

Abstract
Protonation of the Ca2+ ligands of the SR Ca2+-ATPase (SERCA1a) was studied by a combination of rapid scan FTIR spectroscopy and electrostatic calculations. With FTIR spectroscopy, we investigated the pH dependence of C=O bands of the Ca2+-free phosphoenzyme (E2P) and obtained direct experimental evidence for the protonation of carboxyl groups upon Ca2+ release. At least three of the infrared signals from protonated carboxyl groups of E2P are pH dependent with pKa values near 8.3: a band at 1758 cm−1 characteristic of nonhydrogen-bonded carbonyl groups, a shoulder at 1720 cm−1, and part of a band at 1710 cm−1, both characteristic of hydrogen-bonded carbonyl groups. The bands are thus assigned to H+ binding residues, some of which are involved in H+ countertransport. At pH 9, bands at 1743 and 1710 cm−1 remain which we do not attribute to Ca2+/H+ exchange. We also obtained evidence for a pH-dependent conformational change in β-sheet or turn structures of the ATPase. With MCCE on the E2P analog E2(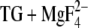 ), we assigned infrared bands to specific residues and analyzed whether or not the carbonyl groups of the acidic Ca2+ ligands are hydrogen bonded. The carbonyl groups of Glu771, Asp800, and Glu908 were found to be hydrogen bonded and will thus contribute to the lower wave number bands. The carbonyl group of some side-chain conformations of Asp800 is left without a hydrogen-bonding partner; they will therefore contribute to the higher wave number band.
), we assigned infrared bands to specific residues and analyzed whether or not the carbonyl groups of the acidic Ca2+ ligands are hydrogen bonded. The carbonyl groups of Glu771, Asp800, and Glu908 were found to be hydrogen bonded and will thus contribute to the lower wave number bands. The carbonyl group of some side-chain conformations of Asp800 is left without a hydrogen-bonding partner; they will therefore contribute to the higher wave number band.
INTRODUCTION
The Ca2+-ATPase (1) of SR performs active transport of Ca2+ from the cytoplasm of muscle cells into the lumen of SR (2–7). The enzyme belongs to the P-type ATPase family and couples the transport of two Ca2+ ions across the SR membrane to the hydrolysis of one molecule of ATP.
The Ca2+-ATPase reaction mechanism is commonly described in terms of an E1/E2 scheme adapted from de Meis and Vianna (8). The scheme is represented in Fig. 1. According to this model two Ca2+ ions bind to the high affinity Ca2+ binding sites of the ATPase in state E1, which releases H+ into the cytoplasm. This is followed by ATP binding and phosphorylation, Ca2E1→Ca2E1ATP→Ca2E1P. The next step is phosphoenzyme conversion, Ca2E1P→E2P. The conversion to E2P is accompanied by release of Ca2+ into the SR and proton uptake from the SR. Hydrolysis of E2P and regeneration of the high affinity Ca2+ binding sites complete the reaction cycle.
FIGURE 1.

Simplified reaction scheme of the Ca2+-ATPase. Only the forward direction under physiological conditions is indicated.
Proton uptake and release reactions lead to proton countertransport. The stoichiometry of proton countertransport is 1–1.5 H+ per transported Ca2+ (9–11). Ca2+ and H+ ions have been indicated to compete for the same sites of the Ca2+-ATPase (12–16) where protons are supposed to be required for stabilization of the ATPase structure by partly neutralizing the negative charge of the empty Ca2+ binding sites (5). The Ca2+ binding sites contain four carboxyl groups (Glu309, Glu771, Asp800, and Glu908), which are candidates for binding the countertransported protons. Proton countertransport stops when the lumenal pH is higher than 8.0, indicating that the pKa of the lumenal H+ binding sites is between ∼7.2 and 7.7 (11,14,15).
Several electrostatic calculations have been performed to identify the acidic Ca2+ ligands that are protonated in the Ca2+-free states. The calculations were done on Ca2E1 (17–19) and different E2 states: E2(TG+BHQ) (20), E2(TG) (18,19), and E2(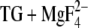 ) (18). The latter is an E2P analog which most closely resembles the product state of the dephosphorylation reaction, i.e., E2 with nonconvalently bound phosphate. The calculations have indicated that Glu309, Glu771, and Asp800 are ionized around physiological pH in Ca2E1. The different calculations disagree regarding the protonation state of Glu908 in Ca2E1. It is protonated up to at least pH 7.5 in continuum electrostatic calculations with fixed side chains (17,20). Our MCCE calculations have yielded a pKa of 3.6 (18). This discrepancy likely arises because our approach allows for side-chain flexibility which seems to make the effective dielectric constant ɛ larger than 20, the maximum value used in the calculations with fixed side chains (17).
) (18). The latter is an E2P analog which most closely resembles the product state of the dephosphorylation reaction, i.e., E2 with nonconvalently bound phosphate. The calculations have indicated that Glu309, Glu771, and Asp800 are ionized around physiological pH in Ca2E1. The different calculations disagree regarding the protonation state of Glu908 in Ca2E1. It is protonated up to at least pH 7.5 in continuum electrostatic calculations with fixed side chains (17,20). Our MCCE calculations have yielded a pKa of 3.6 (18). This discrepancy likely arises because our approach allows for side-chain flexibility which seems to make the effective dielectric constant ɛ larger than 20, the maximum value used in the calculations with fixed side chains (17).
For the E2 states all calculations agree in that Glu771, Asp800, and Glu908 are protonated at pH 6. The calculations agree further that only one of the three residues Glu771, Asp800, and Glu908 titrates between pH 6 and 8. In our calculations, Asp800 titrates with a pKa between 7 and 8 depending on the treatment of water (18), whereas Obara et al. (20) have found that Glu908 titrates with a pKa around 6.5 if ɛ is set to 20. Again we attribute the deviation between the calculations to the different approaches rather than to the different E2 states used. This is because our calculations with the E2(TG) structure—presumed to represent the same state as E2(TG+BHQ)—gave very similar values for Glu771, Asp800, and Glu908 as our calculations with E2(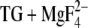 ). Therefore we consider the E2 and E2P analog states equivalent with respect to the protonation state of the acidic Ca2+ ligands. This is supported by infrared spectroscopy: the spectra of carboxyl groups that become protonated upon Ca2+ dissociation are similar for phosphorylated and unphosphorylated ATPase (21).
). Therefore we consider the E2 and E2P analog states equivalent with respect to the protonation state of the acidic Ca2+ ligands. This is supported by infrared spectroscopy: the spectra of carboxyl groups that become protonated upon Ca2+ dissociation are similar for phosphorylated and unphosphorylated ATPase (21).
An important exception to the calculation of similar pKa values in E2(TG) and E2(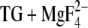 ) is Glu309, where we have calculated a pKa of 4.7 in E2(TG) but a pKa near 8.5 in E2(
) is Glu309, where we have calculated a pKa of 4.7 in E2(TG) but a pKa near 8.5 in E2(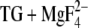 ). The latter value is in accordance with the calculations by Obara et al. (20), which indicate that a small fraction of residues is deprotonated at pH 8 when ɛ is set to 20. The large variation of the pKa of Glu309 arises because Glu309 can adopt a high and a low pKa depending on the local backbone structure, which determines the exposure of the Glu309 side chain to water. The low pKa structure seems to be predominantly adopted, according to our calculations, with the side chain oriented away from the Ca2+ binding sites toward a water-filled channel, as in the E2(TG) structure by Toyoshima and Nomura (22).
). The latter value is in accordance with the calculations by Obara et al. (20), which indicate that a small fraction of residues is deprotonated at pH 8 when ɛ is set to 20. The large variation of the pKa of Glu309 arises because Glu309 can adopt a high and a low pKa depending on the local backbone structure, which determines the exposure of the Glu309 side chain to water. The low pKa structure seems to be predominantly adopted, according to our calculations, with the side chain oriented away from the Ca2+ binding sites toward a water-filled channel, as in the E2(TG) structure by Toyoshima and Nomura (22).
At high pH, all calculations indicate a larger number of protons involved in Ca2+ release than experimentally observed, as discussed previously (18). To clarify the situation we undertake here an infrared spectroscopic investigation on the pH-dependent protonation of acidic residues in E2P.
Time-resolved FTIR spectroscopy allows the detection of ATPase intermediate states in the reaction cycle (23,24). The reaction is started by ATP release from a photolabile caged ATP, which was earlier shown to initiate the Ca2+-ATPase reaction cycle (25). The release of ATP is accompanied by infrared absorbance changes caused by the molecular processes in the ATPase during Ca2+ transport. ATP-induced absorbance changes are not observed in the presence of the Ca2+ chelator EGTA, when the nucleotide binding site is blocked by FITC (26) and when the ATPase is inhibited by TG (27).
In this work we studied protonation of the Ca2+ ligands by a combination of rapid scan FTIR spectroscopy and electrostatic calculations. The aim of the infrared studies was to identify carbonyl bands of protonated carboxyl groups in the infrared spectrum that titrate with the same pKa as the lumenal proton ligands. Thus we investigated the pH dependence of C=O bands of the Ca2+-free phosphoenzyme (E2P), which were studied before only at pH 7.0 (21,23,28,29). At least three of the infrared signals from protonated carboxyl groups of E2P are pH dependent, which makes the previous tentative assignment to H+ binding residues unequivocal. Our results also indicate a pH-dependent conformational change in a β-sheet or turn structure of the ATPase. The MCCE calculations analyzed the hydrogen-bonding pattern of acidic Ca2+ ligands and served to assign the observed infrared bands to specific residues.
MATERIALS AND METHODS
Sample preparation
Ca2+-ATPase from rabbit hind leg and back muscle was prepared in the laboratory of W. Hasselbach by the method of Hasselbach and Makinose (30) and stored at −20°C. ATPase vesicles were dialyzed after thawing and addition of calcium ionophore A23187 in 1H2O or in 2H2O buffer, containing 10 mM Tris-maleate or Tris-diethylamine and 20 μM CaCl2 at the desired pH for 1.5 h. Tris-diethylamine buffer was used for experiments at pH values above 8.0. Measurements in both buffers at pH 7.5 and pH 8.0 have indicated no buffer effect on the spectra. The pH meter reading for 2H2O buffers was corrected by +0.4, according to Glaose and Long (31), to obtain p2H.
Samples for time-resolved infrared spectroscopy were prepared by drying dialyzed SR suspension with added caged ATP, MgCl2, and DTT onto a CaF2 window under N2 flow with subsequent rehydration with 15% DMSO in 1H2O or 2H2O. Approximate concentrations in the samples were 100–150 μg/μl of protein, 100 mM Tris-maleate or Tris-diethylamine, 0.2 mM CaCl2, 5 mM MgCl2, 10 mM caged ATP, 10 mM DTT, 0.5 mg/ml A23187, and 15% DMSO.
FTIR measurements
Time-resolved FTIR measurements of the Ca2E1→E2P reaction were performed at 1°C with a Bruker (Ettlingen, Germany) IFS 66/S spectrometer as described previously (23,29). The resting state of the ATPase in our samples was Ca2E1. The reaction cycle was initiated by photolytic release of ATP from caged ATP. Difference spectra were obtained from spectra before and after the photolysis flash that reflected the difference in absorbance between resting state Ca2E1 and the states that accumulate after ATP release. The reaction was induced up to two times per sample. A total of 6–8 experiments were averaged from experiments on four different samples, two of which were dialyzed on the same day. The averaged time-resolved series of difference spectra was normalized to a standard protein absorbance (amide II absorbance of 0.26) (21). To obtain the resulting Ca2E1→E2P spectrum, those spectra of the time-resolved series of spectra that showed the largest amplitude of the E2P marker band at 1194 cm−1 (23,28,32) were averaged. For this, the time interval 5–30 s was used for all experiments in 1H2O, that of 9–83 s for experiments at p2H 6.0, and that of 5–17 s for the experiments at p2H 7.5 and 9. The amplitude of the 1194 cm−1 band was found to vary slightly for spectra obtained at the different pH values. Thus, they were normalized to that of the spectrum at pH 6.0 using normalization factors between 1 and 1.1. Spectra normalized to 1194 cm−1 are shown in the figures and were used to determine the pKa value of the titrating carboxyl groups.
Previously we have shown spectra of the reactions Ca2E1 → E2P (23,28) and Ca2E1P → E2P (21,29). Here we show Ca2E1 → E2P spectra since the Ca2E1P intermediate could not be clearly resolved at high pH because E2P formation was faster at high pH. Positive bands above 1700 cm−1 are very similar in both types of spectra. These bands are the main focus of this work and are due to C=O vibrations in the E2P state.
Fitting procedure
To determine pKa values from the pH dependence of infrared band amplitudes, we fitted integrated band amplitudes to the equation
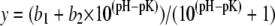 |
using GRAFIT 309b. The 1758 cm−1 band was integrated with respect to a baseline drawn between two averaged data points at both sides of the bands with method E of OPUS 4.0. For the 1710 cm−1 band, method D was used, which integrates with respect to a horizontal line through one baseline point that we placed in the minimum between the 1758 and 1710 cm−1 bands. The error of the pKa value was estimated by varying the method for band integration.
MCCE calculations
MCCE is a hybrid method combining continuum electrostatics and molecular mechanics. References for the methodology and some recent applications can be found in, e.g., (18,19,33–39).
MCCE samples residue ionization and side-chain conformation as a function of pH, which provides pKa values for individual residues. Conformational flexibility is modeled by multiple side-chain conformations which are constructed systematically by rotating rotatable side-chain bonds. For example, multiple conformations of aspartic and glutamic acids are constructed by rotation around the Cα-Cβ, Cβ-Cγ, and the Cα-Cβ, Cβ-Cγ, and Cγ-Cδ bonds, respectively. The ionization states of a side chain in a specific conformation comprise an ionized carboxyl group and two protonated groups, protonated either at the Oδ1 or Oδ2 oxygen for aspartic acids or at the Oɛ1 or Oɛ2 oxygen for glutamic acids. Thus, each residue is represented by a set of structures (commonly called conformers) that differ in side-chain orientation, ionization state, site of protonation, and direction of the OH bond. Conformational and ionization states of all residues are sampled in a Monte Carlo sampling, assuming a Boltzmann distribution of states that yields the occupancy of each residue state as a function of pH.
Calculations were performed with program version “mcce_alpha” and one of the four ATPase molecules in the asymmetric unit (chain A) of the 2.3 Å crystal structure of E2(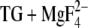 ) (1WPG.pdb) (40) as described (18). Five of the crystal water molecules that are located within 10 Å around Asp800, ligand of both calcium ions, were included in the calculations and treated explicitly. We will discuss only structures occupied to 5% or more. To reduce the high number of structures for each residue in the hydrogen-bond analysis, similar structures with similar hydrogen bonding were combined, and a representative structure is discussed and shown in Fig. 5. These representative structures are given a label which consists of a number and a letter, for example, “structure 1a”. The number denotes a representative structure of the heavy atoms of the side chain, whereas the letter discriminates between different positions of the hydrogen atom.
) (1WPG.pdb) (40) as described (18). Five of the crystal water molecules that are located within 10 Å around Asp800, ligand of both calcium ions, were included in the calculations and treated explicitly. We will discuss only structures occupied to 5% or more. To reduce the high number of structures for each residue in the hydrogen-bond analysis, similar structures with similar hydrogen bonding were combined, and a representative structure is discussed and shown in Fig. 5. These representative structures are given a label which consists of a number and a letter, for example, “structure 1a”. The number denotes a representative structure of the heavy atoms of the side chain, whereas the letter discriminates between different positions of the hydrogen atom.
FIGURE 5.

Acidic Ca2+ ligands with hydrogen-bonding partners in the E2(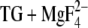 ) state at pH 6. Shown are representative side-chain structures discussed in the text. The respective ligand and interacting residues are colored, other ligands are in black (and without hydrogens except for the hydrogens of the carboxyl groups). For a clearer representation, backbone atoms are not shown, except for Ser767 in Fig. 5 B, since its backbone carbonyl oxygen can be involved in hydrogen bonding to Glu771. Predominantly occupied side-chain structures are solid; less occupied ones are transparent. Black dashes indicate possible hydrogen bonds. M indicates transmembrane helices. In A–C, the cytoplasmic side is on the top of the figure; the lumenal side on the bottom; D shows a top view from the cytoplasmic side. (A) Glu309. Two representative structures are shown: structure 1a (solid) and structure 2a (transparent). The carbonyl oxygen of structure 1a can form a hydrogen bond to Asn796; (B) Glu771. Three representative structures are shown: structure 1a (solid), structure 1b (transparent), and structure 2a (transparent); structures 1a and 1b differ only in hydrogen position. The carbonyl oxygen of all side-chain conformations can form a hydrogen bond to Asn796, only some (structures 1a and 1b) can also form a hydrogen bond to Leu792. The carboxyl hydrogen of some of the side-chain conformations can also form hydrogen bonds to the oxygen of Asn796 (structures 1a and 2a) or Ser767 (structure 1b); (C) Asp800. Two representative side-chain conformations are shown: structure 1a (solid) and structure 2a (transparent). Ser767 can be a hydrogen donor for some Asp800 side-chain conformations (structure 2a). (D) Glu908: Two representative structures are shown: structure 1a (solid) and structure 2a (transparent). More detailed explanations are in the text.
) state at pH 6. Shown are representative side-chain structures discussed in the text. The respective ligand and interacting residues are colored, other ligands are in black (and without hydrogens except for the hydrogens of the carboxyl groups). For a clearer representation, backbone atoms are not shown, except for Ser767 in Fig. 5 B, since its backbone carbonyl oxygen can be involved in hydrogen bonding to Glu771. Predominantly occupied side-chain structures are solid; less occupied ones are transparent. Black dashes indicate possible hydrogen bonds. M indicates transmembrane helices. In A–C, the cytoplasmic side is on the top of the figure; the lumenal side on the bottom; D shows a top view from the cytoplasmic side. (A) Glu309. Two representative structures are shown: structure 1a (solid) and structure 2a (transparent). The carbonyl oxygen of structure 1a can form a hydrogen bond to Asn796; (B) Glu771. Three representative structures are shown: structure 1a (solid), structure 1b (transparent), and structure 2a (transparent); structures 1a and 1b differ only in hydrogen position. The carbonyl oxygen of all side-chain conformations can form a hydrogen bond to Asn796, only some (structures 1a and 1b) can also form a hydrogen bond to Leu792. The carboxyl hydrogen of some of the side-chain conformations can also form hydrogen bonds to the oxygen of Asn796 (structures 1a and 2a) or Ser767 (structure 1b); (C) Asp800. Two representative side-chain conformations are shown: structure 1a (solid) and structure 2a (transparent). Ser767 can be a hydrogen donor for some Asp800 side-chain conformations (structure 2a). (D) Glu908: Two representative structures are shown: structure 1a (solid) and structure 2a (transparent). More detailed explanations are in the text.
Placement of water molecules
To analyze hydrogen bonding of the carboxyl groups of the acidic Ca2+ ligands in E2(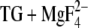 ) (1WPG.pdb) to water, water molecules were filled into protein cavities using the program Dowser (41) (http://hekto.med.unc.edu:8080/HERMANS/software/DOWSER/index.html) for all four ATPase molecules (chains A–D) of the asymmetric unit. The program analyzes the interaction energies and keeps only those water molecules which have a favorable interaction energy that is larger than 42 kJ/mol.
) (1WPG.pdb) to water, water molecules were filled into protein cavities using the program Dowser (41) (http://hekto.med.unc.edu:8080/HERMANS/software/DOWSER/index.html) for all four ATPase molecules (chains A–D) of the asymmetric unit. The program analyzes the interaction energies and keeps only those water molecules which have a favorable interaction energy that is larger than 42 kJ/mol.
RESULTS
Overview over infrared spectra
E2P formation from Ca2E1 was investigated by time-resolved FTIR spectroscopy at different pH values from pH 6.0 to 9.0 with intervals of 0.5 pH units. In Fig. 2 we show the spectra at pH 6.0, 8.0, and 9.0. The other spectra (at pH 6.5, 7.0, 7.5, and 8.5) are omitted in the figure to obtain a clearer representation. For the analysis of the pH dependence of selected bands, all spectra were used (see below).
FIGURE 2.

Infrared difference spectra of the Ca2E1 → E2P reaction. Bands discussed in the text are labeled. (Solid line) Spectra obtained at pH 6.0; (dashed line) pH 8.0; and (dotted line) pH 9.0.
The spectra are shown in the spectral range from 1800 cm−1 to 900 cm−1, which covers the following absorption regions: i), the region of the stretching vibration of C=O groups (ν C=O) of, for example, protonated carboxyl groups and lipids (1800–1700 cm−1); ii), the antisymmetric stretching vibration of unprotonated carboxyl groups (νas COO−) (1610–1536 cm−1); iii), the region of the symmetric stretching vibration of unprotonated carboxyl groups (νs COO−) (1462–1328 cm−1); iv), the amide I mode of the polypeptide backbone, sensitive to secondary structure (1700–1610 cm−1); v), the amide II mode of polypeptide backbone (1580–1520 cm−1) (42,43); and vi), the region of phosphate absorption (<1300 cm−1). Differences between the spectra in the phosphate region are due to hydrolysis of ATP (25,44).
Different bands associated with E2P formation have been tentatively assigned for spectra obtained at pH 7.0 (21,23,28,29). In particular, E2P bands at 1758 and 1710 cm−1 have been assigned to the protonation of at least two carboxyl groups of Asp or Glu residues that might participate in H+ countertransport (28,29). Signals in the amide I region have been tentatively assigned to a conformational change of β-sheet or turn structures (1670–1688 cm−1), β-sheet (1638–1618 cm−1), and α-helical structures (1653 cm−1) (28,29).
E2P accumulation
The band at 1194 cm−1 is a marker band for the E2P state. It appears upon phosphoenzyme conversion (29), has been assigned to the phosphate group of E2P using isotopically labeled ATP (29,32), and indicates the environmental change that is responsible for the change of phosphate chemical specificity upon phosphoenzyme conversion. After normalization of the spectra to a standard protein absorption, the intensity of the 1194 cm−1 band was similar for all spectra, indicating that E2P accumulates at all pH values to a similar extent. Small variations in the amplitude of the 1194 cm−1 band were corrected by normalizing all spectra to the 1194 cm−1 amplitude obtained at pH 6.0. The normalization factors were between 1 and 1.1. These spectra are shown in Figs. 2 and 3 and were used for the titration curve in Fig. 4. The conditions used are known to accumulate E2P to nearly 100% at pH 7.0 as discussed previously (28). From the evaluation of the marker band we conclude that, also at high pH, the accumulation of E2P is close to 100% under our conditions. We attribute this to the presence of DMSO.
FIGURE 3.

Spectral region of protonated carboxyl groups of the Ca2E1 →E2P spectra. (A) Spectra in 1H2O; (B) spectra in 2H2O. (Solid lines) p1H(p2H) 6.0; (dashed lines) p1H 8.0 or p2H 7.5; and (dotted lines) p1H(p2H) 9.0. The inset in A shows a subtraction of a high pH spectrum from a low pH spectrum. The high pH spectrum was the average of the spectra obtained at pH 8.5 and 9.0 and the low pH spectrum the average of the spectra at 6.0, 6.5, 7.0, 7.5, and 8.0. Similar spectra were obtained for all combinations of low and high pH spectra in the subtraction.
FIGURE 4.

C=O band area as a function of pH. (A) Band at 1758 cm−1; (B) band at 1710 cm−1. The solid line is a fit to the data points (see Materials and Methods). pKa values were determined to be 8.3 ± 0.4 for both bands.
C=O bands in 1H2O
The spectra of the Ca2E1→E2P reaction (Fig. 2) reveal distinct differences in the ν C=O region (1800–1700 cm−1) when the pH of the reaction medium increases. This spectral region is shown in Fig. 3 A on an expanded scale for the spectra shown in Fig. 2. At pH 6, three bands at 1758, 1743, and 1710 cm−1 can be identified. At pH 9, the bands at 1758 and 1710 cm−1 are significantly reduced in amplitude. In contrast, the C=O band at 1743 cm−1 does not change with pH and some intensity at 1710 cm−1 remains at high pH. Thus the pH 6 band at 1710 cm−1 seems to be composed of a pH-dependent component and a component that does not titrate between pH 6 and 9. In addition, the 1710 cm−1 band at pH 6 has a shoulder at 1720 cm−1, which is also reduced at high pH. The presence of the shoulder at 1720 cm−1 is obvious in a difference spectrum between the low and high pH spectra, which is shown as an inset in Fig. 3 A. The maximum of this spectrum is at 1705 cm−1.
The pH dependence of the bands at 1758 and 1710 cm −1 is best seen in Fig. 4, where we plotted their band areas against the pH value. The obtained titrations were fitted to determine the pKa value of the H+ binding residues. The best fits were obtained with pKa values of 8.3 ± 0.4 for both bands.
C=O bands in 2H2O
To get additional information for band assignment we recorded the spectra also in 2H2O at p2H 6.0, 7.5, and 9.0. They are shown in Fig. 3 B. The bands at 1758 and 1710 cm−1 in 1H2O (Fig. 3 A) exhibit a 2–6 cm−1 downshift in 2H2O as expected for carboxyl groups (45,46). In contrast, the C=O band at 1743 cm−1 in 1H2O is found at 1746 cm−1 in 2H2O.
Band assignment of the 1758, 1720, and 1710 cm−1 bands
The bands at 1758, 1720, and 1710 cm−1 decrease in amplitude with alkalization of the medium which considerably strengthens the previous tentative assignment (28,29) to protonated carboxyl groups. The pH dependence is similar to that of the apparent pKa of the residues binding lumenal H+ for proton countertransport (7.2–7.7) (11,14,15). Thus we assign the C=O bands at 1758 and 1720 cm−1 and the pH-dependent part of the 1710 cm−1 band to carboxyl groups that bind protons upon Ca2+ release, some or all of which participate in H+ countertransport. The band position at 1758 cm−1 indicates a nonhydrogen-bonded carbonyl group, whereas band positions at 1720 and 1710 cm−1 are characteristic of hydrogen-bonded groups (46). The band at 1710 cm−1, which remains at high pH, is affected by the divalent cation that binds to the phosphorylation site, which suggests that this band originates from the carbonyl group of phosphorylated Asp351 (47).
Band assignment of the 1743 cm−1 band
The band at 1743 cm−1 is neither affected by alkalization (Fig. 3 A) nor does it shift down upon deuteration (Fig. 3 B). The latter does not support an assignment to a carboxyl group but also does not exclude it for the following reasons: i), The group causing this band might not be in contact with the medium and might not exchange its protons with bulk water protons. This would then exclude a role in the proton countertransport of this residue. ii), Downshifts upon deuteration are not observed in all cases; upshifts have even been observed for hydrated carboxyl groups in CCl4 (48) and for the acetic acid dimer in the gas phase (49). Alternative to an assignment to a protonated carboxyl group, the 1743 cm−1 band could also represent a noncarboxyl carbonyl group, for example, from a lipid C=O group that increases its absorption index due to an environmental change. The main lipid band at 1732 cm−1 is hardly shifted in 2H2O in our absorbance spectra (not shown). If the 1743 cm−1 band were related to protonation of Ca2+ ligands, it should appear as a negative band in spectra of Ca2+ binding to the unphosphorylated enzyme because the respective carboxyl groups should become protonated. However, this is not observed (21,50,51) (note that the Ca2+ binding spectrum in Barth et al. (21) is inverted and shown as a Ca2+ release spectrum); nor is a negative band at 1743 cm−1 observed in a series of Ca2+ binding experiments at different pH values (J. Andersson, J. Sun, and A. Barth, unpublished). This also indicates that the band at 1743 cm−1 does not originate from the groups participating in H+/Ca2+ exchange.
Conformational changes
pH-dependent changes are also evident in the amide I region. The 1671/1661 cm−1 difference feature of the pH 6 spectrum is reduced at pH 8 and 9 (see Fig. 3 A). Consequently, low pH minus high pH double difference spectra (not shown) consistently exhibit a broad positive band at 1671 cm−1 and a sharp negative band at 1660 cm−1. The difference between the high and low pH spectra can be explained by a shift of the two pH 6 bands at 1671 and 1661 cm−1 toward each other at high pH, lowering the 1671 cm−1 band position by 0.7 cm−1 and raising that of the 1661 cm−1 band by 2 cm−1 (see Fig. 3 A). The same differences between low and high pH spectra can be observed in 2H2O where p2H 6 bands at 1670 and 1663 cm−1 are reduced at high pH because they have moved closer (see Fig. 3 B). In the low minus high p2H spectrum, this leads to a broad positive band at 1679 cm−1 and a sharp negative band at 1660 cm−1.
The similar band positions of the two bands in 1H2O and 2H2O make an assignment to side chains unlikely since most polar side chains exchange much faster than amide protons (52), in line with experiments showing exchange within several minutes (53,54). Thus we attribute the difference between the low and high pH spectra to a difference in amide I absorption. The band position of both affected bands is characteristic of turns, and that of the 1661 cm−1 band is at the edge of the range for α-helices. We conclude that the turn and probably α-helical segments are affected differently at low and high pH by the transition from Ca2E1 to E2P. The insensitivity to deuteration indicates that the backbone segments involved have stable hydrogen bonds and are rather rigid. The increased overlap of the 1671 and 1661 cm−1 bands at high pH make the spectra of the reaction Ca2E1 → E2P less featureless at high pH. This indicates that the conformations of the two states are more similar at high pH than at low pH.
The low pH minus high pH spectra in 1H2O and 2H2O also exhibit a negative band at 1692 cm−1. This difference is also evident in the spectra shown in Fig. 3 A, where the negative band at 1687 cm−1 in the pH 6.0–7.5 spectra is slightly shifted to 1686 cm−1 in the spectra recorded at pH 8–9 (Fig. 3 A). The negative band appears upon phosphoenzyme conversion and has previously been shown to be composed of at least two components at pH 7 with spectral positions at 1685 cm−1 and at 1693 cm−1 (29), as revealed by the band-narrowing technique fine structure enhancement (55). Fine structure enhancement of the pH series of spectra of this work revealed that the 1685 cm−1 component is present at all pH values, whereas the 1693 cm−1 component is only observed at pH 7.5 and below. This was also observed in 2H2O where a shoulder near 1690 cm−1 in the p2H 6 and 7.5 spectra was missing in the p2H 9 spectra (Fig. 3 B).
The component at 1685 cm−1 has been tentatively attributed to a β-sheet or turn structural changes because it shifts to 1677 cm−1 in 2H2O (29), as also shown in Fig. 3 B (band at 1679 cm−1). The corresponding backbone segment is quite flexible, as indicated by the fast 1H/2H exchange on a timescale of 1 h at pH 6 and 5°C.
For the 1693 cm−1 component, an assignment to β-sheet or turn structural changes of the backbone or to environmental changes around Asn, Gln, or Arg side chains was proposed (29). The apparent insensitivity of the 1693 cm−1 component band position to deuteration (29) (see also Fig. 3 B) makes an assignment to side chains unlikely, as discussed above. Thus, we tentatively assign it to β-sheet or turn structural changes of the backbone. Because this band does not shift upon deuteration on the timescale of our experiments, the backbone segment involved has more stable hydrogen bonds and is more rigid than that giving rise to the 1685 cm−1 component band. Since the negative 1693 cm−1 band in our Ca2E1 → E2P spectra is also observed upon Ca2E1P → E2P conversion (29), the corresponding backbone segment absorbs similarly in Ca2E1 and Ca2E1P but is affected by the formation of E2P.
The observation of the 1693 cm−1 band only at pH 7.5 and below gives rise to two possible interpretations: i), The corresponding backbone segment could absorb at a different wave number in Ca2E1 at high pH, indicating a pH dependency of the Ca2E1 structure. ii), The corresponding backbone segment of Ca2E1 could absorb at 1693 cm−1 at all pH values but not be affected by E2P formation above pH 7.5. This would indicate that Ca2E1 and E2P are more alike at higher pH because the structure of E2P is pH dependent. We conclude that the pH dependency of the 1693 cm−1 band gives evidence of a pH dependency of ATPase structure in the Ca2E1 and/or E2P state, which involves a β-sheet or turn structure with stable hydrogen bonds.
Hydrogen bonding
In the following, we will discuss hydrogen bonding to the side-chain carbonyl oxygens of the four acidic Ca2+ ligands in E2P using the E2(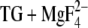 ) structure. The aim is to assign the infrared bands in the carbonyl spectral region. The spectral position of the carbonyl bands is sensitive to the strength of hydrogen bonding to the carbonyl oxygen. That is why we were interested in whether or not the acidic Ca2+ ligands interact with other residues or with water. Even if two atoms do not form a hydrogen bond in the crystal structure, they might come close enough to form a hydrogen bond when side chains move in response to pH. MCCE calculations reveal the movement of side chains in dependence of pH by an alteration of the occupancy of the generated side-chain structures. By analysis of the MCCE results, possible hydrogen donors can be identified. Fig. 5 shows those side-chain structures of the Ca2+ ligands in E2(
) structure. The aim is to assign the infrared bands in the carbonyl spectral region. The spectral position of the carbonyl bands is sensitive to the strength of hydrogen bonding to the carbonyl oxygen. That is why we were interested in whether or not the acidic Ca2+ ligands interact with other residues or with water. Even if two atoms do not form a hydrogen bond in the crystal structure, they might come close enough to form a hydrogen bond when side chains move in response to pH. MCCE calculations reveal the movement of side chains in dependence of pH by an alteration of the occupancy of the generated side-chain structures. By analysis of the MCCE results, possible hydrogen donors can be identified. Fig. 5 shows those side-chain structures of the Ca2+ ligands in E2(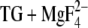 ) that were predominantly occupied in the MCCE calculations. Conformational analysis of Glu309 has been presented elsewhere (18) but without focusing on hydrogen bonding to the carbonyl group.
) that were predominantly occupied in the MCCE calculations. Conformational analysis of Glu309 has been presented elsewhere (18) but without focusing on hydrogen bonding to the carbonyl group.
To analyze hydrogen bonding of the acidic Ca2+ ligands with water, we filled protein cavities with water using the program Dowser (41), which takes into account the interaction energy between the internal water molecules and their environment. We used all four E2(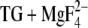 ) chains provided in the data file 1WPG.pdb. The results were slightly different for the four chains, indicating small structural differences between them which are decisive for providing space and favorable interactions for water or not. Dowser seems here to make a conservative suggestion, since not all of the crystal water molecules were reproduced. For example, of the three crystal waters near Asp800, two were reproduced for molecule A but only one for molecules B–D. The text below summarizes our findings on hydrogen bonding to the carbonyl oxygen of the protonated Ca2+ ligands. Table 1 analyzes hydrogen bonding of all significant side-chain structures.
) chains provided in the data file 1WPG.pdb. The results were slightly different for the four chains, indicating small structural differences between them which are decisive for providing space and favorable interactions for water or not. Dowser seems here to make a conservative suggestion, since not all of the crystal water molecules were reproduced. For example, of the three crystal waters near Asp800, two were reproduced for molecule A but only one for molecules B–D. The text below summarizes our findings on hydrogen bonding to the carbonyl oxygen of the protonated Ca2+ ligands. Table 1 analyzes hydrogen bonding of all significant side-chain structures.
TABLE 1.
Side-chain structures of the protonated acidic Ca2+ ligands at pH 6
| Structure | Approximate occupancy | Side-chain structure | Hydroxyl oxygen | Carbonyl oxygen | Hydrogen bond donors to carbonyl oxygen |
|---|---|---|---|---|---|
| Glu309-1a | 60% | Similar to crystal structure | Oɛ2 | Oɛ1 | Nδ2 of Asn796 (N-O distance ∼3.0 Å), Dowser water in chain D |
| Glu309-2a | 40% | Outwards oriented toward a channel to the cytosol | Oxygen closest to backbone oxygen | Water molecules at the bottom of the channel to Glu309 | |
| Glu771-1a | 30% | Carboxyl group rotated ∼50° counterclockwise around the Cγ-Cδ bond when viewed toward the backbone. Structures 1a and 1b differ in the direction of the OH bond | Closest to Oɛ2 | Closest to Oɛ1 | Asn796-Nδ2 (N-O distance ∼2.8–3.1 Å), Leu792-Cδ2 (C-O distance ∼3.3 Å) |
| Glu771-1b | 30% | Closest to Oɛ2 | Closest to Oɛ1 | Asn796-Nδ2 (N-O distance ∼2.8-3.1 Å), Leu792-Cδ2 (C-O distance ∼3.3 Å) | |
| Glu771-2a | 40% | Carboxyl group rotated ∼60° clockwise around the Cγ-Cδ bond when viewed toward the backbone | Closest to Oɛ2 | Closest to Oɛ1 | Asn796-Nδ2 (N-O distance ∼2.8–3.1 Å) |
| Asp800-1a | 48% | Similar to crystal structure | Oδ2 | Oδ1 | Dowser water (O-O distance 2.7 Å) in chains B and C of 1WPG.pdb. This water molecule bridges Asp800 with Glu908. |
| Asp800-1b | 12% | Oδ1 | Oδ2 | Crystal water (O-O distance 2.5 Å), OγH group of ∼30% of Ser767 conformers (O-O distance ∼3.1 Å) | |
| Asp800-2a | 25% | Carboxyl group rotated ∼50o anticlockwise around the Cβ-Cγ bond when viewed toward the backbone | Closest to Oδ1 | Closest to Oδ2 | Crystal water (O-O distance 2.5 Å), OγH group of ∼30% of Ser767 conformers (O-O distance ∼3.6 Å) |
| Asp800-2b | 5% | Closest to Oδ2 | Closest to Oδ1 | Non-linear hydrogen bond to OH of Glu908-2a. | |
| Glu908-1a | 50% | Similar to crystal structure | Oɛ1 | Oɛ2 | Crystal water and Dowser water in chains B and C or two Dowser waters in chains A and D |
| Glu908-1b | 40% | Oɛ2 | Oɛ1 | Crystal water (O-O distance 2.8 Å) in molecules A and D or two Dowser waters in molecules B and C. One of the latter bridges Glu908 with Asp800. | |
| Glu908-2a | 2%* | Rotated around Cα-Cβ and Cβ-Cγ bond | Moved into the cavity near Oɛ2 | Close to Oɛ2 | Water in cavity |
Different side-chain conformations are denoted by i. g. Glu771-1a, which indicates structure 1a of Glu771. The description of side-chain structure is with respect to crystal structure of E2(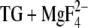 ); the oxygen atoms are labeled as in the crystal structure. Dowser water indicates a water molecule that is placed by the program Dowser. Occupancy is given from MCCE calculations of the E2(
); the oxygen atoms are labeled as in the crystal structure. Dowser water indicates a water molecule that is placed by the program Dowser. Occupancy is given from MCCE calculations of the E2(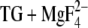 ) structure including the five crystal water molecules close to the calcium ligands.
) structure including the five crystal water molecules close to the calcium ligands.
This structure is occupied 15% in the calculations without explicitly treated water molecules.
Glu309
In the high pKa mode of Glu309 (see Introduction), adopted by a small portion of ATPase molecules, 60% of the side chains adopt structures similar to that of the 1WPG.pdb structure data (structure 1a, Fig. 5 A, solid). The remaining 40% adopt a different conformation in which the Glu309 side chain orients away from the other Ca2+ ligands (structure 2a, Fig. 5 A, transparent). The carbonyl oxygen faces a cavity at the bottom of the channel and is likely in contact with water. In all structures, the carbonyl group is expected to be hydrogen bonded (see Table 1 for details).
Glu771
Glu771 is protonated up to pH 14 (18). Most side chains have a conformation similar to that of the crystal structure except for the carboxyl group, which adopts positions rotated around the Cγ-Cδ bond. None of the adopted structures corresponds exactly to that in the crystal structure. Three main structures (1a, 1b, and 2a) are shown in Fig. 5 B and discussed in Table 1. When viewed toward the backbone they have their carboxyl group rotated by 50° counterclockwise (structures 1a and 1b) and 60° clockwise (structure 2a) with respect to the crystal structure. This does not seem to be in contradiction to the x-ray data because chains B and C of 1WPG.pdb have the Glu771 carboxyl group rotated by ∼30° clockwise and the 60° clockwise rotation is adopted in the E2 structures by Jensen et al. (56).
Our hydrogen-bonding pattern of structure 1a agrees with that calculated previously with fixed side-chain conformation for the E2(TG+BHQ) structure (20). Our analysis shows that other options are also possible. Carbonyl groups of all structures are expected to be hydrogen bonded (Table 1). The hydroxyl group can either hydrogen bond to Asn796 (structures 1a and 2a) or to the backbone carbonyl oxygen of Ser767 (structure 1b), depending on the orientation of the OH bond. A Gln residue at position 771 is likely to undergo interactions with both Asn796 and Ser767. These two hydrogen bonds could lock the enzyme in a specific conformation from which it cannot undergo dephosphorylation of E2P, in line with the inhibiting effects of mutation to Gln (57–60).
Asp800
At pH 6, ∼60% of the Asp800 side chains are protonated and adopt a conformation very similar to that in the crystal structure of E2(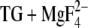 ) (structures 1a and 1b). The dominant structure 1a is shown solid in Fig. 5 C. Ten percent of the Asp800 side chains are unprotonated at pH 6 and also adopt the crystal structure. The remaining ∼30% of the Asp800 side chains are protonated and have their carboxyl group rotated around the Cβ-Cγ axis when compared to the crystal structure (structure 2a, Fig. 5 C, transparent). The carboxyl group of Asp800 shows some rotational freedom in the structures of the different chains in 1WPG.pdb, and a structure close to our structure 2a is adopted in the E2(TG) structures with AMPPCP (56).
) (structures 1a and 1b). The dominant structure 1a is shown solid in Fig. 5 C. Ten percent of the Asp800 side chains are unprotonated at pH 6 and also adopt the crystal structure. The remaining ∼30% of the Asp800 side chains are protonated and have their carboxyl group rotated around the Cβ-Cγ axis when compared to the crystal structure (structure 2a, Fig. 5 C, transparent). The carboxyl group of Asp800 shows some rotational freedom in the structures of the different chains in 1WPG.pdb, and a structure close to our structure 2a is adopted in the E2(TG) structures with AMPPCP (56).
In structure 1a, the oxygen that points toward the cavity with crystal waters is preferentially protonated (see Fig. 5 C and Table 1). The same oxygen atom as in our structure 1a is protonated according to the calculations on the E2(TG+BHQ) structure (20) (see Figs. 6 and 8 of that reference). This preference of protonation can be rationalized in the following way: water is a good hydrogen-bond acceptor but only a moderate hydrogen-bond donor, if the mean distance between a hydrogen-bond donor and an acceptor is taken as a measure for donor and acceptor strength. The OH group of carboxylic acid is a very strong donor, which results in relatively short hydrogen bonds with water acceptors of 1.63 Å between oxygen and hydrogen. In contrast hydrogen bonds between R2C=O and water are considerably longer, 1.90 Å between hydrogen and oxygen, implying that they are weaker (61). This seems to indicate that a COOH group with the choice either to provide the OH group as a hydrogen donor to water or to provide the C=O group as a hydrogen-bond acceptor for water will chose the former.
The carbonyl group of structure 1a (adopted by 48% of all side chains) can hydrogen bond to a water molecule between Asp800 and Glu908. However, the presence of this molecule seems to depend on minute structural changes since Dowser places this water only in two of the four ATPase chains of the E2(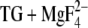 ) structural file. The carbonyl oxygen of structures 1b and 2a (adopted by 37% of all side chains) can interact with water in a nearby cavity and can form a weak hydrogen bond to Ser767. Thus it seems that at least ∼40% of the protonated Asp800 carbonyl groups experience considerable hydrogen bonding. Whether or not structure 1a has a hydrogen-bonded carbonyl group seems to depend on structural fluctuations in E2(
) structural file. The carbonyl oxygen of structures 1b and 2a (adopted by 37% of all side chains) can interact with water in a nearby cavity and can form a weak hydrogen bond to Ser767. Thus it seems that at least ∼40% of the protonated Asp800 carbonyl groups experience considerable hydrogen bonding. Whether or not structure 1a has a hydrogen-bonded carbonyl group seems to depend on structural fluctuations in E2(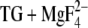 ).
).
Glu908
Glu908 mainly adopts a structure as in the crystal structure of E2(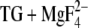 ) (structures 1a and 1b adopted by 90% of all side chains; structure 1a is shown solid in Fig. 5 D). The proton can reside on both carboxyl oxygens at pH 6 with nearly equal probabilities. A second structure (structure 2a, Fig. 5 D, transparent) is adopted by 2% of the side chains in the calculations with explicit water but by 15% in those without explicit water. In structure 2a, the Cγ-Cδ bond and the Cβ-Cγ bond are rotated around the preceding C-C bonds (Fig. 5 D, transparent). The difference between the crystal structure (structures 1a and 1b) and structure 2a can be approximately described by a 45° rotation of the Cγ-CδOO unit around an axis perpendicular to the plane of this unit and running through the middle of the Cγ-Cδ bond. The effect is that one of the oxygens of structure 2a is close to the position of Oɛ2 of structures 1a and 1b and the other has moved into the cavity which is close to Oɛ2 of the crystal structure (= structures 1a and 1b). The carbonyl group of all Glu908 side chains is hydrogen bonded to one or more water molecules.
) (structures 1a and 1b adopted by 90% of all side chains; structure 1a is shown solid in Fig. 5 D). The proton can reside on both carboxyl oxygens at pH 6 with nearly equal probabilities. A second structure (structure 2a, Fig. 5 D, transparent) is adopted by 2% of the side chains in the calculations with explicit water but by 15% in those without explicit water. In structure 2a, the Cγ-Cδ bond and the Cβ-Cγ bond are rotated around the preceding C-C bonds (Fig. 5 D, transparent). The difference between the crystal structure (structures 1a and 1b) and structure 2a can be approximately described by a 45° rotation of the Cγ-CδOO unit around an axis perpendicular to the plane of this unit and running through the middle of the Cγ-Cδ bond. The effect is that one of the oxygens of structure 2a is close to the position of Oɛ2 of structures 1a and 1b and the other has moved into the cavity which is close to Oɛ2 of the crystal structure (= structures 1a and 1b). The carbonyl group of all Glu908 side chains is hydrogen bonded to one or more water molecules.
DISCUSSION
pH-dependent protein conformation
FTIR difference spectra of the Ca2E1 → E2P reaction were obtained at different pH values. Upon alkalization of the reaction medium, we observed spectral changes in the amide I region (Figs. 2 and 3). They indicate a pH dependence of the protein backbone in β-sheets, turns, and probably an α-helix. These structural changes make the spectra of the Ca2E1 → E2P reaction less featureless at higher pH, indicating that the structure of Ca2E1 is more like that of E2P at higher pH.
pH dependence of carbonyl bands in the infrared spectrum
pH-dependent spectral changes were also observed in the spectral region of protonated carboxyl groups (Fig. 3). The pH dependency makes the suggested assignment (21) to protonated carboxyl groups unequivocal.
Bands at 1758 cm−1 and 1710 cm−1 and the shoulder at 1720 cm−1 titrate with a pKa value near 8.3, which is similar to the apparent pKa value of residues binding lumenal H+ for proton countertransport (11,14,15). This similarity of the pKa values supports our interpretation that at least part of the signals at 1758, 1720, and 1710 cm −1 originates from the protonation of carboxyl groups involved in H+ transport (21). These bands have also been observed for Ca2+ binding to the unphosphorylated enzyme (21,50,51,62), indicating that the same residues are protonated in E2 and E2P and that hydrogen bonding to the carbonyl oxygens is similar (21).
The band at 1758 cm− 1 is assigned to nonhydrogen-bonded Asp or Glu side chains, whereas the band at 1710 cm−1 and the shoulder at 1720 cm−1 have vibrational frequencies characteristic of hydrogen-bonded carbonyl groups (46).
Number of carboxyl groups detected by infrared spectroscopy
We observed pH dependence of the two bands at 1758 cm−1 and 1710 cm−1 and the shoulder at 1720 cm−1 (Figs. 3 and 4). This indicates that in the Ca2+ release step three types of carboxyl groups, which can be distinguished by their environment, become protonated at pH 6 but not above pH 8. The simplest interpretation is that three residues are involved. However, this number could be <3, if environmental heterogeneity occurs: a given Asp or Glu residue might experience hydrogen bonds of varying strengths, depending on its side-chain conformation or that of its hydrogen-bonding partner and thus produce several bands in the infrared spectrum. The number of residues causing the pH-dependent infrared bands could be more than three if several groups absorb at the same wave number.
Conformational heterogeneity of the acidic Ca2+ ligands
Our MCCE calculations with E2(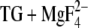 ) indicated that there is conformational heterogeneity of the acidic Ca2+ ligands. This is particularly pronounced for Glu309, as discussed previously (18), but holds also for Glu771, Asp800, and Glu908. The carboxyl groups of Glu771 and Asp800 can rotate around the preceding C-C bond. A minority of Glu908 side chains exhibits more conformational flexibility. There is also considerable heterogeneity as to which oxygen atoms are protonated and regarding the orientation of the O-H bond. Taken together this makes possible different interaction patterns for different side-chain structures. The most occupied structures of the acidic Ca2+ ligands are shown in Fig. 5.
) indicated that there is conformational heterogeneity of the acidic Ca2+ ligands. This is particularly pronounced for Glu309, as discussed previously (18), but holds also for Glu771, Asp800, and Glu908. The carboxyl groups of Glu771 and Asp800 can rotate around the preceding C-C bond. A minority of Glu908 side chains exhibits more conformational flexibility. There is also considerable heterogeneity as to which oxygen atoms are protonated and regarding the orientation of the O-H bond. Taken together this makes possible different interaction patterns for different side-chain structures. The most occupied structures of the acidic Ca2+ ligands are shown in Fig. 5.
Carbonyl bands expected from electrostatic calculations
In the following, we discuss which of the carboxyl groups in the Ca2+ binding sites (Glu309, Glu771, Asp800, and Glu908) contribute to the infrared bands of the reaction Ca2E1 → E2P. Other carboxyl groups are not expected to contribute to the spectra since their pKa is calculated to be 6.1 or lower in E2(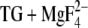 ). Our calculations with E2(TG) and E2(
). Our calculations with E2(TG) and E2(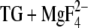 ) (18) have indicated that the pKa of Glu309 can be high (8.4) or low (4.7), depending probably on the local backbone conformation. The infrared results presented in this work are in line with this model since they indicate that the pKa of Glu309 is either below 6 (the lower pH limit of our experiments) or near 8.3 (pKa of the carbonyl bands). Since the pKa of the majority of Glu309 residues is low (4.7) (18), Glu309 is not expected to contribute much to the carbonyl bands above 1700 cm−1. Glu771, Asp800, and Glu908 are expected to contribute to the C=O signals in the infrared spectra because of their lower pKa values in Ca2E1 as compared to those in E2(
) (18) have indicated that the pKa of Glu309 can be high (8.4) or low (4.7), depending probably on the local backbone conformation. The infrared results presented in this work are in line with this model since they indicate that the pKa of Glu309 is either below 6 (the lower pH limit of our experiments) or near 8.3 (pKa of the carbonyl bands). Since the pKa of the majority of Glu309 residues is low (4.7) (18), Glu309 is not expected to contribute much to the carbonyl bands above 1700 cm−1. Glu771, Asp800, and Glu908 are expected to contribute to the C=O signals in the infrared spectra because of their lower pKa values in Ca2E1 as compared to those in E2(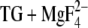 ) in our calculations (18). Thus upon the reaction Ca2E1 → E2P, observed in the infrared experiments, they are expected to protonate. According to other calculations without side-chain flexibility, Glu908 should be excluded from this list because its pKa in E2(TG+BHQ) was calculated to be slightly lower than in Ca2E1 (17,20). These calculations indicate that the number of protons that are exchanged for the two Ca2+ is approximately the same at pH 6 and 8. In contrast, our infrared measurements (measuring the difference in proton binding between Ca2E1 and E2P) and our calculations indicated less exchange at higher pH. According to our calculations, Asp800 is expected to produce pH-dependent protonation signals in our infrared spectra and will contribute to one or several of the pH-dependent protonation bands at 1758, 1720, and 1710 cm−1.
) in our calculations (18). Thus upon the reaction Ca2E1 → E2P, observed in the infrared experiments, they are expected to protonate. According to other calculations without side-chain flexibility, Glu908 should be excluded from this list because its pKa in E2(TG+BHQ) was calculated to be slightly lower than in Ca2E1 (17,20). These calculations indicate that the number of protons that are exchanged for the two Ca2+ is approximately the same at pH 6 and 8. In contrast, our infrared measurements (measuring the difference in proton binding between Ca2E1 and E2P) and our calculations indicated less exchange at higher pH. According to our calculations, Asp800 is expected to produce pH-dependent protonation signals in our infrared spectra and will contribute to one or several of the pH-dependent protonation bands at 1758, 1720, and 1710 cm−1.
Glu908 deprotonates partially above pH 6 in our calculations with E2(TG) and E2(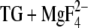 ) (18). The tendency to deprotonate is more pronounced in calculations by Obara et al. (20). Glu908 will therefore generate a protonation signal in our infrared spectra that is slightly smaller at the upper end of the investigated pH range. However, the variation in amplitude of the protonation signal is expected to be smaller than what can reliably be detected.
) (18). The tendency to deprotonate is more pronounced in calculations by Obara et al. (20). Glu908 will therefore generate a protonation signal in our infrared spectra that is slightly smaller at the upper end of the investigated pH range. However, the variation in amplitude of the protonation signal is expected to be smaller than what can reliably be detected.
Inconsistency between calculations and experiments
As discussed previously (18), all calculations seem to return protonation probabilities that are too high at high pH. This inconsistency is further supported by our infrared results: due to the expected protonation signal at high pH, Glu771 and Glu908 might be expected to contribute to the 1743 or the 1710 cm−1 bands at high pH. However, an assignment of the 1743 cm−1 band to a carboxyl group involved in Ca2+/H+ exchange is questionable (see above), and the high pH 1710 cm−1 band has been assigned to Asp351 (47). Thus there is no obvious assignment of a carbonyl band to protonated Glu771 or Glu908 at high pH, and the infrared spectra indicate that none of the acidic Ca2+ ligands is protonated at high pH. Of the several explanations given previously (18), it seems now that the calculations provide pKa values that are too high because of the following reasons:
A pH-dependent conformational change possibly caused by deprotonation of one of the Ca2+ ligands could make the E2P structure, which was obtained at pH 6.1, inappropriate for calculations at high pH. Side-chain flexibility implemented in MCCE reduces but does not eliminate the dependence on the initial structure. In line with this option we obtained evidence of conformational differences between high and low pH structures. Whether or not these affect the pKa values in the Ca2+ binding sites is impossible to deduce from the spectra.
The binding of positive ions other than H+ to the Ca2+ binding site, only partial Ca2+ release due to the higher affinity for Ca2+ at high pH (11,63,64), or nearby hydronium ions could compensate for the negative charge of the unprotonated Ca2+ ligands. This would facilitate the deprotonation of Glu771 and Glu908. The same effect is expected for a more open Ca2+ release pathway in genuine E2P as compared to E2(
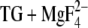 ), which would allow more water molecules to penetrate the transmembrane region.
), which would allow more water molecules to penetrate the transmembrane region.
From our spectra, it is difficult to decide whether none, one, or two Ca2+ are retained at high pH because the relevant spectral region of carboxylate absorption is highly convoluted. If Ca2+ were retained and the Ca2+ coordination by carboxylate groups were the same in high pH E2P as in Ca2E1P, negative bands in the spectral region of carboxylate absorption (∼1570 and ∼1400 cm−1) would be missing in our high pH spectra. This is not observed. All negative bands are present, although with smaller amplitude for some of them. If, on the other hand, Ca2+ were retained with different Ca2+ coordination, new positive bands would be observed at high pH which are characteristic of the absorption of coordinating carboxylate groups in E2P. This also is not observed. The conformational change between Ca2E1 and E2P is largely the same at high and low pH, with only subtle differences observed as discussed above. From the E2P marker band of the phosphate group, it is clear that the phosphate group is in an E2P environment at high and low pH, as discussed above.
Symbiosis of infrared experiments and electrostatic calculations
Because the infrared spectra do not provide evidence for protonated acidic Ca2+ ligands at high pH, we assigned the pH-dependent carboxyl bands to all acidic Ca2+ ligands that are protonated at pH 6: Glu771, Asp800, Glu908, and a small portion of the Glu309 side chains. With the help of the MCCE calculations at pH 6, we further assigned individual bands to specific residues using information regarding hydrogen bonding to their carbonyl oxygens (see Table 1). The carbonyl oxygens of Glu309, Glu771, Asp800, and Glu908 are expected to be hydrogen bonded for all side-chain conformations of Glu309, Glu771, and Glu908 and for at least 40% of the Asp800 side-chain conformations. Thus they will all contribute to the infrared signals at 1720 and 1710 cm−1, which are characteristic of hydrogen-bonded carbonyl groups with a hydrogen-bonding strength similar to that found in water. Some side-chain conformations of Asp800 have no obvious hydrogen bond to the carbonyl oxygen. The high wave number band at 1758 cm−1 is tentatively assigned to those.
CONCLUSIONS
Our infrared experiments and electrostatic calculations demonstrate directly that carboxyl groups become protonated when Ca2+ is released from the phosphoenzyme. Evidence was also obtained for a pH-dependent conformational change of the protein that affects the Ca2E1 → E2P transition. The MCCE calculations helped assign infrared bands to individual acidic Ca2+ ligands. They indicated considerable conformational heterogeneity of the acidic Ca2+ ligands in the Ca2+-free state E2(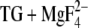 ), which allows for various hydrogen-bonding patterns. Our infrared experiments do not give evidence for protonated Ca2+ ligands at high pH, as indicated by the calculations and explanations for this discrepancy are given above.
), which allows for various hydrogen-bonding patterns. Our infrared experiments do not give evidence for protonated Ca2+ ligands at high pH, as indicated by the calculations and explanations for this discrepancy are given above.
Acknowledgments
We thank Wilhelm Hasselbach (Max-Planck-Institut, Heidelberg) for the gift of Ca2+-ATPase, John E. T. Corrie (National Institute for Medical Research, London) for the preparation of caged compounds, Marilyn Gunner, Junjun Mao, and Yifan Song (City College of New York, New York) for providing the MCCE code and helpful discussions, Christian Weidemüller (Johann Wolfgang Goethe-Universität, Frankfurt) for preparation of Fig. 5, and Ali Boudjema (Stockholm University) for some of the infrared measurements.
This work was supported by Vetenskapsrådet projektbidrag 621-2002-5884, Knut och Alice Wallenbergs Stiftelse bidrag 2002.0115, and postdoctoral grants to J. A. from Carl Tryggers Stiftelse and Wenner-Gren Stiftelserna and the Deutsche Forschungsgemeinschaft (SFB 472).
Abbreviations used: FTIR, Fourier transform infrared; BHQ, 2,5-di-tert-butyl-1,4-dihydroxybenzene; Ca2E1P, ADP-sensitive phosphoenzyme; DMSO, dimethyl sulfoxide; DTT, dithiothreitol; ɛ, dielectric constant; E2(TG), Ca2+-free ATPase with thapsigargin bound; E2(TG+BHQ), Ca2+-free ATPase with thapsigargin and 2,5-di-tert-butyl-1,4-dihydroxybenzene bound; E2P, ADP-insensitive phosphoenzyme; E2(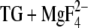 ), the E2P analog structure obtained with
), the E2P analog structure obtained with 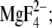 E2(
E2(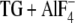 ), the E2P analog with
), the E2P analog with  FITC, fluorescein-5-isothiocyanate; MCCE, multiconformation continuum electrostatics; ν, stretching vibration; νs, symmetric stretching vibration; νas, antisymmetric stretching vibration; SR, sarcoplasmic reticulum; TG, thapsigargin.
FITC, fluorescein-5-isothiocyanate; MCCE, multiconformation continuum electrostatics; ν, stretching vibration; νs, symmetric stretching vibration; νas, antisymmetric stretching vibration; SR, sarcoplasmic reticulum; TG, thapsigargin.
Editor: Janos K. Lanyi.
References
- 1.Hasselbach, W., and M. Makinose. 1961. The calcium pump of the “relaxing granules” of muscle and its dependence on ATP splitting. Biochem. Z. 333:518–528. [PubMed] [Google Scholar]
- 2.McIntosh, D. B. 2000. Portrait of a P-type pump. Nat. Struct. Biol. 7:532–535. [DOI] [PubMed] [Google Scholar]
- 3.Mintz, E., and F. Guillain. 1997. Ca2+ transport by the sarcoplasmic reticulum ATPase. Biochim. Biophys. Acta. 1318:52–70. [DOI] [PubMed] [Google Scholar]
- 4.MacLennan, D. H., and N. M. Green. 2000. Pumping ions. Nature. 405:633–634. [DOI] [PubMed] [Google Scholar]
- 5.Toyoshima, C., and G. Inesi. 2004. Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 73:269–292. [DOI] [PubMed] [Google Scholar]
- 6.Stokes, D. L., and N. M. Green. 2003. Structure and function of the calcium pump. Annu. Rev. Biophys. Biomol. Struct. 32:445–468. [DOI] [PubMed] [Google Scholar]
- 7.Lee, A., and J. East. 2001. What the structure of a calcium pump tells us about its mechanism. Biochem. J. 356:665–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Meis, L., and A. Vianna. 1979. Energy interconversion by the Ca-dependent ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 48:275–292. [DOI] [PubMed] [Google Scholar]
- 9.Chiesi, M. and G. Inesi. 1980. Adenosine 5′-triphosphate dependent fluxes of manganese and hydrogen ions in sarcoplasmic reticulum vesicles. Biochemistry. 19:2912–2928. [DOI] [PubMed] [Google Scholar]
- 10.Levy, D., M. Seigneuret, A. Bluzat, and J. L. Rigaud. 1990. Evidence for proton countertransport by the sarcoplasmic reticulum Ca2+-ATPase during calcium transport in reconstituted proteoliposomes with low ionic permeability. J. Biol. Chem. 265:19524–19534. [PubMed] [Google Scholar]
- 11.Tadini-Buoninsegni, F., G. Bartolommei, M. R. Moncelli, R. Guidelli, and G. Inesi. 2006. Pre-steady state electrogenic events of Ca2+/H+ exchange and transport by the Ca2+-ATPase. J. Biol. Chem. 281:37720–37727. [DOI] [PubMed] [Google Scholar]
- 12.Inesi, G., and T. L. Hill. 1983. Calcium and proton dependence of sarcoplasmic reticulum ATPase. Biophys. J. 44:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin, R. B. 1992. Cooperative proton and calcium binding by sarcoplasmic reticulum ATPase. FEBS Lett. 308:59–61. [DOI] [PubMed] [Google Scholar]
- 14.Yu, X., L. N. Hao, and G. Inesi. 1994. A pK change of acidic residues contributes to cation countertransport in the Ca-ATPase of sarcoplasmic reticulum. Role of H+ in Ca2+-ATPase countertransport. J. Biol. Chem. 269:16656–16661. [PubMed] [Google Scholar]
- 15.Peinelt, C., and H. J. Apell. 2002. Kinetics of the Ca2+, H+, and Mg2+ interaction with the ion-binding sites of the SR Ca-ATPase. Biophys. J. 82:170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butscher, C., M. Roudna, and H. J. Apell. 1999. Electrogenic partial reactions of the SR Ca-ATPase investigated by a fluorescence method. J. Membr. Biol. 168:169–181. [DOI] [PubMed] [Google Scholar]
- 17.Sugita, Y., N. Miyashita, M. Ikeguchi, A. Kidera, and C. Toyoshima. 2005. Protonation of the acidic residues in the transmembrane cation-binding sites of the Ca2+ pump. J. Am. Chem. Soc. 127:6150–6151. [DOI] [PubMed] [Google Scholar]
- 18.Hauser, K., and A. Barth. 2007. Side-chain protonation and mobility in the sarcoplasmic reticulum Ca2+-ATPase: implications for proton countertransport and Ca2+ release. Biophys. J. 93:3259–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hauser, K. 2006. Electrostatic calculations for assignment of IR difference bands to carboxyl groups getting protonated during protein reactions. Biopolymers. 82:430–434. [DOI] [PubMed] [Google Scholar]
- 20.Obara, K., N. Miyashita, C. Xu, I. Toyoshima, Y. Sugita, G. Inesi, and C. Toyoshima. 2005. Structural role of countertransport revealed in Ca2+ pump crystal structure in the absence of Ca2+. Proc. Natl. Acad. Sci. USA. 102:14489–14496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barth, A., W. Mäntele, and W. Kreutz. 1997. Ca2+ release from the phosphorylated and the unphosphorylated sarcoplasmic reticulum Ca2+ ATPase results in parallel structural changes. An infrared spectroscopic study. J. Biol. Chem. 272:25507–25510. [DOI] [PubMed] [Google Scholar]
- 22.Toyoshima, C., and H. Nomura. 2002. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature. 418:605–611. [DOI] [PubMed] [Google Scholar]
- 23.Barth, A., F. von Germar, W. Kreutz, and W. Mäntele. 1996. Time-resolved infrared spectroscopy of the Ca2+-ATPase. The enzyme at work. J. Biol. Chem. 271:30637–30646. [DOI] [PubMed] [Google Scholar]
- 24.Barth, A., and C. Zscherp. 2000. Substrate binding and enzyme function investigated by infrared spectroscopy. FEBS Lett. 477:151–156. [DOI] [PubMed] [Google Scholar]
- 25.Barth, A., W. Mäntele, and W. Kreutz. 1990. Molecular changes in the sarcoplasmic reticulum Ca2+ ATPase during catalytic activity. A Fourier transform infrared (FTIR) study using photolysis of caged ATP to trigger the reaction cycle. FEBS Lett. 277:147–150. [DOI] [PubMed] [Google Scholar]
- 26.Barth, A., W. Mäntele, and W. Kreutz. 1991. Infrared spectroscopic signals arising from ligand binding and conformational changes in the catalytic cycle of sarcoplasmic reticulum Ca2+-ATPase. Biochim. Biophys. Acta. 1057:115–123. [DOI] [PubMed] [Google Scholar]
- 27.Liu, M., and A. Barth. 2003. TNP-AMP binding to the sarcoplasmic reticulum Ca2+-ATPase studied by infrared spectroscopy. Biophys. J. 85:3262–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barth, A., W. Kreutz, and W. Mäntele. 1994. Changes of protein structure, nucleotide microenvironment, and Ca2+ binding states in the catalytic cycle of sarcoplasmic reticulum Ca2+ ATPase: investigation of nucleotide binding, phosphorylation and phosphoenzyme conversion by FTIR difference spectroscopy. Biochim. Biophys. Acta. 1194:75–91. [DOI] [PubMed] [Google Scholar]
- 29.Barth, A. 1999. Phosphoenzyme conversion of the sarcoplasmic reticulum Ca2+-ATPase. Molecular interpretation of infrared difference spectra. J. Biol. Chem. 274:22170–22175. [DOI] [PubMed] [Google Scholar]
- 30.De Meis, L., and W. Hasselbach. 1971. Acetyl phosphate as substrate for calcium uptake in skeletal muscle microsomes. J. Biol. Chem. 246:4759–4763. [PubMed] [Google Scholar]
- 31.Glaose, P. K., and F. A. Long. 1960. Use of glass electrodes to measure acidities in deuterium oxide. J. Phys. Chem. 64:188–190. [Google Scholar]
- 32.Barth, A., and N. Bezlyepkina. 2004. P-O bond destabilization accelerates phosphoenzyme hydrolysis of sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 279:51888–51896. [DOI] [PubMed] [Google Scholar]
- 33.Alexov, E. G., and M. R. Gunner. 1997. Incorporating protein conformational flexibility into the calculation of pH-dependent protein properties. Biophys. J. 72:2075–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Georgescu, R. E., E. G. Alexov, and M. R. Gunner. 2002. Combining conformational flexibility and continuum electrostatics for calculating pKas in proteins. Biophys. J. 83:1731–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hauser, K., J. Mao, and M. R. Gunner. 2004. pH dependence of heme electrochemistry in cytochromes investigated by multiconformation continuum electrostatic calculations. Biopolymers. 74:51–54. [DOI] [PubMed] [Google Scholar]
- 36.Haas, A. H., and C. R. D. Lancaster. 2004. Calculated coupling of transmembrane electron and proton transfer in dihemic quinol:fumerate reductase. Biophys. J. 87:4298–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mao, J., K. Hauser, and M. R. Gunner. 2003. How cytochromes with different folds control heme redox potentials. Biochemistry. 42:9829–9840. [DOI] [PubMed] [Google Scholar]
- 38.Song, Y., J. Mao, and M. R. Gunner. 2006. Electrostatic environment of hemes in proteins: pKas of hydroxyl ligands. Biochemistry. 45:7949–7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim, J., J. Mao, and M. R. Gunner. 2005. Are acidic and basic groups in buried proteins predicted to be ionized? J. Mol. Biol. 348:1283–1298. [DOI] [PubMed] [Google Scholar]
- 40.Toyoshima, C., H. Nomura, and T. Tsuda. 2004. Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature. 432:361–368. [DOI] [PubMed] [Google Scholar]
- 41.Zhang, L., and J. Hermans. 1996. Hydrophilicity of cavities in proteins. Proteins. 24:433–438. [DOI] [PubMed] [Google Scholar]
- 42.Tackett, J. E. 1989. FT-IR characterisation of metal acetates in aqueous solution. Appl. Spectrosc. 43:483–489. [Google Scholar]
- 43.Krimm, S., and J. Bandekar. 1986. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 38:181–367. [DOI] [PubMed] [Google Scholar]
- 44.Thoenges, D., and A. Barth. 2002. Direct measurement of enzyme activity with infrared spectroscopy. J. Biomol. Screen. 7:353–357. [DOI] [PubMed] [Google Scholar]
- 45.Chirgadze, Y. N., O. V. Fedorov, and N. P. Trushina. 1975. Estimation of amino acid residue side chain absorption in the infrared spectra of protein solutions in heavy water. Biopolymers. 14:679–694. [DOI] [PubMed] [Google Scholar]
- 46.Venyaminov, S. Y., and N. N. Kalnin. 1990. Quantitative IR spectrophotometry of peptide compounds in water (H2O) solutions. I. Spectral parameters of amino acid residue absorption bands. Biopolymers. 30:1243–1257. [DOI] [PubMed] [Google Scholar]
- 47.Andersson, J., and A. Barth. 2006. FTIR studies on the bond properties of the aspartyl phosphate moiety of the Ca2+-ATPase. Biopolymers. 82:353–357. [DOI] [PubMed] [Google Scholar]
- 48.Susi, H., D. M. Byler, and W. V. Gerasimowicz. 1983. Vibrational analysis of amino acids: cysteine, serine, β-chloroalanine. J. Mol. Struct. 102:63–79. [Google Scholar]
- 49.Haurie, M., and A. Novak. 1965. Spectres de vibration des molécules CH3COOH, CH3COOD, CD3COOH et CD3COOD. II. Spectres infrarouges et Raman des dimères. J. Chim. Phys. 62:146–157. [Google Scholar]
- 50.Georg, H., A. Barth, W. Kreutz, F. Siebert, and W. Mäntele. 1994. Structural changes of sarcoplasmic reticulum Ca2+ ATPase upon Ca2+ binding studied by simultaneous measurement of infrared absorbance changes and changes of intrinsic protein fluorescence. Biochim. Biophys. Acta. 1188:139–150. [DOI] [PubMed] [Google Scholar]
- 51.Troullier, A., K. Gerwert, and Y. Dupont. 1996. A time-resolved Fourier transformed infrared difference spectroscopy study of the sarcoplasmic reticulum Ca2+-ATPase: kinetics of the high-affinity calcium binding at low temperature. Biophys. J. 71:2970–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Englander, S. W., and N. R. Kallenbach. 1984. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 4:521–655. [DOI] [PubMed] [Google Scholar]
- 53.De Jongh, H. H. J., E. Goormaghtigh, and J.-M. Ruysschaert. 1997. Monitoring structural stability of trypsin inhibitor at the submolecular level by amide-proton exchange using Fourier transform infrared spectroscopy: a test case for more general application. Biochemistry. 36:13593–13602. [DOI] [PubMed] [Google Scholar]
- 54.Vigano, C., M. Smeyers, V. Raussens, F. Scheirlinckx, J. M. Ruysschaert, and E. Goormaghtigh. 2004. Hydrogen-deuterium exchange in membrane proteins monitored by IR spectroscopy: a new tool to resolve protein structure and dynamics. Biopolymers. 74:19–26. [DOI] [PubMed] [Google Scholar]
- 55.Barth, A. 2000. Fine-structure enhancement—assessment of a simple method to resolve overlapping bands in spectra. Spectrochim. Acta [A]. 56:1223–1232. [DOI] [PubMed] [Google Scholar]
- 56.Jensen, A. M. L., T. L. M. Sørensen, C. Olesen, J. V. Møller, and P. Nissen. 2006. Modulatory and catalytic modes of ATP binding by the calcium pump. EMBO J. 25:2305–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andersen, J. P., and B. Vilsen. 1992. Functional consequences of alterations to Glu309, Glu771, and Asp800 in the Ca2+-ATPase of sarcoplasmic reticulum. J. Biol. Chem. 267:19383–19387. [PubMed] [Google Scholar]
- 58.Strock, C., M. Cavagna, W. E. Peiffer, C. Sumbilla, D. Lewis, and G. Inesi. 1998. Direct demonstration of Ca2+ binding defects in sarco-endoplasmic reticulum Ca2+ ATPase mutants overexpressed in COS-1 cells transfected with adenovirus vectors. J. Biol. Chem. 273:15104–15109. [DOI] [PubMed] [Google Scholar]
- 59.Andersen, J. P. 1995. Dissection of the functional domains of the sarcoplasmic reticulum Ca2+-ATPase by site-directed mutagenesis. Biosci. Rep. 15:243–261. [DOI] [PubMed] [Google Scholar]
- 60.Chen, L., C. Sumbilla, D. Lewis, L. Zhong, C. Strock, M. E. Kirtley, and G. Inesi. 1996. Short and long range functions of amino acids in the transmembrane region of the sarcoplasmic reticulum ATPase. J. Biol. Chem. 271:10745–10752. [DOI] [PubMed] [Google Scholar]
- 61.Steiner, T. 2002. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 41:48–76. [DOI] [PubMed] [Google Scholar]
- 62.Buchet, R., I. Jona, and A. Martonosi. 1991. Ca2+ release from caged-Ca2+ alters the FTIR spectrum of sarcoplasmic reticulum. Biochim. Biophys. Acta. 1069:209–217. [DOI] [PubMed] [Google Scholar]
- 63.Verjovski-Almeida, S., and L. De Meis. 1977. pH-induced changes in the reactions controlled by the low- and high-affinity Ca2+-binding sites in sarcoplasmic reticulum. Biochemistry. 16:329–334. [DOI] [PubMed] [Google Scholar]
- 64.Forge, V., E. Mintz, D. Canet, and F. Guillain. 1995. Lumenal Ca2+ dissociation from the phosphorylated Ca2+-ATPase of the sarcoplasmic reticulum is sequential. J. Biol. Chem. 270:18271–18276. [DOI] [PubMed] [Google Scholar]