

Abstract
ATP-binding cassette transporter BtuCD mediating vitamin B12 uptake in Escherichia coli couples the energy of ATP hydrolysis to the translocation of vitamin B12 across the membrane into the cell. Elastic normal mode analysis of BtuCD demonstrates that the simultaneous substrate trapping at periplasmic cavity and ATP binding at the ATP-binding cassette (BtuD) dimer proceeds readily along the lowest energy pathway. The transport power stroke is attributed to ATP-hydrolysis-induced opening of the nucleotide-binding domain dimer, which is coupled to conformational rearrangement of transmembrane domain (BtuC) helices leading to the closing at the periplasmic side and opening at the cytoplasmic gate. Simultaneous hydrolysis of two ATP is supported by the fact that antisymmetric movement of BtuD dimer implying alternating hydrolysis cannot induce effective conformational change of the translocation pathway. A plausible mechanism of translocation cycle is proposed in which the possible effect of the association of periplasmic binding protein BtuF to the transporter is also considered.
INTRODUCTION
ATP-binding cassette (ABC) transporters are membrane protein complexes that use the energy of ATP hydrolysis to facilitate the transmission of various substrates, from small ions and drugs to lipids and proteins across biological membranes (1). Examples of clinically relevant ABC transporters are associated with multidrug resistance in cancer cells (2), cystic fibrosis (3), and antigenic peptide transport (4), etc. In bacteria, ABC importers are mostly involved in nutrient uptake and exporters extrude toxins and harmful substances out of cell (5,6).
Most ABC transporters consist of two transmembrane domains (TMDs) and two cytoplasmic nucleotide-binding domains (NBDs). TMDs form the pathway for substrate translocation with poor sequence similarities reflecting the diversity of the substrates whereas NBDs with ATPase activity are highly conserved among all ABC transporters. Crystal structure studies of isolated NBDs from different ABC transporters (7–19) established that the dimeric arrangement of the NBDs is required for ATP binding and hydrolysis. Recent success in solving the high resolution structures of intact ABC transporters form the basis for our understanding of the molecular mechanism of transport (20–24). BtuCD from Escherichia coli is the ABC importer that mediates the uptake of vitamin B12. The transporter consists of four subunits arranged as two homodimers, one transmembrane dimer BtuC and one nucleotide-binding dimer BtuD. The two membrane-spanning BtuC domains have a total of 20 transmembrane helices forming the translocation pathway, which is accessible to the periplasm and closed at the cytoplasm (20) (Fig. 1 a). In the crystal structure of ATP free transporter (20), the nucleotide-binding subunits BtuD form a nucleotide-sandwich dimer that is similar with the structures observed in MJ0796, the NBD subunit of the Lo1D transporter from Methanococcus jannaschii (12), and MalK, the NBD component of the maltose transporter from E. coli (19). The conserved residues from both BtuD subunits, including P-loop (or Walker A) and LSGGQ signature motifs together form the putative ATP binding site at the NBD dimer interface (Fig. 1 a). The L-loops at the cytoplasmic end of the two BtuC domains interface the TMD part with the NBD part and are expected to play an important role in coupling ATP hydrolysis with transport. Like the majority of bacterial importers, BtuCD employs a specific periplasmic binding protein (PBP) BtuF to selectively bind the vitamin B12 and deliver it to the extracellular gate of the transmembrane domain (25). It is found that interaction of BtuF with the transporter increases the ATPase activity of the cytoplasmic NBDs (26). Both crystal structure study of BtuCD (20) and molecular dynamics simulation (27) suggest that the ATP binding at BtuD dimer triggers the conformational rearrangement of the BtuC dimer and provides the power stroke of substrate translocation across the membrane. However, the newly reported crystal structure of ABC importer HI1470/1 that belongs to the same family of BtuCD demonstrated an inward-facing conformation at TMD and an open conformation at NBD dimer, which implies that the ATP hydrolysis is likely to be responsible for the translocation power stroke. The recent simulation studies of BtuCD (28,29) also examined the possibility of this transport scenario. Nevertheless, a detailed picture of conformational transition and communication between TMDs and NBDs during the whole transport cycle remains elusive.
FIGURE 1.

(a) Ribbon diagram of BtuCD crystal structure. Transmembrane helices TM5, L-loops, Walker A, and signature motifs are indicated. Colors of one-half of the BtuCD are rendered darker than the other. (b) Top view looking down from the periplasmic side of the BtuC dimer. Rotational axes of the two subunits along the two lowest normal mode of isolated BtuC dimer are indicated. (c) Porcupine plot of the lowest normal mode of BtuC dimer (cones denote the direction of the movement of each Cα atom, from the base to the tip, with the length of the cone describing the amplitude of the motion). (d) Porcupine plot of the second lowest normal mode of BtuC dimer.
In this study, we applied coarse-grained elastic normal mode analysis (NMA) on the BtuCD system aiming at simulating the large-scale conformational change during the transport events. The intrinsic flexibility of isolated BtuC and BtuD dimers are evaluated and the conformational movement of intact BtuCD transporter is studied in the context of conformational coupling between BtuC and BtuD. In recent years, many studies have demonstrated the efficiency of the coarse-grained NMA using elastic network models (30–37). The extracted low-frequency modes successfully describe the collective motions and conformational changes of proteins.
The simultaneous closing of NBD dimer and periplasmic gate of TMDs is observed along the lowest normal mode, in agreement with previous MD simulation. However, the conformational change of cytoplasmic gate opening is encoded in a higher frequency mode, in which the opening of NBD dimer upon ATP hydrolysis triggers the closing of the periplasmic side of the pore. The conformational coupling between TMDs and NBDs also supports the simultaneous hydrolysis of two molecules of ATP. The effect of BtuF association on the transporter conformational movement is also discussed.
METHODS
The elastic network model has been shown to be effective in deriving slowest motions of protein dynamics that are almost the same as those obtained from normal mode analysis with empirical atomic force field (30–37). In this coarse-grained model, atomistic structures are simplified as one Cα atom per residue and the Cα atoms that lies within a certain cutoff distance, Rc, to each other, are connected by Hookean springs with the harmonic pairwise potential. As an extension to the simplest Gaussian network model, anisotropic network model (ANM) incorporates the anisotropic effects on fluctuation dynamics. In the ANM method, the pairwise potential is defined as,
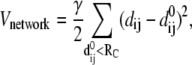 |
(1) |
where dij and 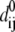 are the separation and equilibrium vectors between residues i and j, respectively. A force constant of 1.0 kcal/mol·Å2 with a cutoff distance Rc of 13 Å was employed in the calculation of vibrational modes, yielding root mean-square fluctuations (RMSF) in excellent agreement with experimental B-factors (data not shown). The stability of the normal mode analysis was also verified within the cutoff distance range of 10 ∼ 15 Å. A component analysis was performed to see how the normal modes of isolated domains contribute to the normal modes of the whole protein. Because in the ANM model crystal structure is presumed as the local minimum on the potential energy surface, the weights of modes of the isolated domain in a certain mode of the whole protein can be derived by direct vector product. For a certain isolated domain A, the weight of its ith vibrational mode in the kth mode of the whole protein is calculated with,
are the separation and equilibrium vectors between residues i and j, respectively. A force constant of 1.0 kcal/mol·Å2 with a cutoff distance Rc of 13 Å was employed in the calculation of vibrational modes, yielding root mean-square fluctuations (RMSF) in excellent agreement with experimental B-factors (data not shown). The stability of the normal mode analysis was also verified within the cutoff distance range of 10 ∼ 15 Å. A component analysis was performed to see how the normal modes of isolated domains contribute to the normal modes of the whole protein. Because in the ANM model crystal structure is presumed as the local minimum on the potential energy surface, the weights of modes of the isolated domain in a certain mode of the whole protein can be derived by direct vector product. For a certain isolated domain A, the weight of its ith vibrational mode in the kth mode of the whole protein is calculated with,
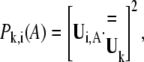 |
(2) |
where 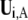 is the ith normal mode vector of domain A and
is the ith normal mode vector of domain A and 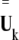 the kth normal mode vector of the whole protein, with the double-line above indicating normalization.
the kth normal mode vector of the whole protein, with the double-line above indicating normalization.
The contribution of overall rigid body rotation and translation of domain A in the kth mode of the whole protein is evaluated as
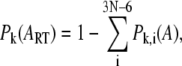 |
(3) |
where N is the number of residues in the isolated domain. By removing the contributions of overall rotation and translation of the isolated domain, 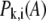 is reevaluated through normalization,
is reevaluated through normalization,
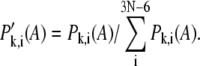 |
(4) |
We aligned the sequences of proteins according to their secondary structures (23) with ClustalX 1.83. The aligned residue pairs whose score in Gonnet Pam250 matrix is >0.5 (that is, the pairs belong to “single” groups or “strong” groups) are picked out. The involvement coefficients were calculated based on these selected residues:
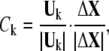 |
(5) |
where 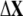 denotes the direction of the conformational change
denotes the direction of the conformational change 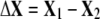 based on the selected residues, and
based on the selected residues, and 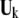 denotes the motions of the selected residues in the kth mode.
denotes the motions of the selected residues in the kth mode.
When the protein consists of two domains, A and B, all the selected residues can be ascribed to the two domains and the above involvement coefficient can be divided into two parts:
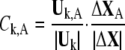 |
(6) |
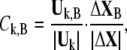 |
(7) |
where 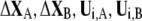 denote the conformational change and normal mode of domain A and B, respectively, based on the selected residues. As a result, the involvement coefficient can be yielded by summing up the coefficients of A and B
denote the conformational change and normal mode of domain A and B, respectively, based on the selected residues. As a result, the involvement coefficient can be yielded by summing up the coefficients of A and B
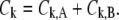 |
The translational vectors TA(1), TA(2), TA(3) and rotational vectors RA(1), RA(2), RA(3) of the domain A can be obtained from the coordinates of the Cα atoms of the corresponding residues. These six vectors are orthogonal to each other and are normalized. Their contributions to the kth mode of the whole protein are evaluated as:
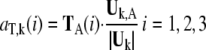 |
(8) |
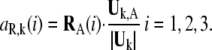 |
(9) |
The contributions of translational and rotational motions to the involvement coefficient of the kth mode are derived by:
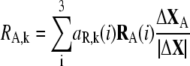 |
(10) |
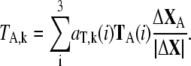 |
(11) |
To model the BtuCDF system, BtuF is docked manually onto BtuC following the method in Borths et al. (25). The interface is formed so that the two conserved negatively charged knobs in BtuF are in close contact with the positively charged pockets in BtuC and the substrate binding site connects well with the putative translocation pathway. To test the reliability of the docking, we chose several docking structures by changing the distance between BtuF and BtuC slightly and did normal mode analysis for each structure. No obvious variations of the eigenvectors and eigenvalues were found among different structures.
RESULTS
It is believed that the conformational changes are communicated between TMD and NBD during the translocation cycle. In this section we first investigate the normal modes of isolated TMD and NBD, which reflect the intrinsic flexibility encoded in their structures. The motions of these isolated parts do contribute to the conformational change of the whole transporter in substrate translocation. Then the conformational changes of the overall transporter are examined by analyzing the normal modes of the intact molecular complex BtuCD in the context of the motional coupling between TMDs and NBDs.
Normal modes of the isolated TMD dimer
The lowest frequency mode of the TMD dimer is essentially the reverse rigid body rotation of the two monomers around respective axes shown in Fig.1, b and c. The axis is approximately parallel with the lipid bilayer along a line passing through residues I198 of TM6, L70 of TM2, L258 of TM8, and V316 of TM10. As the translocation pathway is mainly formed by the opposing TM5 and TM10 helices of the two BtuC subunits (20), the distance between two TM5 helices is taken as a measure of the size of translocation pore. Along this mode, the periplasmic side of the two TM5 helices undergoes large opening and closing motions by tilting of the helices, whereas the cytoplasmic side of the pathway keeps closed. At the same time, the cytoplasmic ends of TM10 helices also keep fixed. All these result in the deviation from total rigid body movement of the two BtuC subunits. The intracelluar L-loops that interface transmembrane domain with nucleotide-binding domain swing with two BtuC subunits back and forth reversely along this mode (see Fig. 1 c). It is worth noticing that the L-loops of the two BtuC move close to each other as the periplasmic side of the translocation pathway opens.
The second lowest normal mode of BtuC dimer can also be basically described as reverse rigid body rotation of the two BtuC subunits around respective axes (Fig. 1, b and d). Different from mode 1, the rotation axis of the BtuC subunits tilts an angle of 25° with the lipid bilayer, and passes through residues W15 of TM1, C279 of TM9, and L70 of TM2. Little distance variation between two TM5 helices can be seen in this mode thereby the diameter of the translocation pore hardly changes. However, the surrounding helices distal from the translocation pore undergo large-scale movements and the extramembrane helix 5a immediately following TM5 swings to restrict the periplasmic entrance of the translocation pore. The cytoplasmic gate of the translocation pathway has a tendency to open although the observed opening in this mode is inconspicuous. In this mode, the L-loops swinging with two BtuC subunits move apart as TM helices pack more closely at the periplasmic end. It is interesting to note that the axes around which the BtuC subunit rotates in the two lowest normal modes cross at L70 of TM2 (Fig. 1 b). In another words, L70 of TM2 helix serves as a pivot in the two lowest frequency conformational movements of TMDs.
Among low-frequency vibrations of TMD dimer, there are two other normal modes, mode 5 and mode 6, affecting the translocation pathway significantly. Mode 5 is in a similar fashion as mode 1, i.e., the periplasmic ends of TM5 undergo large opening and closing motions. But the stretching and twisting of many helices are obvious, reflecting more locate fluctuations than in mode 1. Along mode 6, two BtuC domains rock in a similar way as that of mode 2. However, the opening of the cytoplasmic gate of translocation pathway is much more obvious than in mode 2 since the axis of the rotational motion tilts a larger angle with respect to the lipid bilayer. Overall, the analysis of low-frequency normal modes of BtuC dimer demonstrates that the TM helices are more flexible at its periplasmic side than at its cytoplasmic side. The gate of the translocation pathway is closely packed and keeps rigid in most low-frequency modes. Modes 1 and 5 represent the motion that significantly changes the shape of the transloction pore whereas the motion encoded in modes 2 and 6 tends to open the cytoplasmic gate of the pore.
Normal modes of the isolated NBD dimer
The lowest mode of BtuD dimer is a hinge bending motion between the two monomers, reminiscent of the tweezer-like motion of MALK dimer revealed by several crystal structures (15,19) (Fig. 2 a). The bending hinge of the dimer is located at residues of the D-loop and switch region. Consequently along this mode, the interface of the NBD dimer opens widely at the TMD side and the two composite ATP binding sites open simultaneously. Simultaneous opening of the two ATP binding sites is also found in the second mode of NBD dimer, in which the two monomers rotate reversely along respective axes shown in Fig. 2 b. Residues G33 of P-loop, A76 of S5 near Q-loop and N149 of the loop connecting TM5 helix and Walker B consist of the axis of each monomer. Upon the rotation of two BtuD, the two α-subdomains undergo larger scale movements than the catalytic subdomains, and the dimer interface decreases resulting in simultaneous opening of the two ATP binding sites. Along the third mode, however, the two subunits rotate around the twofold axis of the dimer conversely, resulting in the alternating opening and closing of the two ATP binding sites (Fig. 2 c). The three lowest normal modes of isolated NBD dimer can essentially be attributed to rigid body motions of NBD subunits, and they dominate the NBD dimer motions in low-frequency modes of the intact BtuCD as we will discuss below.
FIGURE 2.

Normal modes of the isolated nucleotide-binding domain BtuD dimer. (a) Porcupine plot of the lowest normal mode. (b) Top view porcupine plot of the second lowest normal mode looking down from the periplasmic side. The axes around which each monomer rotates are indicted. (c) Top view porcupine plot of the third mode looking down from the periplasmic side.
Normal modes of the intact BtuCD
In this case, the criterion that we judge a specific normal mode potentially functional relevant or not is the conformational change of the translocation pathway in TMDs. By examining the low-frequency spectrum of normal modes of intact BtuCD, we can find in Fig. 3 that modes 2, 4, 5, 6, 8, and 9 were excluded from the functional relevant modes as no obvious distance variation between two TM5 helices are found. These modes are further analyzed by projecting them onto the normal modes vector set of isolated TMD and NBD dimers. Table 1 lists the weights of various modes of isolated domains in the mode of the intact BtuCD. This decomposition is quite illuminating. First, we noticed that except mode 5 all the modes that we considered as nonfunctional relevant (modes 2, 4, 6, 8, 9) involve the NBD movement along the third mode of isolated BtuD dimer, i.e., the alternating opening of the ATP binding sites. This antisymmetric movement of two NBD subunits with respect to the twofold axis of the transporter cannot induce any meaningful conformational change of the translocation pathway in BtuC dimer. This is not surprising since all the effective modes related to shape changes in translocation pathway are all symmetric motion of the two TMD subunits with respect to the twofold axis. All the symmetric movements of TMD subunits couple with the symmetric motion in the NBD dimer through the L-loops. These data strongly suggests that the two ATP binding sites bind and hydrolyze two ATP at the same time, and the former “alternating” mechanism of ATP hydrolysis (38) seems unlikely at least in the case of BtuCD. Excluding the nonfunctional related modes, the left low-frequency modes may contribute to some extent to the translocation event of BtuCD. Table1 shows that the two lowest normal modes of isolated NBD and TMD dimers dominate the motion of their counterparts in the low-frequency modes of intact BtuCD. There are exceptions, however, in mode 5, in which the TMD movements are dominated by the sixth and fifth mode of isolated TMD. In the following section, we will discuss these modes in detail.
FIGURE 3.

Distance variations of the opposing TM5 helices of BtuC dimer along low-frequency normal modes. The distances between two TM5 helices are represented by the distances between corresponding Cα atoms. The variation values are the deviations of TM5 distances in the end structure of each mode from those in the crystal structure. The vibrational directions in all the modes are chosen such that the nucleotide-binding site at BtuD dimer is opening.
TABLE 1.
Weights of normal modes of isolated TMDs and NBDs in the low-frequency modes of intact BtuCD complex
| TMD
|
NBD
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Mode (k) | P′vib,k(i)* | PRT,k† | P′vib,k(i)* | PRT,k† | ||||
| 1 | 82.9(1) | 10.1(2) | 1.7(6) | 60.1 | 86.2(2) | 8.2(4) | 2.7(11) | 86.8 |
| 2 | 48.8(9) | 10.1(4) | 8.4(7) | 88.1 | 73.0(3) | 19.2(5) | 1.2(20) | 89.0 |
| 3 | 58.6(1) | 36.2(2) | 2.7(6) | 2.7 | 68.7(1) | 21.4(2) | 6.0(10) | 33.3 |
| 4 | 49.5(4) | 21.4(11) | 7.8(9) | 73.7 | 71.5(3) | 19.2(5) | 2.3(9) | 46.9 |
| 5 | 49.2(6) | 39.0(2) | 1.3(10) | 39.1 | 64.0(1) | 17.8(2) | 11.7(4) | 30.1 |
| 6 | 75.6(3) | 20.2(4) | 0.6(2) | 15.4 | 91.9(3) | 3.3(9) | 1.7(5) | 37.6 |
| 7 | 47.7(2) | 20.3(1) | 14.5(5) | 1.4 | 45.4(2) | 42.8(1) | 2.4(8) | 4.9 |
| 8 | 64.4(4) | 11.9(7) | 10.4(11) | 23.1 | 84.5(3) | 6.5(9) | 5.1(6) | 41.7 |
| 9 | 65.4(3) | 24.4(4) | 2.9(9) | 3.0 | 92.0(3) | 4.2(9) | 1.1(2) | 7.7 |
| 10 | 95.2(5) | 2.8(1) | 0.4(4) | 0.4 | 79.8(2) | 7.6(1) | 4.5(4) | 4.0 |
Values after renormalization by removing the overall rotation and translation of TMD or NBD dimer. The numbers in parentheses are the indices of the normal modes of isolated TMD or NBD dimer.
The contribution of overall rotation and translation of isolated TMD or NBD dimer.
In Table 1, the weights of normal modes of isolated TMDs or NBDs in the modes of whole BtuCD complex are evaluated by excluding the translational and rotational motions of TMD or NBD dimer as a whole from the projection basis set. The weights of these translational and rotational rigid body movements of TMD and NBD dimer are listed separately. We can see that the lowest normal mode of BtuCD contains large portion of overall rigid body movements of TMD and NBD dimer, up to 60% for TMDs and 87% for NBDs. This rigid body movement can be mainly described as relative rotation of TMD and NBD dimers about the twofold axis of the whole complex, resulting in the change of the angle between the long axes of TMD dimer and NBD dimer. In addition to the overall rigid body movement of TMDs relative to NBDs, the lowest frequency mode of the intact BtuCD can be considered as a composite of the first mode of isolated TMDs and the second mode of isolated NBDs (Table 1). The two TMD subunits rotate along the opposite direction leading to the pore opening at the periplasmic side while the two NBDs rotate along the respective axis to open the ATP binding sites simultaneously. Looking from the side face of the whole transporter, the two halves of the BtuCD each of which includes a BtuC and a BtuD subunit are rotating reversely with the pivot locating near the cytoplasmic gate of the translocation pathway. The movement of TMD is coupled to the motion of NBD through the back and forth swinging of the L-loops. As the cytoplasmic gate of TMD serves as a pivot of the rotation, there is no evidence at all of gate opening along this motion. The main characteristic of this mode is the simultaneous opening and closing of the periplasmic side of the pore and the two ATP binding sites at NBDs.
Unlike mode 1, the contribution of overall rigid body translation and rotation of TMD or NBD dimer in the third mode of BtuCD is much less. The two lowest modes of isolated TMD dimer dominate the motion of the transmembrane helices. However, the helices exhibit more flexibility than those in mode 1, and motions in the TMD part cannot be mainly described as rigid body movements. Along this mode, the surrounding helices far removed from the translocation pore are bending outwards whereas the two TM5 helices of each subunit approach each other leading to the contract of the periplasmic side of the translocation pore (Fig. 4 b). The motion of NBD dimer is basically a combination of the lowest two normal modes of isolated NBDs in a way that the two BtuD domains rotate reversely along mode 2 and at the same time approach each other in a hinge bending manner of mode 1. As a result, the two ATP binding sites open while the distance between the centers of mass of the two BtuDs decreases slightly. Generally, the translocation pathway contracts at the periplasmic side upon the opening of the two ATP binding sites at NBDs. We also noticed that in this mode the vibrational amplitude of the TMD part is much larger than that of the NBD part. In addition, the cytoplasmic gate of the translocation pathway remains occluded.
FIGURE 4.

Normal modes of the intact BtuCD complex. (a) Porcupine plot of the lowest normal mode of BtuCD. (b) Porcupine plot of the third normal mode of BtuCD. (c) Porcupine plot of the seventh normal mode of BtuCD.
In mode 7, we also find the domination of the two lowest normal modes of isolated TMD and NBD dimers. Similar with mode 3, the translocation pathway formed by the two TM5 helices contracts at the periplasmic side along with the opening of the ATP binding sites at NBDs in this mode (Fig. 4 c). However, the surrounding TM helices are less flexible than those in the third mode. Therefore there is no expansion of the surrounding helices together with the pore contraction. The movement of nucleotide-binding domains in this mode is also a combination of the two lowest normal modes of isolated BtuD dimer (Table 1), but this combination is in a “coherent” way that the hinge bending and converse rotation of two NBDs open and close the two ATP binding sites simultaneously resulting in the obvious separation of the centers of mass of two BtuD monomers. The distinct feature of mode 7 different from mode 3 is that with the opening of the two NBD subunits the L-loops exhibit lateral movement outwards leading to the expansion of the cytoplasmic side of the BtuC dimer (Fig. 4 c). The cytoplasmic gate thereby has a tendency of opening, although the amplitude of movement at the gate is still small compared with that at the periplasmic side.
The effect of the BtuF capping
The translocation cycle is triggered by the binding of the periplasmic binding protein (PBP) BtuF to the periplasmic side of BtuC. Although it is not sure whether the association of BtuF with BtuCD is retained all through the translocation cycle, BtuF is shown to affect the ATPase activity of BtuCD (26). Therefore we examined the effect of BtuF association on the conformational dynamics of BtuCD. The interaction between BtuF and BtuC is attributed to interprotein salt bridges formed by E72 and E202 of BtuF and R56, R59, and R295 of BtuC. According to the crystal structure, the salt-bridge forming sites match pretty well between BtuF and BtuCD (25). To examine the effect of the BtuF association, we modeled the system by docking BtuF onto BtuCD and redid the normal mode analysis of the BtuCD-F complex, finding that the order and main feature of most low-frequency modes are conserved (data not shown). This is especially true for the lowest mode, which is conserved more than 90%. However, the third mode of BtuCD disappeared in low-frequency spectrum of the complex. This can be comprehended by noticing that in mode 3 of BtuCD the periplasmic side of the TMD helices experience large-scale movement resulting in the large distance variation between the R56, R59, and R295 residues of two BtuC subunits that contribute to the salt bridges between BtuF and BtuCD. Therefore the capping of BtuF confines the periplasmic motion thereby moving mode 3 to higher frequency region.
Comparison with HI1740/1 structure
The HI1740/1 transporter with an inward facing conformation is of the same family with BtuCD and shares 24 and 33% sequence identity to BtuD and BtuC, respectively (23). Therefore it may serve as a reference structure of an intermediate in the transport cycle of BtuCD. We calculated the involvement coefficients of the normal modes of BtuCD in the putative conformational change between BtuCD and HI1470/1. The results listed in Table 2 demonstrate that the lowest normal mode has the largest involvement coefficient and some low-frequency modes including mode 10, 14, 3, and 7, also have significant contributions. Looking at the involvement coefficients in more detail by decomposing them into contributions corresponding to TMD and NBD parts, we found that modes 1, 10, 3, and 7 contribute more in the conformational change of the TMD part whereas mode 14 contributes mostly in the conformational change of the NBD part. In addition, in modes 10 and 3 the coefficients in TMD and NBD parts have different signs, which means that these modes contribute along different directions to the conformational change of TMD and NBD parts, respectively. In another words, the conformational coupling between the TMD and NBD parts in these modes does not conform with the structure difference between BtuCD and HI1470/1 although the total involvement coefficients are large. We also noticed that mode 1 has significant contribution to conformational change in NBD, but almost all the contributions come from the overall rotational movement of NBD dimer instead of conformational change within the NBD dimer. Overall, among the few normal modes with large involvement coefficients, modes 10 and 3 exhibit wrong coupling relationship between TMD and NBD. Modes 1 and 7 contribute largely to the conformational change of the TMD part and mode 14 contributes mostly to NBD movement.
TABLE 2.
Some large global and local involvement coefficients associated with the BtuCD to HI1470/1 transition based on the normal modes of BtuCD
| Mode (k) | Ck* | CTMD† | CNBD† | TTMD‡ | TNBD‡ | RTMD§ | RNBD§ |
|---|---|---|---|---|---|---|---|
| 1 | 0.4072 | 0.2868 | 0.1205 | 0.0009 | 0.0013 | 0.0850 | 0.1270 |
| 10 | 0.2849 | 0.2989 | −0.0144 | 0.0011 | −0.0019 | 0.0039 | −0.0011 |
| 14 | −0.2832 | −0.0369 | −0.2453 | −0.0011 | 0.0006 | −0.0234 | 0.0273 |
| 3 | −0.2667 | −0.3170 | 0.0505 | 0.0035 | 0.0034 | 0.0153 | 0.0358 |
| 7 | 0.2625 | 0.2547 | 0.0054 | 0.0009 | 0.0029 | −0.0310 | −0.0254 |
| 47 | 0.1933 | 0.1300 | 0.0594 | 0.0001 | −0.0005 | 0.0164 | 0.0102 |
| 17 | 0.1836 | 0.0255 | 0.1605 | 0.0026 | 0.0028 | 0.0192 | −0.0019 |
| 25 | 0.1732 | 0.1531 | 0.0203 | 0.0002 | 0.0013 | 0.0098 | −0.0115 |
| 19 | 0.1601 | 0.0619 | 0.0994 | −0.0056 | −0.0088 | −0.0052 | −0.0078 |
| 22 | 0.1590 | 0.2313 | −0.0727 | 0.0052 | 0.0050 | −0.0057 | 0.0112 |
| 92 | 0.1270 | 0.1154 | 0.0088 | −0.0009 | −0.0005 | 0.0111 | 0.0049 |
| 49 | −0.1050 | −0.0399 | −0.0633 | 0.0005 | 0.0010 | −0.0130 | −0.0111 |
| 58 | 0.1037 | 0.0397 | 0.0618 | 0.0004 | −0.0010 | 0.0070 | 0.0317 |
| 62 | −0.1034 | −0.0827 | −0.0211 | 0.0002 | 0.0004 | 0.0016 | −0.0088 |
| 26 | 0.1031 | 0.0724 | 0.0344 | 0.0014 | 0.0005 | 0.0026 | −0.0106 |
Global involvement coefficients.
Local involvement coefficients of TMD or NBD parts.
Isolated translational components of overall TMD or NBD in the involvement coefficients.
Isolated rotational components of overall TMD or NBD in the involvement coefficients.
The contributions from modes 1 and 7 are predicted and in accord with the above normal mode analysis of BtuCD. The contribution of mode 14 is not considered in the former discussion because this mode does not induce obvious conformational change at the translocation pathway. This mode is dominated by the movement of NBD dimer along the fourth modes of isolated NBD dimer, in which the helical subdomain of each NBD monomer rotates relative to its catalytic subdomain. Experimentally, the hinge rotation of helical subdomain relative to the catalytic subdomain was observed among the crystal structures of isolated NBDs, in which the rotation angle ranges from 5° to 25° (8–10,14,15). We calculated the involvement coefficients of isolated NBD dimer associated with the transition from BtuD to the ATP binding conformation of MalK (1Q12) or MJ0796 (1L2T), both cases in which the two lowest normal modes of BtuD dimer contribute mostly to the conformational change. On the other hand, by projecting the normal modes of isolated BtuD dimer onto the structure difference of BtuCD and HI1470/1, we found the most involved normal modes are the fourth and seventh. Therefore, in addition to the rigid body tweezers-like movement of the entire NBD monomers, the hinge rotation of helical domain contributes significantly to the conformational change of NBD dimer between BtuCD and HI1470/1.
DISCUSSION
The nucleotide-free crystal structure of BtuCD (20) is considered as the resting state at the beginning of a translocation cycle. As indicated by the lowest normal mode, with the reverse rotation of the two halves of BtuCD the transporter undergoes simultaneous opening and closing motions at the periplasmic side of the pore and the ATP binding sites. The closing of the BtuD subunits will of course benefit the ATP binding and thereby hydrolysis. Experimental evidence shows that the presence of free-state BtuF can increase the ATPase activity of BtuCD (26). This may be rationalized that the forming of the BtuF-BtuCD complex prevents the large-scale motion of the periplasmic side and even the dissociation of BtuCD dimer.
As a translocation cycle begins, BtuF bound with substrate docks to the periplasmic side of BtuCD. The opening motion of the two lobes of BtuF releases vitamin B12 to the cavity at the periplasmic side of the transporter. Crystal structure of BtuCD shows that the cavity opening to the periplasmic side is of sufficient size to accommodate much of a vitamin B12 molecule (20). According to the lowest normal mode of BtuCD, the simultaneous closing of the periplasmic cavity and the NBD dimer will occur readily, consequently resulting in the ATP binding. As shown in the previous section, the docking of BtuF hardly changes the lowest mode of BtuCD at all.
The following question is what kind of conformational change is induced exactly at the TMD region upon ATP binding. One possibility is that with the pore closing at the periplasmic side the cytoplasmic gate opens and the translocation of vitamin B12 is achieved as proposed in the literature (20,28). However, by examining the conformational dynamics of BtuCD, we found the above mechanism unlikely. First, the normal mode analysis of BtuCD reveals that the gate region of the transporter is quite rigid on the whole, and from the conformational change along the lowest mode of BtuCD we can hardly see any tendency of gate opening. Along this mode, the two halves of BtuCD rotate reversely about the pivot at the cytoplasmic gate of translocation pathway. Serving as a pivot, the gate is essentially rigid and will not open along the direction of NBD dimer closing. In another words, the nature of the lowest normal mode destines that dramatic conformational change at the gate will not occur along this lowest energy pathway. Secondly, one may argue that with the closing of NBD dimer, the conformational change may alter the shape of potential energy surface thereby altering the nature of the lowest normal mode. This may occur in large-scale conformational movement, but in this case the conformational change induced by NBD closing is moderate. This can be seen in the comparison of the ATP-binding sites of the BtuD dimer and Rad50, showing that the distance between the ABC signature and the P-loop of opposing BtuCD subunits is ∼4 Å longer than that in Rad50 (20). Therefore we tend to speculate that with ATP binding at the NBD dimer, the vitamin B12 molecule is trapped into the periplasmic cavity of the translocation pathway rather than transported to the cytoplasmic side.
According to the above reasoning, the dramatic conformational change of translocation pathway and the substrate transport will be the result of ATP hydrolysis. This is also consistent with the recent crystal structure of HI1470/1 (23), the translocation pathway of which exhibits an inward facing conformation whereas the NBD dimer adopts a more open conformation than in BtuCD. Therefore upon ATP hydrolysis, the NBD dimer opens inducing the occlusion of periplasmic side of the translocation pathway and opening of the cytoplasmic gate. This kind of motional coupling between NBD and TMD can be found in both modes 3 and 7 of BtuCD. These are not the lowest frequency modes of intact BtuCD, but in Table 1 we can find that in mode 3 the motion of NBD is dominated by the lowest normal mode of isolated NBD dimer and in mode 7 the lowest two modes of isolated NBD contribute equally. As ATP hydrolysis occurs at the interface of NBD dimer, it is reasonable to expect that the released energy has a large chance to trigger the ATP binding site opening along the low energy pathway of BtuD dimer and thereby induce the collective motion of whole transporter along mode 3 or mode 7. Based on this, we predict that mode3 and mode7 may contribute largely to the conformational change upon ATP hydrolysis. However, we cannot exclude the possibility that the BtuF association is retained during the translocation process, which will suppress the conformational change depicted in mode 3. If this is the case, mode 7 is assumed to contribute mostly to the conformational change in the substrate translocation step. Another noticeable feature of mode 7 is the lateral movement of the L-loops outwards, consequently expanding the cytoplasmic side of BtuCD and tending to open the gate. We can see that the L-loops indeed play a key role in the communication between TMD and NBD of BtuCD. The fashion of L-loop movement directly determines what kind of conformational changes in TMD and NBD are coupled together.
There is a long debated question about the number of ATP molecules hydrolyzed in each power stroke of translocation. The alternating mechanism proposed that ATP is hydrolyzed alternately at the two binding sites of NBD dimer (38). In this mechanism, the NBD dimer is undergoing antisymmetric wobbling upon hydrolysis. In our normal mode analysis, it is found that the antisymmetric movement of NBD dimer cannot induce effective conformational change of the translocation pathway in BtuCD. Thus it is more likely that two ATP molecules are hydrolyzed simultaneously to achieve the inward opening of the translocation pathway and to transport the substrate across membrane. It is difficult to determine whether one or two ATPs are hydrolyzed per transport cycle. Experimental evidences supporting both models are reported. The early work of growth yields measurements in bacteria suggesting that only one ATP is used to transport one substrate (39), whereas a more recent report of OpuA transporter suggests that two ATPs are needed (40). The recent crystal structure of HI1470/1 with an inward opening translocation pathway may represent a posthydrolysis structure with symmetric homodimer conformation (23).
Comparison with the inward-facing structure of HI1470/1 (23) also provides insights into the translocation mechanism of BtuCD. The calculated involvement coefficients show that mode 1 and mode 7 contribute largely to the conformational change of TMD from the outward-facing to inward-facing and the coupling between TMD and NBD of these modes also conforms to the structure difference between BtuCD and HI1470/1. However, the movement of NBD part in mode 1 is almost pure overall rotation of NBD dimer relative to TMD. This rotation changes the angel between the long axes of NBD and TMD dimers but not the conformation of NBD itself. Therefore, mode 1 may contribute partly to the conformational change of TMD whereas mode 7 may contribute to the coupling between NBD and TMD upon ATP hydrolysis. It is worth noting, however, that the conformational coupling of TMD and NBD parts dictated in the experimental structures of BtuCD and HI1470/1 does not conform to any of the normal mode very well. The conformational change of NBD dimer is best represented by mode 14, in which the NBD dimer vibrates along a higher frequency mode including helical subdomain movement and no obvious conformational change of TMD part is found. This strongly suggests that between the conformations of BtuCD and HI1470/1 there exists another intermediate state, which is also a posthydrolysis structure without the subdomain displacement at NBD. Obviously, comprehensive understanding of the detailed transport mechanism needs further biochemical and structural characterizations to catch more snapshots along the transport cycle.
So far, a plausible framework of transport mechanism can be sketched and is depicted in Fig. 5. In a first step, the association of BtuF bound with substrate to BtuCD promotes the binding of two ATP molecules at the NBD interface, and vitamin B12 is trapped into the cavity at the same time. The conformational change in this step is moderate and proceeds readily. In a second step, upon hydrolysis of two ATP molecules and simultaneous opening of two nucleotide-binding sites at NBD dimer, the periplasmic side of the pathway occludes and the cytoplasmic gate opens, consequently pushing vitamin B12 toward the cytoplasmic side. TMD experiences larger conformational change in this step and we presume that the NBD dimer experiences a tweezers-like motion to open the two ATP binding sites simultaneously. Thirdly, the ADP dissociation presumably causes the helical subdomain displacement accompanied by further opening of the cytoplasmic gate of the translocation pathway resulting in the release of substrate to the cytoplasm. At last, the gate-opening conformation of the transporter may return to the resting state upon ATP association to the catalytic subdomain of NBD and a new cycle of translocation begins
FIGURE 5.

A proposed mechanism of vitamin B12 transport cycle. ATP binding induces NBD closure. ATP hydrolysis triggers NBD opening that coincides with opening of translocation pathway to cytoplasm and closing to periplasm. See text for detailed description.
CONCLUSIONS
The elastic normal mode analysis in this work reveals the intrinsic conformational motions of isolated TMD and NBD dimers and motional communication between them in BtuCD transporter. The contraction of the periplasmic side of the translocation pathway is found to induce the closing of the ATP binding sites at the NBD readily along the lowest normal mode. However, the opening of the cytoplasmic gate of the translocation pore is predicted to be triggered by ATP-hydrolysis-induced conformational rearrangement encoded in another low-frequency mode of BtuCD. The simultaneous hydrolysis of two molecules of ATP is suggested according to the conformational coupling between the TMD and NBD parts of BtuCD complex.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 20473019), Natural Science Foundation of Shanghai Science & Technology Committee (04JC14016), Shanghai Education Committee (04SG05), National Major Basic Research Program of China (2003CB615807), and Shanghal Leading Academic Discipline Project (B108). J.M. thanks support by the Welch Foundation.
Editor: Ron Elber.
References
- 1.Higgins, C. F. 1992. ABC transporters—from microorganisms to man. Annu. Rev. Cell Biol. 8:67–113. [DOI] [PubMed] [Google Scholar]
- 2.Gottesman, M. M., and S. V. Ambudkar. 2001. Overview: ABC transporters and human disease. J. Bioenerg. Biomembr. 33:453–458. [DOI] [PubMed] [Google Scholar]
- 3.Sheppard, D. N., and M. J. Welsh. 1999. Structure and function of the CFTR chloride channel. Physiol. Rev. 79:S23–S45. [DOI] [PubMed] [Google Scholar]
- 4.Lankat-Buttgereit, B., and R. Tampe. 2002. The transporter associated with antigen processing: function and implications in human diseases. Physiol. Rev. 82:187–204. [DOI] [PubMed] [Google Scholar]
- 5.Boos, W., and J. M. Lucht. 1996. In Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology, Vol. 1. F. C. Neidhardt and R. Curtiss 3rd, editors. American Society for Microbiology, Washington DC. 1175–1209.
- 6.Nikaido, H. 1994. Prevention of drug access to bacterial targets: permeability barriers and active efflux. Science. 264:382–388. [DOI] [PubMed] [Google Scholar]
- 7.Hung, L. W., I. X. Wang, K. Nikaido, P. Q. Liu, G. F. L. Ames, and S. H. Kim. 1998. Crystal structure of the ABC-binding subunit of an ABC transporter. Nature. 396:703–707. [DOI] [PubMed] [Google Scholar]
- 8.Diederichs, K., J. Diez, G. Greller, C. Muller, J. Breed, C. Schnell, C. Vonrhein, W. Boos, and W. Welte. 2000. Crystal structure of MALK, the ATPase subunit of the trehalose/maltose ABC transporter of the archaeon Thermococcus litoralis. EMBO J. 19:5951–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaudet, R., and D. C. Wiley. 2001. Structure of the ATPase domain of human TAP1: the transporter associated with antigen processing. EMBO J. 20:4964–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan, Y. R., S. Blecker, O. Martsinkevich, L. Millen, P. J. Thomas, and J. Hunt. 2001. The crystal structure of the MJ0796 ATP-binding cassette. J. Biol. Chem. 276:32313–32321. [DOI] [PubMed] [Google Scholar]
- 11.Karpowich, N., O. Martsinkevich, L. Millen, Y. Yuan, P. L. Dai, K. MacVey, P. J. Thomas, and J. F. Hunt. 2001. Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure. 9:571–586. [DOI] [PubMed] [Google Scholar]
- 12.Smith, P. C., N. Karpowich, L. Millen, J. E. Moody, J. Rosen, P. J. Thomas, and J. F. Hunt. 2002. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell. 10:139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmitt, L., H. Benabdelhak, M. A. Blight, I. B. Holland, and M. T. Stubbs. 2003. Crystal structure of the nucleotide-binding domain of the ABC-transporter haemolysin B: identification of a variable region within ABC helical domains. J. Mol. Biol. 330:333–342. [DOI] [PubMed] [Google Scholar]
- 14.Verdon, G., S. V. Albers, B. W. Dijkstra, A. J. Driessen, and A. M. Thunnissen. 2003. Crystal structure of the ATPase subunit of the glucose ABC transporter from Sulfolobus solfataricus: nucleotide-free and nucleotide-bound conformations. J. Mol. Biol. 330:343–358. [DOI] [PubMed] [Google Scholar]
- 15.Chen, J., G. Lu, J. Lin, A. L. Davison, and F. A. Quiocho. 2003. A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol. Cell. 12:651–661. [DOI] [PubMed] [Google Scholar]
- 16.Lewis, H. A., S. G. Buchanan, S. K. Burley, K. Conners, M. Dickey, M. Dorwart, R. Fowler, X. Gao, W. B. Guggino, W. A. Hendrickson, J. F. Hunt, M. C. Kearins, et al. 2004. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 23:282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ose, T., T. Fujie, M. Yao, N. Watanabe, and I. Tanaka. 2004. Crystal structure of the ATP-binding cassette of multisugar transporter from Pyrococcus horikoshii OT3. Proteins. 57:635–638. [DOI] [PubMed] [Google Scholar]
- 18.Scheffel, F., U. Demmer, E. Warkentin, A. Hulsmann, E. Schneider, and U. Ermler. 2005. Structure of the ATPase subunit CysA of the putative sulfate ATP-binding cassette (ABC) transporter from Alicyclobacillus acidocaldarius. FEBS Lett. 579:2953–2958. [DOI] [PubMed] [Google Scholar]
- 19.Lu, G., J. M. Westbrooks, A. L. Davidson, and J. Chen. 2005. ATP hydrolysis is required to reset the ATP-binding cassette dimer into the resting-state conformation. Proc. Natl. Acad. Sci. USA. 102:17969–17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Locher, K. P., A. T. Lee, and D. C. Rees. 2002. The E-coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 296:1091–1098. [DOI] [PubMed] [Google Scholar]
- 21.Dawson, R. J. P., and K. P. Locher. 2006. Structure of a bacterial multidrug ABC transporter. Nature. 443:180–185. [DOI] [PubMed] [Google Scholar]
- 22.Dawson, R. J. P., and K. P. Locher. 2007. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 581:935–938. [DOI] [PubMed] [Google Scholar]
- 23.Pinkett, H. W., A. T. Lee, P. Lum, K. P. Locher, and D. C. Rees. 2006. An inward-facing conformation of a putative metal-chelate-type ABC transporter. Science. 315:373–377. [DOI] [PubMed] [Google Scholar]
- 24.Hollenstein, K., D. C. Frei, and K. P. Locher. 2007. Structure of an ABC transporter in complex with its binding proteins. Nature. 446:213–216. [DOI] [PubMed] [Google Scholar]
- 25.Borths, E. L., K. P. Locher, A. T. Lee, and D. C. Rees. 2002. The structure of Escherichia coli BtuF and binding to its cognate ATP binding cassette transporter. Proc. Natl. Acad. Sci. USA. 99:16642–16647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borths, E. L., B. Poolman, R. N. Hvorup, K. P. Locher, and D. C. Rees. 2005. In vitro functional characterization of BtuCD-F, the Escherichia coli ABC transporter for vitamin B-12 uptake. Biochemistry. 44:16301–16309. [DOI] [PubMed] [Google Scholar]
- 27.Oloo, E. O., and D. P. Tieleman. 2004. Conformational transitions induced by the binding of MgATP to the vitamin B12 ATP-binding cassette (ABC) transporter BtuCD. J. Biol. Chem. 279:45013–45019. [DOI] [PubMed] [Google Scholar]
- 28.Sonne, J., C. Kandt, G. H. Peters, F. Y. Hansen, M. O. Jensen, and D. P. Tieleman. 2007. Simulation of the coupling between nucleotide binding and transmembrane domains in the ABC transporter BtuCD. Biophys. J. 92:2727–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivetac, A., J. D. Campbell, and M. S. Sansom. 2007. Dynamics and function in a bacterial ABC transporter: simulation studies of the BtuCDF system and its components. Biochemistry. 46:2767–2778. [DOI] [PubMed] [Google Scholar]
- 30.Tirion, M. M. 1996. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 77:1905–1908. [DOI] [PubMed] [Google Scholar]
- 31.Halioglu, T., I. Bahar, and B. Erman. 1997. Gaussian dynamics of folded proteins. Phys. Rev. Lett. 79:3090–3093. [Google Scholar]
- 32.Bahar, I., A. R. Atilgan, and B. Erman. 1997. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 2:173–181. [DOI] [PubMed] [Google Scholar]
- 33.Doruker, P., R. L. Jernigan, and I. Bahar. 2002. Dynamics of large proteins through hierarchical levels of coarse-grained structures. J. Comput. Chem. 23:119–127. [DOI] [PubMed] [Google Scholar]
- 34.Atilgan, A. R., S. R. Durell, R. L. Jernigan, M. C. Demirel, O. Keskin, and I. Bahar. 2001. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma, J. 2004. New advances in normal mode analysis of supermolecular complexes and applications to structural refinement. Curr. Protein Pept. Sci. 5:119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma, J. 2005. Usefulness and limitations of normal mode analysis in modeling dynamics of biomolecular complexes. Structure. 13:373–380. [DOI] [PubMed] [Google Scholar]
- 37.Bahar, I., and A. J. Rader. 2005. Coarse-grained normal mode analysis in structural biology. Curr. Opin. Struct. Biol. 15:586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Senior, A. E., M. K. Al-Shawi, and I. L. Urbatsch. 1995. The catalytic cycle of P-glycoprotein. FEBS Lett. 377:285–289. [DOI] [PubMed] [Google Scholar]
- 39.Ferenci, T., W. Boos, and M. Schwartz. 1977. Energy-coupling of the transport system of Escherichia coli dependent on maltose-binding protein. Eur. J. Biochem. 75:187–193. [DOI] [PubMed] [Google Scholar]
- 40.Patzlaff, J. S., T. van der Heide, and B. Poolman. 2003. The ATP/substrate stoichiometry of the ATP-binding cassette (ABC) transporter OpuA. J. Biol. Chem. 278:29546–29551. [DOI] [PubMed] [Google Scholar]