. 2003. Joule heating during solid tissue electroporation. Med. Biol. Eng. Comput. 41:215–219. [DOI] [PubMed] [Google Scholar]
- 24.Kotnik, T., D. Miklavcic, and L. M. Mir. 2001. Cell membrane electropermeabilization by symmetrical bipolar rectangular pulses. Part II. Reduced electrolytic contamination. Bioelectrochemistry. 54:91–95. [DOI] [PubMed] [Google Scholar]
- 25.Kotnik, T., L. M. Mir, K. Flisar, M. Puc, and D. Miklavcic. 2001. Cell membrane electropermeabilization by symmetrical bipolar rectangular pulses. Part I. Increased efficiency of permeabilization. Bioelectrochemistry. 54:83–90. [DOI] [PubMed] [Google Scholar]
- 26.Kotnik, T., G. Pucihar, M. Rebersek, D. Miklavcic, and L. M. Mir. 2003. Role of pulse shape in cell membrane electropermeabilization. Biochim. Biophys. Acta. 1614:193–200. [DOI] [PubMed] [Google Scholar]
- 27.Braet, K., S. Aspeslagh, W. Vandamme, K. Willecke, P. E. Martin, W. H. Evans, and L. Leybaert. 2003. Pharmacological sensitivity of ATP release triggered by photoliberation of inositol-1,4,5-trisphosphate and zero extracellular calcium in brain endothelial cells. J. Cell. Physiol. 197:205–213. [DOI] [PubMed] [Google Scholar]
- 28.Braet, K., K. Paemeleire, K. D'Herde, M. J. Sanderson, and L. Leybaert. 2001. Astrocyte-endothelial cell calcium signals conveyed by two signalling pathways. Eur. J. Neurosci. 13:79–91. [PubMed] [Google Scholar]
- 29.Vandamme, W., K. Braet, L. Cabooter, and L. Leybaert. 2004. Tumour necrosis factor alpha inhibits purinergic calcium signalling in blood-brain barrier endothelial cells. J. Neurochem. 88:411–421. [DOI] [PubMed] [Google Scholar]
- 30.el-Fouly, M. H., J. E. Trosko, and C. C. Chang. 1987. Scrape-loading and dye transfer. A rapid and simple technique to study gap junctional intercellular communication. Exp. Cell Res. 168:422–430. [DOI] [PubMed] [Google Scholar]
- 31.Meda, P. 2001. Assaying the molecular permeability of connexin channels. Methods Mol. Biol. 154:201–224. [DOI] [PubMed] [Google Scholar]
- 32.Deleze, J., B. Delage, O. Hentati-Ksibi, F. Verrecchia, and J. C. Herve. 2001. Fluorescence recovery after photobleaching. Methods Mol. Biol. 154:313–327. [DOI] [PubMed] [Google Scholar]
- 33.Dakin, K., Y. Zhao, and W. H. Li. 2005. LAMP, a new imaging assay of gap junctional communication unveils that Ca2+ influx inhibits cell coupling. Nat. Methods. 2:55–62. [DOI] [PubMed] [Google Scholar]
- 34.Bond, S. L., J. F. Bechberger, N. K. Khoo, and C. C. Naus. 1994. Transfection of C6 glioma cells with connexin32: the effects of expression of a nonendogenous gap junction protein. Cell Growth Differ. 5:179–186. [PubMed] [Google Scholar]
- 35.Zhu, D., S. Caveney, G. M. Kidder, and C. C. Naus. 1991. Transfection of C6 glioma cells with connexin 43 cDNA: analysis of expression, intercellular coupling, and cell proliferation. Proc. Natl. Acad. Sci. USA. 88:1883–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puc, M., S. Corovic, K. Flisar, M. Petkovsek, J. Nastran, and D. Miklavcic. 2004. Techniques of signal generation required for electropermeabilization. Survey of electropermeabilization devices. Bioelectrochemistry. 64:113–124. [DOI] [PubMed] [Google Scholar]
- 37.Leybaert, L., A. de Meyer, C. Mabilde, and M. J. Sanderson. 2005. A simple and practical method to acquire geometrically correct images with resonant scanning-based line scanning in a custom-built video-rate laser scanning microscope. J. Microsc. 219:133–140. [DOI] [PubMed] [Google Scholar]
- 38.Cooper, C. D., and P. D. Lampe. 2002. Casein kinase 1 regulates connexin-43 gap junction assembly. J. Biol. Chem. 277:44962–44968. [DOI] [PubMed] [Google Scholar]
- 39.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–685. [DOI] [PubMed] [Google Scholar]
- 40.Musil, L. S., and D. A. Goodenough. 1991. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J. Cell Biol. 115:1357–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Musil, L. S., and D. A. Goodenough. 1993. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell. 74:1065–1077. [DOI] [PubMed] [Google Scholar]
- 42.Gehl, J. 2003. Electroporation: theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol. Scand. 177:437–447. [DOI] [PubMed] [Google Scholar]
- 43.Krassowska, W., and P. D. Filev. 2007. Modeling electroporation in a single cell. Biophys. J. 92:404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tieleman, D. P. 2004. The molecular basis of electroporation. BMC Biochem. 5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gabriel, B., and J. Teissie. 1999. Time courses of mammalian cell electropermeabilization observed by millisecond imaging of membrane property changes during the pulse. Biophys. J. 76:2158–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tekle, E., R. D. Astumian, W. A. Friauf, and P. B. Chock. 2001. Asymmetric pore distribution and loss of membrane lipid in electroporated DOPC vesicles. Biophys. J. 81:960–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weaver, J., and Y. Chizmadzhev. 1996. Theory of electroporation: a review. Bioelectrochem. Bioenerg. 41:135–160. [Google Scholar]
- 48.Dupont, G., L. Combettes, and L. Leybaert. 2007. Calcium dynamics: spatio-temporal organization from the subcellular to the organ level. Int. Rev. Cytol. 261:193–245. [DOI] [PubMed] [Google Scholar]
- 49.Peracchia, C. 2004. Chemical gating of gap junction channels; roles of calcium, pH and calmodulin. Biochim. Biophys. Acta. 1662:61–80. [DOI] [PubMed] [Google Scholar]