

Abstract
The synthesis of mixed backbone oligodeoxynucleotides (18-mers) consisting of positively charged guanidinium linkages along with negatively charged phosphodiester linkages is carried out. The use of a base labile-protecting group for guanidinium linkage offers a synthetic strategy similar to standard oligonucleotide synthesis. The nuclease resistance of the oligodeoxyribonucleotides capped with guanidinium linkages at 5′ and 3′ ends are reported. The hybridization properties and sequence specificity of binding of these deoxynucleic guanidine/DNA chimeras with complementary DNA or RNA are described.
Keywords: antisense/hybrid duplex/cationic oligonucleotide/DNA capping/exonuclease
The use of antisense oligodeoxyribonucleotides (ODNs) to regulate gene products requires the development of modified ODNs possessing the properties of enhanced cellular uptake, nuclease resistance, and sequence specific hybridization to complementary RNAs. Numerous DNA structural analogues with modified heterocycle, sugar, and phosphodiester backbone moieties have been synthesized (1–3). Substantial progress has been made toward successful backbone modifications by using phosphorus and non-phosphorus groups (4, 5). A number of modifications or replacements of phosphodiester linkages such as 2′-fluoro-N-3′-P5′-phosphoramidates (6), 3′-thioformacetals (7, 8), 2′-O-Me methylene(methylimino) (9), 2′-O-Me amide (10), 2′-O-methylribonucleoside methylphosphonate (11), and peptide nucleic acid (PNA) (12) have been shown to complement with DNA and RNA with similar or higher stability while maintaining the sequence specificity. Except for the 2′-fluoro-N3′-P5′-phosphoramidates, these analogues are neutral and thus eliminate the electrostatic repulsion of negative charges present in natural DNA and RNA. An alternative approach, involves replacement of anionic phosphodiester groups by cationic linkages (13–17) or the use of oligonucleotides conjugated with positively charged groups to provide zwitterionic DNA analogues (18–20). These ODNs show increased binding with complementary DNA or RNA. Conceptually, replacement of anionic phosphodiester linkage by neutral or positively charged linkages can modulate the net charge of antisense ODN complex and thereby may enhance its antisense properties (4).
Our ongoing research in this area is focused on the development of deoxynucleic guanidine (DNG) in which the negatively charged —O—(PO2−)—O— backbone of DNA is replaced by positively charged, achiral —NH—C(⩵NH2+)—NH— linkage (15–17) to provide very stable complexes (21, 22). As DNG is positively charged, it binds effectively to target DNA or RNA because the repulsive electrostatic effects in double-stranded DNA (dsDNA) would be replaced by attractive electrostatic interactions in DNG:DNA or DNG:RNA duplexes. On the other hand, if electrostatic binding between polycationic and polyanionic structures becomes more significant than the specific interactions between heterocyclic bases, then binding becomes nonspecific and independent of complementary base pairing. To overcome this possible limitation, we propose to synthesize mixed backbone ODNs, to reduce the net positive charge of poly(DNG). For this purpose, we have synthesized fully protected guanidinium-linked dinucleoside incorporable into oligonucleotide (23). This allows us to insert positive point charges into otherwise negatively charged ODNs providing a net reduced charge to DNG/DNA chimeras (Fig. 1) and rendering increased binding ability.
Figure 1.

Structure of internucleoside phosphodiester (DNA) and guanidium linkage (DNG/DNA). ∗ indicates guanidinium linkage in DNG/DNA chimeras 9–11.
In this paper we report solid phase synthesis of chimeric guanidinium/phosphodiester (DNG/DNA) oligonucleotides. The thermal stability of duplexes and sequence-specific binding of these DNG/DNA chimeras to their complementary DNA or RNA strands are reported. The resistance of synthesized DNG/DNA chimeras to nucleolytic hydrolysis by exonuclease I has been studied. Importantly, DNA capped with guanidinium linkages proves to be stable to exonucleases.
MATERIALS AND METHODS
General.
1H, 13C, and 31P NMR spectra were recorded at 400, 50, and 161.9 MHz, respectively; chemical shifts are reported in δ (ppm) relative to CHCl3 (7.24 ppm) and DMSO-d6 (2.49 ppm), and for 31P NMR are given from H3PO4 as an internal standard. IR spectra were measured by using neat samples over NaCl plate for liquids and using the KBr technique for solids. Mass spectra were obtained by using either fast atom bombardment (FAB) or electrospray ionization (ESI) conditions. TLC was carried out on aluminium-backed silica gel 60 (F254) 0.25-mm plates (Merck). Column chromatography was performed by using silica gel 230–400 mesh from ICN.
Acetylisothiocyanate (Compound 3).
Potassium thiocyanate was finely powdered and dried in an oven at 100°C for 24 hr. To a suspension of dried potassium thiocyanate (15.71 g, 161.7 mmol) in anhydrous benzene (15 ml), acetyl chloride (10 ml, 140.6 mmol) was added. The suspension was refluxed for 5 hr at 90°C and was allowed to cool to room temperature (RT). Reaction was monitored by gas chromatography, which showed the disappearance of starting material. The supernatant was decanted and then purified by vacuum distillation. The first fraction boiling at 60°C was discarded. The second fraction boiling at 90°C contained the pure product. Yield was 5.8 g (40.7%). The IR spectrum had peaks at 1965, 1740, 1230, and 1145 cm−1. 1H NMR (CDCl3) 2.37, S, CH3. This singlet in isothiocyanate moved upfield compared with CH3COCl (2.66 p). 13C NMR (CDCl3) 165.3, CO; 146.9, NCS, and 27.7, CH3. HRCI. m/z 101.12 calculated for C3H3NOS (M + H)+ 102.1361.
N-Acetyl-N′-(3′-deoxythymidine-3′-yl)-5′-O-(4-methoxyphenyl)-diphenyl methyl thiourea (Compound 4).
The solution of 5′-O-MMTr-3′-amino-3′-deoxythymidine 2 (1.02 g, 2 mmol) in anhydrous dicholoromethane (15 ml) was cooled in ice bath. To this solution, acetylisothiocyanate 3 (0.185 ml, 2.2 mmol) was added slowly. The reaction mixture was allowed to attain RT and stirred for 3 hr. Reaction was monitored by TLC, evaporated to dryness, and then purified by column chromatography by using CH2Cl2:MeOH solvent system. Yield was 0.760 g (62%). TLC (CH2Cl2:MeOH, 95:5) Rf = 0.35; IR (KBr) spectrum had peaks at 3330, 3200, 2106, 1678, 1473, 1277, and 1085 cm−1. 1H NMR (400 MHz DMSO-d6): δ (ppm) 1.5 (s, 3H, CH3 Thiourea), 2.0 (s, 3H, CH3 Thym), 2.25–2.35 (m, 2H, 2′ and 2′′ H2), 3.2–3.4 (m, 2H, 5′ and 5′′ H2), 3.8 (s, 3H, —OCH3 MMTr), 4.1 (m, 1H, 4′H), 5.1 (m, 1H, 3′H), 6.2 (t, 1H, 1′H), 6.9 (d, 2H, MMTr), 7.2–7.4 (m, 12H, MMTr), 7.6 (s, 1H, 6-H), 10.8 (d, 1H, exch NH Thiourea), 11.28 (s, 1H, exch NH Thiourea), and 11.34 (s, 1H, exch NH Thym). (FAB) m/z 614.2119, calculated for C33H34N4O6S (M + H)+ 615.2277.
N′-Acetyl-N-(3′-deoxythymidine-3′-yl)-N′′-(5′-deoxythymidine-5′-yl)guanidine (Compound 7).
To solution of 4 (0.400 g, 0.65 mmol) and 5′-amino-5′-deoxythymidine (6) (0.156 g, 0.65 mmol) in anhydrous dimethylformamide (DMF; 5 ml) was added anhydrous triethylamine (0.22 ml, 1.62 mmol) and HgCl2 (0.211 gm, 0.78 mmol). Reaction mixture was allowed to stir overnight, when black precipitate (ppt) of Hg2S separates out. Reaction was monitored by TLC and filtered over celite, and filtrate was evaporated to dryness under reduced pressure resulting in a oily residue. This oil was then purified by silica gel column chromatography by using CH2Cl2:MeOH solvent system. Pure compound elutes at 10% MeOH. Yield was 0.365 g (68%). TLC (CH2Cl2:MeOH, 9:1) Rf = 0.4; 1H NMR (400 MHz DMSO-d6): δ (ppm) 1.4 (s, 3H, CH3 Guan), 1.7 (s, 3H, CH3 Thym), 1.8 (s, 3H, CH3 Thym), 2–2.4 (m, 4H, 2 × 2′-H2), 3.2–3.4 (m, 4H, 2 × 5′H2), 3.75 (s, 3H, —O—CH3 MMTr), 3.8 (m, 1H, 4′H), 3.95 (m, 1H, 4′H), 4.15 (m, 1H, 3′H), 4.8 (m, 1H, 3′H), 5.4 (m, 1H, exch, 3′-OH), 6.2 (t, 1H, 1′H), 6.4(t, 1H, 1′H), 6.9 (d, 2H, MMTr), 7.2–7.5 (m, 12H, MMTr), 7.6 (s, 1H, 6-H), 10.3 (s, 1H, exch, NH Thym), 11.4 (2s, 2H, NH Guan). High-resolution mass spectrometry (FAB) m/z 822.3384, calculated for C43H48N7O10 (M + H)+ 822.3479.
Preparation of Amidite Building Block (Compound 1).
To a solution of 7 (0.6 g, 0.73 mmol) in anhydrous dichloromethane (10 ml), diisopropylethylamine (0.5 ml, 2.92 mmol) and [Chloro(diisopropylamino)-β-cyanoethoxyphosphine] was added. Reaction mixture was kept stirring at RT for 3 hr. Reaction was monitored by TLC and evaporated to dryness under vacuum. This amidite was coevaporated several times over anhydrous acetonitrile and was used without further purification. 31P NMR (DMSO-d6): δ (ppm), 147.7 and 148.5. (FAB) m/z 1022.10, calculated for C52H64N9O11P (M + H)+ 1021.4462.
Oligonucleotide Synthesis and Purification.
RNA oligomers were obtained from the Integrated DNA Technologies (Coralville, IA). All of the oligodeoxynucleotides were synthesized on 1.3 μmol scale on a Pharmacia GA Plus DNA synthesizer by using CPG support and base protected 5′-O-(4, 4′-dimethoxytrityl)deoxyribonucleoside-3[-O-(diisopropylamino)-β-cyanoethylphosphoramidite] monomers and phosphoramidite dimer 1. Standard synthesis cycle with an extended coupling time (15 min) was used during coupling of modified phosphoramidite dimer 1; coupling efficiency of >95% was observed for this step. The final trityl was kept on for purification purpose, and oligonucleotides were deprotected with NH4OH at 60°C. Oligonucleotides containing guanidinium linkages were subjected to longer NH4OH treatment (48 hr, 60°C) so that guanidinium deprotection is complete. All oligonucleotides were purified by RP-HPLC on preparative Alltech C-8 RP column. Solvent system used was solvent A: 0.1 M triethylammoniumacetate, pH 7.0 and solvent B: CH3CN. The gradient used was 0–2 min, 15% B; 2–12 min, 25% B, and 12–45 min, 25% B at a flow rate of 4 ml/min. The HPLC-purified oligonucleotides were then detritylated by using 1 ml of 80% acetic acid (1 hr, RT), evaporated to dryness under reduced pressure, redissolved in double distilled water, and further purified by size exclusion chromatography.
Thermal Denaturation Studies.
The concentrations of nucleotide solutions were determined by using the extinction coefficients (per mol of nucleotide) calculated according to nearest neighbor approximation for the DNA. (24). ɛ260 M−1 cm−1; 9–12, 40880; 13, 50750; 15, 50751, and 16, 50750. All experiments were conducted in 10 mM Na2HPO4 buffer pH 7.1, and ionic strength was adjusted with NaCl (0, 10, and 100 mM). The final concentration of oligonucleotide strands was 3 μM. The solutions were heated to 95°C for 5 min and allowed to cool to RT slowly before being stored at 4°C overnight. Spectrophotometric measurements were performed at 260 nm on a Cary 1E UV/vis spectrophotometer equipped with temperature-programming and a thermal-melting software package, using 1-cm path length quartz cuvettes at a heating rate of 0.5°C/min over the range of 10–80°C. Dry nitrogen gas was flushed in the spectrophotometer chamber to prevent moisture condensation at temperatures below 15°C. Melting temperatures were taken as the temperature of half dissociation and were obtained from first derivative plots. The Tm values are accurate to ± 0.5°C over reported values.
Stoichiometry of Binding.
The stoichiometry of binding was determined by the method of continuous variation (25). Solutions containing the different molar ratios of DNG/DNA 10 and complementary DNA 13 were heated to 90°C and allowed to cool slowly to 15°C. The total concentration of duplex was always 3 μm. The pH was maintained at 7.1 with 10 mM Na2HPO4 buffer while the ionic strength was held constant at 10 mM with NaCl. The A at 260 nm of each solution was measured by Cary 1E UV/vis spectrophotometer.
Base Composition Analysis.
The base composition analysis of modified oligonucleotides were confirmed by enzymatic hydrolysis (26). Oligonucleotides 9–12 (0.2 A254 unit) were dissolved in 20 mM Tris⋅HCl (200 μl, pH 8.9) and treated with snake venom phosphodiesterase (0.02 unit) and alkaline phosphatase (0.8 unit) at 37°C for 12 hr. This hydrolyzate (30 μl) was analyzed on analytical C-8 RP-HPLC column and eluted by using 0.5%/min gradient of CH3CN in 0.1 M TEAA, pH 7.0 for 50 min at a flow rate of 1 ml/min. The retention times for (1) standard nucleosides were dC, 4.59 min; T, 7.12 min; dG, 7.77 min; dA, 11.47 min, and for dinucleoside containing guanidinium linkage 8, 14.27 min (2) for enzymatic hydrolyzate of 9 were dC, 4.58 min; T, 7.13 min; dG, 7.77 min; dA, 11.69 min, and TgT 8, 14.13 min. The corresponding peak areas were used to calculate the base composition of oligonucleotides.
Exonuclease I Digestion Experiment.
Oligonucleotides 9–12 (0.2 OD) with phosphodiester or guanidinium linkages were treated with exonuclease I (500 units) in 10 mM Tris⋅HCl, pH 9.0 (0.2 ml) containing 50 mM KCl. Reaction mixtures were analyzed on C-8 RP-HPLC column by using 0.5%/min gradient of CH3CN in 0.1 M tetraethylammonium acetate, pH 7.0, for 50 min at a flow rate of 1 ml/min. Time points were 0, 1, 2, 6, and 12 hr for DNG/DNA oligonucleotides (9-11) and only 0 and 1 hr for phosphodiester oligomer 12 because this was completely digested by that time.
RESULTS AND DISCUSSION
Synthesis.
For incorporation of guanidinium internucleoside linkages into ODN, we synthesized the phosphoramidite 1 (Scheme 1 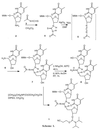 ). The guanidinium linkage of 1 remained protected during ODN synthesis and can be deprotected at the end of the synthesis to give a positively charged guanidinium linkage. There are reports of the synthesis of N-substituted guanidinium internucleoside linkages that are neutral and cannot be deblocked to give charged guanidinium (27, 28). The synthesis of 1 involves coupling of the 5′-amino group of 5′-amino-5′-deoxythymidine 6 with in situ-generated carbodiimide 5, obtained from reaction of the acetyl-protected thiourea 4 with mercury (II) in the presence of Triethylamine (29). The acetyl protected thiourea 4 was synthesized by using 5′-O-monomethoxytrityl-3′-amino-3′-deoxythymidine 2 and acetylisothiocyanate 3, in dichloromethane. The electron withdrawing nature of acetyl group on thiourea of 4 activates the carbodiimide intermediate 5, facilitating the attack by the 5′-amine, and then this acetyl group acts as a protecting group on the resulting guanidinium linkage of dinucleoside 7.
). The guanidinium linkage of 1 remained protected during ODN synthesis and can be deprotected at the end of the synthesis to give a positively charged guanidinium linkage. There are reports of the synthesis of N-substituted guanidinium internucleoside linkages that are neutral and cannot be deblocked to give charged guanidinium (27, 28). The synthesis of 1 involves coupling of the 5′-amino group of 5′-amino-5′-deoxythymidine 6 with in situ-generated carbodiimide 5, obtained from reaction of the acetyl-protected thiourea 4 with mercury (II) in the presence of Triethylamine (29). The acetyl protected thiourea 4 was synthesized by using 5′-O-monomethoxytrityl-3′-amino-3′-deoxythymidine 2 and acetylisothiocyanate 3, in dichloromethane. The electron withdrawing nature of acetyl group on thiourea of 4 activates the carbodiimide intermediate 5, facilitating the attack by the 5′-amine, and then this acetyl group acts as a protecting group on the resulting guanidinium linkage of dinucleoside 7.
The acetyl protection of internucleoside guanidinium remains stable to conditions required for DNA solid-phase synthesis, and the acetyl is removed during usual final deprotection conditions of DNA synthesis. To confirm deprotection of guanidinium linkage, 7 was treated with 35% ammonium hydroxide at 55°C for 45 hr followed by detritylation to provide the deprotected guanidinium dinucleoside 8, in quantitative yield (as determined by HPLC and characterized by FAB MS [(M + H)+ = 508]). The phosphotylation of 7, using [chloro(diisopropylamino)-β-cyanoethoxyphosphine] provided the final phosphoramidite 1, which exhibited the characteristic 31P NMR signals at 147.7 and 148.5 ppm. Thus, the ability to protect the guanidinium linkage with an acetyl function and remove this function at completion of synthesis of DNG/DNA chimera makes it suitable for standard automated solid phase synthesis.
The guanidinium linkage was incorporated into ODNs 9–11 (Fig. 1) by using Pharmacia GA Plus DNA synthesizer. The “standard synthesis cycle” with an extended time (15 min) was used during the coupling of phosphoramidite 1. After completion of the synthesis, ODNs 9–11 were purified by RP-HPLC and then detritylated and again purified by size exclusion. Electrospray mass spectroscopic analysis for ODN 5′-T*TAGGGT*TA-3′ (C92H113N39O46P6, 2686.9) indicated the expected masses for the doubly charged C92H113N39O46P6 (M + 2H)2+: 1343.4 and triply charged C92H113N39O46P6 (M + 3H)3+: 895.4 species confirming the presence of modified moiety into purified DNG/DNA chimera.
To further insure that the guanidinium-linked nucleobases have survived the synthetic chemistry of solid phase synthesis by phosphoramidite approach, enzymatic hydrolysis of 9–11 were carried out by using snake venom phosphodiesterase followed by alkaline phosphatase (26). The enzyme digest was analyzed by RP-HPLC, which indicated the presence of dinucleoside 8, with guanidinium internucleoside linkage. The corresponding peak areas showed the correct base composition of ODNs 9–11.
Duplex Formation by DNG/DNA Chimeras.
The DNG/DNA chimeric ODNs 9–11 are 18-mers (Fig. 1) with either three (at 5′, 3′, and center), two (at 5′ and 3′) or one (at center) guanidinium linkages. The duplexes were formed between DNG/DNA chimeric ODNs 9–11 with complementary DNA 13 or RNA 15. The 18-mer ODNs containing all four nucleobases are non self-complementary and form only antiparallel duplexes. Before performing thermal denaturation experiments the stoichiometry of binding of DNG/DNA chimeric ODN and DNA was determined by the method of continuous variation (25) to generate mixing curves of the absorbance vs. mol fraction of 10 and 13 (Fig. 2). Increasing mol fraction of 13 to the 10 (in 10 mM Na2HPO4, pH 7.1 at 15°C) lowered the UV A at 260 nm an inflection point at 0.5 mol fraction indicated the formation of expected 1:1 stoichiometry for DNG/DNA:DNA duplex (10:13).
Figure 2.

Job plot (continuous variation method) of 10 with 13 at 3 μm total concentration, at λ = 260 nm and 15°C.
Tm values for unmodified DNA:DNA (12:13) and DNA:RNA (12:15) duplexes were determined. These Tm values serve for comparative purposes with the Tm values of DNG/DNA chimeras (Table 1). As expected, the Tm was observed to increase with increase in salt concentration with DNA:DNA and DNA:RNA. The duplexes of DNG/DNA chimera with complementary DNA or RNA showed increase in A at 260 nm upon increasing temperature and showed the characteristic sigmoidal melting pattern (Fig. 3). Incorporation of three or one guanidinium linkages (ODN 9 and 11) has no effect on hybridization properties with complementary DNA 13, when there is no salt (cf. Exps. 3 and 5 with 1). As salt concentration was increased (0–100 mM NaCl) stability of duplexes 9:13 and 11:13 was found to decrease when compared with DNA:DNA (cf. Exps. 3 and 5 with 1). The duplex of DNG/DNA chimeras 9 and 11 with complementary RNA 15 showed destabilization at all salt concentrations (cf. Exps. 6 and 8 with 2). When there are two guanidinium linkages, as in the duplex of ODN 10 with complementary DNA 13, and in absence of salt, there is 2°C and 2.8°C increase in Tm compared with DNA:DNA (12:13) and DNA:RNA (12:15) hybrid, respectively (cf. Exp. 4 with 1 and 2). In absence of salt, the duplex of DNG/DNA chimera 10 with complementary RNA 15 exhibits similar stability as does the DNA:RNA duplex (compare Exp. 7 with 2). With increasing salt concentration the duplexes of 10 with complementary DNA as well as RNA exhibit a decrease in Tm (cf. Exp. 4 with 1 and Exp. 7 with 2).
Table 1.
Duplex melting* temperatures (Tm°C)
| Exp no. | Duplex | No NaCl | 10 mM NaCl | 100 mM NaCl |
|---|---|---|---|---|
| 1 | 12:13 | 34.8 | 48.6 | 58.5 |
| 2 | 12:15 | 34.0 | 49.7 | 59.9 |
| 3 | 9:13 | 34.8 | 46.6 | 53.5 |
| 4 | 10:13 | 36.8 | 48.6 | 57.5 |
| 5 | 11:13 | 34.8 | 47.6 | 56.5 |
| 6 | 9:15 | 30.1 | 41.8 | 50.9 |
| 7 | 10:15 | 34.2 | 45.9 | 57.1 |
| 8 | 11:15 | 31.2 | 43.9 | 54.0 |
| 9 | 12:14 | — | 39.5 | — |
| 10 | 12:16 | — | 45.5 | — |
| 11 | 9:14 | — | 35.5 | — |
| 12 | 10:14 | — | 38.5 | — |
| 13 | 11:14 | — | 37.6 | — |
| 14 | 9:16 | — | 30.5 | — |
| 15 | 10:16 | — | 35.5 | — |
| 16 | 11:16 | — | 34.6 | — |
Buffer used: 10 mM Na2HPO4, pH 7.1 with differant NaCl concentration (0–100 mM).
Figure 3.

Plots of A260 vs. t°C., for (○) duplex 12:13 and (▵) 10:13 in buffer 10 mM Na2HPO4, pH 7.1 with differant NaCl concentration. (0–100 mM).
Our thermal denaturation results show that incorporation of guanidinium internucleoside linkages into ODNs, either separated by a few nucleotide units (as in 9) or at the center position (11), in absence of salt, has no effect on duplex stability. On the other hand, incorporating guanidinium linkages at 5′ and 3′ ends (10) shows stabilization of duplex with complementary DNA as well as RNA (with no salt). This shows the importance of the position of guanidinium linkages in ODN. As expected, increasing salt concentration (0–100 mM NaCl) destabilizes duplexes consisting of DNG/DNA chimeras 9–11, with complementary DNA as well as RNA (cf. Exps. 3–5 with 1 and Exps. 6–8 with 2). This salt effect is opposite to that seen with DNA:DNA (12:13) and DNA:RNA (12:15) duplexes, where increasing salt concentration provides stability by masking the electrostatic opposition of negative charges. Decreasing salt concentration allows the positively charged guanidinium of DNG/DNA chimera to become intimately salt paired with negatively charged phosphodiester of complementary DNA or RNA. Thus, duplexes of DNG/DNA with DNA and RNA are stabilized by lowering of the ionic strength.
Sequence Specificity.
To study the sequence specificity of binding of DNG/DNA chimera with complementary DNA or RNA, DNG/DNA chimeric ODN was allowed to form duplex with complementary DNA 14 or RNA 16 containing one base mismatch (T/U instead of dA/A) at the center and then the stability of the duplex was monitored by thermal denaturation. The duplexes 12:14 (DNA:DNA) and 12:16 (DNA:RNA) exhibit (Table 1) 9.1°C and 4.2°C decrease in Tm, respectively, in comparison to fully complementary 12:13 and 12:15 duplexes (cf. Exp. 9 with 1 and 10 with 2). Approximately the same mismatch discrimination was observed for DNG/DNA:DNA hybrid (ΔTm −10 to −11°C, cf. Exps. 11–13 with 3–5) whereas for DNG/DNA:RNA hybrid base mismatch, discrimination was almost double (ΔTm −9 to −11°C, cf. Exps. 14–16 with 6–8). This clearly demonstrates that binding of DNG/DNA chimera with complementary DNA and RNA is sequence specific.
Stability of DNG/DNA Toward Exonuclease.
ODNs capped with guanidinium internucleoside linkages are expected to be resistant to the cleavage by exonucleases. To investigate this DNG/DNA, ODNs 9–11 were subjected to nucleolytic digestion by exonuclease I. The hydrolyzate was then analyzed by RP-HPLC. The control ODN 12 was found to be completely hydrolyzed after 1 hr of incubation. The DNG/DNA chimera 9 and 10 were found to be absolutely stable toward exonuclease 1 digestion even after 12 hr of incubation. The DNG/DNA chimera 11, which contains only one guanidinium linkage at the center of ODN, was found to be partially hydrolyzed after 1 hr (at 0 hr ODN 11, on RP-HPLC gave Rt 32.8 min, and after 1 hr Rt 29.7 min) and there was no further hydrolysis even after 12 hr (Rt 29.7 min). This clearly shows that DNG/DNA ODNs 9 and 10 having guanidinium linkages at 5′ and 3′ ends are completely stable to exonuclease 1. The partial hydrolysis of ODN 11, having guanidinium at the center indicates that phosphodiester linkages around guanidinium are stable to exonuclease cleavage. Further investigations are in progress.
Conclusions.
In this paper, we successfully have demonstrated the insertion of cationic internucleoside guanidinium linkage in place of negative phosphodiester linkages in DNA. For this purpose, we have used standard phosphoramidite chemistry and automated solid phase synthesis. The DNG/DNA chimera 10, with two terminal positive charges, showed enhanced binding to complementary DNA and RNA in absence of salt. The binding of 10, with complementary DNA at 10 mM NaCl (close to physiological conditions), was similar to unmodified DNA:DNA. The binding of DNG/DNA chimeras is highly sequence specific. The 5′ and 3′ capping of the DNA with gaunidinium linkage provides protection to hydrolysis by exonuclease 1. The lower net negative charge of DNG/DNA chimera, arising from insertion of positive guanidinium linkages, may assist the cellular uptake of these ODNs.
Acknowledgments
This work was initiated under support by the Office of Naval Research (N00014-96-1-01232). We gratefully acknowledge continuing support by the National Institutes of Health (3 R37 DK09171–3451).
ABBREVIATIONS
- ODNs
oligodeoxyribonucleotides
- DNG
deoxynucleic guanidine
- Rt
retention time
- MMTr
monomethoxytrityl
References
- 1.Uhlmann E, Peyman A. Chem Rev (Washington, DC) 1990;90:543–584. [Google Scholar]
- 2.Cook P D. Anti-Cancer Drug Des. 1991;6:585–607. [PubMed] [Google Scholar]
- 3.De Mesmaeker A, Haner R, Martin P, Moser H E. Acc Chem Res. 1995;28:366–374. [Google Scholar]
- 4.Sanghvi Y S, Cook P D. In: Nucleosides and Nucleotides as Antitumor and Antiviral Agents. Chu C K, Baker D C, editors. New York: Plenum; 1993. p. 311. [Google Scholar]
- 5.Milligan J F, Matteucci M D, Martin C J. J Med Chem. 1993;36:1923–1937. doi: 10.1021/jm00066a001. [DOI] [PubMed] [Google Scholar]
- 6.Gryaznov S. Nucleosides Nucleotides. 1997;16:899–905. [Google Scholar]
- 7.Jones R J, Lin K-Y, Milligan J F, Wadwani S, Matteucci M D. J Org Chem. 1993;58:2983–2991. [Google Scholar]
- 8.Rice J S, Gao X. Biochemistry. 1997;36:399–411. doi: 10.1021/bi961760i. [DOI] [PubMed] [Google Scholar]
- 9.Sanghvi Y S, Swayze E E, Peoc’h D, Bhat B, Dimock S. Nucleosides Nucleotides. 1997;16:907–916. [Google Scholar]
- 10.De Mesmaeker A, Lesueur C, Bevierre M, Waldner A, Fritsch V, Wolf R M. Angew Chem Int Ed Engl. 1996;35:2790–2794. [Google Scholar]
- 11.Kean J M, Kipp S A, Miller P S, Kulka M, Aurelian L. Biochemistry. 1995;34:14617–14620. doi: 10.1021/bi00045a001. [DOI] [PubMed] [Google Scholar]
- 12.Nielsen P, Egholm M, Berg R H, Buchardt O. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- 13.Letsinger R L, Singman C N, Histand G, Salunkhe M. J Am Chem Soc. 1988;110:4470–4471. [Google Scholar]
- 14.Fathi R, Huang Q, Coppola G, Delaney W, Teasdale R, Krieg A M, Cook A F. Nucleic Acids Res. 1994;22:5416–5424. doi: 10.1093/nar/22.24.5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dempcy R O, Browne K, A, Bruice T C. J Am Chem Soc. 1995;117:6140–6141. [Google Scholar]
- 16.Dempcy R O, Luo J, Bruice T C. Proc Natl Acad Sci USA. 1996;93:4326–4330. doi: 10.1073/pnas.93.9.4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linkletter B, Bruice T C. Bioorg Med Chem Lett. 1998;8:1285–1290. doi: 10.1016/s0960-894x(98)00228-5. [DOI] [PubMed] [Google Scholar]
- 18.Barawkar D A, Rajeev K G, Kumar V A, Ganesh K N. Nucleic Acids Res. 1996;24:1229–1237. doi: 10.1093/nar/24.7.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hashimoto H, Nelson M G, Switzer C J. J Am Chem Soc. 1993;115:7128–7134. [Google Scholar]
- 20.Schmid N, Behr J P. Tetrahedron Lett. 1995;36:1447–1450. [Google Scholar]
- 21.Blasko A, Dempcy R O, Minyat E E, Bruice T C. J Am Chem Soc. 1996;118:7892–7899. [Google Scholar]
- 22.Blasko A, Minyat E E, Dempcy R O, Bruice T C. Biochemistry. 1997;36:7821–7831. doi: 10.1021/bi970064v. [DOI] [PubMed] [Google Scholar]
- 23.Barawkar D A, Linkletter B, Bruice T C. Bioorg Med Chem Lett. 1998;8:1517–1520. doi: 10.1016/s0960-894x(98)00251-0. [DOI] [PubMed] [Google Scholar]
- 24.Gray D M, Hung S-H, Johnson K H. Methods in Enzymology. Vol. 246. New York: Academic; 1995. p. 19. [DOI] [PubMed] [Google Scholar]
- 25.Job P. Ann Chim (Rome) 1928;9:113–203. [Google Scholar]
- 26.Connoly B A. Methods in Enzymology. New York: Academic; 1992. pp. 36–53. [Google Scholar]
- 27.Vandendriessche F, Aerschot A V, Voortmans M, Janssen G, Busson R, Overbeke A V, Bossche W V, Hoogmartens J, Herdewijn P. J Chem Soc Perkin Trans. 1993;1:1567–1575. [Google Scholar]
- 28.Pannecouque C, Wigerinck P, Areschot V A, Herdewijn P. Tetrahedron Lett. 1992;33:7609–7612. [Google Scholar]
- 29.Robinson S, Roskamp E J. Tetrahedron. 1997;53:6697–6705. [Google Scholar]